

Abstract
The discovery of powerful genetic targets has spurred clinical development of gene therapy approaches to treat patients with malignant brain tumors. However, lack of success in the clinic has been attributed to the inability of conventional gene vectors to achieve gene transfer throughout highly disseminated primary brain tumors. Here, we demonstrate ex vivo that small nanocomplexes composed of DNA condensed by a blend of biodegradable polymer, poly(β-amino ester) (PBAE), with PBAE conjugated with 5 kDa polyethylene glycol (PEG) molecules (PBAE-PEG) rapidly penetrate healthy brain parenchyma and orthotopic brain tumor tissues in rats. Rapid diffusion of these DNA-loaded nanocomplexes observed in fresh tissues ex vivo demonstrated that they avoided adhesive trapping in the brain owing to their dense PEG coating, which was critical to achieving widespread transgene expression throughout orthotopic rat brain tumors in vivo following administration by convection enhanced delivery. Transgene expression with the PBAE/PBAE-PEG blended nanocomplexes (DNA-loaded brain-penetrating nanocomplexes, or DNA-BPN) was uniform throughout the tumor core compared to nanocomplexes composed of DNA with PBAE only (DNA-loaded conventional nanocomplexes, or DNA-CN), and transgene expression reached beyond the tumor edge, where infiltrative cancer cells are found, only for the DNA-BPN formulation. Finally, DNA-BPN loaded with anti-cancer plasmid DNA provided significantly enhanced survival compared to the same plasmid DNA loaded in DNA-CN in two aggressive orthotopic brain tumor models in rats. These findings underscore the importance of achieving widespread delivery of therapeutic nucleic acids within brain tumors and provide a promising new delivery platform for localized gene therapy in the brain.
Keywords: Biodegradable polymer, DNA nanocomplex, Non-viral gene therapy, Tumor tissue barrier, Convection enhanced delivery
1. Introduction
Glioblastoma (GBM) is the most common and aggressive form of primary brain tumor [1]. GBM is characterized by a heterogeneous tumor microenvironment and diffuse tumor cell infiltration throughout the brain [2]. The standard of care is surgical resection followed by radiation and intensive chemotherapy, but this aggressive multimodal therapeutic regimen only marginally improves median survival time [3]. Further, the standard regimen is associated with poor patient life quality due to its toxic nature. Development of a well-tolerated and effective therapeutic option that may replace or complement the current frontline therapy is sorely needed.
Recent genetic association studies have unraveled linkages between specific genetic mutations and glioma sub-phenotypes, leading to the identification of common molecular pathways that are critical for the maintenance, progression and/or recurrence of brain tumors [4,5]. In addition, innovations in plasmid engineering and gene editing techniques [6] make it possible to have long-term and/or tumor-specific transgene expression. To this end, gene therapy has emerged as a potent therapeutic opportunity to potentially arrest or even reverse the progression of this devastating disease [7,8]. Encouragingly, intracranial administration of viral and non-viral gene vectors has yielded promising outcomes in several preclinical studies and has been well tolerated in Phase I clinical trials [9]. However, subsequent clinical trials have failed to demonstrate significant therapeutic benefits in humans. Inadequate efficacy has been primarily attributed to poor distribution of therapeutic transgene expression achieved with the gene delivery systems tested to date [7,10–13]. Of note, invasive cancer cells that migrate away from the bulk of the tumor mass into the healthy brain parenchyma are difficult to access, and thus, are primarily responsible for tumor relapse [14].
Convection-enhanced delivery (CED) provides a continuous pressure-driven flow of fluid into brain tissue from an implanted catheter. Thus, CED has been widely explored in an attempt to increase the penetration of therapeutics within the brain parenchyma [15]. CED of a retroviral replicating vector carrying a suicide transgene enhanced survival in an orthotopic rodent U87MG glioma model compared to an intratumoral bolus injection of the same vector [16]. However, most preclinical and clinical studies have shown that CED exerted limited effect on the distribution of common vectors [17–20] and, thus, provided marginal improvement at best in anti-tumor efficacy. These failures are likely due to the limited ability of conventional gene vectors to avoid steric and/or adhesive trapping [21,22] at the point of administration, which prevents them from spreading with the CED infusate through the brain extracellular space.
We have previously demonstrated that synthetic DNA nanocomplexes possessing small particle diameters (~50 nm) and non-adhesive surface coatings (requiring an exceptionally dense polyethylene glycol (PEG) corona) are capable of efficiently penetrating healthy rodent brain parenchyma ex vivo and in vivo[23,24]. However, whether these DNA-loaded brain penetrating nanocomplexes (DNA-BPN) can also efficiently spread within brain tumor tissue has yet to be determined. Compared to healthy brain parenchyma, brain tumor tissues possess a more heterogeneous microenvironment with varying cellular density and collagen content [25,26], as well as necrotic areas [27]. We formulated DNA-BPN composed of a blend of a promising biodegradable polymer, poly(β-amino ester) (PBAE) [23], with PEG conjugated to two terminal ends of PBAE (PBAE-PEG), and evaluated vector distribution and transgene expression in two aggressive orthotopic brain tumor models.
2. Results and discussion
2.1. Formulation and characterization of DNA-loaded nanocomplexes
Plasmid DNA were condensed either with PBAE (Fig. 1A) alone or a blend of PBAE with PBAE-PEG (Fig. 1B) to yield DNA-loaded conventional nanocomplexes (PBAE-CN) or brain-penetrating nanocomplexes (PBAE-BPN), respectively [23]. The hydrodynamic diameters of PBAE-CN and PBAE-BPN were 94 ± 5.8 nm and 52 ± 2.6 nm, respectively (Fig. 1C, Table S1). PBAE-BPN had near neutral surface charge (ζ-potential = −3.6 ± 0.4 mV) whereas PBAE-CN possessed highly positive surface charge (ζ-potential = 26.9 ± 1.1), thus confirming the dense PEG corona on PBAE-BPN that masked the cationic (i.e. amine-rich) PBAE core (Fig. 1D, Table S1). The size and morphology of PBAE-CN and PBAE-BPN were confirmed by transmission electron microscopy (Fig. 1E). We previously demonstrated that nanoparticles as large as 70 nm in diameter efficiently penetrated orthotopically-established 9L gliosarcoma (GS) if the particle surface was passivated with a dense PEG corona [28]. Surface plasmon resonance analysis has also revealed that such nanoparticles resist adhesion of brain extracellular matrix (ECM) molecules on their surfaces [29].
Fig. 1.
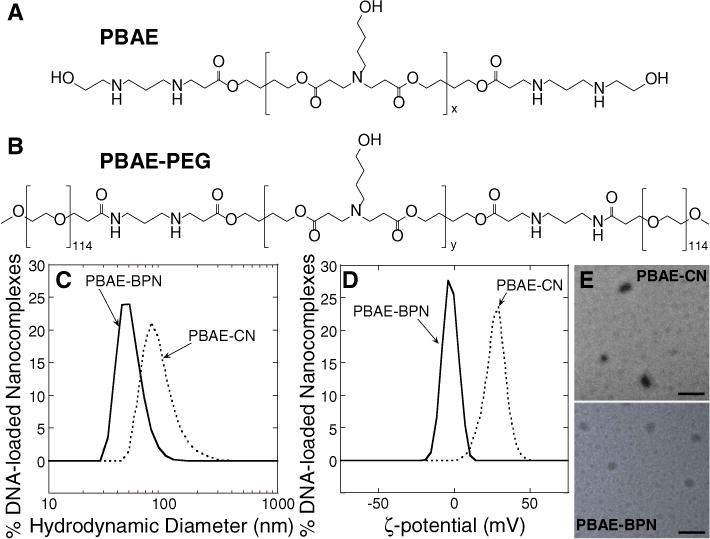
Chemical structures of polymers and physicochemical properties of DNA-loaded nanocomplexes. Chemical structures of (A) PBAE (x = 19–20) and (B) PBAE-PEG (y = 13–14) polymers. (C) Hydrodynamic diameters and (D) ζ-potentials of PBAE-CN and PBAE-BPN. (E) Representative transmission electron micrographs of PBAE-CN and PBAE-BPN. Scale bars = 200 nm.
Based on these findings, densely PEGylated PBAE-BPN were expected to diffuse within brain tumor tissue without being significantly hindered. However, physicochemical properties of nanoparticles are often altered in physiological environments [30]. We thus evaluated colloidal stability of PBAE-BPN and PBAE-CN in artificial cerebrospinal fluid (aCSF). While PBAE-CN instantaneously aggregated following the incubation in aCSF to 310 ± 35 nm, the size of PBAE-BPN remained unchanged in aCSF (53 ± 1.2 nm), suggesting that PBAE-BPN retained the physicochemical properties required for efficient brain penetration. This is in good agreement with our previous finding where a dense PEG coverage prevented polyethylenimine (PEI)- and poly-L-lysine (PLL)-based DNA-BPN from aggregating for up to 24 h in aCSF, whereas non-PEGylated and/or conventionally PEGylated (i.e. lower PEG density) counterparts lost colloidal stability shortly after the exposure to aCSF [24,31].
