

Abstract
Computational drug repositioning methods can scalably nominate approved drugs for new diseases, with reduced risk of unforeseen side effects. The majority of methods eschew individual‐level phenotypes despite the promise of biomarker‐driven repositioning. In this study, we propose a framework for discovering serendipitous interactions between drugs and routine clinical phenotypes in cross‐sectional observational studies. Key to our strategy is the use of a healthy and nondiabetic population derived from the National Health and Nutrition Examination Survey, mitigating risk for confounding by indication. We combine complementary diagnostic phenotypes (fasting glucose and glucose response) and associate them with prescription drug usage. We then sought confirmation of phenotype‐drug associations in unidentifiable member claims data from the Aetna Insurance company using a retrospective self‐controlled case analysis approach. We identify bupropion as a plausible glucose lowering agent, suggesting that surveying otherwise healthy individuals in cross‐sectional studies can discover new drug repositioning hypotheses that have applicability to longitudinal clinical practice.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Observational studies are emerging as ways to search for repositioning candidates, yet are fraught with bias and do not consider quantitative phenotypes.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ That it is possible to use health monitoring surveys and longitudinal administrative population databases coupled with continuous phenotypes to search for and replicate new repositioning candidates.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ We present a novel approach for drug repositioning that harnesses health monitoring surveys and multiple clinical trait phenotypes to avoid confounding bias and increase specificity of evidence for repositioning discovery.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ Our method enhances the repositioning process using quantitative phenotyping from humans, potentially closing the gap between computational and clinical drug repositioning.
Identifying new indications for previously approved drugs, known as drug repositioning, is an attractive alternative to the traditional drug discovery paradigm, as previously approved drugs have substantially lower risk of unforeseen adverse events.1 Computational drug repositioning builds on this premise by prescreening for promising repositioning candidates, with current methods primarily relying on molecular data2, 3, 4, 5 and/or the biological literature.6, 7, 8, 9, 10 Although these methods have been successful in predicting plausible repositioning candidates, a key challenge in computational repositioning is to provide direct evidence of candidate efficacy in humans, rather than relying on surrogate biomarkers or indirect evidence.
One alternative is to consider a single or a few quantitative phenotypes' association with drug prescription history. In doing so, one can not only be certain that the phenotypes chosen are clinically relevant to a disease of interest, but also readily access effect sizes for power considerations in future clinical studies. Although such a strategy is appealing, even a study limited to a single disease may be confounded due to shared disease etiology,11 off‐label drug usage,12 and variable effects of drugs due to disease severity. We propose a novel framework in which the association between drugs and quantitative phenotypes is assessed in a noninstitutionalized population who do not have the target disease for repositioning.
To demonstrate the potential of this strategy, we search for putative modulators of glycemic health in a normoglycemic and US‐representative population of participants from the 2005–2012 National Health and Nutrition Examination Survey (NHANES). We evaluated associations between 115 prevalent drugs and 2 diabetes diagnostic phenotypes, fasting blood glucose and glucose after following a 2‐hour oral glucose tolerance challenge (or glucose response). By combining findings from two glycemic phenotypes, we identified a single potential antidiabetic candidate associated with lower glycemic phenotypes, the antidepressant bupropion. Notably, other commonly used antidepressants did not show multimodal antidiabetic potential. To replicate the association, we designed a retrospective self‐controlled study in a normoglycemic cohort derived from unidentifiable member claims data provided by Aetna Insurance, and again verified that bupropion, but not other commonly prescribed antidepressants, was associated with lower levels of fasting blood glucose after exposure to the drug.
