

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
The oral PI3K-δ,γ inhibitor duvelisib demonstrated clinical activity and a favorable safety profile in patients with CTCL and PTCL.
Duvelisib induced cell-autonomous killing of TCL lines and reprogrammed PTCL-associated macrophages in vivo.
Abstract
Duvelisib (IPI-145) is an oral inhibitor of phosphatidylinositol 3-kinase (PI3K)–δ/γ isoforms currently in clinical development. PI3K-δ/γ inhibition may directly inhibit malignant T-cell growth, making duvelisib a promising candidate for patients with peripheral (PTCL) or cutaneous (CTCL) T-cell lymphoma. Inhibition of either isoform may also contribute to clinical responses by modulating nonmalignant immune cells. We investigated these dual effects in a TCL cohort from a phase 1, open-label study of duvelisib in patients with relapsed or refractory PTCL (n = 16) and CTCL (n = 19), along with in vitro and in vivo models of TCL. The overall response rates in patients with PTCL and CTCL were 50.0% and 31.6%, respectively (P = .32). There were 3 complete responses, all among patients with PTCL. Activity was seen across a wide spectrum of subtypes. The most frequently observed grade 3 and 4 adverse events were transaminase increases (40% alanine aminotransferase, 17% aspartate aminotransferase), maculopapular rash (17%), and neutropenia (17%). Responders and nonresponders had markedly different changes in serum cytokine profiles induced by duvelisib. In vitro, duvelisib potently killed 3 of 4 TCL lines with constitutive phospho-AKT (pAKT) vs 0 of 7 lines lacking pAKT (P = .024) and exceeded cell killing by the PI3K-δ–specific inhibitor idelalisib. Administration of duvelisib to mice engrafted with a PTCL patient-derived xenograft resulted in a shift among tumor-associated macrophages from the immunosuppressive M2-like phenotype to the inflammatory M1-like phenotype. In summary, duvelisib demonstrated promising clinical activity and an acceptable safety profile in relapsed/refractory TCL, as well as preclinical evidence of both tumor cell–autonomous and immune-mediated effects. This trial was registered at www.clinicaltrials.gov as #NCT01476657.
Visual Abstract
Introduction
T-cell lymphomas (TCLs) include >20 different non-Hodgkin lymphoma subtypes that are often divided into either cutaneous TCL (CTCL) or peripheral TCL (PTCL).1 The latter are associated with a very poor prognosis and include PTCL not otherwise specified (NOS), angioimmunoblastic TCL (AITL), anaplastic large-cell lymphoma (ALCL), hepatosplenic TCL, and others.2 Patients with newly diagnosed PTCL are most commonly treated with combination chemotherapy regimens such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) often followed by consolidation with high-dose chemotherapy and stem cell transplantation; however, the majority of patients either do not achieve a complete response (CR) or relapse after completion of therapy.3 Patients with relapsed PTCL have a dismal prognosis with a median overall survival (OS) of <6 months.4 Multiple agents have been approved by the US Food and Drug Administration for relapsed PTCL but have response rates <30% and median progression-free survival (PFS) <4 months.5-7 The anti-CD30 antibody drug conjugate brentuximab vedotin is an exception, with a very high response specific to ALCL, which universally expresses CD30 in all tumor cells.8
Many patients with CTCL follow a more indolent course and can be adequately managed with skin-directed therapy. Disease progression frequently necessitates systemic therapy, which is similarly inadequate as the available treatments for PTCL. Mycosis fungoides (MF) is the most common type of CTCL; commonly used systemic agents for this disease achieve responses in ∼30% to 60% of patients, although these responses are generally not durable.9-13 New and more active agents with distinct mechanisms of action are clearly needed for patients with TCL.
Phosphatidylinositol 3-kinase (PI3K) is a lipid kinase involved in intracellular signal transduction. Four catalytic subunits of PI3K exist in human cells (α, β, δ, and γ). The PI3K-δ and PI3K-γ isoforms are preferentially expressed in leukocytes and extensively modulate both innate and adaptive immune function.14-17 Multiple pathways mediated by PI3K-δ and/or PI3K-γ contribute to survival, proliferation, and differentiation of malignant hematopoietic cells through tumor cell–autonomous effects. At the same time, cancer cells can modulate the tumor microenvironment through juxta-, para-, and endocrine effects on nonmalignant stromal and immune cells that involve PI3K signaling.18-20
Recent studies have suggested that PI3K-γ may also suppress antitumor immune responses involving innate and adaptive effector cells. PI3K-γ signaling functions through C/EBPβ as a key inhibitor of phagocytosis by tumor-associated macrophages (TAMs).21 In this state, TAMs negatively regulate effector T and natural killer (NK) cells by secreting soluble immunosuppressive factors and expressing membrane-bound immune checkpoint molecules such as PD ligand 1 (PDL1).22 Selective inhibition of PI3K-γ in solid tumor models can induce an immunostimulatory transcriptional program and M1 macrophage phenotype that restores CD8+ T-cell activation.21,23
Thus, there are at least 3 different mechanisms through which PI3K-δ,γ inhibition could be active against lymphoid malignancies. The first involves blocking of mitogenic and survival signaling within the tumor cell (ie, cell autonomous). The second involves blocking of mitogenic and survival signaling induced by factors within the tumor microenvironment, including cytokines, chemokines, and juxtacrine interactions. Finally, inhibition of PI3K-δ, PI3K-γ, or both together could activate antilymphoma immune responses.
Duvelisib (also known as IPI-145) is an oral, dual inhibitor of PI3K-δ and PI3K-γ. In preclinical models of B-cell lymphomas and chronic lymphocytic leukemia, duvelisib had potent activity that was greater than inhibition of either isoform alone.24-30 The effects of duvelisib on T-cell malignancies remain completely unexplored. To begin defining the extent and mechanisms of duvelisib activity in these diseases, we analyzed both preclinical models and a cohort of patients with relapsed and refractory PTCL and CTCL treated in a larger phase 1 trial.