2.2. In vitro transfection efficiency of DNA-loaded nanocomplexes
We next assessed in vitro gene transfer efficacy of PBAE-CN and PBAE-BPN carrying plasmid DNA encoding luciferase protein. For comparison, transfection efficiencies of other conventional DNA-loaded nanocomplexes based on PEI (PEI-CN) and PLL (PLL-CN) were also evaluated. PEI is the most widely explored cationic polymer for gene delivery applications in vitro and in preclinical settings [32], including brain gene transfer studies [33,34], and has been tested in clinical trials for gene therapy of several cancers [35,36]. The PLL-CN used here is composed of DNA condensed into nanocomplexes using block copolymers of a 30-mer PLL connected to PEG via a cysteine residue; this DNA nanocomplex was well tolerated by cystic fibrosis patients [37] and demonstrated promising results in rodent brain [38,39].
Two different types of malignant rat gliomas, including F98 GBM and 9L GS cells, were selected to assess in vitro transfection efficiency and to establish orthotopic brain tumor models for subsequent ex vivo and in vivo studies. F98 GBM forms highly aggressive tumors that mimic many of the hallmarks of human GBM, including a highly invasive pattern of growth, overexpression of protein-based tumor markers and resistance to many chemotherapeutics and irradiation [40–42]. We also note that while being less invasive compared to F98-based models, orthotopic models established with 9L GS have been widely used in the preclinical setting to determine the potential of novel therapeutic approaches to be tested in clinical trials [43,44]. Indeed, clinical trials for Gliadel® [45–47] and OncoGel™ [48–50] were initiated based on pre-clinical evaluation in the 9L GS model.
We discovered that the in vitro transfection efficiencies of PBAE-BPN in F98 GBM and 9L GS cells, as well as differentiated mouse hippocampal neuronal cells (HT22), were significantly lower than those achieved using PBAE-CN (p < 0.05; Fig. S1). This result is in agreement with previous findings that PEGylation reduces in vitro transfection efficiency of non-viral gene delivery systems by reducing cellular uptake and/or intracellular processing [51,52]. However, despite the dense PEGylation, PBAE-BPN exhibited a similar or significantly higher in vitro transfection efficiency compared to non-PEGylated PEI-CN and conventionally PEGylated PLL-CN in rat brain tumors cells (Figs. S1A, B). This result is likely due to the biodegradable nature of PBAE that may facilitate intracellular release of plasmid DNA [53], unlike DNA-loaded nanocomplexes formulated with non-biodegradable polymers such as PEI. The transfection efficiency of PLL-CN was comparable to or only marginally improved from carrier-free plasmid DNA control (Fig. S1).
Importantly, in vitro transfection efficiency of PBAE-BPN was at least two orders of magnitude higher in rapidly dividing F98 GBM or 9L GS cells than in slowly dividing neuronal HT22 cells (Fig. S1). The differentiation state-dependent doubling rates may at least partially explain the differences in in vitro transfection efficiency. Mitosis transiently opens the nuclear membrane, a significant obstacle to intracellular gene delivery, thereby allowing nucleic acid payloads to enter the cell nucleus. However, it should be also noted that transfection efficiencies of gene delivery systems are highly variable depending on the type of cells being transfected, regardless of their differentiation states. In addition, it is possible that specific physicochemical properties of PBAE-BPN may have provided greater gene transfer to tumor cells over non-cancerous cells via a mechanism yet to be elucidated. Likewise, it has been previously shown that non-PEGylated DNA nanocomplexes formulated with a specific PBAE polymer provided preferential transgene expression in brain tumor-initiating cells compared to primary human neuronal progenitor cells [54,55].
2.3. Diffusion of DNA-loaded nanocomplexes in healthy brain tissue and in brain tumors ex vivo
We investigated whether the favorable physicochemical properties and colloidal stability of PBAE-BPN (Table S1) resulted in their rapid diffusion in brain tissues ex vivo. Using multiple particle tracking (MPT) [56], we first confirmed our previous finding [23] that PBAE-BPN efficiently penetrated the healthy rat brain parenchyma, whereas the diffusion-mediated penetration of PBAE-CN was significantly hindered (Fig. 2). We then repeated this study with brain tumor tissues that were orthotopically established with F98 GBM or 9L GS cells. Similar to our observation with healthy tissues, PBAE-BPN traversed up to a few microns within 20 s in both tumor tissue types ex vivo, whereas PBAE-CN exhibited highly confined trajectories (Fig. 2A). Overall, the median mean squared displacements (MSD; a measure of particle diffusion rate [56]) of PBAE-BPN at the timescale of 1 s were 33-fold, 10-fold, and 38-fold greater than those of PBAE-CN in healthy, F98 GBM, and 9L GS tissues, respectively (p < 0.05) (Fig. 2B). The higher diffusion rates of PBAE-BPN compared to PBAE-CN are most likely attributed to the non-adhesive surfaces on PBAE-BPN that prevent nanocomplex-tissue adhesive interactions [22,24,28], as well as the excellent colloidal stability that prevents PBAE-BPN from aggregating to sizes larger than the mesh spacings of brain tissues [23,24,57].
Fig. 2.
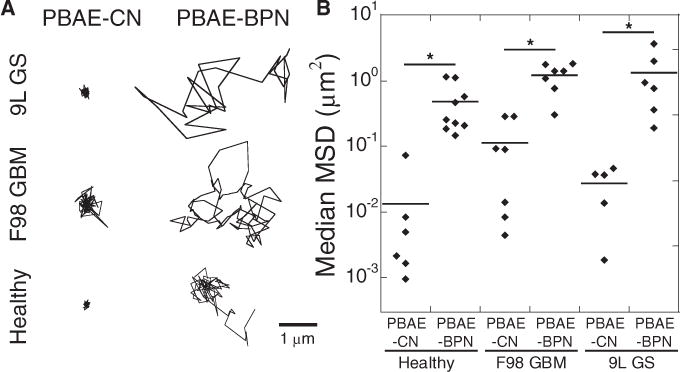
Ex vivo diffusion of DNA-loaded nanocomplexes in healthy and tumor-bearing rat brain tissues. (A) Representative nanocomplex trajectories (scale bar = 1 μm) and (B) median MSD dot plot of nanocomplexes in various rat brain tissues, including healthy brain, orthotopically established F98 GBM and 9L GS tumor tissues, with n ≥ 500 nanocomplexes tracked for each experiment. Individual dots indicate different brain tissue samples. Differences are statistically significant (*p < 0.05).
The difference in median MSD of PBAE-CN was not statistically significant in healthy tissues versus in two different tumor tissues, suggesting that the diffusion of PBAE-CN was similarly hindered in all three different tissues tested. Likewise, the difference of median MSD in healthy and two tumor tissues was not statistically significant for PBAE-BPN. This is in contrast to our previous finding that diffusion of densely PEGylated polystyrene (PS) beads as large as 100 nm in diameter is significantly greater in healthy rat brain parenchyma than in 9L GS tissues [28], presumably attributed to substantially higher cell density and greater collagen content in brain tumors than in normal brain tissues [25]. The discrepancy is likely due to the smaller particle sizes of PBAE-BPN (~50 nm) compared to the PEGylated PS beads used in the previous study. Although the pore size may be smaller in some tumor tissues compared to normal brain tissue, the degrees of steric obstruction experienced by non-adhesive nanoparticles are expected to be comparable in both tissues if the particle sizes are sufficiently smaller than the mesh spacings. In support of this hypothesis, smaller differences in diffusion rates in healthy brain versus 9L GS tissues were observed with 70 nm PEGylated PS compared to their 100 nm particle counterparts [28], implying that the difference may be even smaller for smaller (< 70 nm) PEGylated PS.
2.4. In vivo distribution and transgene expression of DNA-loaded nanocomplexes in healthy brain tissue and in brain tumors in rats
It is critical to confirm whether ex vivo diffusion behaviors of DNA-loaded nanocomplexes correlate with their distribution volume and subsequent transgene expression in vivo. We thus administered PBAE-CN and PBAE-BPN into healthy brain tissues or brain tumor tissues in rats via CED. To simultaneously monitor the DNA-loaded nanocomplex distribution and transgene expression, DNA-loaded nanocomplexes were formulated with Cy5-labeled plasmid DNA encoding green fluorescence protein (GFP). We first confirmed our previous finding that PBAE-BPN achieves greater distribution and volume of transgene expression in healthy rat brain compared to PBAE-CN (Figs. 3A, C). The distribution of both PBAE-BPN and PBAE-CN overlapped with the volumes of transgene expression, suggesting that at least a portion of cells within the tissues reached by each nanocomplex type were transfected. Importantly, we confirmed that there was no fluorescence bleed-through between two different color channels at the microscopic setup we employed. We next evaluated in vivo distribution of PBAE-BPN and PBAE-CN, as well as volume of transgene expression in orthotopic F98 rat tumors following CED. Similar to the observations with healthy rat brain tissues, PBAE-BPN exhibited greater distribution and volume of transgene expression in the tumor tissues compared to PBAE-CN (Figs. 3B, D). Quantitatively, the volume of transgene expression achieved by PBAE-BPN was 6- and 5-fold greater compared to PBAE-CN in healthy tissues and tumor rat brain tissues following CED, respectively (Fig. 3E).
Fig. 3.