METHODS
Cross‐sectional study cohort
The cross‐sectional study cohort was derived from a combination of four independent waves of the continuous NHANES: the 2005–2006, 2007–2008, 2009–2010, and the 2011–2012 surveys.22 NHANES is a cross‐sectional survey conducted by the United States Centers for Disease Control and Prevention, wherein a large number of participants are recruited to answer a number of questions pertaining to their medical, psychosocial, and sociodemographic histories. A random subset of respondents also receive extensive anthropometric and laboratory testing, including a variety of routine clinical measures. For this study, several variables were obtained for each respondent, including: (1) self‐reported history of diabetes (field DIQ010 from the respective DIQ questionnaire datasets); (2) fasting blood glucose and fasting time, as well as glucose taken at 2‐hours postoral glucose tolerance test (field LBXGLU, PHASFSTHR, and LBXGLT, respectively, from the respective GLU laboratory datasets); (3) self‐reported prescription drug usage at the time of interview (including generic drug names and drug category, as defined in the Lexicon Plus database, Cerner Multum; see Figure 1 a); (4) routine demographic information, including age (field RIDAGEYR), sex (field RIAGENDR), race/ethnicity (field RIDRETH1), and body mass index (BMI; field BMXBMI); and (5) history of chronic disease, including hypertension (field BPQ020), coronary heart disease (field MCQ160c), and kidney disease (as ascertained using field KIQ022). Respondents without complete information for any of these fields were excluded from further analysis. To obtain a nondiabetic final cohort for association testing, respondents were filtered to include those with no reported history of diabetes, no use of antidiabetic medications at the time of interview, and normal glycemic status (fasting blood glucose less than 100 mg/dL according to American Diabetes Association guidelines13).
Figure 1.
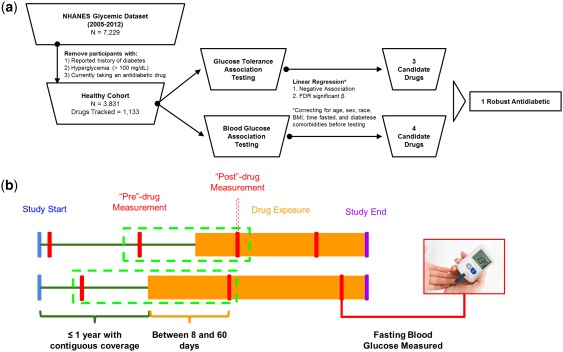
Quantitative‐phenotype based repositioning overview. (a) The National Health and Nutrition Examination Survey (NHANES; cross‐sectional) quantitative‐phenotype based repositioning workflow. (b) Conceptual diagram of claims data‐based replication efforts. FDR, false discovery rate.
Drug‐phenotype association in the cross‐sectional study cohort
Linear regression was performed to individually test the association of usage of 1,133 drugs (mean [95% confidence interval (CI)] drugs per participant 1.16 [1.11–1.22]) and either fasting blood glucose or blood glucose taken at 2‐hours postoral glucose tolerance test. All testing was performed controlling for age, sex, race, BMI, number of hours fasted, and history of hypertension, coronary heart disease, or chronic kidney disease. Variables were chosen to include a variety of cardiometabolic health‐modifying demographics and comorbidities associated with poor cardiometabolic health (Saydah et al.15 2017); all controlling variables were included in all regression models regardless of significance. Regression coefficients and significance were estimated using the survey package in R software to account for the stratified design of NHANES.23 To avoid erroneous associations, drugs with <12 prescribed individuals were removed from further analysis (see Austin & Steyerberg17 for rationale). For the remaining 115 drugs, regression coefficients and significance were obtained and corrected for multiple testing using the false discovery rate (FDR) method.25 Drugs with loosely significant (FDR <0.3), negative associations with both fasting blood glucose and blood glucose following a 2‐hour postoral glucose tolerance test were considered candidate antidiabetic agents (Figure 1 a). To provide additional confidence in the results of the association analysis and the loose FDR cutoff, simulation studies were performed as follows: (1) drug use labels were permuted randomly 50 times for numbers of exposed individuals ranging from 12–200 individuals; (2) survey‐adjusted regressions were performed as above; and (3) the resulting absolute effect sizes were compared to FDR significant drugs.
