

Abstract
The unmet medical need of providing evidence‐based pharmacotherapy for pregnant women is recognized by the regulatory bodies. Physiologically based pharmacokinetic (PBPK) modeling offers an attractive platform to quantify anticipated changes in the pharmacokinetics (PKs) of drugs during pregnancy. Recent publications applying a pregnancy PBPK module to the prediction of maternal and fetal exposure of drugs are summarized. Future opportunities to use PBPK models to predict breast milk exposure and assess human fetotoxicity risks are presented.
CURRENT STATUS AND NEED FOR CLINICAL STUDIES IN PREGNANT WOMEN
The treatment of medical conditions during pregnancy is often necessary, whether to improve the mothers’ quality of life or to treat diseases that threaten their health, the fetus’ normal development, or the lives of both.1 Pregnant women and their fetuses are orphan populations with respect to the safety and efficacy of drugs. In the United States, over 89% of women use at least one prescription or over‐the‐counter medication during pregnancy,2 despite the fact that 70% of the medications approved in the United States between 2000 and 2010 have no human pregnancy data and 98% have insufficient published data to determine the teratogenic risk in humans.3 At a multinational level across Europe, North and South America, and Australia, a cross‐sectional, web‐based survey4 found that 81.2% of pregnant women reported the use of at least one medication. Almost 70% of the women reported the treatment of an acute or short‐term illness and 17% reported treatments for chronic or long‐term illnesses. Overall, the most frequently used medications in pregnancy, either prescription or over‐the‐counter, were taken to treat acute conditions that may be exacerbated by pregnancy, such as headaches, general aches and pain, gastric reflux, cold symptoms, nausea, and urinary tract infections. The most common chronic conditions for which medications were administered include allergy, asthma, thyroid conditions, and depression, or pregnancy‐related conditions, such as gestational diabetes and hypertension.4 The treatment of pre‐existing or self‐limiting chronic conditions including viral (e.g., human immunodeficiency virus (HIV)), fungal, or bacterial infections, solid organ transplantation, smoking cessation, and epilepsy are also common.
Pregnant women are rarely included in clinical trials for numerous practical, legal, and ethical reasons. Most drugs are prescribed to pregnant women off‐label; the dose administered is usually based on the recommended dose for men or nonpregnant women combined with the clinician's judgment. One case in point is the pharmacotherapy of pregnant HIV‐infected women. Prescribing pregnant HIV‐infected women with combination antiretroviral treatment is critical for the prevention of mother‐to‐child transmission of the virus, and adequate maternal exposure to antiretroviral drugs is required for the maximal reduction of viral load and the reduced risk of viral transmission. Reductions in maternal exposure to these drugs during pregnancy may lead to subtherapeutic levels, as shown for a number of drugs, including atazanavir, indinavir, ritonavir, darunavir, and efavirenz,5, 6, 7, 8 eventually leading to virologic failure and/or drug resistance. Historically, for several antiviral drugs (e.g., tenofovir, darunavir, and efavirenz) there has been a significant lag time, ranging from 8–15 years between the initial US Food and Drug Administration (FDA) approval of a drug and the time when pregnancy data first became available (personal communications with Drs Stein Schalkwijk and Angela Colbers, Radboud University Medical Center, Nijmegen, The Netherlands). Alternative, mechanism‐driven approaches for designing appropriate dosing regimens, and, importantly, to shorten the lag time for newer treatment options (e.g., dolutegravir, elvitegravir, and cobicistat) are highly desirable. This review highlights a case example that demonstrates the utility of physiologically based pharmacokinetic (PBPK) model predictions in selecting the most optimal dose for darunavir/ritonavir, taking into account both maternal and fetal drug exposure.9, 10
The intentional or unintentional maternal exposure to drugs in early pregnancy is often accompanied by concerns regarding potential teratogenic effects, as illustrated by the well‐known case of thalidomide in the mid‐20th century. Fetal drug exposure in later trimesters may also lead to complications, as drugs may interfere with fetal growth or result in the acute interference of key homeostatic processes in the unborn child. Examples include intra‐uterine exposure to opioid analgesics, resulting in postpartum withdrawal symptoms and respiratory depression in the newborn.11, 12 There are also cases in which maternal drug treatment is prescribed to treat or prevent disease in the unborn child. Challenging examples of the latter are the titration of the mother with anti‐arrhythmic agents to reach an adequate fetal drug exposure to treat fetal cardiac arrhythmia13 and the maternal treatment with antiretroviral agents to prevent maternal‐to‐child‐transmission of the HIV virus at the time of delivery.14
The FDA also recognizes that clinicians need more information to make appropriate dosing decisions for their pregnant patients. The FDA's Pregnancy and Lactation Labeling Rule (PLLR), which took effect on 30 June 2015, requires that all prescription drug and biological products remove the pregnancy letter categories A, B, C, D, and X from their product labeling over the next 2–4 years.15 The PLLR rule further specifies that in Section “8.1 Pregnancy” the drug's labeling “must also contain relevant information, if it is available, to help health care providers make prescribing decisions and counsel women about the use of the drug during pregnancy; this could include information on disease‐associated maternal and/or embryo/fetal risk, dose adjustments during pregnancy and the postpartum period, maternal and fetal/neonatal adverse reactions, etc.” The PLLR rule, defined above, should also be applied to Section “8.2 Lactation,” which is intended to provide information about using the drug while breastfeeding, including the amount of drug present in breast milk and the potential effects on the breastfed infant. In the absence of pharmacokinetic (PK) data from clinical pregnancy and lactation studies, alternative approaches, such as PBPK modeling and simulation, may inform this additional dosing information.