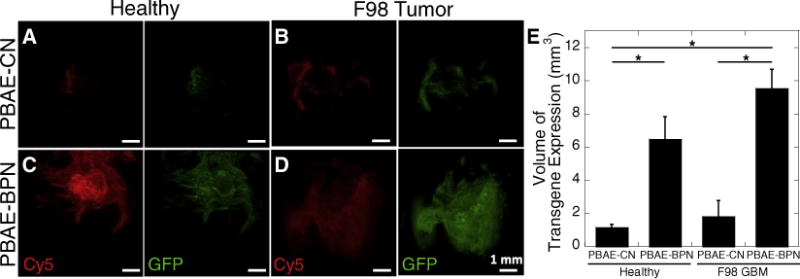
In vivo distribution of DNA-loaded nanocomplexes and transgene expression in healthy and tumor-bearing rat brain tissues following CED. Representative 3D reconstructed confocal images of (A, B) PBAE-CN and (C, D) PBAE-BPN in (A, C) healthy brain and (B, D) orthotopically established F98 GBM tumor tissues, showing distribution of nanocomplexes (Cy5; red) and transgene expression (GFP; green). Scale bars = 1 mm. (E) Image-based quantification of volume of GFP transgene expression in healthy rat brain and F98 tumor tissues. Data represents the mean ± SEM (n = 4–6 per group). Differences are statistically significant (*p < 0.05). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Although difference in overall volume of transgene expression mediated by PBAE-CN in healthy tissues versus tumor tissues was not statistically significant (Fig. 3E), the transgene expression appeared to span a greater inclusive area in tumors than in healthy brain tissue (Figs. 3A, B). This is most likely attributed to the heterogeneous microenvironment of the F98 GBM tissue, specifically the necrotic regions through which PBAE-CN distributed relatively unhindered (Fig. S2A). Transgene expression was also apparent near necrotic regions following CED of PBAE-BPN (Fig. S2B). However, while the transgene expression mediated by PBAE-CN was confined to the close vicinity of necrotic regions (Fig. S2A), PBAE-BPN provided widespread distribution of transgene expression far away from those regions (Fig. S2B). The transgene expression mediated by PBAE-BPN was found even beyond the edge of the tumor tissue (Fig. S2B), suggesting that cells at the infiltrative tumor edge that are responsible for tumor recurrence [14] may be targeted by PBAE-BPN. In accordance with ex vivo diffusion behavior (Fig. 2B), the volume of in vivo distribution and transgene expression following CED of PBAE-BPN was similar in healthy and tumor tissues (Fig. 3E). This finding suggests that PBAE-BPN possess particle diameters smaller than the average pore size in the ECM of either tissue type and that they do not adhere strongly to the ECM in either.
We next evaluated in vivo uptake by, and subsequent transgene expression in, individual cells following CED of PBAE-CN or PBAE-BPN. Consistent with findings in the whole tissue-based study (Fig. 3), PBAE-BPN exhibited greater cellular uptake and transgene expression, as compared to PBAE-CN, in both healthy brain cells (Figs. 4A, B) and orthotopic F98 GBM cells (Figs. 4C, D). PBAE-CN were found in 20% or less of the cells within healthy brain tissue and tumor tissue. In contrast, PBAE-BPN were found in 50% or more of cells in healthy brain tissue and in tumor tissue in the brain (Fig. 4E). The percentage of cells with reporter GFP transgene expression tightly correlated with the cellular uptake data (Fig. 4F). We also quantified the overall levels of in vivo transgene expression following CED of unlabeled PBAE-BPN and PBAE-CN carrying plasmid DNA encoding GFP, using a western blot analysis. We found that PBAE-BPN provided significantly greater overall GFP expression compared to PBAE-CN in both healthy brain and brain tumor tissue (Fig. 4G).
Fig. 4.
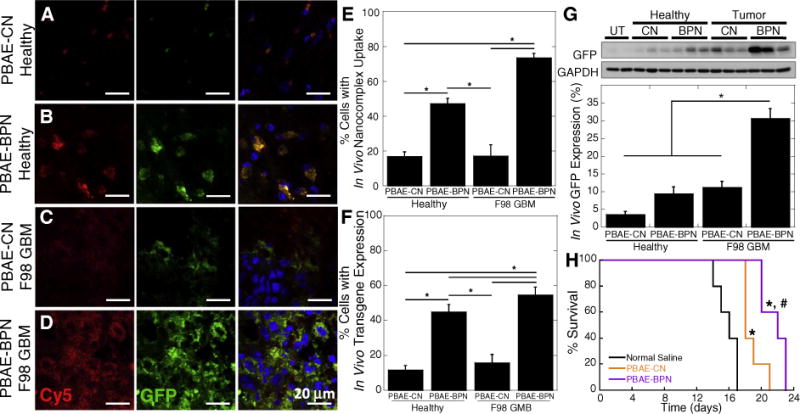
In vivo cellular uptake, transgene expression and therapeutic effect in healthy and/or F98 GBM-bearing rat brain tissues following CED of DNA-loaded nanocomplexes. Representative fluorescence microscopic images of (A, C) PBAE-CN and (B, D) PBAE-BPN in (A, B) healthy brain and (C, D) orthotopically established F98 GBM tumor tissues, showing cellular uptake of nanocomplexes (Cy5; red) and transgene expression (GFP; green). The DAPI (blue) staining represents cell nuclei. Scale bars = 20 μm. Image-based quantification of cells with (E) nanocomplex uptake and (F) GFP transgene expression. Data represents mean ± SEM (n = 4). Differences are statistically significant (*p < 0.001) (G) Western blot-based quantification of overall GFP transgene expression normalized to the untreated (UT) control. Data represents the mean ± SEM (n = 8). Difference is statistically significant (*p < 0.01). (H) Kaplan-Meier survival curve of an orthotopic F98 GBM model following CED of nanocomplexes carrying pTK or normal saline and intraperitoneal GCV treatments (n = 5 per group). Differences are statistically significant compared to groups treated with normal saline (*p < 0.01) and PBAE-CN (#p < 0.05). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
PBAE-BPN provided greater in vivo gene transfer efficacy in orthotopic F98 GBM than in healthy rat brain (Figs. 4F, G), which was consistent with the in vitro observations (Fig. S1). This finding may suggest that PBAE-BPN provide widespread yet cancer-favoring transgene expression following CED. The ability to preferentially transfect cancer cells over normal cells in the brain may improve the safety profile of cancer gene therapy, particularly if the gene therapy is de-signed to produce a toxic product.
2.5. Anti-cancer efficacy of DNA-loaded nanocomplexes
Based on the widespread reporter transgene expression achieved in vivo (Figs. 3, 4), we sought to investigate whether CED of PBAE-BPN may provide enhanced therapeutic efficacy in highly aggressive orthotopic models of rat brain tumors based on F98 GBM or 9L GS. We started by evaluating the ability of widely explored anti-cancer therapeutic genes, including plasmid DNA constructs encoding either the suicide herpes simplex virus type-1 thymidine kinase protein (HSV-tk) [13,58–60] or the tumor suppressor p53 protein [61], to mediate the killing of cancer cells in vitro. HSV-tk converts a systemically administered prodrug, ganciclovir (GCV), into a metabolite called deoxyguanosine monophosphate, which is further phosphorylated within the cell to a highly toxic nucleotide analog [62]. The cancer cells expressing HSV-tk are capable of inducing the death of neighboring untransfected cancer cells via a bystander effect; the mechanism involves transfer of toxic metabolites of GCV through intercellular gap junctions [63]. We found that both F98 GBM and 9L GS cells were highly sensitive to the HSV-tk suicide gene therapy followed by GCV treatment (Figs. S3A, B). In contrast, p53 gene therapy effectively reduced the viability of 9L GS cells, but not F98 GBM cells (Figs. S3C, D). This is likely due to the presence of wild-type p53 in F98 GBM [64], whereas p53 is mutated in 9L GS [40].
We first tested in vivo anti-cancer efficacy following the CED of PBAE-BPN carrying plasmid DNA encoding HSV-tk and subsequent intraperitoneal GCV administration. For this study, we used the F98 GBM orthotopic model since the bystander effect in 9L GS models may involve an anti-tumor immune response [40]. HSV-tk plasmid DNA delivered by PBAE-CN (p < 0.01) and PBAE-BPN (p < 0.01) each provided a significantly improved survival compared to untreated controls; however, the improvement in survival was significantly greater with PBAE-BPN than with PBAE-CN (p < 0.05) (Fig. 4H).
We next investigated whether the in vitro tumor killing effect mediated by p53 gene transfer in 9L GS cells (Fig. S3D) may translate to in vivo anti-cancer efficacy using a 9L GS orthotopic rat brain tumor model. We first confirmed that PBAE-BPN provided widespread distribution throughout the orthotopic 9L GS tissue beyond the tumor edge, whereas the distribution of co-infused PBAE-CN was confined to the vicinity of administration site (p < 0.01; Figs. 5A, B). Accordingly, PBAE-BPN carrying plasmid DNA encoding wild-type p53 exhibited more widespread transgene expression compared to the dose-matched PBAE-CN carrying the identical plasmid DNA, as determined by immunohistochemistry (Fig. 5C). Similar to the study with F98 GBM model (Fig. 4H), we found that CED of p53 plasmid DNA delivered via PBAE-CN (p < 0.01) or PBAE-BPN (p < 0.00001) resulted in enhanced survival in an orthotopic model of 9L GS rat brain tumors compared to untreated controls (Fig. 5D). Again, PBAE-BPN provided improved anti-cancer efficacy, in terms of survival, as compared to PBAE-CN (p < 0.01), suggesting that the superior distribution pattern and/or overall level of transgene expression plays a significant role in therapeutic outcomes.
Fig. 5.