Longitudinal validation cohort and self‐controlled analyses
The longitudinal validation cohorts were derived from unidentifiable member claims data from Aetna Insurance, spanning 8 years (2008–2016) with over 50 million lives in all 50 states. To obtain drug‐specific nondiabetic cohorts, the following restrictions were made: (1) patients did not have any instance of diabetes diagnosis (International Classification of Disease (ICD)‐9 codes 250–250.93) and were not prescribed any antidiabetic medications during their entire subscription to Aetna Insurance (any of the following: metformin, Glucophage, Glumetza, Glyburide, Glipizide, Glimepiride, Repaglinide, Nateglinide, Rosiglitazone, Pioglitazone, Sitagliptin, Saxagliptin, Linagliptin, Liraglutide, Canagliflozin, Dapagliflozin, Insulin glulisine, Insulin lispro, Insulin aspart, Insulin glargine, Insulin detemir, Insulin isophane, Dapagliflozin, Sulfonylureas, Meglitinides, Thiazolidinediones, DPP 4 inhibitors, Byetta, Victoza, Invokana, Farxiga, Apidra, Novolog, Humalog, Lantus, Levemir, Humulin N, or Novolin N); (2) patients were prescribed one of the study drugs (bupropion, escitalopram, or gabapentin); (3) patients were enrolled in an insurance plan for at least 1 year before the first prescription date for the respective study drug; (4) patients had a fasting blood glucose measurement up to 1 year before starting the respective study drug (“pre” measurement), and between 8 and 60 days after starting (“post” measurement; Figure 1 b). Time points were chosen to enable the collection of at least two glucose measurements (typically taken annually, per the American Diabetes Association recommendations), and for individuals to have reached steady‐state concentrations of study drug (typically ∼1 week) while still minimizing time‐dependent variation. Prevalence of three chronic diseases were ascertained for all patients fitting these criteria by searching for billing codes for hypertension (ICD‐9 codes 401–405), coronary heart disease (ICD‐9 codes 410–414.9), and chronic kidney disease (ICD‐9 codes 585–585.9).
Following cohort creation, self‐controlled analysis was performed for each dose of each study drug using a paired t test between the preglucose and postglucose measurements, as previously described.19 More recent methods have proposed additional corrections to the simple paired t test method; however, these methods examine longer time periods of exposure (which allows for additional confounders to accumulate), and a population containing cases in addition to healthy controls.26 In contrast, the method described here relies on a very short analysis window. Dosage forms of study drugs with <198 participants were removed from further analysis due to power considerations (assuming a small effect size, Cohen's d equal to 0.2, requiring power of 80% or greater, see ref. 27 for details). Study drugs with significant (t test P value < 0.05) and negative associations (improved glucose response) with the predrug to postdrug regimes were considered replicated agents. For the purposes of validation, multiple testing correction was not required due to the small number of independent tests (three total), and, furthermore, would not alter the ascertainment of significance.
RESULTS
Association of bupropion with complementary diabetes phenotypes in a normoglycemic population
To develop a nondiabetic cohort from NHANES, we began with all participants in the 4 surveys considered, totaling 40,790 participants. We first excluded participants without complete demographic, chronic disease history, and glycemic (glucose and tolerance testing) information resulting in a subcohort of 7,229 participants. These participants had similar demographics as compared to the full cohort in terms of sex, race (black, Hispanic, and other race), and chronic disease burden (Z‐test for proportions, P > 0.1). The subcohort did differ significantly in terms of age (∼10 years older on average, two‐sample t test, P < 0.05), and BMI (two‐sample t test, P < 0.05); however, these did not impact chronic disease rates (see Supplementary Table S1 for demographics of full and subcohorts). We then excluded participants with a reported history of diabetes, abnormal fasting blood glucose (including diabetes and prediabetes, according to the American Diabetes Association guidelines, ≥100 mg/dL), and who were currently prescribed an antidiabetic drug, to obtain a final NHANES‐derived cross‐sectional cohort of 3,831 participants with putatively normoglycemic status (see Table 1 for demographic characteristics of the cohort).
