

Abstract
Cervical artery dissection (CeAD), a mural hematoma in a carotid or vertebral artery, is a major cause of ischemic stroke in young adults although relatively uncommon in the general population (incidence of 2.6/100,000 per year)1. Minor cervical traumas, infection, migraine and hypertension are putative risk factors1–3, and inverse associations with obesity and hypercholesterolemia are described3,4. No confirmed genetic susceptibility factors have been identified using candidate gene approaches5. We performed genome-wide association studies (GWAS) in 1,393 CeAD cases and 14,416 controls. The rs9349379[G] allele (PHACTR1) was associated with lower CeAD risk (odds ratio (OR) = 0.75, 95% confidence interval (CI) = 0.69–0.82; P = 4.46 × 10−10), with confirmation in independent follow-up samples (659 CeAD cases and 2,648 controls; P = 3.91 × 10−3; combined P = 1.00 × 10−11). The rs9349379[G] allele was previously shown to be associated with lower risk of migraine and increased risk of myocardial infarction6–9. Deciphering the mechanisms underlying this pleiotropy might provide important information on the biological underpinnings of these disabling conditions.
We organized an international initiative with the aim of collecting clinical data and DNA for the largest possible number of CeAD cases to identify genetic susceptibility loci. We obtained 942 CeAD cases from the Cervical Artery Dissections and Ischemic Stroke Patients (CADISP) study (CADISP-1: 170 Finns and 772 non-Finnish Europeans)10. An additional 451 CeAD cases of European origin were recruited specifically for the CADISP-genetics project at European and US centers (CADISP-2). These collections provided a total of 1,393 CeAD cases of European ancestry for the GWAS (discovery) phase. All CeAD cases were ascertained through departments of neurology specialized in stroke care (Supplementary Figs. 1–3 and Supplementary Note; ClinicalTrials.gov identifier NCT00657969)11. DNA from 14,416 controls was available (287 Finns and 14,129 non-Finnish Europeans). We obtained 659 additional CeAD cases that could not be included in the GWAS because of the timing of their inclusion or the availability of only limited amounts of DNA. These cases were used along with 2,648 controls for follow-up of a small number of GWAS results. Finally, to examine the disease specificity of the genetic associations, we recruited 583 individuals with an ischemic stroke attributable to other causes (non-CeAD ischemic stroke: 162 Finns and 421 non-Finnish Europeans) who had similar age, sex and geographical origin characteristics as the CeAD cases. The clinical characteristics of the research subjects are shown in Table 1.
Table 1.
Baseline clinical characteristics of the study subjects
| CeAD cases
|
Non-CeAD ischemic stroke |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| GWAS
|
Follow-up studies |
||||||||
| CADISP-1 | CADISP-2 | ||||||||
| n | 942 | 659 | 583 | ||||||
| Mean age (s.d.) | 44.2 | (10.0) | 44.6 | (10.5) | 43.6 | (9.4) | 44.6 | (10.5) | |
| Men, n (%) | 544 | (57.7) | 264 | (58.5) | 387 | (58.7) | 257 | (61.2) | |
| Location | Carotida, n (%) | 603 | (64.0) | 253 | (60.7)b | 375 | (56.9) | – | |
| Vertebral, n (%) | 305 | (32.4) | 147 | (35.3)b | 256 | (38.8) | – | ||
| Both, n (%) | 33 | (3.5) | 17 | (4.0)b | 26 | (3.9) | – | ||
| Clinical presentation | Cerebral ischemiac, n (%) | 729 | (77.4) | 333 | (79.5)b | 544 | (82.5) | – | |
| Ischemic stroke, n (%) | 606 | (64.3) | 281 | (67.1)b | 508 | (77.1) | – | ||
CeAD, cervical artery dissection.
Internal carotid artery dissection (one additional case had a common carotid artery dissection in CADISP-1).
Information on dissection site and associated ischemia was missing for 34 and 32 cases, respectively, in CADISP-2.
Cerebral ischemia corresponds to ischemic stroke or transient ischemic attack (including transient monocular blindness).