Methods
Eligibility criteria
A phase 1, open-label study was conducted at 8 study sites in the United States to evaluate the safety, pharmacokinetics, pharmacodynamics, and clinical activity of duvelisib in adult patients (≥18 years of age) with relapsed advanced hematologic malignancies. Patients with a diagnosis of mature cutaneous or noncutaneous TCL (excluding lymphoblastic lymphoma) who had progressed during, were refractory to, intolerant of, or ineligible for established therapy, or had a disease for which there is no established therapy were eligible to enroll in the TCL expansion cohort. There was no limit as to the maximum number of prior regimens. Key reasons for exclusion from the study included a diagnosis of overt leptomeningeal or central nervous system lymphoma; ongoing treatment with chronic immunosuppressive agents; inadequate hepatic function (aspartate aminotransferase [AST] or alanine transaminase [ALT] >2.5 × upper limit of normal [ULN]; bilirubin >1.5 × ULN); inadequate renal function (serum creatinine >1.5 × ULN); HIV infection; chronic hepatitis or other chronic liver disease. During the study, medications or foods known to be strong or moderate inhibitors or inducers of cytochrome P450 (CYP)3A were prohibited. Institutional review boards at each of the study sites approved the study protocol. This study was conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent prior to treatment.
Study treatment
The study (#NCT01476657; https://clinicaltrials.gov/ct2/show/NCT01476657) employed a dose-escalation scheme with disease-specific expansion cohorts at and below the maximum tolerated dose (MTD31). Patients with TCL were enrolled between June 2012 and January 2014, and 27 (77%) were treated at the MTD of oral duvelisib 75 mg twice daily on a 28-day cycle. The other 8 patients received 25 mg (n = 1), 50 mg (n = 1), 60 mg (n = 4), or 100 mg (n = 2) twice daily. Treatment was continuous until intolerance, progression, or discontinuation by physician or patient choice. The majority of patients were treated after a protocol amendment required standard prophylaxis for Pneumocystis jiroveci pneumonia. All patients with TCL who consented to this protocol amendment received Pneumocystis prophylaxis concomitant with duvelisib treatment. Herpes simplex virus (HSV) prophylaxis was recommended per protocol and was implemented at investigator discretion.
Efficacy and safety assessments
Efficacy was evaluated using computed tomography (CT) scans in TCL patients with radiologically measurable disease and by modified severity weighted assessment tool in CTCL patients at cycles 3, 5, and 7; every third cycle until cycle 19; and every sixth cycle thereafter until disease progression. 18-FDG–positron emission tomography (PET) scans were performed in patients with PET avid disease at cycle 1, day 22 and at the discretion of investigators. Response was determined by investigators using the International Working Group32 and International Society for Cutaneous Lymphomas consensus criteria33 in PTCL and CTCL, respectively.
To assess safety, adverse event (AE) monitoring and laboratory testing were performed at all clinic visits. Severity was graded using the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. AE relationship to duvelisib treatment was based on investigator assessment. Safety summaries include all patients who received at least 1 dose of duvelisib.
Proliferation, cell cycle, and apoptosis assays
Proliferation assays were performed in a 384-well format, using a JANUS Automated Workstation (PerkinElmer) and CellTiter-Glo Luminescent Cell Viability reagent (Promega) at 0 hours and 72 hours as previously described.34 Each data point represents quadruplicates, and experiments were repeated at least twice. Detection of apoptosis and cell cycle analysis were performed as previously described.35
Phosphoproteomics
TCL lines were seeded in triplicates of 107 cells in culture media. Either dimethyl sulfoxide or 1 µM duvelisib was added, and cells were incubated at 37°C for 24 hours. Cells were then washed twice in cold phosphate-buffered saline and pelleted at 300g for 5 minutes. Supernatants were aspirated and cell pellets snap frozen. The targeted phosphoproteomic assay P100 utilizes a set of 96 phosphopeptide probes to assess reduced-representation phosphosignatures measured by quantitative targeted mass spectrometry. Sample processing and analysis for P100 were performed as described.36
In vivo model
The patient-derived xenograft (PDX) model DFTL-7802437 was injected into 6 NSG mice (106 cells per mouse) via tail vein injection. Upon engraftment with >0.2% hCD45/hCD2+ cells in peripheral blood, mice were randomized and treatment was initiated with either duvelisib (10 mg/kg, daily, by mouth) or vehicle for 7 days (n = 3 per arm). Mice were euthanized, and spleens were harvested on day 8. Spleen cells were stained with hCD45 (clone HI30, V500; BD Bioscience), mouse F4/80 (Pacific Blue, clone BM8; Ebioscience), mouse CD206 (MMR) (phycoerythrin, Biolegend), mouse I-A/I-E (major histocompatibility complex [MHC] class II) (clone M5/114.15.2, PerCP/Cy5.5), and mouse CD11b (clone M1/70, allophycocyanin; Biolegend).
See supplemental Data (available on the Blood Web site) for addition information.
Results
Patient characteristics at baseline and study disposition
A dose-escalation, phase 1 clinical trial of duvelisib in patients with hematologic malignancies (#NCT01476657) identified 75 mg duvelisib twice daily as the MTD.31 We describe 35 patients with TCL enrolled in the dose-escalation phase or as part of a TCL-specific expansion cohort, of which 28 (77%) were treated at the MTD.