Table 1.
Demographic breakdown of NHANES (cross‐sectional) and unidentifiable member claims data from Aetna Insurance (longitudinal) cohorts
| Claims dataa | ||||
|---|---|---|---|---|
|
NHANES Nondiabetic |
Bupropion (150 mg SR) |
Escitalopram (10 mg) |
Gabapentin (300 mg) |
|
| No. of patients | 3,381 | 378 | 199 | 547 |
| Age, mean [95% CI] | 42.5 [41.7–43.2] | 56.4 [55.2–57.6] | 58.5 [56.3–60.7] | 64.6 [63.5–65.7] |
| % Female [95% CI] | 0.58 [0.57–0.6] | 0.7 [0.65–0.75] | 0.72 [0.66–0.78] | 0.71 [0.67–0.75] |
| Race | – | – | – | |
| % White [95% CI] | 0.75 [0.73–0.77] | |||
| % Black [95% CI] | 0.11 [0.09–0.12] | |||
| % Latino [95% CI] | 0.08 [0.07–0.10] | |||
| % Other race [95% CI] | 0.06 [0.04–0.08] | |||
| BMI, mean [95% CI] | 27 [26.7–27.2] | – | – | – |
| Hypertension, % diagnosed | 0.2 [0.19–0.22] | 0.53 [0.48–0.58] | 0.53 [0.46–0.6] | 0.69 [0.65–0.73] |
| Coronary heart disease, % diagnosed | 0.01 [0.01–0.02] | 0.13 [0.1–0.17] | 0.17 [0.11–0.22] | 0.23 [0.2–0.27] |
| Chronic kidney disease, % diagnosed | 0.01 [0.009–0.02] | 0.05 [0.03–0.07] | 0.06 [0.02–0.09] | 0.12 [0.09–0.15] |
BMI, body mass index; CI, confidence interval; NHANES, National Health and Nutrition Examination Survey; SR, sustained release.
Race and BMI information is not available for nonidentifiable Aetna Insurance claims data.
Using the normoglycemic cohort, we performed comprehensive association testing between prescription drug use and either fasting blood glucose or blood glucose following an oral glucose tolerance test, adjusting for age, sex, race, and BMI. Of the 115 prescription drugs with power to detect an association (from 1,133 total drugs tracked), 16 drugs had significant associations with either fasting blood glucose or blood glucose following an oral glucose tolerance test. Even with a lenient significance cutoff of FDR <0.3, all but one drug‐trait association fell within the 95th percentile of absolute effect sizes in simulation studies (see Methods section, Supplementary Figure S1). Five of the 16 significant drugs had concordant, negative associations with both glycemic measures (Table 2); however, only the antidepressant bupropion was significantly and negatively associated with both glycemic phenotypes (survey‐corrected linear regression β <0, FDR <0.3, Figure 1 a). Bupropion was associated with −2.3 mg/dL (95% CI [−4.2, −0.4]) lower fasting blood glucose, and −9.6 mg/dL (95% CI [−15.7, −3.5]) lower blood glucose following an oral glucose tolerance test. Notably, we did not observe significant differences in age, BMI, or chronic disease burden between those exposed to bupropion and those not exposed (full demographic differences between exposed and unexposed participants are available in Supplementary Table S2).
Table 2.