DNA samples were genotyped on an Illumina Human610-Quad or Human660W-Quad BeadChip. We performed imputation to the non-monomorphic SNPs described in the HapMap 2 and 1000 Genomes Project (August 2010 release) CEU (European-ancestry) panels. CADISP-1 and CADISP-2 data were analyzed separately because genotyping was carried out on different platforms (Supplementary Table 1). Moreover, because Finnish participants had a distinct ancestral origin (Supplementary Fig. 4), CADISP-1 was divided into CADISP-1 non-Finnish and CADISP-1 Finnish studies. We performed a fixed-effects inverse variance–weighted meta-analysis after applying a genomic control correction to each of the three GWAS results (CADISP-1 non-Finnish, CADISP-1 Finnish and CADISP-2; Supplementary Note). The quantile-quantile plot for the CeAD GWAS is shown in Supplementary Figure 5. We observed no overall inflation of P values or evidence for significant population stratification (genomic inflation factor λ = 1.032).
A Manhattan plot of the meta-analysis association results for genotyped SNPs is shown in Supplementary Figure 6 (and in Supplementary Fig. 7 by substudy). The evidence of association with CeAD reached genome-wide significance for two SNPs at two loci. These were rs9349379[G] in intron 2 of PHACTR1 on chromosome 6p24.1-p23 (OR = 0.75, 95% CI = 0.69–0.82; P = 4.46 × 10−10) and rs11172113[C] in intron 1 of LRP1 on chromosome 12q13.3 (OR = 0.78, 95% CI = 0.71–0.85; P = 4.22 × 10−8). Overall, we found 77 SNPs (at 51 loci) with association P < 1 × 10−4 (Supplementary Table 2) and 6 SNPs (at 5 loci) with association P < 1 × 10−5 (Table 2).
Table 2.
SNPs yielding the most significant associations with CeAD in the GWAS
| SNPa | Chr. | Position (bp) | EA | Nearest geneb | Stage | EAF | OR (95% CI) | P |
|---|---|---|---|---|---|---|---|---|
| rs12402265 | 1 | 59,463,190 | A | FGGY | CADISP-1 non-Finnish | 0.26 | 1.21 (1.07–1.36) | 2.70 × 10−3 |
| CADISP-1 Finnish | 0.28 | 1.16 (0.87–1.54) | 0.315 | |||||
| CADISP-2 | 0.28 | 1.28 (1.10–1.49) | 1.30 × 10−3 | |||||
| Meta-analysis | 1.23 (1.13–1.35) | 6.12 × 10−6 | ||||||
| rs6741522 | 2 | 185,544,143 | A | ZNF804A | CADISP-1 non-Finnish | 0.14 | 1.33 (1.15–1.54) | 1.99 × 10−4 |
| CADISP-1 Finnish | 0.10 | 1.32 (0.85–2.05) | 0.230 | |||||
| CADISP-2 | 0.13 | 1.38 (1.14–1.66) | 1.21 × 10−3 | |||||
| Meta-analysis | 1.34 (1.19–1.50) | 5.65 × 10−7 | ||||||
| rs6820391 | 4 | 54,109,453 | A | LNX1 | CADISP-1 non-Finnish | 0.29 | 1.21 (1.07–1.36) | 2.33 × 10−3 |
| CADISP-1 Finnish | 0.29 | 1.15 (0.86–1.54) | 0.345 | |||||
| CADISP-2 | 0.28 | 1.26 (1.09–1.47) | 2.69 × 10−3 | |||||
| Meta-analysis | 1.23 (1.12–1.35) | 6.35 × 10−6 | ||||||
| rs9349379 | 6 | 13,011,943 | G | PHACTR1 | CADISP-1 non-Finnish | 0.40 | 0.76 (0.68–0.86) | 8.90 × 10−6 |
| CADISP-1 Finnish | 0.45 | 0.84 (0.63–1.10) | 0.216 | |||||
| CADISP-2 | 0.40 | 0.73 (0.62–0.84) | 3.39 × 10−5 | |||||
| Meta-analysis | 0.75 (0.69–0.82) | 4.46 × 10−10 | ||||||
| rs11172113 | 12 | 55,813,550 | C | LRP1 | CADISP-1 non-Finnish | 0.38 | 0.77 (0.68–0.86) | 1.66 × 10−5 |
| CADISP-1 Finnish | 0.39 | 0.90 (0.69–1.18) | 0.456 | |||||
| CADISP-2 | 0.40 | 0.77 (0.67–0.89) | 7.61 × 10−4 | |||||
| Meta-analysis | 0.78 (0.71–0.85) | 4.22 × 10−8 | ||||||
| rs1466535c | 12 | 55,820,737 | A | LRP1 | CADISP-1 non-Finnish | 0.32 | 0.75 (0.66–0.85) | 1.06 × 10−5 |
| CADISP-1 Finnish | 0.28 | 1.11 (0.83–1.49) | 0.473 | |||||
| CADISP-2 | 0.34 | 0.80 (0.68–0.93) | 4.04 × 10−3 | |||||
| Meta-analysis | 0.80 (0.73–0.88) | 2.07 × 10−6 |
Only results for genotyped SNPs with P < 1 × 10−5 are presented here (for all associations with P < 1 × 10−4, see Supplementary Table 2). Chr., chromosome; EA, effect allele (minor allele); EAF, effect allele frequency in controls. Alleles and chromosomal positions were identified on the basis of the plus strand of the NCBI Build 36 reference genome.