Patient demographics and characteristics at baseline are summarized in Table 1 for the 35 patients with TCL. The PTCL population was composed of 16 patients with PTCL-NOS (n = 6), AITL (n = 3), subcutaneous panniculitis-like TCL (SPTCL; n = 3), ALCL (n = 2), enteropathy-associated TCL (EATL; n = 1), and NKTCL (n = 1). Prior systemic therapies included CHOP or CHOP-like combinations (n = 13, 81%), other combination chemotherapy (n = 7, 48%), and single agents (eg, pralatrexate, brentuximab; n = 12, 75%). Prior treatment with histone deacetylase (HDAC) inhibitors was reported in 4 patients (25%). Two patients (12.5%) had undergone prior autologous stem cell transplantation and none had undergone prior allogeneic stem cell transplantation.
Table 1.
Patient demographics and baseline characteristics
| Demographics and baseline characteristics | PTCL (n = 16) | CTCL (n = 19) | All TCL (n = 35) |
|---|---|---|---|
| Demographics | |||
| Age (y), median (range) | 70 (34-86) | 64 (48-81) | 64 (34-86) |
| Sex, male, n (%) | 8 (50) | 8 (42) | 16 (46) |
| Baseline disease status | |||
| Baseline disease stage, n (%) | Stage IE: 1 (6) | Stage IB: 5 (26) | ≥Stage III: 25 (71) |
| Stage III: 5 (31) | Stage II, IIB: 4 (21) | ||
| Stage IV: 10 (63) | Stage III, IIIA: 3 (16) | ||
| Stage IV, IVA: 7 (37) | |||
| Eastern Cooperative Oncology Group score, 0 /1/2 | 6%/69%/25% | 21%/68%/11% | 14%/69%/17% |
| No. of prior systemic therapies, median (range) | 2.5 (1-7) | 6 (2-10) | 4 (1-10) |
| Years from initial diagnosis, median (range) | 2.9 (0.6-8.9) | 5.1 (0.4-24.9) | 4.1 (0.4-24.9) |
| Months from most recent systemic therapy, median (range) | 1.7 (0.4-24.8) | 0.8 (0.2-2.9) | 1.1 (0.2-24.8) |
| International Prognostic Index score (poor, 3-5 factors), n (%) | 10 (63) | 8 (42) | 18 (51) |
The CTCL population was composed of 19 patients with MF (n = 9), MF with large-cell transformation (n = 4), Sézary syndrome (n = 5), and primary cutaneous ALCL (n = 1). Prior systemic therapies taken by >50% included bexarotene (n = 18, 95%), HDAC inhibitors (n = 15, 79%), and other single agents (n = 14, 74%).
Efficacy and imaging correlates
In the PTCL population, the median duration of duvelisib treatment was 11.3 weeks (range: 1.9-95.6 weeks), with 50% receiving ≥4 cycles and 38% receiving ≥6 cycles. One patient with PTCL continues on therapy at >50 months. Three additional PTCL patients who discontinued treatment continued in follow-up at the time of data analysis. In the CTCL population, the median duration of duvelisib treatment was 12.6 weeks (range: 2-60 weeks), with 58% of patients treated with duvelisib for ≥4 cycles and 26% of patients treated for ≥6 cycles. The overall response rate (ORR) was 50.0% in the PTCL population and 31.6% in the CTCL population (P = .32 by 2-sided Fisher’s exact test; Table 2). Among PTCL patients (Figure 1A), there were 3 CRs and 5 PRs. All CRs occurred in <2 months (range: 1.8-1.9 months) and all PRs in <4 months (range: 1.6-3.5 months). Responses were seen across the spectrum of PTCL subtypes with CRs in patients with EATL, AITL, and PTCL-NOS, and PRs in those with SPTCL (n = 2), ALCL, AITL, and PTCL-NOS. The duration of response in the PTCL population ranged from 1.8 to 17.3 months with median PFS of 8.3 months and median OS of 8.4 months (supplemental Figure 1). Two patients who achieved CR and PR completed >12 months on treatment.
Table 2.
Summary of responses
| Response category | PTCL (n = 16) | CTCL (n = 19) |
|---|---|---|
| ORR (CR + PR) | ||
| ORR, n (%) | 8 (50.0) | 6 (31.6) |
| 95% Confidence interval (CI) | (24.7, 75.3) | (12.6, 56.6) |
| Best overall response, n (%) | ||
| CR | 3 (18.8) | 0 |
| PR | 5 (31.3) | 6 (31.6) |
| Stable disease | 1 (6.3) | 6 (31.6) |
| Progressive disease | 6 (37.5) | 6 (31.6) |
| Unknown | 1 (6.3) | 1 (5.3) |
| Time to response, median months (range) | 1.9 (1.6-3.5) | 2.4 (1.6-3.8) |
| PFS, median months (95% CI) | 8.3 (1.4, NR) | 4.5 (1.0-13.8) |
| OS, median months (95% CI) | 8.4 (4.3, NR) | NR (18.6, NR) |
The Clopper-Pearson CI is presented. Patients with no postbaseline response assessment are represented as having an “unknown” response. Percentages are based on the number of patients in each indication.
NR, not reached at the time of this analysis; PR, partial response.
Figure 1.