NHANES (cross‐sectional) results for drugs associated (FDR <0.3) with decreased fasting glucose and improved glucose tolerance
| Drug name |
Participants prescribed (of 3,831) |
Observed effect on fasting glucose, mg/dL [95% CI] | FDR |
Observed effect on glucose tolerance, mg/dL [95% CI] |
FDR |
|---|---|---|---|---|---|
| Bupropion | 39 | −2.3 [−4.2, −0.4] | 0.29 | −9.6 [−15.7, −3.5] | 0.11 |
| Escitalopram | 42 | −1.9 [−3.7, 0] | 0.34 | −13.5 [−22.3, −4.7] | 0.11 |
| Gabapentin | 30 | −2.6 [−4.6, −0.6] | 0.24 | −4.9 [−28, 18.2] | 0.93 |
| Levothyroxine | 196 | −1.5 [−2.5, −0.5] | 0.17 | −1.4 [−6.1, 3.3] | 0.91 |
| Trimaterene | 48 | −2.1 [−3.9, −0.3] | 0.29 | −7.3 [−17.2, 2.7] | 0.54 |
CI, confidence interval; FDR, false discovery rate; NHANES, National Health and Nutrition Examination Survey.
In addition to bupropion, we selected escitalopram and gabapentin for replication in longitudinal claims data, which had stronger negative associations than bupropion with blood glucose following an oral glucose tolerance test and fasting glucose, respectively. We reasoned that gabapentin and escitalopram would serve as negative controls and demonstrate whether multiple complementary associations are required for glycemic status improvement in a longitudinal setting. Escitalopram is an antidepressant like bupropion and would further serve as a control for confounding by indication (i.e., whether improvement in depression status, rather than use of drug, directly drives the effect).
Replication in a retrospective self‐controlled study
To replicate the association of bupropion with improved fasting blood glucose, we performed a self‐controlled study of fasting blood glucose using unidentifiable member claims data from Aetna Health Insurance, Inc., containing information from 50 million individuals over 9 years. We extracted three nondiabetic populations with exposure either to bupropion or to one of two control antidepressants, duloxetine, and escitalopram (see Table 1 for demographic characteristics of each drug‐exposed cohort). Age, sex, and chronic disease burden (as ascertained by hypertension, coronary heart disease, and chronic kidney disease status) were not significantly different between drug‐exposed cohorts (analysis of variance, P > 0.1). Within each drug‐exposed cohort, we identified individuals with a fasting glucose measurement up to a year before being exposed (glucose measurements are typically performed on an annual basis13), and within 2 months after being exposed (with a buffer of 8 days to reach steady‐state drug concentration). Within each drug, we selected dosage forms with at least 198 individuals for well‐powered association testing. For bupropion, the only dosage form with sufficient individuals was 150 mg sustained release with 383 individuals (of 11 total dosage forms), for escitalopram only 10 mg was powered with 199 individuals (of 3 total dosage forms), and for gabapentin only 300 mg was powered with 547 individuals (of 6 total dosage forms). We note here that, although there are substantial differences in the raw doses of drug in this analysis, the dosage forms presented in this study all represent the recommended initial dose for each given formulation. We found that only bupropion 150 mg sustained release was associated with significantly decreased fasting blood glucose (mean difference −1.92 mg/dL, 95% CI [−2.97, −0.87], P < 0.0005, see Table 3).
Table 3.
Unidentifiable member claims data from Aetna Insurance (longitudinal) replication analysis of selected drugs
| Drug name (dosage) | Sample size |
Mean difference in fasting glucose, mg/dL [95% CI] |
P value |
|---|---|---|---|
| Bupropion (150 mg SR) | 378 | −1.92 [−2.97, −0.87] | <0.0005 |
| Escitalopram (10 mg) | 199 | 0.08 [−1.31, 1.47] | N.S. |
| Gabapentin (300 mg) | 547 | 0.24 [−0.62, 1.10] | N.S. |
CI, confidence interval; N.S., not significant; SR, sustained release.
DISCUSSION
In this study, we describe preliminary results from a novel quantitative phenotype‐based drug repositioning methodology. Our methodology uses a combination of complementary quantitative phenotypes to efficiently reduce the number of potential repositioning candidates. Our method enables straightforward follow‐up in prospective investigations and provides estimation of the population sizes required to detect modulation of disease‐relevant phenotypes by a candidate drug. Furthermore, we perform all association testing in a nondiseased and otherwise nationwide representative population to avoid common sources of confounding.