rs9349379, rs11172113, rs1466535 and rs6820391 are intronic; rs6741522 and rs12402265 are intergenic.
The names for the nearest genes are shown according to the Human Gene Organization (HUGO) Gene Nomenclature System.
rs1466535 is in LD with rs11172113 (r2 = 0.72).
We restricted further analyses to loci with association P < 1 × 10−5. Two of these SNPs showed nominally significant heterogeneity in effect according to dissection site (P = 6.49 × 10−3 for rs1466535 (LRP1) and P = 0.038 for rs6820391 (LNX1)), with stronger associations for carotid than vertebral dissection (Table 3). None of these markers were associated with non-CeAD ischemic stroke (Supplementary Table 3).
Table 3.
SNPs yielding the most significant associations with CeAD and their proxies in GWAS and follow-up studies
| GWAS meta-analysis (n = 1,393/14,416) |
Follow-up meta-analysis (n = 659/2,648) |
Overall meta-analysis (n = 2,052/17,064) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr. | EA | EAF | Gene | CeAD site | Phet CeAD siteb | OR (95% CI) |
P | OR (95% CI) |
P | OR (95% CI) |
P | Phet |
| rs12402265a | 1 | A | 0.27 | FGGY | All | 0.70 | 1.23 (1.13–1.35) |
6.12 × 10−6 | 1.21 (1.04–1.40) |
0.012 | 1.23 (1.14–1.33) |
2.30 × 10−7 | 0.83 |
| Carotid | 1.23 (1.10–1.38) |
2.95 × 10−4 | 1.40 (1.17–1.68) |
2.52 × 10−4 | 1.28 (1.16–1.41) |
5.46 × 10−7 | 0.24 | ||||||
| Vertebral | 1.27 (1.10–1.47) |
1.36 × 10−3 | 1.05 (0.85–1.31) |
0.64 | 1.20 (1.06–1.35) |
3.47 × 10−3 | 0.16 | ||||||
| rs6741522 | 2 | A | 0.13 | ZNF804A | All | 0.64 | 1.34 (1.19–1.50) |
5.65 × 10−7 | 1.09 (0.91–1.31) |
0.36 | 1.26 (1.15–1.39) |
2.29 × 10−6 | 0.06 |
| Carotid | 1.37 (1.19–1.57) |
1.17 × 10−5 | 1.05 (0.83–1.32) |
0.68 | 1.27 (1.13–1.44) |
7.33 × 10−5 | 0.06 | ||||||
| Vertebral | 1.32 (1.10–1.60) |
3.53 × 10−3 | 1.24 (0.96–1.61) |
0.10 | 1.29 (1.11–1.51) |
8.92 × 10−4 | 0.69 | ||||||
| rs6820391 | 4 | A | 0.29 | LNX1 | All | 0.038 | 1.23 (1.12–1.35) |
6.35 × 10−6 | 1.28 (1.10–1.47) |
9.28 × 10−4 | 1.24 (1.15–1.34) |
2.36 × 10−8 | 0.68 |
| Carotid | 1.30 (1.16–1.45) |
3.88 × 10−6 | 1.29 (1.08–1.55) |
4.84 × 10−3 | 1.30 (1.18–1.43) |
6.41 × 10−8 | 0.97 | ||||||
| Vertebral | 1.09 (0.94–1.26) |
0.28 | 1.23 (1.00–1.52) |
0.049 | 1.13 (1.00–1.28) |
0.042 | 0.33 | ||||||
| rs9349379 | 6 | G | 0.40 | PHACTR1 | All | 0.52 | 0.75 (0.69–0.82) |
4.46 × 10−10 | 0.81 (0.71–0.94) |
3.91 × 10−3 | 0.77 (0.72–0.83) |
1.00 × 10−11 | 0.36 |
| Carotid | 0.76 (0.68–0.85) |
1.27 × 10−6 | 0.76 (0.64–0.90) |
1.79 × 10−3 | 0.76 (0.69–0.83) |