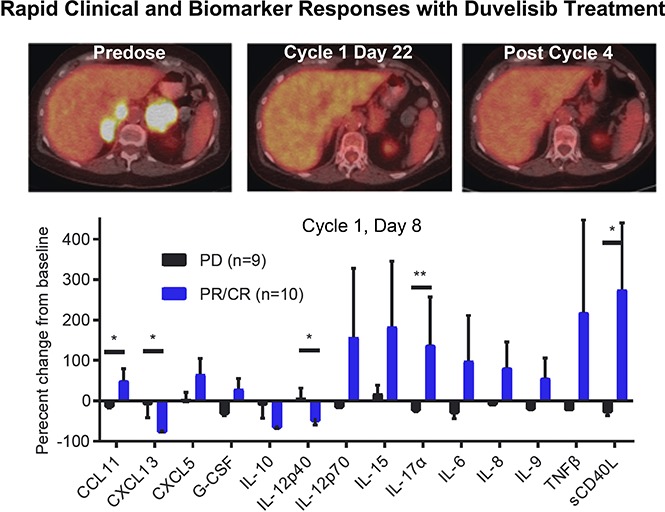
Results from the phase 1 trial of duvelisib among patients with TCL. (A) Plot of the best response and time on treatment in PTCL (left) and CTCL (right) patients. “Other” indicates ALCL (2 patients), EATL, and NK-TCL; LCT-MF, mycosis fungoides with large-cell transformation; SS, Sézary syndrome. (B) Pretreatment and posttreatment PET-CT imaging in a patient with PTCL and PR to duvelisib. (C) Serum factors measured at baseline and cycle 1 day 8 (C1D8) in patients treated with duvelisib and the change from baseline in cytokine levels were calculated and compared for the patient groups based on response. Error bars indicate standard deviation. *P < .05; **P < .01 by Mann-Whitney U test.
Among patients with CTCL (Figure 1A), all 6 PRs occurred within 4 months of beginning treatment (range: 1.6-3.8 months). Responses were observed in 1 patient with Sézary syndrome, 1 patient with stage IV large-cell transformation, and in 4 patients with MF (2 stage IB and 2 stage IIB), including 2 patients with folliculotropic MF. Duration of response ranged from 0.7 to 10.1 months. Median PFS was 4.5 months for patients with CTCL. At both 6 months and 12 months, the estimated probability of being progression-free for patients with CTCL was 26.5%. (supplemental Figure 1). The median OS was not reached with an estimated probability of survival of 89.5% at 6 months, 78.9% at 12 and 18 months, and 72.9% at 24 months (supplemental Figure 1).
PET-CT was examined on cycle 1, day 22 in 8 PTCL and 2 CTCL patients. All 3 CRs were confirmed by PET-CT. Of 6 patients with standardized uptake value reductions, 5 (83%) achieved a subsequent CR or PR on duvelisib, whereas all 4 patients without standardized uptake value reduction had disease progression (P = .048 by 2-sided Fisher’s exact test). The sequence of PET-CT imaging indicative of response is exemplified between predose, cycle 1, day 22, and after cycle 4 in a 75-year-old woman with relapsed AITL (Figure 1B).
Safety results
Reasons for treatment discontinuation in the PTCL population were progression of disease (n = 7), AEs (n = 6), death because of disease progression (n = 1), and death because of an unknown cause after the patient refused further treatment (n = 1). In the CTCL population, reasons for treatment discontinuation were progression of disease (n = 10), AEs (n = 7), death because of HSV pneumonia (n = 1), and voluntary withdrawal from the study (n = 1). At the time of this data analysis, 8 patients with CTCL, including 3 whose disease progressed, continued in follow-up. Treatment-emergent AEs (TEAEs) that occurred in ≥15% of PTCL or CTCL patients while receiving duvelisib are shown in Table 3. Treatment modifications, including interruptions and/or dose reductions, most commonly occurred in patients with ALT/AST increased (12 patients, 34%) and maculo-papular rash, diarrhea, and pyrexia (3 each, 9%). AEs that led to treatment discontinuation in >1 patient were ALT/AST increased in 4 (21%) CTCL patients and 2 (13%) PTCL patients and colitis in 1 (5%) CTCL and 1 (6%) PTCL patient.
Table 3.
TEAEs in ≥15% of patients
| TEAE | Total (N = 35) | ||
|---|---|---|---|
| All grades, n (%) | Grade 3, n (%) | Grade 4, n (%) | |
| Any AE | 34 (97) | 16 (46) | 8 (23) |
| Hematologic | |||
| Neutropenia | 7 (20) | 3 (9) | 3 (9) |
| Investigations | |||
| ALT/AST increased | 20 (57) | 11 (31) | 3 (9) |
| Blood alkaline phosphatase increased | 6 (17) | 0 | 0 |
| Blood creatinine increased | 6 (17) | 0 | 0 |
| Nonhematologic | |||
| Pyrexia | 13 (37) | 0 | 0 |
| Cough | 12 (34) | 0 | 0 |
| Diarrhea | 11 (31) | 0 | 0 |
| Fatigue | 11 (31) | 3 (9) | 0 |
| Nausea | 9 (26) | 0 | 0 |
| Weight decrease | 9 (26) | 0 | 0 |
| Headache | 8 (23) | 0 | 0 |
| Maculopapular rash | 8 (23) | 6 (17) | 0 |
| Chills | 6 (17) | 0 | 0 |
| Constipation | 6 (17) | 0 | 0 |
| Dyspnea | 6 (17) | 3 (9) | 1 (3) |
| Pneumonia* | 8 (23) | 5 (14) | 1 (3) |
| Wheezing | 6 (17) | 0 | 0 |
Includes all events of lung infections, viral and bacterial pneumonia, including 1 event of HSV pneumonia (grade 5).
New grade 3 or 4 hematologic laboratory abnormalities emerging in ≥10% of TCL patients while receiving duvelisib included decreased lymphocytes (13 patients, 37%) and neutrophils (10 patients, 29%). Lymphopenia was not reported as an AE. Severe infections of any type were observed in 10 (29%) patients, including 7 pneumonia/lung infections and 3 skin infections. There were no cases of P jiroveci pneumonia or other fungal infections among TCL patients.