To demonstrate our complementary phenotype‐based approach, we predicted repositioning candidates with the potential to modulate diabetes health using a 3,831 person nondiabetic cohort in NHANES. Fasting blood glucose and glucose following an oral glucose tolerance test capture related but distinct etiological components of diabetes health, and impairment in either test implies distinct disease etiology (hepatic and muscular insulin resistance, respectively).14 By combining these two phenotypes, we identified a single candidate, the antidepressant bupropion, associated with improved glycemic status across multiple etiological pathways in diabetes.
Bupropion is well‐known as a treatment for obesity comorbid with diabetes, both alone15, 16 and in combination with naltrexone,17 as well as a monotherapy for comorbid depression and diabetes.18 However, what was unclear from the previous studies is the degree to which the observed glycemic effects were caused by improvement in BMI or depression, which subsequently led to improvement in glycemic status (confounding by indication). In contrast, we explicitly address confounding by adiposity, depression status, and glycemic status: (1) by explicitly adjusting for BMI and cardiometabolic disease status in all associations; (2) by testing another commonly used antidepressant for associations with improved glycemic health; and (3) by testing for associations in a nondiabetic and normoglycemic cohort, decreasing the likelihood of more complex confounding scenarios (e.g., statins lower low‐density lipoprotein levels, which, in turn, lowers heart attack and stroke risk simultaneously). Although we explicitly control for several key cardiometabolic factors in our models, we note here that there are likely additional hidden confounders that may influence the observed effects, including prescription patterns and unascertained disease status. We, therefore, urge further follow‐up clinical studies that explicitly control for these and other known variables that may be possible confounders to the extent possible.
Another key benefit of our method is the ability to design clinical studies derived from our potential discoveries; we demonstrated this benefit by performing a retrospective self‐controlled study using unidentifiable member claims data from Aetna Insurance. By designing an experiment by which patients serve as their own control, we avoid time‐invariant confounding.19 We successfully replicated that bupropion alone among the drugs considered is associated with lower fasting blood glucose. Neither escitalopram nor gabapentin, which were more strongly associated with improved fasting glucose and glucose tolerance than bupropion in NHANES, respectively, showed a significant impact on fasting blood glucose in the self‐controlled study. This result underscores the importance of combining multiple quantitative phenotypes to achieve high specificity in repositioning candidates and to enable future clinical trials.
Although we have discussed the potential of combining complementary quantitative phenotypes for drug repositioning, we do note that it has some limitations. Chief among these is the requirement that nondiseased individuals are assayed for quantitative phenotypes. Whereas common diseases by necessity have associated routine diagnostics, for example, fasting blood glucose and glucose tolerance for diabetes or lipid levels and blood pressure for cardiovascular disease, repositioning for rarer diseases may require nonstandard tests. We expect that this limitation will diminish over time with the development of birth cohorts (e.g., Avon Longitudinal Study of Parents and Children (ALSPAC)20), and large biobanking initiative (e.g., UK Biobank21 among others), most of which include clinical phenotyping of all participants to complement a variety of “omic measurements.” Although this may not be the case in follow‐up replication studies, whereas ascertainment bias (i.e., some tests are prescribed preferentially to those suspected of having a disease) may skew results. It is important in future studies, therefore, to contextualize diagnostic testing as either routine preventative screening, or high‐risk screening. A second potential limitation is the requirement for multiple complementary quantitative phenotypes that are associated with the disease of interest for repositioning. For diseases in which such phenotypes are not available, further biomarker identification may be required before using our repositioning strategy. We note that any quantitative phenotype‐based methodology would likely require disease‐associated phenotypes before producing meaningful and interpretable results. Last, because we assess all associations between drugs and phenotypes in a nondiseased population, it will be important to verify any repositioning candidates that arise from this method in patients with diseases, and especially so for cardiometabolic diseases like diabetes, in which insulin sensitivity differs dramatically between healthy individuals and those with disease. We, therefore, recommend follow‐up studies of bupropion's effect on glycemic status in a diabetic population.