8.24 × 10−9 | 0.96 | ||||||
| Vertebral | 0.72 (0.62–0.84) |
1.38 × 10−5 | 0.92 (0.75–1.13) |
0.42 | 0.79 (0.70–0.88) |
6.32 × 10−5 | 0.06 | ||||||
| rs11172113 | 12 | C | 0.39 | LRP1 | All | 0.061 | 0.78 (0.71–0.85) |
4.22 × 10−8 | 0.93 (0.81–1.07) |
0.34 | 0.82 (0.76–0.89) |
3.03 × 10−7 | 0.03 |
| Carotid | 0.74 (0.66–0.83) |
1.14 × 10−7 | 0.88 (0.75–1.05) |
0.16 | 0.78 (0.71–0.86) |
1.90 × 10−7 | 0.08 | ||||||
| Vertebral | 0.89 (0.77–1.03) |
0.11 | 0.99 (0.81–1.21) |
0.93 | 0.92 (0.82–1.04) |
0.17 | 0.39 | ||||||
| rs1466535a | 12 | A | 0.32 | LRP1 | All | 6.49 × 10−3 | 0.80 (0.73–0.88) |
2.07 × 10−6 | 0.92 (0.80–1.07) |
0.30 | 0.83 (0.77–0.90) |
4.94 × 10−6 | 0.10 |
| Carotid | 0.73 (0.65–0.82) |
2.50 × 10−7 | 0.85 (0.71–1.01) |
0.07 | 0.76 (0.69–0.84) |
1.17 × 10−7 | 0.18 | ||||||
| Vertebral | 0.97 (0.84–1.12) |
0.68 | 0.99 (0.80–1.22) |
0.92 | 0.98 (0.87–1.10) |
0.69 | 0.87 | ||||||
Chr., chromosome; EA, effect allele (minor allele); EAF, effect allele frequency (in controls from the GWAS); OR, odds ratio; CI, confidence interval; Phet, P value for heterogeneity between the discovery GWAS and the follow-up sample.
These SNPs were imputed (HapMap 2) in the Maryland follow-up sample with R2 = 0.93 (rs1466535) and 1 (rs12402265).
The P value of association with the dissection site in a case-only analysis.
We also identified two loci that were not included in the GWAS genotyping panel but reached genome-wide significance when imputed: rs2163474 in CCDC102B on chromosome 18q22.1 (OR = 1.78, 95% CI = 1.50–2.11; P = 3.86 × 10−11) and rs9915775 on chromosome 17q21.1 (OR = 3.39, 95% CI = 2.21–5.19; P = 1.97 × 10−8). Neither showed significant heterogeneity in effect by dissection site (P = 0.72 and 0.24, respectively), nor were they associated with non-CeAD ischemic stroke (Supplementary Table 3). We removed rs9915775 from further consideration because of its low estimated minor allele frequency (MAF = 0.01) and relatively poor imputation quality (R2 = 0.59).
We examined 6 SNP markers at the 5 most significantly associated loci from the genotyped panel (P < 1 × 10−5 in the GWAS) in the 659 CeAD case and 2,648 control samples available for follow-up (Supplementary Table 4). In addition, we selected two SNPs from the imputed locus at 18q22.1 for genotyping in follow-up samples (Supplementary Table 5). We also genotyped two proxies for two of the most significantly associated SNPs from the GWAS meta-analysis (rs12215208, R2 = 0.39 with rs9349379; rs6761601, R2 = 0.67 with rs6741522).