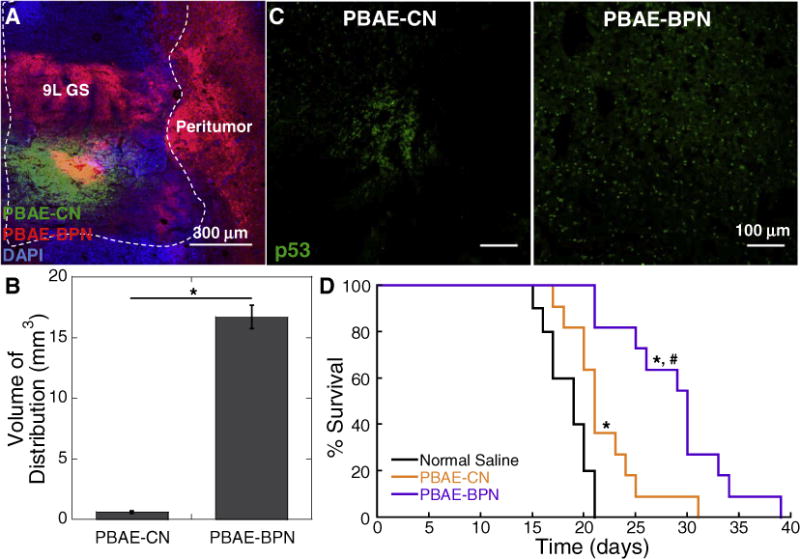
In vivo distribution, transgene expression and therapeutic effect in 9L GS-bearing rat brain tissues following CED of DNA-loaded nanocomplexes. (A) Representative image showing the distribution of PBAE-CN (Cy3; green) and PBAE-BPN (Cy5; red) in the 9L GS-bearing rat brain. DAPI (blue) staining indicates cell nuclei and co-localization of PBAE-CN and PBAE-BPN is depicted in yellow. Dashed line delineates the boundary between healthy and 9L GS tumor tissues. Scale bar = 300 μm. (B) Image-based quantification of volume of nanocomplex distribution in 9L GS-bearing rat brain tissues. Data represents the mean ± SEM (n = 3 per group). Difference is statistically significant (*p < 0.01). (C) Representative fluorescence images demonstrating p53 transgene expression mediated by CED of PBAE-CN (left) and PBAE-BPN (right). Scale bar = 100 μm. (D) Kaplan-Meier survival curve of an orthotopic 9L GS model following CED of nanocomplexes carrying plasmid DNA encoding p53 (i.e. pC53-SN3) or normal saline (n = 10–11 per group). Differences are statistically significant compared to groups treated with normal saline (*p < 0.01) and PBAE-CN (#p < 0.00001). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
In a recent next generation sequencing study exploring genetic variations in gliomas from 121 different patients, p53 mutation was found in approximately 40% and 80% of the entire and recurrent GBM cases, respectively [65]. Importantly, human high-grade gliomas with p53 mutation were found less responsive to 28 different chemotherapeutics, including carmustine—the active pharmaceutical ingredient in Gliadel®—than gliomas with wild-type p53 [66]. Likewise, p53 mutation has been found to be associated with resistance to temozolomide [67], the standard of care chemotherapeutic agent used in glioma patients. Thus, widespread transgene expression of wild-type p53 throughout the tumor tissue, achieved by CED of PBAE-BPN, may enhance the therapeutic efficacy of current chemotherapies or increase the repertoire of effective chemotherapeutics previously dropped due to the generation of drug resistance.
3. Conclusion
We have demonstrated the benefit of biodegradable DNA-loaded nanocomplexes engineered to achieve widespread and high-level transgene expression in brain tumors in vivo (i.e. PBAE-BPN), leading to enhanced survival in two different models of rats bearing aggressive orthotopic brain tumors. Specifically, we show that anti-cancer genes delivered via PBAE-BPN offer significantly improved survival of brain tumor-bearing rats compared to rats treated with conventional DNA-loaded nanocomplexes that are unable to efficiently penetrate brain tumor tissues. These findings suggest that systems that provide more widespread transgene expression in the brain may be more effective in gene therapy-based treatment of brain tumors. Further validation in human-derived brain tumors in animals and, eventually, human clinical trials will be needed to fully determine the utility of the approach described. Importantly, the PBAE-BPN platform can be used to deliver any gene and, thus, is synergistic with efforts geared to identify powerful new genetic targets as well as with efforts to create plasmid DNA that provide long-term gene expression and/or tumor-specific transgene expression.
4. Materials and methods
4.1. Materials and reagents
The reagents were purchased from different companies as follows: 1,4-butanediol diacrylate and 4-amino-1-butanol from Alfa Aesar (Ward Hill, MA); 1,3-diaminopropane (C1) from Sigma-Aldrich (St. Louis, MO); 2-(3-aminopropylamino)ethanol (C5) from Oakwood Chemical (West Columbia, SC); methoxy-PEG-succinimidyl succinate from JenKem Technology (Plano, TX). Solvents were purchased as follows: anhydrous ethyl ether from Fischer Scientific (Pittsburgh, PA); dimethyl sulfoxide anhydrous (DMSO) and tetrahydrofuran (THF) from Sigma-Aldrich.
4.1.1. Polymer synthesis
PBAE polymers (P1) were synthesized using a two-step Michael addition reaction as previously described [23,54,57,68–73]. Briefly, 1,4-butanediol diacrylate and 4-amino-1-butanol were reacted at molar ratios of 1.1:1 and 1.2:1 for 24 h at 90 °C to yield P1 of 6 kDa and 4 kDa, respectively. The resulting polymers were precipitated using cold ether, dried under vacuum and lyophilized. The molecular weight (MW) was estimated using nuclear magnetic resonance (NMR) and gel permeation chromatography (GPC). Subsequently, 6 kDa P1 was dissolved in THF at 100 mg/ml and 30 molar excess of a selected capping group (C5; [23]) was added. The reaction was undertaken while stirring for 4–5 h at room temperature. The end capped PBAE polymer (P1C5) was crashed out in cold ether, and subsequently dried under vacuum and lyophilization. To synthesize PEGylated PBAE polymers (i.e. PBAE-PEG), 4 kDa P1 was similarly capped with C1, and the resulting P1C1 and 2.05 molar equivalents of 5 kDa methoxy-PEG-succinimidyl succinate were transferred to a glass viral, vacuumed and purged with nitrogen. The mixture of reactants was dissolved in THF and reacted at room temperature for 16 h. The PBAE-PEG polymers were washed with cold ether and dried under vacuum. PEG conjugation was confirmed by NMR. All Polymers were dissolved in DMSO at 100 mg/ml and stored at −20 °C for further use.
4.2. Nuclear magnetic resonance
1H NMR spectra of the polymer intermediates and final products dissolved in deuterated methanol (MeOH-d4; Cambridge Iso. Lab., Inc., Andover, MA) were recorded with a Bruker spectrometer (500 MHz; Billerica, MA). The MeOH peak was used as an internal standard and 1H chemical shifts are reported in ppm (δ). Data were processed using the MestRe-C software (Mestrelab Research, Escondido, CA).
4.3. Gel permeation chromatography
Polymer MW was also measured by GPC using a Waters Breeze System and three Styragel Columns (7.8 × 300 mm) in series HR 1, HR 3, and HR 4 (Waters, Milford, MA). A solution of 95% THF, 5% DMSO and 0.1 M piperidine was used as a mobile phase and the samples were eluted at a flow rate of 1 ml/min.
4.4. DNA-loaded nanocomplex formulation and characterization
A plasmid DNA encoding enhanced GFP (pEGFP) and HSV-tk (pSELECT-zeo-HSV1tk; pTK) were purchased from Clontech Laboratories Inc. (Mountainview, CA) and InvivoGen (San Diego, CA), respectively. The plasmid DNA encoding luciferase (pd1GL3-RL) and p53 (pC53-SN3; [74]) were provided by Drs. Alexander M. Klibanov (M.I.T.) and Bert Vogelstein (Johns Hopkins University), respectively. The gene expression of the aforementioned plasmid DNA is regulated by a cytomegalovirus promoter. Fluorescent labeling of plasmid DNA with Cy3 or Cy5 fluorophores was performed using the Mirus Label IT® Tracker™ Intracellular Nucleic Acid Localization Kit (Mirus Bio, Madison, WI). Transformation of plasmid DNA into E. coli DH5α competent bacterial cells were conducted using a heat shock method. Following the bacterial expansion in LB media, plasmid DNA was purified using EndoFree Plasmid Giga Kit (QIAGEN, Valencia, CA), as per manufacturer’s protocol.
Non-PEGylated PBAE-CN was formulated as previously reported [23,54,57,68,69,72,75]. PBAE-BPN were formulated using a protocol we have recently established [23]. Briefly, 5 volumes of labeled or unlabeled plasmid DNA (0.1 mg/ml) were added drop-wise to 1 volume of polymer solution, consisting of a mixture of PBAE and PBAE-PEG at a w/w ratio of 2:3 based on PBAE mass and at a PBAE to plasmid DNA ratio of 60:1 w/w. Using a 0.1 M hydrochloric acid solution, both the DNA and the polymer solutions were adjusted to pH 6.0. Following formulation, DNA-loaded nanocomplexes were washed with 3 volumes of ultrapure water, and concentrated to 1 mg/ml of plasmid DNA using Amicon® Ultra Centrifugal Filters (100,000 MWCO, Millipore Corp., Billerica, MA) as measured using the Quant-iT™ PicoGreen® dsDNA Assay Kit (Life Technologies, Grand Island, NY). PEI-CN and PLL-CN were formulated as previously described [21,37,76].