PHYSIOLOGICAL CHANGES DURING PREGNANCY
Pregnancy is associated with a multitude of temporal physiological and metabolic changes,16 and these changes in maternal physiology can have a direct effect on drug absorption, distribution, metabolism, and excretion (ADME). Various pregnancy‐related hemodynamic changes lead to an increase in plasma volume (up to 50%) and a decrease in plasma protein binding, which can both alter the apparent volume of distribution of drugs. Serum albumin and alpha 1 acid glycoprotein concentrations decrease by up to 31% and 19% during late pregnancy, respectively.17 There is a marked 33% increase in cardiac output during pregnancy. The change in maternal hepatic enzyme activity is cytochrome P450 (CYP) isoform‐specific. For example, the metabolism of drugs catalyzed by selective CYP isoenzymes (e.g., CYP3A4, CYP2D6, and CYP2C9) and uridine diphosphate glucuronosyltransferase (UGT) isoenzymes (e.g., UGT1A4 and UGT1A1) is increased during pregnancy. In contrast, CYP1A2 and CYP2C19 activity is decreased during pregnancy.16 The magnitude of pregnancy‐related induction in drug‐metabolizing enzyme activities can be as high as 300%, as suggested by a study of the kinetics of lamotrigine, a substrate drug of UGT1A4.18 The causative mechanisms of hepatic CYP activity changes are believed to be regulated by rising concentrations of various hormones (e.g., estradiol, progesterone, or growth hormones).19 The renal excretion of unchanged drugs is increased during pregnancy owing to an increase in the glomerular filtration rate and also possibly to an increase in renal secretion via transporters. Currently, there is evidence relating to the pregnancy effect on renal transporter organic cation transporter (OCT) and intestinal/renal transporter P‐glycoprotein, based on metformin20 and digoxin data.21 Future studies should address other important renal and hepatic transporters, such as organic anion transporters (OAT) and organic anion‐transporting polypeptides (OATPs) 1B1 and 1B3.
PBPK MODELING AND SIMULATION
PBPK modeling and simulation offers a viable alternative to clinical trials for this vulnerable patient population. Regulatory agencies, such as the FDA and the European Medicines Agency, have adopted PBPK modeling and simulation to facilitate the review of Investigational New Drug and New Drug Application submissions to address a variety of clinical issues, including the assessment of the effect of intrinsic or extrinsic factors on drug PKs.22, 23, 24 In these applications, PBPK modeling is primarily used to facilitate the decision making process on whether, when, and how to conduct a clinical pharmacology study, and, more importantly, to inform drug labeling to support dosing recommendations.25 Increasingly, the regulatory incentives and challenges in conducting and obtaining clinical data in special populations have significantly contributed to the increased use of PBPK models in pediatric and organ impairment populations.23, 26
Under the overarching umbrella of systems pharmacology, PBPK models map drug movements in the body to a physiologically realistic compartmental structure using sets of differential equations.25 This approach has the advantage of integrating both physiological parameters (e.g., time‐varying changes in maternal weight, individual organ volumes/blood flows, cardiac output, glomerular filtration rate, and drug‐metabolizing enzyme activities) that are important for ADME processes and drug‐specific parameters (e.g., physicochemical and in vitro metabolism/transport characteristics). A number of pregnancy PBPK models have been reported for pregnant animals and humans in the past 3 decades, which have mainly been applied in the risk assessment of environmental chemicals.27, 28 Although many of the existing models have considered parameters, such as developmental change of distribution volume, blood flow rate in the maternal body, and the growth of the placental‐fetal unit on disposition of environmental chemicals, few have modeled the absorption, metabolism, and elimination of drugs.
In 2013, the Simcyp Population‐based Simulator offered the first pregnancy module designed to model the kinetics of drugs by incorporating time‐varying physiology of the virtual population.17, 29, 30 More recently, other software providers have started to offer a pregnancy module.31, 32 Since then, a number of publications have provided independent verification and application of a pregnancy module to study drugs in the pregnant population. The tested drugs covered a wide spectrum of clearance mechanisms, including several major isoforms of CYP enzymes (CYP3A4, 2D6, 1A2, 2B6, 2C9, and 2C19),9, 33, 34 and renal clearance.34, 35 These examples followed the best practice approach in PBPK modeling of drug PKs in special populations, demonstrating the performance of drug models for the nonpregnant subjects, verifying the key model components using independent clinical PK and drug‐drug interaction data, before predicting the drug PKs in the pregnant subjects (Figure 1). Justifications on the model assumptions of inclusion of pregnancy‐induced changes that are relevant to major ADME processes of the test drug were provided. These model application examples not only helped us gain confidence in the predictive performance of the respective model components, but also provided information relating to less‐established pregnancy effects on physiology, such as the effect of pregnancy on drug transporter activities.
Figure 1.