Serious AEs were reported for 20 patients (57%), with the following events in ≥3 patients: pneumonias/lung infections (7 patients, 20%), colitis (4 patients, 11%), and pyrexia (3 patients, 9%). There were 3 on-treatment deaths, which included 2 PTCL patients whose disease progressed and 1 CTCL patient who died because of HSV pneumonia that was attributed to treatment by the investigator. The latter was a 77-year-old who received 9 prior treatment regimens over 17 years, including bexarotene, chemotherapy, interferon, total skin electron beam therapy, and denileukin diftitox. She did not receive HSV prophylaxis. A purulent left axillary soft tissue infection, culture-positive for Staphylococcus aureus and HSV, was reported concurrent with the lung infection. Lymphocyte counts were normal throughout duvelisib treatment with a grade 1 decrease noted at the time of the event.
Early biomarkers to predict response
We assessed serum factors as exploratory response biomarkers by evaluating the serum levels of 72 cytokines, chemokines, and matrix metalloproteinases at baseline and on cycle 1, day 8 in 19 patients. The evaluation group consisted of 9 patients with progressive disease (PD) on duvelisib and 10 with either a PR or CR. To avoid confounding effects that could result from slow disease progression, patients with stable disease were excluded from this analysis. There were marked differences in the serum factor changes noted between responders (PR or CR) and nonresponders (PD; Figure 1C). Most notably, responders had statistically significant increases in serum levels of the chemotactic cytokine CCL1, the proinflammatory cytokine interleukin 17α (IL-17α), and soluble CD40L (Figure 1C). At the same time, responders had statistically significant decreases in the IL-12 inhibitory subunit β (p40) and the chemokine CXCL13, which is known to be produced by follicular helper-like PTCLs including AITL (Figure 1C).
Duvelisib activity in TCL lines
To begin dissecting possible mechanisms for duvelisib activity, we quantified the expression of PI3K isoforms across a panel of 11 TCL lines representing 6 distinct subtypes. PI3K-α, PI3K-δ, and PI3K-γ were appreciably expressed in 7, 10, and all 11 lines, respectively (Figure 2A). Only 4 lines had constitutive phosphorylation of AKT (pAKT) at serine 473, an activating site phosphorylated by PI3K (Figure 2A).
Figure 2.

Duvelisib induces cell-autonomous and -nonautonomous effects in TCL cell lines. (A) Expression pattern of the p110 isoforms α, δ, and γ; AKT; and phosphorylation status of AKT at Ser473 across 11 T- and NK-cell lymphoma cell lines. (B) Example of GR50 and GRmax calculations for duvelisib in OCI-LY13.2 cells. The day 0 value is indicated by the green box. (C) GR50 and GRmax values for the 11 lines treated with the indicated agents. Missing data bars indicate GR50 >10 µM. (D) Annexin V/7-AAD staining after 48 hours incubation with dimethyl sulfoxide (DMSO) or 1 µM duvelisib.
Next, we assessed the isoform-specific dependences of PTCL cell line models using duvelisib, the PI3K-δ–specific inhibitor idelalisib (CAL-101), and the pan-PI3K inhibitor copanlisib (BAY 80-6946). We used the metrics GR50 (50% growth reduction compared with day 0) and GRmax (maximum effect compared with day 0) to assay response (Figure 2B), as these parameters are agnostic to differences in growth rate across cell lines.37 Unlike 50% inhibitory concentration, GRmax distinguishes cell killing from growth inhibition, where a value of 1 indicates no effect of drug compared with vehicle, 0 indicates no change compared with day 0 (ie, growth is equivalent to death), and a −1 indicates 100% death (ie, same as analyte with no cells).
Duvelisib potently induced cell death in 3 of 4 lines with constitutive pAKT compared with 0 of 7 lines that lack pAKT (P = .024 by 2-sided Fisher’s test; Figure 2A,C). Sensitivity was not dependent on TP53 as OCI-Ly13.2 is known to harbor TP53 loss.38 Duvelisib induced approximately twofold more killing than idelalisib or copanlisib in 2 of the 3 lines, as assessed by GRmax (Figure 2C). In line with the GRmax values, duvelisib induced apoptosis in all sensitive cell lines (Figure 2D). Among lines that lack pAKT, copanlisib had broad cytostatic activity (Figure 2C).
To further gain insights into the mechanism(s) of response and resistance to duvelisib, we used the targeted phosphoproteomic approach “P100.” This assay characterizes the effects of inhibitor treatment on representative phosphorylation targets that characterize common signaling pathways, including PI3K/mTOR. We established a phosphoproteomic signature by comparing duvelisib-induced changes in phosphorylation within 3 sensitive cell lines (OCI-Ly13.2, DERL-2, and Karpas-384) vs 3 resistant cell lines (SMZ-1, OCI-Ly12, and MAC-2A). This “duvelisib signature” was queried against the P100 database of signatures established by cataloging phosphoproteomic effects from treatment of diverse cell lines with a range of inhibitors (Figure 3A).36 The subsequent enrichment analysis identified a significant (false discovery rate < 0.1) positive enrichment of the duvelisib signature with perturbation sets that include PI3K and cyclin-dependent kinase inhibitors (CDKi’s; Figure 3B; supplemental Table 1), indicating the expected on-target effect of duvelisib. This was also consistent with immunoblotting, which showed decreases in phosphorylation of both AKT and GSK3β induced by duvelisib in OCI-Ly13.2 cells (Figure 3C).
Figure 3.

Phosphoproteomic signature of duvelisib. (A) Workflow of the P100 targeted phosphoproteomic assay and data analysis. (B) Perturbation set enrichment analysis. Each column represents the indicated class of compounds. Each horizontal line in a column represents a single compound from that class. The ranked positions of connectivity compared with the duvelisib-sensitive or -resistant profile is shown for each compound in a class. q-values were computed using the unweighted preranked GSEA algorithm. BRDi, bromodomain inhibitors; ddPKi, DNA-dependent protein kinase inhibitors; genKi, general kinase inhibitor; p450i, cytochrome p450 inhibitor; PI3Ki, PI3 kinase inhibitor; SIRT1a, SIRT1 activator. (C) Immunoblot of the duvelisib-sensitive cell line OCI-LY13.2 and duvelisib-resistant cell lines SMZ1 after treatment with 1 µM duvelisib for 6 hours and 72 hours. (D) Quantification of cell cycle phases by Hoechst33342 staining and flow cytometry. (E) Dose response matrix and isobologram of 8 TCL cells lines treated with duvelisib in combination with the HDAC inhibitor romidepsin.