The hydrodynamic diameters as well as polydispersity index (PDI) and ζ-potentials of DNA-loaded nanocomplexes were measured by dynamic light scattering (DLS; in ultrapure water) and laser Doppler anemometry (in 10 mM NaCl solution at pH 7.0), respectively, using a Nanosizer ZS90 (Malvern Instruments, Southborough, MA). The size and morphology of DNA-loaded nanocomplexes were also confirmed by transmission electron microscopy (Hitachi H7600, Japan). Nanocomplex stability was assessed by monitoring the change in hydrodynamic diameters and PDI in aCSF (Harvard Apparatus, Holliston, MA) at 37 °C.
4.5. Cell culture
Rat tumor cells, including F98 GBM and 9L GS, and mouse hippocampal neuronal cells (i.e. HT22) were provided by Prof. Betty Tyler and Dr. Seulki Lee, respectively. Cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen Corp., Carlsbad, CA) supplemented with 10% heat inactivated fetal bovine serum (Invitrogen Corp.) and 1% penicillin/streptomycin (Invitrogen Corp.). Cells at 70–80% confluence were re-seeded onto 24- and 96-well plates to evaluate transgene expression and cytotoxicity in vitro, respectively.
4.6. In vitro transfection
F98 GBM, 9L GS or HT22 cells were seeded at an initial density of 5.0 × 104 cells/well onto 24-well plates and incubated at 37 °C for 24 h. Subsequently, different types of DNA-loaded nanocomplexes carrying 1 μg pd1GL3-RL plasmid DNA were added at a plasmid DNA concentration of 1 μg/well. After 5 h of incubation, the culture media were replaced with fresh media. After 48 h of additional incubation, media was removed and 0.5 ml of 1× Reporter Lysis Buffer (Promega, Madison, WI) was added. Cells were subjected to three freeze-and-thaw cycles to achieve complete lysis. The luciferase activity in the relative light units (RLU) was measured using a standard Luciferase Assay Kit (Promega) and a 20/20n luminometer (Turner Biosystems, Sunnyvale, CA). The RLU were normalized to the total protein amount measured by a Bicinchoninic Acid (BCA) Protein Assay Kit (Thermo scientific, Rockford, IL).
4.7. In vitro cell viability
F98 GBM and 9L GS cells were seeded at an initial density of 5.0 × 103 cells/well onto 96-well plates and incubated at 37 °C for 24 h. To assess the combined cytotoxic effect of pTK and GCV, we treated the tumor cells with PBAE-CN carrying a pTK plasmid DNA at 0.2 μg DNA/well and incubated for 24 h. Subsequently, the culture media were replaced with fresh media with different concentrations of GCV ranging from 10−4 to 102 μg/ml and the cells were incubated for additional 24 h. Cells treated solely with different concentrations of GCV but without PBAE-CN carrying pTK served as a control. Likewise, to assess the tumor suppressive effect of p53, we treated the tumor cells with PBAE-CN carrying pC53-SN3 at 0.2 μg plasmid DNA/well. For both experiments, cell viability was assessed using the Dojindo Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc., Rockville, MD) 48 h after the addition of PBAE-CN. Absorbance at 450 nm was measured spectrophotometrically using the Synergy Mx Multi-Mode Microplate Reader (Biotek, Instruments Inc. Winooski, VT), and the cell viability was normalized to untreated cells.
4.8. Animal studies
We used female Fischer 344 rats (6–8 weeks old; Harlan Laboratories, Frederick, MD) for in vivo experiments. Inbred rats were chosen over outbred strains due to the impact that genetic variation may have on transgene expression [77]. Animals were treated in accordance with the guidelines and policies of the Johns Hopkins University Animal Care and Use Committee. Surgical procedures were performed using standard sterile surgical techniques. Animals were anesthetized using a mixture of ketamine (75 mg/kg) and xylazine (7.5 mg/kg), as previously described [78]. A midline scalp incision was made to expose the coronal and sagittal sutures and a burr whole was drilled 3 mm lateral to the sagittal suture and 0.5 mm posterior to the bregma. Following the administration of the tumor cell suspension or DNA-loaded nanocomplex solution, the skin was closed using biodegradable sutures (Polysorb™ Braided Absorbable Sutures 5–0) and bacitracin was applied.
4.9. Orthotopic tumor inoculation
Orthotopic tumor cell inoculation was performed as previously described [78]. Briefly, 1 × 105 F98 GBM or 9L GS cells in 10 μl of phosphate buffer saline were administered over 5 min at a depth of 3.5 mm using a Hamilton Neuro Syringe (Hamilton, Reno, NV) mounted on a small animal stereotactic frame (Stoelting, Wood Dale, Il). To establish brain tumor models mimicking high-grade gliomas characterized by rapid tumor growth rates, we orthotopically inoculated rat brains with the maximum numbers of tumor cells, similar to the calculations used as the basis of clinical translation [48,49].
4.10. Multiple particle tracking
MPT was used to measure MSD of fluorescently labeled DNA-loaded nanocomplexes ex vivo in normal rat brain slices or orthotopically established F98 tumor slices as previously reported [22]. Briefly, rat brains were harvested from healthy (8–10 weeks) or F98 GBM-bearing rats (10 days after tumor inoculation), incubated in aCSF for 10 min and sliced using a Zivic brain matrix slicer (Zivic Instruments, Pittsburgh, PA). The resulting 1.5 mm coronal slices were placed on custom-made slides. Subsequently, we injected 0.5 μl of fluorescently labeled nanocomplex solution into the cerebral cortex at a depth of 1 mm using a 50 μl Hamilton Neuro Syringe (Hamilton, Reno, NV) mounted on a stereotactic frame. Nanocomplex trajectories were recorded over 20 s at an exposure time of 66.7 ms by an Evolve 512 EMCCD camera (Photometrics, Tucson, AZ) mounted on an inverted epifluorescence microscope (Axio Observer D1; Carl Zeiss, Hertfordshire, UK) equipped with a 100×/1.46 NA oil-immersion objective. Movies were analyzed with a custom-made automated particle tracking MATLAB script to extract x, y-coordinates of nanocomplex centroids over time and calculate the MSD of individual nanocomplexes as a function of timescale [56]. Median MSD was determined based on the measured MSD of individual nanocomplexes; individual MSD values were not ensemble-averaged given their inherently heterogeneous, non-Gaussian distribution [56,79].
4.11. Convection enhanced delivery
The CED into healthy rat striatum was performed as previously described [23,24]. Briefly, a 33 gauge 50 μl Hamilton Neuro Syringe was mounted onto Chemyx Nanojet Injector Module (Chemyx, Stafford, TX), which was held on a small animal stereotactic frame. The catheter was lowered to a depth of 3.5 mm and a 20 μl solution of DNA-loaded nanocomplexes at a 1 mg/ml plasmid DNA concentration in normal saline was administered at a rate of 0.33 μl/min. The CED into the orthotopically established F98 GBM and 9L GS was similarly conducted, but the catheter was lowered at a depth of 2.5 mm for targeting the tumor core.
4.12. Imaging analysis
To assess the distributions of locally infused DNA-loaded nanocomplexes and transgene expression, we administered a solution of PBAE-CN or PBAE-BPN carrying 95% pEGFP and 5% Cy5-labeled plasmid DNA at a total DNA concentration of 1 mg/ml into the brains of healthy or orthotopic brain tumor-bearing (i.e. 10 days after the inoculation) rats via CED. Animals were sacrificed 48 h after the administration.
Harvested brains were fixed in 4% paraformaldehyde overnight and incubated in a gradient of sucrose solutions prior to cryosection. Tissues were sectioned into 50 μm coronal slices using a Leica CM 1905 cryostat (Leica Biosystems, Buffalo Grove, IL). Slices were stained with 4′,6-diamidino-2-phenylindole (DAPI; Molecular Probes, Eugene, OR) to visualize cell nuclei, and imaged for the fluorescence using a confocal LSM 710 microscope (Carl Zeiss) under 10× and 63× magnification. We carefully optimized the settings to avoid background fluorescence based on the microscopy of untreated control rat brains. Importantly, laser power, pinhole, gain, offset and digital gain were selected individually for each magnification employed and maintained constant throughout the study.
To quantify the volume of nanocomplex distribution as well as the transgene expression, we used a custom-made MATLAB script that subtracted the background fluorescence and thresholded the fluorescent intensities to 10% of the maximum intensity of each image (10×). Of note, fluorescence in the white matter tract generated by backflow was excluded from the analysis. Every other 50 μm slice within 2 mm of the injection plane was imaged. The area of transgene expression in each slice was integrated to calculate the total volume of transgene expression. To reconstruct three-dimensional images of nanocomplex distribution and transgene expression, we stacked and aligned the acquired images using Metamorph ® Microscopy Automation & Image Analysis Software (Molecular Devices, CA).
We subsequently utilized the function spots of Imaris ® Software (Bitplane, CT) to quantify the percentage of the cells that took up DNA-loaded nanocomplexes as well as the cells with successful GFP transgene expression shown in each image (63×). This was achieved by examining the co-localization of DAPI with the Cy5 or GFP signal for nanocomplexes or transgene expression, respectively.