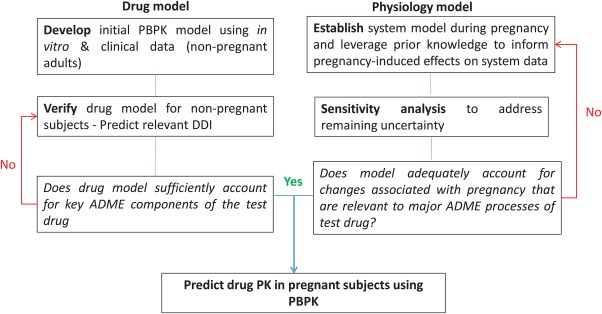
The best practice approach to physiologically based pharmacokinetic (PBPK) model development and application in pregnant women. ADME, absorption, distribution, metabolism, and excretion; DDI, drug‐to‐drug interaction; PK, pharmacokinetic.
For example, CYP3A activity is induced by up to 99% during the third trimester, as informed by a midazolam PK study.21 One study36 explored whether a previously developed PBPK model29 could quantitatively predict PK profiles of CYP3A‐metabolized drugs during the third trimester, and discern the site of CYP3A induction (i.e., liver, intestine, or both). The PBPK model successfully predicted midazolam, nifedipine, and indinavir disposition during the third trimester. A sensitivity analysis suggested that CYP3A induction in the third trimester is most likely hepatic and not intestinal. CYP1A2 enzyme activity, as measured by caffeine salivary clearance, is suppressed by up to 65% in the third trimester. A PBPK model incorporating this decrease in 1A2 activity predicted a modest increase (25%) in theophylline exposure during pregnancy, which was consistent with the observed data.30 For CYP isoforms, such as CYP2B6, in which PK data obtained from the dosing of a sensitive probe drug are lacking, an in vitro to in vivo extrapolation (IVIVE) approach was utilized to predict the pregnancy‐related induction effect from in vitro human hepatocyte incubations with clinically relevant concentrations of estradiol. A PBPK model accounting for a CYP2B6 induction of up to 90% as well as known pregnancy effects on other clearance pathways, including CYP3A, CYP2C19, and renal clearance, successfully predicted methadone disposition during the second trimester and the third trimester as compared with the observed data.33 Additional performance verification utilizing efavirenz data7 would be helpful to further verify the pregnancy‐induced effect on CYP2B6.
PBPK modeling of renally excreted antiretroviral drugs, including tenofovir, emtricitabine, and lamivudine in pregnant women, have shown that the maximum clearance increases were ∼30%, which are in agreement with the observed data.35 Importantly, the authors showed that for all three drugs, both renal filtration and renal secretion of drugs increased during pregnancy, the latter being approximated by pregnancy‐induced changes in renal plasma flow. Tenofovir is mainly a substrate for OAT1, whereas emtricitabine and lamivudine are both substrates for OCT2. Additional sensitivity analysis results suggest that there seems to be insignificant changes in renal transporter activity during pregnancy, including OCT2 and OAT1, or alternatively, the kinetics of these three drugs were not sensitive markers for the respective renal transporters likely due to a more significant secretion clearance attributable to passive diffusion. In another study, PBPK modeling of oseltamivir carboxylate, a drug predominantly eliminated by renal OAT1 and OAT3, in pregnant women, informed pregnancy‐induced changes in OAT.37 The sensitivity analysis revealed that OAT‐mediated renal clearance for oseltamivir carboxylate needed to be increased by ∼100% to recover oseltamivir carboxylate PK during pregnancy. One can explore PK changes of other OAT‐substrate drugs (e.g., ciprofloxacin) in pregnant women.38
Despite these aforementioned successes in the application of PBPK modeling in the pregnant population, several challenges still exist. First, although there is adequate literature evidence documenting the key physiological, biochemical, and metabolic changes occurring during pregnancy, the granularity of information (e.g., the gestation age‐dependent changes especially during early gestation) and the impact on the overall PK outcome has not always been sufficiently verified to warrant incorporating all these changes in the model.27 For example, the changes in hepatic enzyme activity are typically informed by probe‐drug studies. For the majority of the probe‐drug studies conducted in pregnant women, data are obtained only in the third trimester as some probe drugs cannot be safely administered to pregnant women during early gestation if they are not used for therapeutic purposes. Opportunistic PK studies in pregnant women are also likely restricted to the phase of pregnancy during which the patients are afflicted with the disease. In any case, extrapolating the magnitude of change in CYP enzyme activity from one trimester to other trimesters may be difficult. Similar issues also need to be addressed for several intestinal, renal, and hepatic transporters such as P‐glycoprotein (P‐gp), OATP1B1/3, OCT2, and OAT1/3. Second, the system data used to populate the current pregnancy PBPK models mainly came from healthy pregnant women with singleton births,17 and, as a result, the potential effects on drug PKs due to concomitant diseases are not considered. Finally, an added complexity is that there are potential pharmacodynamic (PD) changes during pregnancy, such as a higher degree of insulin resistance exhibited by the women with gestational diabetes, and immunosuppression during pregnancy that may impact the PD response to antibiotics.39 Because the efficacy of drugs will be influenced by alteration in the PK and the PD of drugs, the latter, when and if readily quantified, should be included as another component to guide dose adjustment in pregnancy.