First, we calculated association statistics for the follow-up samples, using a threshold of P = 5.00 × 10−3 to assign significance (Bonferroni correction for ten SNPs; a conservative threshold because of the existence of positive linkage disequilibrium (LD) between some markers); irrespective of significance, we calculated combined association test statistics using meta-analysis and evaluated evidence of heterogeneity between the GWAS and follow-up data (Table 3 and Supplementary Table 5). Second, we applied a Bayesian approach to evaluate the significance of the associations using Wakefield’s approximate Bayes factor (Online Methods)12. Asymptotic Bayes factor (ABF) and Bayesian false discovery probability (BFDP) values are provided in Supplementary Tables 6 and 7.
rs9349379 (in PHACTR1), which provided the strongest evidence of association in the GWAS data, showed significant association with CeAD in the follow-up sample using the Bonferroni-corrected P-value threshold described above (P = 5.00 × 10−3), and the result from the meta-analysis of the discovery and follow-up studies maintained the finding of genome-wide significance (OR = 0.77, 95% CI = 0.72–0.83; P = 1.00 × 10−11) (Fig. 1 and Supplementary Fig. 8). Application of the Wakefield Bayesian approach yielded an ABF of 1.70 × 10−10. Another SNP in PHACTR1, rs12215208, selected as a proxy (P = 2.52 × 10−5 in the GWAS), exhibited a nominally significant association in the follow-up samples (P = 0.015), but the combined GWAS and follow-up data did not show genome-wide significant association (Supplementary Table 5). In contrast, we found no evidence of association in follow-up samples at rs11172113 in LRP1, the second genotyped marker that had genome-wide significant association in the GWAS (Supplementary Fig. 8).
Figure 1.
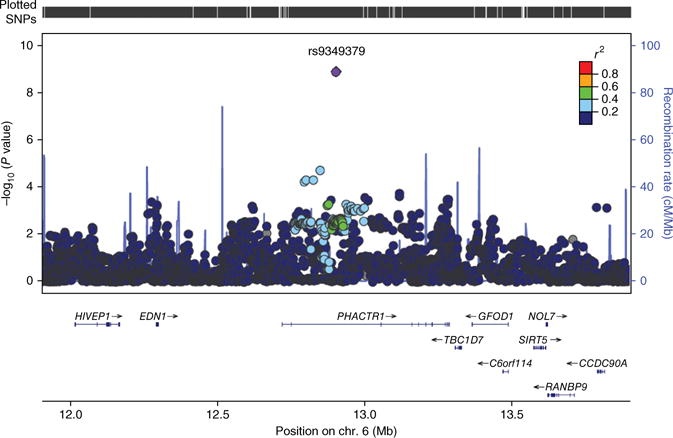
Regional association plot centered on rs9349379 (PHACTR1). Regional plot for associations in the region centered on rs9349379, drawn using LocusZoom software49. All SNPs, on the basis of 1000 Genomes Project imputed results (dots), are plotted with their GWAS meta-analysis association P values against their genomic position. The color of the dots represents the LD between SNPs. The purple line represents the estimated recombination rates. Genes and exons are shown as dark blue arrows and vertical lines, respectively.
Of the other markers that we examined, one met the criteria for significance in the follow-up samples: rs6820391 in LNX1 on chromosome 4q12 gave P = 9.28 × 10−4 in the follow-up samples and reached genome-wide significance in the combined meta-analysis (OR = 1.24, 95% CI = 1.15–1.34; P = 2.36 × 10−8) (Supplementary Fig. 8). However, as the Wakefield Bayesian approach yielded an ABF of 3.25 × 10−6, we consider this association to be only suggestive, requiring further studies for confirmation. Data for the imputed 18q22.1 locus provided no evidence in favor of association in the follow-up collection (P > 0.15; Supplementary Table 5).
In sensitivity analyses, stratifying on sex, migraine status and recent cervical trauma status did not modify the genetic associations with CeAD for the top loci (Supplementary Tables 8–10). We observed a nominally significant association of the rs9349379 (PHACTR1) risk allele for CeAD with a younger age of onset (Supplementary Table 11). The effect of rs9349379 was more marked in individuals with CeAD without ischemia, whereas the effect of rs6820391 (LNX1) was more marked in individuals with CeAD with ischemia, although the confidence intervals largely overlapped and P values were similar (Supplementary Table 12). Secondary analyses of association performed separately for carotid and vertebral artery dissections did not yield any genome-wide significant association (Supplementary Table 13).