For assessing the distribution of DNA-loaded nanocomplexes in an orthotopic 9L GS following CED, we administered a solution of Cy3-labeled PBAE-CN and Cy5-labeled PBAE-BPN at a plasmid DNA concentration of 500 μg/ml for each nanocomplex type (i.e. 1 mg/ml overall plasmid DNA concentration). The rat brains were harvested, processed and imaged as described above.
4.13. Western blot analysis
To examine the overall levels of transgene expression achieved by CED of DNA-loaded nanocomplexes, we similarly administered either PBAE-CN or PBAE-BPN carrying pEGFP at a plasmid DNA concentration of 1 mg/ml. The animals were sacrificed 48 h following CED and brain tissues were harvested and lysed using 1 ml of Tissue Protein Extraction reagent (T-PER; Thermo Scientific, Pittsburgh, PA). Protein concentration was determined using the BCA assay and samples were diluted to a concentration of 1.3 μg/μl. Subsequently, the sampling buffer (10% glycerol, 2% SDS, 62.5 mM Tris–HCl and 2% β-mercaptoethanol, pH 6.8) was added and samples were boiled at 95 °C for 10 min. Samples were resolved using SDS–polyacrylamide gel electrophoresis (PAGE) and proteins were transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA) using a wet electroblotting system (Bio-Rad). The membrane was blocked with 3% bovine serum albumin in a mixture of Tris-buffered saline and Tween 20 (TBST; 10 mM Tris-Cl, pH 8.0, 150 mM NaCl and 0.5% Tween-20) and incubated overnight at 4 °C with primary antibodies. Specifically, anti-GFP (B-2): sc-9996 (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (6C5): sc-32233 (Santa Cruz Biotechnology) were used for the detection of GFP transgene expression and a housekeeping protein (i.e. GAPDH), respectively. Immunoblots were visualized by an enhanced chemiluminescence method. Quantification of western blot results was conducted using the gel analysis tool of ImageJ Software (NIH, Bethesda, MD) [80].
4.14. Anti-cancer efficacy studies
We compared the therapeutic effects following CED of PBAE-CN and PBAE-BPN carrying pTK. We randomized the rats bearing orthotopic F98 GBM into 3 groups to be treated with PBAE-CN, PBAE-BPN or normal saline. Six days after the tumor inoculation, a 20 μl solution of PBAE-CN or PBAE-BPN carrying pTK at 1 mg/ml plasmid DNA concentration was administered via CED (Day 6 in Fig. 4H). Rats received normal saline served as an untreated control. GCV was administered intraperitoneally at a dose of 25 mg/kg twice a day for a total of 10 days.
In parallel, we examined the therapeutic effects following CED of PBAE-CN and PBAE-BPN carrying plasmid DNA encoding p53 (i.e. pC53-SN3). At 5 and 10 days after the tumor inoculation, rats were treated with PBAE-BPN or PBAE-CN carrying pC53-SN3 at 1 mg/ml plasmid DNA concentration or normal saline (days 5 and 10 in Fig. 5D). For assessing the distribution of p53 transgene expression, rat brains were harvested 2 days after the treatment, processed to immunohistochemically stain with Alex Fluor 647-labeled anti-p53 (FL-393): sc-6243 (Santa Cruz Biotechnology) and imaged using a confocal LSM 710 microscope.
Animals were monitored twice a day for physical deficiencies and weight loss. Rats were sacrificed when > 25% weight loss from their original weights or sign of distress was observed.
4.15. Statistical analysis
Statistical analysis between two groups was conducted using a two-tailed Student’s t-test assuming unequal variances. If multiple comparisons were involved, one-way analysis of variance, followed by post hoc test, was employed, using SPSS 18.0 software (SPSS Inc., Chicago, IL). The differences in the survival of rats were analyzed using the log-rank test. Differences were determined to be statistically significant at p < 0.05.
Supplementary Material
Acknowledgments
The funding was provided by the National Institutes of Health (R01CA164789, R01EB020147, R01CA197111, R01CA204968 and P30EY001765), Focused Ultrasound Foundation and W.W. Smith Charitable Trust (J.S.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx. doi.org/10.1016/j.jconrel.2017.07.009.
References
- 1.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 2.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN. An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sathornsumetee S, Rich JN. Designer therapies for glioblastoma multiforme. Ann N Y Acad Sci. 2008;1142:108–132. doi: 10.1196/annals.1444.009. [DOI] [PubMed] [Google Scholar]
- 6.van Gaal EV, Hennink WE, Crommelin DJ, Mastrobattista E. Plasmid engineering for controlled and sustained gene expression for nonviral gene therapy. Pharm Res. 2006;23:1053–1074. doi: 10.1007/s11095-006-0164-2. [DOI] [PubMed] [Google Scholar]
- 7.Costa PM, Cardoso AL, Custodia C, Cunha P, Pereira de Almeida L, Pedroso de Lima MC. MiRNA-21 silencing mediated by tumor-targeted nanoparticles combined with sunitinib: a new multimodal gene therapy approach for glioblastoma. J Control Release. 2015;207:31–39. doi: 10.1016/j.jconrel.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Dent P, Yacoub A, Park M, Sarkar D, Shah K, Curiel DT, Grant S, Fisher PB. Searching for a cure: gene therapy for glioblastoma. Cancer Biol Ther. 2008;7:1335–1340. doi: 10.4161/cbt.7.9.6408. [DOI] [PubMed] [Google Scholar]
- 9.Tobias A, Ahmed A, Moon KS, Lesniak MS. The art of gene therapy for glioma: a review of the challenging road to the bedside. J Neurol Neurosurg Psychiatry. 2013;84:213–222. doi: 10.1136/jnnp-2012-302946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwami K, Natsume A, Wakabayashi T. Gene therapy for high-grade glioma. Neurol Med Chir (Tokyo) 2010;50:727–736. doi: 10.2176/nmc.50.727. [DOI] [PubMed] [Google Scholar]
- 11.Lang FF, Bruner JM, Fuller GN, Aldape K, Prados MD, Chang S, Berger MS, McDermott MW, Kunwar SM, Junck LR, Chandler W, Zwiebel JA, Kaplan RS, Yung WK. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol. 2003;21:2508–2518. doi: 10.1200/JCO.2003.21.13.2508. [DOI] [PubMed] [Google Scholar]
- 12.Walther W, Stein U. Viral vectors for gene transfer: a review of their use in the treatment of human diseases. Drugs. 2000;60:249–271. doi: 10.2165/00003495-200060020-00002. [DOI] [PubMed] [Google Scholar]
- 13.Smitt PS, Driesse M, Wolbers J, Kros M, Avezaat C. Treatment of relapsed malignant glioma with an adenoviral vector containing the herpes simplex thymidine kinase gene followed by ganciclovir. Mol Ther. 2003;7:851–858. doi: 10.1016/s1525-0016(03)00100-x. [DOI] [PubMed] [Google Scholar]
- 14.Giese A, Bjerkvig R, Berens ME, Westphal M. Cost of migration: invasion of malignant gliomas and implications for treatment. J Clin Oncol. 2003;21:1624–1636. doi: 10.1200/JCO.2003.05.063. [DOI] [PubMed] [Google Scholar]
- 15.Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci U S A. 1994;91:2076–2080. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin D, Zhai Y, Gruber HE, Ibanez CE, Robbins JM, Kells AP, Kasahara N, Forsayeth J, Jolly DJ, Bankiewicz KS. Convection-enhanced delivery improves distribution and efficacy of tumor-selective retroviral replicating vectors in a rodent brain tumor model. Cancer Gene Ther. 2013;20:336–341. doi: 10.1038/cgt.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voges J, Reszka R, Gossmann A, Dittmar C, Richter R, Garlip G, Kracht L, Coenen HH, Sturm V, Wienhard K, Heiss WD, Jacobs AH. Imaging-guided convection-enhanced delivery and gene therapy of glioblastoma. Ann Neurol. 2003;54:479–487. doi: 10.1002/ana.10688. [DOI] [PubMed] [Google Scholar]
- 18.MacKay JA, Deen DF, Szoka FC., Jr Distribution in brain of liposomes after convection enhanced delivery; modulation by particle charge, particle diameter, and presence of steric coating. Brain Res. 2005;1035:139–153. doi: 10.1016/j.brainres.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 19.Kenny GD, Bienemann AS, Tagalakis AD, Pugh JA, Welser K, Campbell F, Tabor AB, Hailes HC, Gill SS, Lythgoe MF, McLeod CW, White EA, Hart SL. Multifunctional receptor-targeted nanocomplexes for the delivery of therapeutic nucleic acids to the brain. Biomaterials. 2013;34:9190–9200. doi: 10.1016/j.biomaterials.2013.07.081. [DOI] [PubMed] [Google Scholar]