DARUNAVIR/RITONAVIR CASE EXAMPLE
Recently, Colbers et al.9 used PBPK modeling to delineate the potential impact of drug transporters on darunavir PK and to identify current knowledge gaps that limit the accurate prediction of darunavir/ritonavir exposure in pregnancy. In addition, PBPK modeling was used to quantitatively assess the impact of drug‐drug interactions. For example, ritonavir, a strong CYP3A4 inhibitor, is used as a booster to intentionally increase the exposure of antiretroviral drugs that are metabolized by CYP3A4, including darunavir. Initially, the authors used a well‐stirred liver model incorporating the Michaelis‐Menten constant (K m) and the maximum rate of metabolite formation (V max) for CYP3A4‐mediated darunavir metabolism, and the simulations overestimated the total exposure of darunavir when compared with the clinical data. These results indicate the involvement of active transport in the hepatic disposition of darunavir. In the absence of definitive transport data, the authors derived a hepatic uptake component and an efflux component using sensitivity analysis. Subsequently, simulations using a permeability‐limited liver model, including hepatic uptake mediated by OATPs and efflux mediated by P‐gp followed by enterohepatic circulation resulted in an acceptable recovery of darunavir/ritonavir exposure. The ritonavir model incorporated in vitro CYP3A4 inactivation parameters, as well as competitive inhibition constants for hepatic OATP2B1 and P‐gp. For the dosing regimens of 600/100 mg darunavir/ritonavir twice daily (b.i.d.) and 800/100 mg once daily (q.d.), the predicted PK parameters were within a twofold range of the observed data in the nonpregnant subjects. For the pregnant subjects, the predicted decreases in the area under the curve (AUC) values during pregnancy for the b.i.d. and q.d. regimens were 27% and 41%, respectively, consistent with the observed decreases of 17–22% and 33%. These results informed the selection of alternative dosages (800/100 mg b.i.d. and 900/100 mg b.i.d.) that are being investigated in clinics, the outcome of which will allow further verification of the model. This case example nicely illustrates the utility of PBPK model predictions in choosing the most optimal dose in a complex case, when the disposition of the drug is determined by the interplay of hepatic uptake/efflux transporters and hepatic CYP3A4, the combined inhibitory effects from ritonavir, as well as the pregnancy effects on all these processes.
MODELING PLACENTAL TRANSFER AND FETAL EXPOSURE OF DRUGS
Understanding the relationship between external dose and fetal exposure of drugs is a key step in understanding potential harmful or beneficial effects of maternal drug treatment for the unborn child. Although the extent and rate of placental drug transfer is highly dependent upon the physicochemical properties of the drug,40 this also varies considerably between species.41 The latter reflects the significant variation in placental anatomy and hemodynamics between human and different animals. In addition, potential differences in the expression of placental drug transporters and drug metabolizing enzymes, as well as their substrate specificities, can contribute to this phenomenon. Hence, PBPK modeling, based on IVIVE approaches of data obtained with human tissues and cells, is a relevant approach for predicting the rate and extent of the placental transfer of drugs in pregnant women.
PBPK models that adequately describe the placental transfer of drugs should ideally include a description of the gestational changes in maternal physiology that impact drug disposition, incorporate changes that occur in placental physiology, as well as include a description of changes in fetal drug disposition and clearance over time. Zhang et al.42 described such a feto‐pregnancy‐PBPK model in which a placenta, amniotic fluid compartment, and fetal organs that are relevant to drug disposition were added, the model also took into account typical characteristics of the fetal circulation. Gestational age‐dependent changes in fetal, placental, and maternal physiological parameters during the second half of pregnancy were described using empirically derived polynomial equations.42 Using the model, the authors elegantly demonstrated that cord to maternal plasma ratios, even at distribution equilibrium, do not indicate fetal drug exposure relative to that in the mother, which is due to the time‐dependent distributional kinetics of drugs across the placenta. In addition, they used sensitivity analysis to assess the possible impact of gestational age‐dependent changes in placental metabolism and transport processes on placental transfer. To verify performance of the model for predicting the placental passage of only passively diffusing drugs, the authors populated the model using midazolam as a calibrator drug, and subsequently verified the model by adequately predicting the fetal exposure of two other passively diffusing drugs, theophylline and zidovudine.42 For antiretroviral agents, similar approaches for incorporating placental transfer and drug disposition in lumped fetal compartments have been described by De Sousa Mendes et al.43 and Schalkwijk et al.10 These authors use ex vivo placenta perfusion data obtained from term human placentas in a bottom‐up approach to parameterize the placental passage model parameters with clearance values or rate constants for maternal to fetal transfer and vice versa. In these reports, the predictions of fetal exposure at term were also successfully verified against clinical drug levels measured in cord blood samples.
Specifically, Schalkwijk et al.10 conducted a follow‐up investigation of the darunavir study by Colbers et al.6, 9 with the aim of incorporating placental transfer into the established pregnancy PBPK model of darunavir to simulate fetal darunavir exposure at term. The authors determined maternal‐to‐fetal and fetal‐to‐maternal darunavir/ritonavir placental clearance with an ex vivo human cotyledon perfusion model. These placental transfer clearance values were then used to link a feto‐placental unit to the maternal PBPK model. For model qualification, simulated maternal and fetal PK profiles were compared with clinical data obtained during the third trimester and at the time of delivery. The verified model was then used to simulate fetal PK profiles at different maternal darunavir/ritonavir dosing regimens. For darunavir 600/100 mg b.i.d., the predicted fetal maximum plasma concentration, trough concentration, time to maximum plasma concentration, and half‐life were 1.1, 0.57 mg/L, 3, and 21 hours, respectively. Because the predicted fetal trough concentration is higher or around the half‐maximal effective darunavir concentration for a resistant virus (0.55 mg/L), these results support the theory that the fetal exposure after oral maternal darunavir given at the standard dosages is therapeutic.