As suggested by the enrichment of CDKi’s, duvelisib treatment resulted in dose- and time-dependent G1 cell cycle arrest in sensitive lines (Figure 3D). These p100 findings correlated with immunoblotting that showed a reduction of Cyclin D1 protein upon treatment of OCI-LY13.2 cells (Figure 3C). The strongest enrichment was for a class of general pleiotropic kinase inhibitors typified by staurosporine, which is frequently cytotoxic in cell-based assays. In contrast, perturbation sets defined by bromodomain inhibitors were enriched by the resistant profile (Figure 3B; supplemental Table 1); this suggests an epigenetic mechanism could mitigate the effects of PI3K inhibition in resistant lines. In support of this, there was synergistic activity between duvelisib and the HDAC inhibitor romidepsin in several TCL lines tested (Figure 3E) despite the complete lack of cytotoxic activity of single-agent duvelisib in multiple lines (Figure 2C).
Duvelisib induces macrophage repolarization in vivo
We previously reported a large repository of PDX of human leukemias and lymphomas established in NOD.Scid.IL2Rγ−/− (NSG) mice.36 Although these mice lack NK, T, and B cells, they have functional innate immunity, which allows for the study of macrophage and neutrophil responses.40 To assess the effects of duvelisib on macrophages within the in vivo PTCL microenvironment, we engrafted an AITL PDX that lacks constitutive pAKT (DFTL-78024) by tail-vein injection into mice (supplemental Figure 2).
Upon engraftment (>0.2% human CD45/CD2+ cells in peripheral blood), we randomized mice to receive duvelisib or vehicle for 7 days and then eutahnized the animals on day 8 (Figure 4A). Duvelisib treatment of 7 days resulted in a nonsignificant reduction of spleen size (Figure 4B). Although the total number of CD11b+/F4/80+ macrophages within involved spleens did not differ between mice receiving duvelisib and mice receiving vehicle (Figure 4C), polarization of these macrophages was dramatically affected. Specifically, duvelisib-treated spleens involved with human AITL had ∼50% fewer macrophages harboring an M2 (CD206+MHC class II−) phenotype and nearly 50% more macrophages harboring an M1 (MHC class II+CD206−) phenotype (Figure 4D). These changes in macrophage polarization are essentially identical to findings using exactly the same markers after treatment with a PI3K-γ–specific inhibitor in mouse models of melanoma and breast cancer.23 Of note, NSG mice that were not engrafted with AITL cells had very few M2 macrophages in the spleen, and treatment with duvelisib had no effect on either spleen size or macrophage polarization (supplemental Figure 2).
Figure 4.

Duvelisib changes TAM polarization in vivo. (A) Schematic of the in vivo experiment for macrophage polarization in the presence of an AITL PDX. (B) Spleen sizes and spleen weights of mice engrafted with DFTL-78024 and treated with vehicle or duvelisib. Quantification of total macrophages by F4/80 and CD11b staining (C) and macrophage polarization by CD206 and MHC-II staining (D) in vehicle- or duvelisib-treated animals. Statistics: unpaired Student t test.
Discussion
We report the phase 1 results of single-agent duvelisib among patients with TCLs. The safety profile was consistent with the overall study population.31 Severe transaminase elevations and colitis, which have also been described as potentially immune-mediated toxicities with the PI3K-δ–specific inhibitor idelalisib,41-43 were observed in this cohort and were the only events resulting in treatment discontinuation in >1 patient. Serious infections, including 1 fatal pneumonia, were reported. The majority of patients received Pneumocystis prophylaxis, and no cases of P jiroveci pneumonia occurred. Correlative analyses using pretreatment factors such as prior therapies or on-treatment factors such as lymphopenia, neutropenia, or T-cell subset alterations may be informative for characterizing infection risk in larger clinical trials of duvelisib, where the number of patients and events would optimally permit a robust investigation.
The ORRs, CR rates and PFS with duvelisib treatment were longer among patients with PTCL than those with CTCL. These differences did not achieve statistical significance, so their implications are unclear. A more robust characterization of differential efficacy for duvelisib within individual TCL subtypes will require larger clinical trials. Optimally, these trials will include biomarker assessments to help refine patient selection.
We undertook multiple approaches to develop such biomarkers for future trials of PI3K-δ/γ inhibition. First, we noted that baseline pAKT was present in all cell lines that were killed by duvelisib but none of the lines intrinsically resistant to this agent. Correlating patient tumor pAKT expression and duvelisib response would further inform the utility of pAKT (or a proteomic or transcriptional surrogate) as a predictive biomarker. Of note, transcriptional profiling of PTCLs has identified a poor-risk subset of PTCL-NOS that is characterized by GATA3 overexpression and significant enrichment of PI3K-associated pathway gene expression.44 Given the observations noted with cell line experiments, determining the extent to which GATA3-associated tumors align with constitutive pAKT may further characterize the potential activity of duvelisib in this particular TCL subset.