We did not find any association with CeAD for (i) SNPs reported to be associated with CeAD in candidate gene association studies13–15; (ii) SNPs within a 100-kb window from the start and end of COL3A1, the gene harboring causal mutations for vascular Ehlers-Danlos syndrome, a rare etiology of CeAD16; and (iii) published genome-wide susceptibility SNPs for intracranial aneurysms17–19 and for thoracic aortic aneurysms and dissection20 (Supplementary Tables 14–17).
Our most significant association with CeAD was for rs9349379 (PHACTR1), which has been associated with myocardial infarction and coronary calcifications in various ancestry groups6,7,21–26, with effects in the opposite direction of that in CeAD. Two other susceptibility loci for myocardial infarction showed significant associations with CeAD in our data set with effects in the opposite direction of those for CeAD (rs2023938 (HDAC9)7 and rs9982601 (SLC5A3, MRPS6 and KCNE2)6), and one susceptibility locus for myocardial infarction at 9p21 was associated with CeAD with the same direction of effect (rs3217992 (CDKN2B-AS1)7) (Supplementary Table 18). No significant association was found between rs9349379 and non-CeAD ischemic stroke in our data set, although such an association was recently described in a larger sample for the ischemic stroke subtype secondary to large artery atherosclerosis (LAA-IS), where the effect was in the opposite direction to the one we observed for CeAD27. Another variant predisposing to LAA-IS (rs11984041 (HDAC9)28,29; R2 = 1 with rs2023938) also displayed an inverse association with CeAD (Supplementary Table 19). Opposite effects of the same genetic variant on different diseases have been described elsewhere30 and suggest either that the same region harbors different causal variants or that the same causal variant has biological effects with opposite implications for each disease. The vascular risk factor profile3,4, young age of occurrence1 and heterogeneous echostructure of carotid arteries in individuals with CeAD31 all suggest that atherosclerosis is not a predisposing condition for CeAD, in contrast with aortic dissection32. With aging and arteriosclerosis, the increased synthesis and reduced degradation of extracellular matrix components, as well as increasing collagen and elastin cross-links, could be hypothesized to make the arterial walls of cervical arteries more resistant to tears33,34, as could arterial wall calcifications, thus rendering the artery more stiff and resistant to the shear forces of lateral rotation and hyperextension that contribute to CeAD35.
Migraine is more common in individuals with CeAD than in the community1,2, and vascular mechanisms are thought to have a key role in the pathophysiology of this disease36. Recently, rs9349379 (PHACTR1) and rs11172113 (LRP1) were identified as migraine susceptibility SNPs8,9,37. Moreover, one additional migraine risk variant (rs13208321 in FHL5)9 was associated with CeAD at P = 6.80 × 10−4. All these SNPs showed effects in the same direction as those for CeAD (Supplementary Table 20). Interestingly, associations with these shared variants were most significant for migraine without aura9, the migraine subtype most commonly associated with CeAD38.
PHACTR1 is in a highly conserved genomic region, suggesting that it has a crucial involvement in biological processes39, but its function is poorly understood. Experimental studies identified a pivotal role for the PHACTR1 protein in vascular tube formation and actin polymerization, suggesting that it possibly has a role in angiogenic processes40,41. Upregulation of PHACTR1 by transforming growth factor (TGF)-β has been described in breast cancer cell lines42, potentially pointing to a connection with the TGF-β signaling pathway, which is also implicated in genetic predisposition to migraine9 and has a key role in Marfan and Loeys-Dietz syndromes, two inherited connective tissue disorders causing aortic dissection43,44. In silico functional annotation obtained from a wide array of published and unpublished expression quantitative trait locus (eQTL) data sets (Supplementary Table 21) provides some support for a functional effect of CeAD-associated SNPs in the 6p24.1-p23 locus on PHACTR1 expression in whole blood and cerebellum cells (although the SNPs in the databases are in relatively weak LD with rs9349379)45,46.
Although the follow-up analysis did not provide additional support for association with markers in LRP1, this locus remains of interest because of its association with migraine and abdominal aortic aneurysm, with effects in the same direction as in CeAD (Supplementary Table 17)47.