The advantage of using ex vivo placental perfusion data for model parameterization is that the data intrinsically captures both passive as well as active transfer processes acting on the drug.44, 45, 46 Despite these promising results, prediction of fetal exposure at earlier gestational ages is still difficult. To allow the prediction of the placental transfer of drugs, mechanistic PK data on these placental processes also needs to be established, but generating data via ex vivo placenta perfusion at earlier gestational ages is technically challenging and not very well established.47 An alternative approach could be to generate in vitro kinetic parameters (Km, Vmax, or intrinsic clearance values) of drugs in recombinant transporter and drug metabolizing enzyme overexpression systems, as well as passive permeability via in vitro measurements in cellular monolayers. For successful IVIVE of placental clearance from these in vitro kinetic parameters, further knowledge of the abundance of specific placental enzymes and the abundance of transporters in the membranes of placental syncytiotrophoblasts and fetal capillary endothelial cells is required. To move this approach forward, the measurement of gestational age‐dependent changes in the expression levels of placental enzymes is required to parameterize feto‐pregnancy‐PBPK models accordingly. Finally, it is important that sufficient and high quality clinical data for validation purposes becomes available. In particular, this includes the well‐timed collection of cord and maternal blood samples, as was also noted by others, but we would like to stress that this should include gathering clinical data on placental drug concentrations as well.
MODELING BREAST MILK EXPOSURE OF DRUG
In response to the PLLR rule calling for prescribing information on the predicted amount of drug appearing in breastmilk, Abduljalil et al.48 developed a dynamic lactation model consisting of three compartments, mammary blood, the tissue itself, and the milk. The model incorporates physiological parameters, including the tissue volume and blood flow, milk pH, and the milk intake, along with individual physiological parameters and their variability. The model was integrated within the Simcyp Simulator using a first order absorption model and the Rodgers & Rowland49 method to predict the partition of drugs between tissues and plasma. In addition, the elimination kinetics of administered drugs were predicted using IVIVE. The kinetics of single‐dose alprazolam (0.5 mg) and caffeine (200 mg) were predicted and the outcomes were compared with the reported data. The model prediction of milk and plasma AUC0–24h values were within twofold of the observations. In another application, the lactation PBPK model was used to predict the concentration of tramadol in breastfeeding mothers who are extensive metabolizers of CYP2D6.50 The results were in agreement with the clinical observations. The model was then used to predict tramadol concentration in the plasma and milk for poor and ultrarapid metabolizer subpopulations. The infant daily doses were calculated after maternal intake of the drug. These encouraging results demonstrated that the three‐compartmental model for mammary gland coupled with the PBPK model is useful to explore scenarios in which clinical observations are not available to predict the amount of drug ingested by the neonate. Although the disposition of these compounds is mainly governed by passive permeability, future work will focus on extending the model to predict the concentration and distribution of compounds whose kinetics involve drug transporters.
FUTURE OPPORTUNITIES – A MECHANISTIC FRAMEWORK FOR PREDICTING HUMAN FETOTOXICITY RISK
We believe that with the established base of knowledge relating to the profound physiological changes during pregnancy and the significant advances in PBPK approaches that have been applied in special populations (such as pediatric populations), the PBPK approach during pregnancy offers an attractive tool to quantify anticipated changes in the PK of drugs during pregnancy. Conducting trials in silico before executing them in vivo can also be helpful in optimizing the design of a first‐in‐pregnancy PK study, including the prioritization of the study period, the sample size, and the dose selection.
The coupled maternal‐fetal physiology imposes additional concerns about both efficacy and safety of treatment. To assess fetal exposure to drugs administered to the mother, the established maternal PBPK model has recently been expanded to include a fully physiological fetal model to allow the quantitative predictions of fetal systemic and tissue exposures.51 This is the first step toward providing a mechanistic framework for applying a PBPK feto‐maternal model to predict human fetotoxicity risk from preclinical species (Figure 2). In contrast with existing PBPK feto‐maternal models in the literature in which many of the longitudinal changes of physiological parameters were largely based on scaling rat data, the model by Abduljalil et al.51 in 2017, was fully parameterized with human physiology data with respect to tissue growth, blood flow, binding proteins, and drug metabolizing enzymes together with the associated variability with these parameters, which are necessary in order to broaden its application particularly in drug discovery and development for pregnant women. For example, the PBPK platform can provide a means to estimate therapeutic concentrations of drugs (e.g., antiretrovirals or antiarrhythmics treating in utero conditions of the fetus).
Figure 2.