- 20.Writer MJ, Kyrtatos PG, Bienemann AS, Pugh JA, Lowe AS, Villegas-Llerena C, Kenny GD, White EA, Gill SS, McLeod CW, Lythgoe MF, Hart SL. Lipid peptide nanocomplexes for gene delivery and magnetic resonance imaging in the brain. J Control Release. 2012;162:340–348. doi: 10.1016/j.jconrel.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang F, Mastorakos P, Mishra MK, Mangraviti A, Hwang L, Zhou J, Hanes J, Brem H, Olivi A, Tyler B, Kannan RM. Uniform brain tumor distribution and tumor associated macrophage targeting of systemically administered dendrimers. Biomaterials. 2015;52:507–516. doi: 10.1016/j.biomaterials.2015.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nance EA, Woodworth GF, Sailor KA, Shih TY, Xu Q, Swaminathan G, Xiang D, Eberhart C, Hanes J. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci Transl Med. 2012;4:149ra119. doi: 10.1126/scitranslmed.3003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mastorakos P, Song E, Zhang C, Berry S, Park HW, Kim YE, Park JS, Lee S, Suk JS, Hanes J. Biodegradable DNA nanoparticles that provide widespread gene delivery in the brain. Small. 2015;12:678–685. doi: 10.1002/smll.201502554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mastorakos P, Zhang C, Berry S, Oh Y, Lee S, Eberhart CG, Woodworth GF, Suk JS, Hanes J. Highly PEGylated DNA nanoparticles provide uniform and widespread gene transfer in the brain. Adv Healthc Mater. 2015 doi: 10.1002/adhm.201400800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gritsenko PG, Ilina O, Friedl P. Interstitial guidance of cancer invasion. J Pathol. 2012;226:185–199. doi: 10.1002/path.3031. [DOI] [PubMed] [Google Scholar]
- 26.Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, di Tomaso E, Brown EB, Izumi Y, Campbell RB, Berk DA, Jain RK. Role of tumor-host in-teractions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proc Natl Acad Sci U S A. 2001;98:4628–4633. doi: 10.1073/pnas.081626898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hambardzumyan D, Bergers G. Glioblastoma: defining tumor niches. Trends Cancer. 2015;1:252–265. doi: 10.1016/j.trecan.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nance E, Zhang C, Shih TY, Xu Q, Schuster BS, Hanes J. Brain-penetrating nanoparticles improve paclitaxel efficacy in malignant glioma following local ad-ministration. ACS Nano. 2014;8:10655–10664. doi: 10.1021/nn504210g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider CS, Perez JG, Cheng E, Zhang C, Mastorakos P, Hanes J, Winkles JA, Woodworth GF, Kim AJ. Minimizing the non-specific binding of nanoparticles to the brain enables active targeting of Fn14-positive glioblastoma cells. Biomaterials. 2015;42:42–51. doi: 10.1016/j.biomaterials.2014.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for im-proving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99(Pt A):28–51. doi: 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berry S, Mastorakos P, Zhang C, Song E, Patel H, Suk JS, Hanes J. Enhancing intracranial delivery of clinically relevant non-viral gene vectors. RSC Adv. 2016;48:41665–41674. doi: 10.1039/C6RA01546H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patnaik S, Gupta KC. Novel polyethylenimine-derived nanoparticles for in vivo gene delivery. Expert Opin Drug Deliv. 2013;10:215–228. doi: 10.1517/17425247.2013.744964. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Lei Y, Xie C, Lu W, Wagner E, Xie Z, Gao J, Zhang X, Yan Z, Liu M. Retro-inverso CendR peptide-mediated polyethyleneimine for intracranial glioblastoma-targeting gene therapy. Bioconjug Chem. 2014;25:414–423. doi: 10.1021/bc400552t. [DOI] [PubMed] [Google Scholar]
- 34.do Hwang W, Son S, Jang J, Youn H, Lee S, Lee D, Lee YS, Jeong JM, Kim WJ, Lee DS. A brain-targeted rabies virus glycoprotein-disulfide linked PEI nanocarrier for delivery of neurogenic microRNA. Biomaterials. 2011;32:4968–4975. doi: 10.1016/j.biomaterials.2011.03.047. [DOI] [PubMed] [Google Scholar]
- 35.Gofrit ON, Benjamin S, Halachmi S, Leibovitch I, Dotan Z, Lamm DL, Ehrlich N, Yutkin V, Ben-Am M, Hochberg A. DNA based therapy with diphtheria toxin-A BC-819: a phase 2b marker lesion trial in patients with intermediate risk nonmuscle invasive bladder cancer. J Urol. 2014;191:1697–1702. doi: 10.1016/j.juro.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 36.Anwer K, Kelly FJ, Chu C, Fewell JG, Lewis D, Alvarez RD. Phase I trial of a formulated IL-12 plasmid in combination with carboplatin and docetaxel chemotherapy in the treatment of platinum-sensitive recurrent ovarian cancer. Gynecol Oncol. 2013;131:169–173. doi: 10.1016/j.ygyno.2013.07.081. [DOI] [PubMed] [Google Scholar]
- 37.Konstan MW, Davis PB, Wagener JS, Hilliard KA, Stern RC, Milgram LJ, Kowalczyk TH, Hyatt SL, Fink TL, Gedeon CR, Oette SM, Payne JM, Muhammad O, Ziady AG, Moen RC, Cooper MJ. Compacted DNA nanoparticles administered to the nasal mucosa of cystic fibrosis subjects are safe and demonstrate partial to complete cystic fibrosis transmembrane regulator reconstitution. Hum Gene Ther. 2004;15:1255–1269. doi: 10.1089/hum.2004.15.1255. [DOI] [PubMed] [Google Scholar]
- 38.Harmon BT, Aly AE, Padegimas L, Sesenoglu-Laird O, Cooper MJ, Waszczak BL. Intranasal administration of plasmid DNA nanoparticles yields successful transfection and expression of a reporter protein in rat brain. Gene Ther. 2014;21:514–521. doi: 10.1038/gt.2014.28. [DOI] [PubMed] [Google Scholar]
- 39.Fletcher AM, Kowalczyk TH, Padegimas L, Cooper MJ, Yurek DM. Transgene expression in the striatum following intracerebral injections of DNA nanoparticles encoding for human glial cell line-derived neurotrophic factor. Neuroscience. 2011;194:220–226. doi: 10.1016/j.neuroscience.2011.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barth RF, Kaur B. Rat brain tumor models in experimental neuro-oncology: the C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J Neuro-Oncol. 2009;94:299–312. doi: 10.1007/s11060-009-9875-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biston MC, Joubert A, Adam JF, Elleaume H, Bohic S, Charvet AM, Esteve F, Foray N, Balosso J. Cure of Fisher rats bearing radioresistant F98 glioma treated with cis-platinum and irradiated with monochromatic synchrotron X-rays. Cancer Res. 2004;64:2317–2323. doi: 10.1158/0008-5472.can-03-3600. [DOI] [PubMed] [Google Scholar]
- 42.Rhines LD, Sampath P, Dolan ME, Tyler BM, Brem H, Weingart J. O6-benzylguanine potentiates the antitumor effect of locally delivered carmustine against an intracranial rat glioma. Cancer Res. 2000;60:6307–6310. [PubMed] [Google Scholar]
- 43.Sipos EP, Tyler B, Piantadosi S, Burger PC, Brem H. Optimizing interstitial delivery of BCNU from controlled release polymers for the treatment of brain tumors. Cancer Chemother Pharmacol. 1997;39:383–389. doi: 10.1007/s002800050588. [DOI] [PubMed] [Google Scholar]
- 44.Tamargo RJ, Myseros JS, Epstein JI, Yang MB, Chasin M, Brem H. Interstitial chemotherapy of the 9L gliosarcoma: controlled release polymers for drug delivery in the brain. Cancer Res. 1993;53:329–333. [PubMed] [Google Scholar]
- 45.Chowdhary SA, Ryken T, Newton HB. Survival outcomes and safety of carmustine wafers in the treatment of high-grade gliomas: a meta-analysis. J Neuro-Oncol. 2015;122:367–382. doi: 10.1007/s11060-015-1724-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brem H, Piantadosi S, Burger PC, Walker M, Selker R, Vick NA, Black K, Sisti M, Brem S, Mohr G, et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-brain Tumor Treatment Group. Lancet. 1995;345:1008–1012. doi: 10.1016/s0140-6736(95)90755-6. [DOI] [PubMed] [Google Scholar]
- 47.Brem H, Mahaley MS, Jr, Vick NA, Black KL, Schold SC, Jr, Burger PC, Friedman AH, Ciric IS, Eller TW, Cozzens JW, et al. Interstitial chemotherapy with drug polymer implants for the treatment of recurrent gliomas. J Neuro-Oncol. 1991;74:441–446. doi: 10.3171/jns.1991.74.3.0441. [DOI] [PubMed] [Google Scholar]
- 48.Tyler B, Fowers KD, Li KW, Recinos VR, Caplan JM, Hdeib A, Grossman R, Basaldella L, Bekelis K, Pradilla G, Legnani F, Brem H. A thermal gel depot for local delivery of paclitaxel to treat experimental brain tumors in rats. J Neurosurg. 2010;113:210–217. doi: 10.3171/2009.11.JNS08162. [DOI] [PubMed] [Google Scholar]
- 49.Vellimana AK, Recinos VR, Hwang L, Fowers KD, Li KW, Zhang Y, Okonma S, Eberhart CG, Brem H, Tyler BM. Combination of paclitaxel thermal gel depot with temozolomide and radiotherapy significantly prolongs survival in an experimental rodent glioma model. J Neuro-Oncol. 2013;111:229–236. doi: 10.1007/s11060-012-1014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DuVall GA, Tarabar D, Seidel RH, Elstad NL, Fowers KD. Phase 2: a dose-escalation study of OncoGel (ReGel/paclitaxel), a controlled-release formulation of paclitaxel, as adjunctive local therapy to external-beam radiation in patients with inoperable esophageal cancer. Anti-Cancer Drugs. 2009;20:89–95. doi: 10.1097/CAD.0b013e3283222c12. [DOI] [PubMed] [Google Scholar]