We assessed the effects of duvelisib on both serum factors within treated patients and macrophages within a PDX model. Consistent with a previously reported disease model using a PI3K-γ–specific inhibitor,23 a short course of duvelisib treatment can promote transition in tumor infiltration from M2 macrophages (tumor promoting) to M1 macrophages (immune stimulatory). The near absence of CD206+ M2 macrophages in unengrafted NSG mouse spleens suggests that human AITL either recruits these macrophages, repolarizes non-M2 macrophages in situ, or both, and that treatment with duvelisib reverts that phenotype. In patients, the M2 to M1 change could promote an immunostimulatory environment that activates effector T cells. This is consistent with the significant increase in IL-17α, which is secreted by activated T cells, among responders to duvelisib. It should be noted that the serum factor modulations on cycle 1, day 8 occur significantly prior to the documentation of CR (range: 1.8-1.9 months) or PR (range: 1.6-3.5 months), supporting the notion that cytokine changes may be an early indicator of response. Further validation is needed to determine whether the serum cytokine levels applied at an early time point during treatment with duvelisib can predict response with adequate predictive value.
Multiple potential mechanisms of resistance revealed by the phosphoproteomic analysis may inform rational combination strategies in the future. Based on the promising clinical activity and manageable safety profile of duvelisib in this single-agent trial and the evidence of in vitro synergy with romidepsin, we have initiated a phase 1 combination study of duvelisib with either romidepsin or bortezomib in patients with PTCL or CTCL (#NCT02783625). In this study, we have included a lead-in phase of single-agent duvelisib within a subset of patients on the combination trial solely to acquire serial biopsies. Analysis of these specimens will be essential for validating the serum cytokine effects, the cell line phosphoproteomics, and the murine macrophage reprogramming within treated patients. Therefore, we established a multicolor immunofluorescense assay that allows us to assess PI3K activation marks like p-AKT and markers of macrophage polarization. Additionally, a phase 2 study to further evaluate the clinical efficacy and safety of duvelisib monotherapy in relapsed and refractory PTCL patients is planned.
Finally, we have attempted to use cell line, in vivo xenograft, and clinical data to create a more comprehensive assessment of duvelisib activity and mechanisms. There are several limitations to our study, including the small number of patients treated, the limited utility of assessing drug response in cell lines, and the immunodeficient nature of NSG mice. However, we believe that future studies attempting to interrogate multiple platforms can provide novel insights that are otherwise not achievable by solely assessing clinical response.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the study investigators, coordinators, nurses, and patients and their families for their contributions; Barry Turnbull for statistical assistance and Veristat for combined term analysis; and Acumen Medical Communications for writing and editing assistance. John Welle and Jeffrey L. Friedman of Paginae Inc. provided graphical and editorial support, respectively.
This work was supported by Leukemia and Lymphoma Society Specialized Center of Research (#7011-16) and Deutsche Forschungsgemeinschaft (fellowship grant KO 4627/2-1) (R.K.) and by the National Institutes of Health, Library of Integrated Network-Based Cellular Signatures Program (U54 HG008097) (J.D.J.). Infinity Pharmaceuticals provided financial support for this study. This research was funded in part through the National Institutes of Health, National Cancer Institute Cancer Center (support grant P30 CA008748).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.M.H., R.K., and A.M. designed and performed research, analyzed data, and wrote the manuscript: P.P., Y.O., M.P., S.N.M., K.A., M.D., and P.M. designed and performed research; A.O., J.D.J., and F.M.F. designed and performed research and analyzed data; H.S., J.S., P.K., J.C.A., and V.K. designed research and analyzed data; D.W. analyzed data and wrote the manuscript; and D.M.W. designed research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: S.M.H. has received research funding/grant support from Celgene, Millennium Pharmaceuticals, Seattle Genetics, Spectrum Pharmaceuticals, and Infinity Pharmaceuticals, as well as consulting/honorarium from Celgene, Millennium Pharmaceuticals, Seattle Genetics, Infinity, and Spectrum Pharmaceuticals. D.M.W. has received research funding/grant support from Novartis, AbbVie, AstraZeneca, Aileron, Roche, and Infinity Pharmaceuticals, as well as consulting/honorarium from Novartis, Roche, Seattle Genetics, Dragonfly, and Infinity Pharmaceuticals. P.P. has received research funding/grant support from Celgene, Millennium Pharmaceuticals, Seattle Genetics, Galderma, Innate Pharma, and Infinity Pharmaceuticals. Y.O. declares having received honorarium from BMS and Takeda and research funding from Infinity Pharmaceuticals, Rhizen, and Curis. H.S., P.K., V.K., K.A., M.D., and J.S. are former employees of Infinity Pharmaceuticals. F.M.F. has received consulting fees and funding for a clinical study from Infinity Pharmaceuticals and has research funding from Celgene, Spectrum, and Millennium Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Steven M. Horwitz, Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10065; e-mail: horwitzs@mskcc.org; and David M. Weinstock, Dana-Farber Cancer Institute, 450 Brookline Ave, Dana 510B, Boston, MA 02130; e-mail: dweinstock@partners.org.