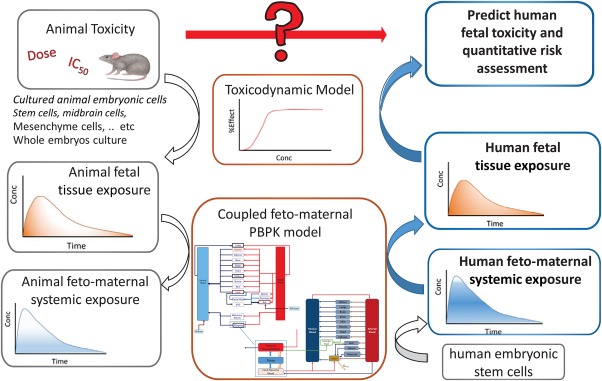
A mechanistic framework for applying a physiologically based pharmacokinetic (PBPK) feto‐maternal model for predicting human fetotoxicity risk from preclinical species described in the following steps. (1) Perform fetotoxicity studies in preclinical animals and establish exposure‐toxicity relationships. (2) Construct a coupled feto‐maternal PBPK model that accounts for gestational age related changes in the physiology and placenta to describe the relationship between fetal tissue and systemic concentrations together with maternal systemic concentration. (3) Use the constructed PBPK model to find the toxic doses that can cause feto‐toxic drug levels after accounting for species differences in physiological/biochemical parameters (number of placentas, weight, blood flow, transporters, and enzyme expression). (4) The human PBPK model can also be refined if in vitro and/or in vivo human data are available. (5) The human PBPK model that predicts the systemic exposure in fetus and mother can be used to predict the local fetal tissue concentration. (6) Fetal local tissue concentration can be linked to the predicted toxicity after accounting for any potential species differences in the toxicodynamic model. This figure is adapted from the publication by Abduljalil et al.51 Conc, concentration; IC50, half‐maximal inhibitory concentration.
Animal models remain an important source of information on maternal‐fetal toxicity. However, the specific types of developmental effects caused by xenobiotics may vary widely between animal models and humans due to biological differences, and this can pose a problem in translating the toxicological observations from animals to humans. Application of the PBPK feto‐maternal model can complement the traditional fetotoxicity studies in preclinical animals to identify the toxic doses that can cause feto‐toxic drug levels after accounting for species differences in physiological and biochemical parameters. The human PBPK model that predicts the systemic exposure in the fetus and mother can be used to simulate the local fetal tissue concentration, the latter can be linked to the predicted toxicity after accounting for any potential species differences in the toxicodynamic model. This approach now has special relevance in relation to recent FDA regulatory guidance on drugs in pregnancy and lactation,15 which emphasized the need to distinguish between risks based on human vs. animal data findings or between differences in frequency, severity, and type of fetal developmental toxicities. We believe that PBPK feto‐maternal models will be highly desirable to support fetal exposure assessment. Once verified, these models have the potential to better translate risk assessment in animals to human scenarios, and to help elucidate appropriate dosing information for pregnant women and their unborn children.
Conflict of Interest
Alice Ban Ke and Khaled Abduljalil are employees of Simcyp Limited (a Certara company).
Source of Funding
No funding was received for this work.
Author Contributions
A.B.K., R.G., and K.A. wrote the manuscript.
References
- 1. Wyszynski, D.F. & Shields, K.E. Frequency and type of medications and vaccines used during pregnancy. Obstet. Med. 9, 21–27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mitchell, A.A. et al Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. Am. J. Obstet. Gynecol. 205, 51.e51–51.e58 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adam, M.P. , Polifka, J.E. & Friedman, J.M. Evolving knowledge of the teratogenicity of medications in human pregnancy. Am. J. Med. Genet. C Semin. Med. Genet. 157C, 175–182 (2011). [DOI] [PubMed] [Google Scholar]
- 4. Lupattelli, A. et al Medication use in pregnancy: a cross‐sectional, multinational web‐based study. BMJ Open 4, e004365 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mirochnick, M. , Best, B.M. & Clarke, D.F. Antiretroviral pharmacology: special issues regarding pregnant women and neonates. Clin. Perinatol. 37, 907–927 (2010). [DOI] [PubMed] [Google Scholar]
- 6. Colbers, A. et al Pharmacokinetics of total and unbound darunavir in HIV‐1‐infected pregnant women. J. Antimicrob. Chemother. 70, 534–542 (2015). [DOI] [PubMed] [Google Scholar]
- 7. Olagunju, A. et al Pharmacogenetics of pregnancy‐induced changes in efavirenz pharmacokinetics. Clin. Pharmacol. Ther. 97, 298–306 (2015). [DOI] [PubMed] [Google Scholar]