- 51.Mishra S, Webster P, Davis ME. PEGylation significantly affects cellular uptake and intracellular trafficking of non-viral gene delivery particles. Eur J Cell Biol. 2004;83:97–111. doi: 10.1078/0171-9335-00363. [DOI] [PubMed] [Google Scholar]
- 52.Ogris M, Steinlein P, Carotta S, Brunner S, Wagner E. DNA/polyethylenimine transfection particles: influence of ligands, polymer size, and PEGylation on internalization and gene expression. AAPS PharmSci. 2001;3:E21. doi: 10.1208/ps030321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagner E. Polymers for nucleic acid transfer-an overview. Adv Genet. 2014;88:231–261. doi: 10.1016/B978-0-12-800148-6.00008-0. [DOI] [PubMed] [Google Scholar]
- 54.Guerrero-Cazares H, Tzeng SY, Young NP, Abutaleb AO, Quinones-Hinojosa A, Green JJ. Biodegradable polymeric nanoparticles show high efficacy and specificity at DNA delivery to human glioblastoma in vitro and in vivo. ACS Nano. 2014;8:5141–5153. doi: 10.1021/nn501197v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mangraviti A, Tzeng SY, Kozielski KL, Wang Y, Jin Y, Gullotti D, Pedone M, Buaron N, Liu A, Wilson DR, Hansen SK, Rodriguez FJ, Gao GD, DiMeco F, Brem H, Olivi A, Tyler B, Green JJ. Polymeric nanoparticles for nonviral gene therapy extend brain tumor survival in vivo. ACS Nano. 2015;9:1236–1249. doi: 10.1021/nn504905q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schuster BS, Ensign LM, Allan DB, Suk JS, Hanes J. Particle tracking in drug and gene delivery research: state-of-the-art applications and methods. Adv Drug Deliv Rev. 2015;91:70–91. doi: 10.1016/j.addr.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mastorakos P, da Silva AL, Chisholm J, Song E, Choi WK, Boyle MP, Morales MM, Hanes J, Suk JS. Highly compacted biodegradable DNA nanoparticles capable of overcoming the mucus barrier for inhaled lung gene therapy. Proc Natl Acad Sci U S A. 2015;112:8720–8725. doi: 10.1073/pnas.1502281112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shand N, Weber F, Mariani L, Bernstein M, Gianella-Borradori A, Long Z, Sorensen AG, Barbier N. A phase 1-2 clinical trial of gene therapy for recurrent glioblastoma multiforme by tumor transduction with the herpes simplex thymidine kinase gene followed by ganciclovir. GLI328 European-Canadian Study Group. Hum Gene Ther. 1999;10:2325–2335. doi: 10.1089/10430349950016979. [DOI] [PubMed] [Google Scholar]
- 59.Klatzmann D, Valery CA, Bensimon G, Marro B, Boyer O, Mokhtari K, Diquet B, Salzmann JL, Philippon J. A phase I/II study of herpes simplex virus type 1 thymidine kinase “suicide” gene therapy for recurrent glioblastoma. Study Group on Gene Therapy for Glioblastoma. Hum Gene Ther. 1998;9:2595–2604. doi: 10.1089/hum.1998.9.17-2595. [DOI] [PubMed] [Google Scholar]
- 60.Immonen A, Vapalahti M, Tyynela K, Hurskainen H, Sandmair A, Vanninen R, Langford G, Murray N, Yla-Herttuala S. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Mol Ther. 2004;10:967–972. doi: 10.1016/j.ymthe.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 61.Vecil GG, Lang FF. Clinical trials of adenoviruses in brain tumors: a review of Ad-p53 and oncolytic adenoviruses. J Neuro-Oncol. 2003;65:237–246. doi: 10.1023/b:neon.0000003653.45635.32. [DOI] [PubMed] [Google Scholar]
- 62.Hamel W, Magnelli L, Chiarugi VP, Israel MA. Herpes simplex virus thymidine kinase/ganciclovir-mediated apoptotic death of bystander cells. Cancer Res. 1996;56:2697–2702. [PubMed] [Google Scholar]
- 63.Mesnil M, Piccoli C, Tiraby G, Willecke K, Yamasaki H. Bystander killing of cancer cells by herpes simplex virus thymidine kinase gene is mediated by connexins. Proc Natl Acad Sci U S A. 1996;93:1831–1835. doi: 10.1073/pnas.93.5.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schlegel J, Piontek G, Kersting M, Schuermann M, Kappler R, Scherthan H, Weghorst C, Buzard G, Mennel H. The p16/Cdkn2a/Ink4a gene is frequently deleted in nitrosourea-induced rat glial tumors. Pathobiology. 1999;67:202–206. doi: 10.1159/000028073. [DOI] [PubMed] [Google Scholar]
- 65.Zacher A, Kaulich K, Stepanow S, Wolter M, Kohrer K, Felsberg J, Malzkorn B, Reifenberger G. Molecular diagnostics of gliomas using next generation sequencing of a glioma-tailored gene panel. Brain Pathol. 2016;27:146–159. doi: 10.1111/bpa.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bredel M. Anticancer drug resistance in primary human brain tumors. Brain Res Brain Res Rev. 2001;35:161–204. doi: 10.1016/s0165-0173(01)00045-5. [DOI] [PubMed] [Google Scholar]
- 67.Danhier F, Messaoudi K, Lemaire L, Benoit JP, Lagarce F. Combined anti-Galectin-1 and anti-EGFR siRNA-loaded chitosan-lipid nanocapsules decrease temozolomide resistance in glioblastoma: in vivo evaluation. Int J Pharm. 2015;481:154–161. doi: 10.1016/j.ijpharm.2015.01.051. [DOI] [PubMed] [Google Scholar]
- 68.Akinc A, Anderson DG, Lynn DM, Langer R. Synthesis of poly(beta-amino ester)s optimized for highly effective gene delivery. Bioconjug Chem. 2003;14:979–988. doi: 10.1021/bc034067y. [DOI] [PubMed] [Google Scholar]
- 69.Anderson DG, Akinc A, Hossain N, Langer R. Structure/property studies of polymeric gene delivery using a library of poly(beta-amino esters) Mol Ther. 2005;11:426–434. doi: 10.1016/j.ymthe.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 70.Keeney M, Ong SG, Padilla A, Yao Z, Goodman S, Wu JC, Yang F. Development of poly(beta-amino ester)-based biodegradable nanoparticles for nonviral delivery of minicircle DNA. ACS Nano. 2013;7:7241–7250. doi: 10.1021/nn402657d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zugates GT, Peng W, Zumbuehl A, Jhunjhunwala S, Huang YH, Langer R, Sawicki JA, Anderson DG. Rapid optimization of gene delivery by parallel end-modification of poly(beta-amino ester)s. Mol Ther. 2007;15:1306–1312. doi: 10.1038/mt.sj.6300132. [DOI] [PubMed] [Google Scholar]
- 72.Zugates GT, Tedford NC, Zumbuehl A, Jhunjhunwala S, Kang CS, Griffith LG, Lauffenburger DA, Langer R, Anderson DG. Gene delivery properties of end-modified poly(beta-amino ester)s. Bioconjug Chem. 2007;18:1887–1896. doi: 10.1021/bc7002082. [DOI] [PubMed] [Google Scholar]
- 73.Zhou D, Gao Y, Aied A, Cutlar L, Igoucheva O, Newland B, Alexeeve V, Greiser U, Uitto J, Wang W. Highly branched poly(beta-amino ester)s for skin gene therapy. J Control Release. 2016;244(Pt B):336–346. doi: 10.1016/j.jconrel.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 74.Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249:912–915. doi: 10.1126/science.2144057. [DOI] [PubMed] [Google Scholar]
- 75.Greenland JR, Liu H, Berry D, Anderson DG, Kim WK, Irvine DJ, Langer R, Letvin NL. Beta-amino ester polymers facilitate in vivo DNA transfection and adjuvant plasmid DNA immunization. Mol Ther. 2005;12:164–170. doi: 10.1016/j.ymthe.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 76.Suk JS, Kim AJ, Trehan K, Schneider CS, Cebotaru L, Woodward OM, Boylan NJ, Boyle MP, Lai SK, Guggino WB, Hanes J. Lung gene therapy with highly compacted DNA nanoparticles that overcome the mucus barrier. J Control Release. 2014;178C:8–17. doi: 10.1016/j.jconrel.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu Y, Liggitt HD, Dow S, Handumrongkul C, Heath TD, Debs RJ. Strain-based genetic differences regulate the efficiency of systemic gene delivery as well as expression. J Biol Chem. 2002;277:4966–4972. doi: 10.1074/jbc.M110285200. [DOI] [PubMed] [Google Scholar]
- 78.Recinos VR, Tyler BM, Bekelis K, Sunshine SB, Vellimana A, Li KW, Brem H. Combination of intracranial temozolomide with intracranial carmustine improves survival when compared with either treatment alone in a rodent glioma model. Neurosurgery. 2010;66:530–537. doi: 10.1227/01.NEU.0000365263.14725.39. [DOI] [PubMed] [Google Scholar]
- 79.Feinstein AR. Principles of Medical Statistics. Chapman and Hall/CRC; 2001. [Google Scholar]
- 80.Elliott SL, Cullen CF, Wrobel N, Kernan MJ, Ohkura H. EB1 is essential during Drosophila development and plays a crucial role in the integrity of chordotonal mechanosensory organs. Mol Biol Cell. 2005;16:891–901. doi: 10.1091/mbc.E04-07-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.