References
- 1.Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019-5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K. Peripheral T-cell lymphoma. Blood. 2011;117(25):6756-6767. [DOI] [PubMed] [Google Scholar]
- 3.Moskowitz AJ, Lunning MA, Horwitz SM. How I treat the peripheral T-cell lymphomas. Blood. 2014;123(17):2636-2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mak V, Hamm J, Chhanabhai M, et al. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: spectrum of disease and rare long-term survivors. J Clin Oncol. 2013;31(16):1970-1976. [DOI] [PubMed] [Google Scholar]
- 5.O’Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coiffier B, Pro B, Prince HM, et al. Romidepsin for the treatment of relapsed/refractory peripheral T-cell lymphoma: pivotal study update demonstrates durable responses. J Hematol Oncol. 2014;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol. 2012;30(18):2190-2196. [DOI] [PubMed] [Google Scholar]
- 9.Duvic M, Talpur R, Ni X, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109(1):31-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109-3115. [DOI] [PubMed] [Google Scholar]
- 11.Prince HM, Duvic M, Martin A, et al. Phase III placebo-controlled trial of denileukin diftitox for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(11):1870-1877. [DOI] [PubMed] [Google Scholar]
- 12.Whittaker SJ, Demierre MF, Kim EJ, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(29):4485-4491. [DOI] [PubMed] [Google Scholar]
- 13.Prince HM, Kim YH, Horwitz SM, et al. ; ALCANZA study group. Brentuximab vedotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): an international, open-label, randomised, phase 3, multicentre trial. Lancet. 2017;390(10094):555-566. [DOI] [PubMed] [Google Scholar]
- 14.Clayton E, Bardi G, Bell SE, et al. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med. 2002;196(6):753-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okkenhaug K, Bilancio A, Farjot G, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002;297(5583):1031-1034. [DOI] [PubMed] [Google Scholar]
- 16.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329-341. [DOI] [PubMed] [Google Scholar]
- 17.Fung-Leung WP. Phosphoinositide 3-kinase delta (PI3Kδ) in leukocyte signaling and function. Cell Signal. 2011;23(4):603-608. [DOI] [PubMed] [Google Scholar]
- 18.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66(2):605-612. [DOI] [PubMed] [Google Scholar]
- 19.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674. [DOI] [PubMed] [Google Scholar]
- 20.Schmid MC, Avraamides CJ, Dippold HC, et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kγ, a single convergent point promoting tumor inflammation and progression. Cancer Cell. 2011;19(6):715-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaneda MM, Messer KS, Ralainirina N, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539(7629):437-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27(4):462-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Henau O, Rausch M, Winkler D, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539(7629):443-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reif K, Okkenhaug K, Sasaki T, Penninger JM, Vanhaesebroeck B, Cyster JG. Cutting edge: differential roles for phosphoinositide 3-kinases, p110gamma and p110delta, in lymphocyte chemotaxis and homing. J Immunol. 2004;173(4):2236-2240. [DOI] [PubMed] [Google Scholar]
- 25.Winkler DG, Faia KL, DiNitto JP, et al. PI3K-δ and PI3K-γ inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem Biol. 2013;20(11):1364-1374. [DOI] [PubMed] [Google Scholar]
- 26.Faia K, White K, Proctor J, et al. High throughput in vitro combination sensitivity screen in hematologic malignancies with the phosphoinositide-3 kinase (PI3K-)δ,γ inhibitor, duvelisib [abstract]. J Clin Oncol. 2015. Abstract 8559. [Google Scholar]
- 27.Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603-3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peluso M, Faia K, Winkler D, et al. Duvelisib (IPI-145) inhibits malignant B-cell proliferation and disrupts signaling from the tumor microenvironment through mechanisms that are dependent on PI3K-delta and PI3K-gamma [abstract]. Blood. 2014;124(21):328.24894774 [Google Scholar]
- 29.Balakrishnan K, Peluso M, Fu M, et al. The phosphoinositide-3-kinase (PI3K)-delta and gamma inhibitor, IPI-145 (Duvelisib), overcomes signals from the PI3K/AKT/S6 pathway and promotes apoptosis in CLL. Leukemia. 2015;29(9):1811-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gunderson AJ, Kaneda MM, Tsujikawa T, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov. 2016;6(3):270-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flinn IW, O'Brien S, Kahl B, et al. Duvelisib, a novel oral dual inhibitor of PI3K-δ,γ, is clinically active in advanced hematologic malignancies. Blood. 2018;131(8):877-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheson BD, Pfistner B, Juweid ME, et al. ; International Harmonization Project on Lymphoma. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579-586. [DOI] [PubMed] [Google Scholar]
- 33.Olsen EA, Whittaker S, Kim YH, et al. ; Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. Clinical end points and response criteria in mycosis fungoides and Sézary syndrome: a consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol. 2011;29(18):2598-2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weigert O, Lane AA, Bird L, et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J Exp Med. 2012;209(2):259-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobson C, Kopp N, Layer JV, et al. HSP90 inhibition overcomes ibrutinib resistance in mantle cell lymphoma. Blood. 2016;128(21):2517-2526. [DOI] [PubMed] [Google Scholar]
- 36.Abelin JG, Patel J, Lu X, et al. Reduced-representation phosphosignatures measured by quantitative targeted MS capture cellular states and enable large-scale comparison of drug-induced phenotypes. Mol Cell Proteomics. 2016;15(5):1622-1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Townsend EC, Murakami MA, Christodoulou A, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice [published correction appears in Cancer Cell 2016;30(1):183]. Cancer Cell. 2016;29(4):574-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hafner M, Niepel M, Chung M, Sorger PK. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat Methods. 2016;13(6):521-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang H, Benchimol S, Minden MD, Messner HA. Alterations of p53 and c-myc in the clonal evolution of malignant lymphoma. Blood. 1994;83(2):452-459. [PubMed] [Google Scholar]
- 40.Aparicio S, Hidalgo M, Kung AL. Examining the utility of patient-derived xenograft mouse models. Nat Rev Cancer. 2015;15(5):311-316. [DOI] [PubMed] [Google Scholar]
- 41.Lampson BL, Kasar SN, Matos TR, et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood. 2016;128(2):195-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Louie CY, DiMaio MA, Matsukuma KE, Coutre SE, Berry GJ, Longacre TA. Idelalisib-associated enterocolitis: clinicopathologic features and distinction from other enterocolitides. Am J Surg Pathol. 2015;39(12):1653-1660. [DOI] [PubMed] [Google Scholar]
- 43.Weidner AS, Panarelli NC, Geyer JT, et al. Idelalisib-associated colitis: histologic findings in 14 patients. Am J Surg Pathol. 2015;39(12):1661-1667. [DOI] [PubMed] [Google Scholar]
- 44.Iqbal J, Wright G, Wang C, et al. ; Lymphoma Leukemia Molecular Profiling Project and the International Peripheral T-cell Lymphoma Project. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123(19):2915-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.