- 8. Kreitchmann, R. et al Pharmacokinetics of an increased atazanavir dose with and without tenofovir during the third trimester of pregnancy. J. Acquir. Immune Defic. Syndr. 63, 59–66 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Colbers, A. , Greupink, R. , Litjens, C. , Burger, D. & Russel, F.G. Physiologically based modelling of darunavir/ritonavir pharmacokinetics during pregnancy. Clin. Pharmacokinet. 55, 381–396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schalkwijk, S. et al Prediction of fetal darunavir exposure by integrating human ex‐vivo placental transfer and physiologically based pharmacokinetic modeling. Clin. Pharmacokinet. (2017). Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huybrechts, K.F. et al Risk of neonatal drug withdrawal after intrauterine co‐exposure to opioids and psychotropic medications: cohort study. BMJ 358, j3326 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aaronson, J. , Abramovitz, S. , Smiley, R. , Tangel, V. & Landau, R. A survey of intravenous remifentanil use for labor analgesia at academic medical centers in the United States. Anesth. Analg. 124, 1208–1210 (2017). [DOI] [PubMed] [Google Scholar]
- 13. Martin‐Suarez, A. et al Pharmacokinetics and dosing requirements of digoxin in pregnant women treated for fetal supraventricular tachycardia. Expert Rev. Clin. Pharmacol. 10, 911–917 (2017). [DOI] [PubMed] [Google Scholar]
- 14. Colbers, A. , Greupink, R. & Burger, D. Pharmacological considerations on the use of antiretrovirals in pregnancy. Curr. Opin. Infect. Dis. 26, 575–588 (2013). [DOI] [PubMed] [Google Scholar]
- 15. Sahin, L. , Nallani, S.C. & Tassinari, M.S. Medication use in pregnancy and the pregnancy and lactation labeling rule. Clin. Pharmacol. Ther. 100, 23–25 (2016). [DOI] [PubMed] [Google Scholar]
- 16. Ke, A.B. , Rostami‐Hodjegan, A. , Zhao, P. & Unadkat, J.D. Pharmacometrics in pregnancy: an unmet need. Annu. Rev. Pharmacol. Toxicol. 54, 53–69 (2014). [DOI] [PubMed] [Google Scholar]
- 17. Abduljalil, K. , Furness, P. , Johnson, T.N. , Rostami‐Hodjegan, A. & Soltani, H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin. Pharmacokinet. 51, 365–396 (2012). [DOI] [PubMed] [Google Scholar]
- 18. Fotopoulou, C. et al Prospectively assessed changes in lamotrigine‐concentration in women with epilepsy during pregnancy, lactation and the neonatal period. Epilepsy Res. 85, 60–64 (2009). [DOI] [PubMed] [Google Scholar]
- 19. Zhang, Z. , Farooq, M. , Prasad, B. , Grepper, S. & Unadkat, J.D. Prediction of gestational age‐dependent induction of in vivo hepatic CYP3A activity based on HepaRG cells and human hepatocytes. Drug Metab. Dispos. 43, 836–842 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eyal, S. et al Pharmacokinetics of metformin during pregnancy. Drug Metab. Dispos. 38, 833–840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hebert, M.F. et al Effects of pregnancy on CYP3A and P‐glycoprotein activities as measured by disposition of midazolam and digoxin: a University of Washington specialized center of research study. Clin. Pharmacol. Ther. 84, 248–253 (2008). [DOI] [PubMed] [Google Scholar]
- 22. Luzon, E. , Blake, K. , Cole, S. , Nordmark, A. , Versantvoort, C. & Berglund, E.G . Physiologically based pharmacokinetic modeling in regulatory decision‐making at the European Medicines Agency. Clin. Pharmacol. Ther. (2016). Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 23. Wagner, C. et al Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin. Pharmacokinet. 54, 117–127 (2015). [DOI] [PubMed] [Google Scholar]
- 24. Wagner, C. , Pan, Y. , Hsu, V. , Sinha, V. & Zhao, P. Predicting the effect of CYP3A inducers on the pharmacokinetics of substrate drugs using physiologically based pharmacokinetic (PBPK) modeling: an analysis of PBPK submissions to the US FDA. Clin. Pharmacokinet. 55, 475–483 (2016). [DOI] [PubMed] [Google Scholar]
- 25. Jamei, M. Recent advances in development and application of physiologically‐based pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Curr. Pharmacol. Rep. 2, 161–169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mehrotra, N. et al Role of quantitative clinical pharmacology in pediatric approval and labeling. Drug Metab. Dispos. 44, 924–933 (2016). [DOI] [PubMed] [Google Scholar]
- 27. Lu, G. , Abduljalil, K. , Jamei, M. , Johnson, T.N. , Soltani, H. & Rostami‐Hodjegan, A. Physiologically‐based pharmacokinetic (PBPK) models for assessing the kinetics of xenobiotics during pregnancy: achievements and shortcomings. Curr. Drug Metab. 13, 695–720 (2012). [DOI] [PubMed] [Google Scholar]
- 28. Corley, R.A. , Mast, T.J. , Carney, E.W. , Rogers, J.M. & Daston, G.P. Evaluation of physiologically based models of pregnancy and lactation for their application in children's health risk assessments. Crit. Rev. Toxicol. 33, 137–211 (2003). [DOI] [PubMed] [Google Scholar]
- 29. Gaohua, L. , Abduljalil, K. , Jamei, M. , Johnson, T.N. & Rostami‐Hodjegan, A. A pregnancy physiologically based pharmacokinetic (p‐PBPK) model for disposition of drugs metabolized by CYP1A2, CYP2D6 and CYP3A4. Br. J. Clin. Pharmacol. 74, 873–885 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ke, A.B. , Nallani, S.C. , Zhao, P. , Rostami‐Hodjegan, A. , Isoherranen, N. & Unadkat, J.D. A physiologically based pharmacokinetic model to predict disposition of CYP2D6 and CYP1A2 metabolized drugs in pregnant women. Drug Metab. Dispos. 41, 801–813 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dallmann, A. et al Physiologically based pharmacokinetic modeling of renally cleared drugs in pregnant women. Clin. Pharmacokinet. 56, 1525–1541 (2017). [DOI] [PubMed] [Google Scholar]