Conflict of Interest
The authors declare that they have no conflicting interests related to the work presented herein.
Supporting information
Supplementary Figure S1 Simulation studies demonstrate robust signal for observed drug‐trait associations even at a loose significance cutoff. (a) Simulated values for absolute effect size in fasting blood glucose (boxplots, outliers hidden for clarity), and observed absolute effect sizes for significant drugs (red points). (b) As in a, with glucose following an oral glucose tolerance test.
Supplementary Table S1 Demographic breakdown of full NHANES cohort (2005–2006, 2007–2008, 2009–2010, and the 2011–2012 surveys) and subcohort with complete demographic information
Supplementary Table S2 Demographic differences between exposed and unexposed participants in nondiabetic cohort by drug
Source of Funding
A.S.B. and D.R. were supported by a National Institutes of Health (NIH) Training grant from the National Human Genome Research Institute (NHGRI), T32HG002295‐13. C.J.P. is supported by a National Institute of Environmental Health Sciences (NIEHS) R00 ES023504, R21 ES025052, National Science Foundation Big Data Spoke (1636870), and a gift from Agilent Technologies.
Author Contributions
C.J.P., A.S.B., and D.R. wrote the manuscript. C.J.P. and A.S.B. designed the research. A.S.B. and D.R. performed the research. A.S.B. and D.R. analyzed the data.
References
- 1. Rodriguez‐Esteban, R. A drug‐centric view of drug development: how drugs spread from disease to disease. PLoS Comput. Biol. 12, e1004852 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lamb, J. et al The Connectivity Map: using gene‐expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006). [DOI] [PubMed] [Google Scholar]
- 3. Gottlieb, A. , Stein, G.Y. , Ruppin, E. & Sharan, R. PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol. 7, 496 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang, H. , Nguyen, T. , Ibrahim, S. , Shantharam, S. , Yue, Z. & Chen, J.Y. DMAP: a connectivity map database to enable identification of novel drug repositioning candidates. BMC Bioinformatics 16(suppl. 13), S4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown, A.S. , Kong, S.W. , Kohane, I.S. & Patel, C.J. ksRepo: a generalized platform for computational drug repositioning. BMC Bioinformatics 17, 78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qu, X.A. , Gudivada, R.C. , Jegga, A.G. , Neumann, E.K. & Aronow, B.J. Inferring novel disease indications for known drugs by semantically linking drug action and disease mechanism relationships. BMC Bioinformatics 10(suppl. 5), S4 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheung, W.A. , Ouellette, B.F. & Wasserman, W.W. Quantitative biomedical annotation using medical subject heading over‐representation profiles (MeSHOPs). BMC Bioinformatics 13, 249 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kissa, M. , Tsatsaronis, G. & Schroeder, M. Prediction of drug gene associations via ontological profile similarity with application to drug repositioning. Methods 74, 71–82 (2015). [DOI] [PubMed] [Google Scholar]
- 9. Patchala, J. & Jegga, A.G. Concept modeling‐based drug repositioning. AMIA Jt Summits Transl. Sci. Proc. 2015, 222–226 (2015). [PMC free article] [PubMed] [Google Scholar]
- 10. Brown, A.S. & Patel, C.J. MeSHDD: literature‐based drug‐drug similarity for drug repositioning. J. Am. Med. Inform. Assoc. 24, 614–618 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu, H. et al Validating drug repurposing signals using electronic health records: a case study of metformin associated with reduced cancer mortality. J. Am. Med. Inform. Assoc. 22, 179–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jin, G. & Wong, S.T. Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 19, 637–644 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Centers for Disease Control and Prevention (CDC) & National Center for Health Statistics (NCHS) National Health and Nutrition Examination Survey Data, 2005–2012. <http://www.cdc.gov/nchs/nhanes/nhanes_questionnaires.htm>.