- 32. Xia, B. , Heimbach, T. , Gollen, R. , Nanavati, C. & He, H. A simplified PBPK modeling approach for prediction of pharmacokinetics of four primarily renally excreted and CYP3A metabolized compounds during pregnancy. AAPS J. 15, 1012–1024 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ke, A.B. , Nallani, S.C. , Zhao, P. , Rostami‐Hodjegan, A. & Unadkat, J.D. Expansion of a PBPK model to predict disposition in pregnant women of drugs cleared via multiple CYP enzymes, including CYP2B6, CYP2C9 and CYP2C19. Br. J. Clin. Pharmacol. 77, 554–570 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jogiraju, V.K. , Avvari, S. , Gollen, R. & Taft, D.R. Application of physiologically based pharmacokinetic modeling to predict drug disposition in pregnant populations. Biopharm. Drug Dispos. 38, 426–438 (2017). [DOI] [PubMed] [Google Scholar]
- 35. De Sousa Mendes, M. et al Physiologically‐based pharmacokinetic modeling of renally excreted antiretroviral drugs in pregnant women. Br. J. Clin. Pharmacol. 80, 1031–1041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ke, A.B. , Nallani, S.C. , Zhao, P. , Rostami‐Hodjegan, A. & Unadkat, J.D. A PBPK model to predict disposition of CYP3A‐metabolized drugs in pregnant women: verification and discerning the site of CYP3A induction. CPT Pharmacometrics Syst. Pharmacol. 1, e3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hsu, V. , Grimstein, M. & Zhao, P. Leveraging potential changes in renal transporter activity to predict drug pharmacokinetics during pregnancy using PBPK modeling. ASCPT 2016 Annual Meeting; San Diego, CA; 8–12 March 2016.
- 38. Grimstein, M. , Hsu, V. & Zhao, P. PBPK modeling of ciprofloxacin – knowledge extension by confirming the effect of intrinsic and extrinsic patient factors on renal OAT activities. ASCPT 2016 Annual Meeting; San Diego, CA; 8–12 March 2016.
- 39. Fischer, J.H. et al Influence of body weight, ethnicity, oral contraceptives, and pregnancy on the pharmacokinetics of azithromycin in women of childbearing age. Antimicrob. Agents Chemother. 56, 715–724 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eliesen, G.A.M. et al Editor's highlight: Placental disposition and effects of crizotinib: an ex vivo study in the isolated dual‐side perfused human cotyledon. Toxicol. Sci. 157, 500–509 (2017). [DOI] [PubMed] [Google Scholar]
- 41. Carter, A.M. Animal models of human placentation–a review. Placenta 28 (suppl A), S41–S47 (2007). [DOI] [PubMed] [Google Scholar]
- 42. Zhang, Z. , Imperial, M.Z. , Patilea‐Vrana, G.I. , Wedagedera, J. , Gaohua, L. & Unadkat, J.D. Development of a novel maternal‐fetal physiologically based pharmacokinetic model I: insights into factors that determine fetal drug exposure through simulations and sensitivity analyses. Drug Metab. Dispos. 45, 920–938 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. De Sousa Mendes, M. et al A physiologically‐based pharmacokinetic model to predict human fetal exposure for a drug metabolized by several CYP450 pathways. Clin. Pharmacokinet. 56, 537–550 (2017). [DOI] [PubMed] [Google Scholar]
- 44. Baumann, M.U. et al Regulation of human trophoblast GLUT1 glucose transporter by insulin‐like growth factor I (IGF‐I). PLoS One 9, e106037 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pollex, E. , Lubetsky, A. & Koren, G. The role of placental breast cancer resistance protein in the efflux of glyburide across the human placenta. Placenta 29, 743–747 (2008). [DOI] [PubMed] [Google Scholar]
- 46. Porter, C. et al Certolizumab pegol does not bind the neonatal Fc receptor (FcRn): consequences for FcRn‐mediated in vitro transcytosis and ex vivo human placental transfer. J. Reprod. Immunol. 116, 7–12 (2016). [DOI] [PubMed] [Google Scholar]
- 47. Brownbill, P. et al An international network (PlaNet) to evaluate a human placental testing platform for chemicals safety testing in pregnancy. Reprod. Toxicol. 64, 191–202 (2016). [DOI] [PubMed] [Google Scholar]
- 48. Abduljalil, K. , Johnson, T. & Jame, M. Development and integration of a dynamic lactaion model within a full PBPK model. American Conference on Pharmacometrics (ACoP)8 meeting; Fort Lauderdale, Florida; 15–18 October 2017.
- 49. Rodgers, T. & Rowland, M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 95, 1238–1257 (2006). [DOI] [PubMed] [Google Scholar]
- 50. Abduljalil, K. , Johnson, T. & Jamei, M. Application of physiologically‐based pharmacokinetic model to predict tramadol concentration in human milk. Population Approach Group Europe (PAGE) 26th annual meeting; Budapest, Hungary; 6–9 June 2017. Abstract 7087.
- 51. Abduljalil, K. , Johnson, TN. & Rostami‐Hodjegan, A. Fetal physiologically‐based pharmacokinetic models: systems information on fetal biometry and gross composition. Clin. Pharmacokinet. (2017). Epub ahead of print. [DOI] [PubMed] [Google Scholar]