- 14. American Diabetes Association . Diagnosis and classification of diabetes mellitus. Diabetes Care 34(suppl. 1), S62–S69 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saydah, S.H. , Gregg, E.W. , Kahn, H.S. & Ali, M.K. Mortality associated with less intense risk‐factor control among adults with diabetes in the United States. Prim. Care Diabetes (2017). [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 16. Lumley, T. Analysis of complex survey samples. J. Stat. Softw. 9, 1–19 (2004). [Google Scholar]
- 17. Austin, P.C. & Steyerberg, E.W. The number of subjects per variable required in linear regression analyses. J. Clin. Epidemiol. 68, 627–636 (2015). [DOI] [PubMed] [Google Scholar]
- 18. Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300 (1995). [Google Scholar]
- 19. Piantadosi, S. Crossover Designs Clinical Trials. 515–527 (John Wiley & Sons, Inc, Hoboken, NJ, 2005). [Google Scholar]
- 20. Kuang, Z. , Thomson, J. , Caldwell, M. , Peissig, P. , Stewart, R. & Page, D. Computational drug repositioning using continuous self‐controlled case series. KDD 2016, 491–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosner, B. Fundamentals of Biostatistics (Rosner, Fundamentals of Biostatics) (Duxbury Press, Duxbury, MA, 2010). [Google Scholar]
- 22. Nathan, D.M. , Davidson, M.B. & DeFronzo, R.A. Impaired fasting glucose and impaired glucose tolerance. Diabetes <http://care.diabetesjournals.org/content/30/3/753.short> (2007). [DOI] [PubMed] [Google Scholar]
- 23. Serretti, A. & Mandelli, L. Antidepressants and body weight: a comprehensive review and meta‐analysis. J. Clin. Psychiatry 71, 1259–1272 (2010). [DOI] [PubMed] [Google Scholar]
- 24. Hollander, P. et al Effects of naltrexone sustained‐release/bupropion sustained‐release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care 36, 4022–4029 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Plodkowski, R.A. , Nguyen, Q. , Sundaram, U. , Nguyen, L. , Chau, D.L. & St Jeor, S. Bupropion and naltrexone: a review of their use individually and in combination for the treatment of obesity. Expert Opin. Pharmacother. 10, 1069–1081 (2009). [DOI] [PubMed] [Google Scholar]
- 26. Lustman, P.J. , Williams, M.M. , Sayuk, G.S. , Nix, B.D. & Clouse, R.E. Factors influencing glycemic control in type 2 diabetes during acute‐ and maintenance‐phase treatment of major depressive disorder with bupropion. Diabetes Care 30, 459–466 (2007). [DOI] [PubMed] [Google Scholar]
- 27. Pembrey, M. ; ALSPAC Study Team. The Avon Longitudinal Study of Parents and Children (ALSPAC): a resource for genetic epidemiology. Eur. J. Endocrinol. 151(suppl. 3), U125–U129 (2004). [DOI] [PubMed] [Google Scholar]
- 28. Sudlow, C. et al UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 Simulation studies demonstrate robust signal for observed drug‐trait associations even at a loose significance cutoff. (a) Simulated values for absolute effect size in fasting blood glucose (boxplots, outliers hidden for clarity), and observed absolute effect sizes for significant drugs (red points). (b) As in a, with glucose following an oral glucose tolerance test.
Supplementary Table S1 Demographic breakdown of full NHANES cohort (2005–2006, 2007–2008, 2009–2010, and the 2011–2012 surveys) and subcohort with complete demographic information
Supplementary Table S2 Demographic differences between exposed and unexposed participants in nondiabetic cohort by drug