CeAD is relatively uncommon (~2.6/100,000 cases per year)48, and the current study is by far the largest genetic study of CeAD thus far (2,052 CeAD cases). Nevertheless, we might have lacked power to detect and replicate some associations, especially for loci showing heterogeneity in effect according to dissection site (Supplementary Table 22). We deliberately chose to focus on the discovery of genetic variants with overarching effects on CeAD risk, as we were underpowered to identify genetic variants underlying carotid or vertebral dissection exclusively. Future studies on larger samples are warranted to explore specific genetic susceptibility factors for carotid and vertebral artery dissections and to determine whether the 12q13.3 (LRP1), 4q12 (LNX1) and 18q22.1 (CCDC102B) loci are associated with CeAD.
In summary, we identified one previously unreported genome-wide significant risk locus for CeAD at PHACTR1 and additional highly suggestive loci requiring confirmation in future independent studies. PHACTR1 is also a major susceptibility locus for myocardial infarction and migraine. Understanding the mechanisms by which this locus appears to influence key vascular functions could have major applications for the treatment of these severe and disabling conditions.
URLs
CADISP, http://www.cadisp.com/; International Stroke Genetics Consortium, http://www.strokegenetics.org/.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Study population
CeAD cases
We included 942 CeAD cases in the CADISP study in 2004–2009 (CADISP-1: 170 Finns and 772 non-Finnish Europeans)10. An additional 451 CeAD cases of European origin were recruited in 2008–2010, exclusively for the CADISP-genetics project, in some CADISP centers and additional European and US centers (CADISP-2). In total, 1,393 CeAD cases of European ancestry were available. All CeAD cases were ascertained through departments of neurology specialized in stroke care. Inclusion criteria, recruiting centers and participant selection are described in Supplementary Figures 1–3 and the Supplementary Note11.
Individuals with non-CeAD ischemic stroke
As most individuals with CeAD sustained a cerebral ischemia, we planned to test whether genetic variants associated with CeAD were specific for CeAD, possibly through a predisposing vasculopathy, and did not confer generalized susceptibility to cerebral ischemia in young adults that could also predispose to other subtypes of ischemic stroke. We recruited 583 individuals with an ischemic stroke attributable to other causes (non-CeAD ischemic stroke: 162 Finns and 421 non-Finnish Europeans), frequency matched on the basis of age, sex and geographical origin to the CADISP-1 CeAD cases (Supplementary Fig. 3 and Supplementary Note).
Control populations
DNA for 14,416 controls was available. Most controls (n = 14,203: 74 Finns and 14,129 non-Finnish Europeans) were selected from an anonymized control genotype database at the CNG (Centre National de Génotypage) to match cases for ancestry, on the basis of the distribution of eigenvectors (Supplementary Fig. 4 and Supplementary Note)50. In addition, 213 Finnish controls were recruited (Supplementary Note). Although controls were not screened for CeAD, given the low disease incidence in the community, a misclassification bias is unlikely.
Genotyping, quality control filters and imputation
DNA samples were genotyped on an Illumina Human610-Quad or Human660W-Quad BeadChip (Supplementary Table 1 and Supplementary Note). After quality control (Supplementary Note), we used 472,862 autosomal SNPs and 10,029 X-chromosomal SNPs for analyses. We performed genotype imputation to the non-monomorphic SNPs described in the HapMap 2 and 1000 Genomes Project (August 2010 release) CEU panels (Supplementary Note). Only SNPs with an imputation score of R2 > 0.3 and a MAF of >0.01 were used for analysis.
Follow-up studies
We sought to replicate our strongest association signals in an independent sample comprising 659 CeAD cases, mostly drawn from existing databases of ischemic stroke (recruited through neurology departments specialized in stroke care according to the same inclusion criteria as in the discovery GWAS), and 2,648 controls, all of European ancestry (Supplementary Table 4). Some of these (85 CeAD cases and 998 controls) had already been genotyped elsewhere on a genome-wide chip (Supplementary Table 1 and Supplementary Note). In addition, DNA samples from 238 CeAD cases were genotyped across the genome, and 1,584 controls were genotyped on a custom chip including the SNPs yielding the most significant associations in the GWAS (Supplementary Table 1 and Supplementary Note). Finally, DNA samples from 391 additional CeAD cases and 162 controls, recruited in the same centers as other individuals included in the discovery or follow-up analyses, were genotyped for the top 5 genotyped loci and the top imputed locus (1–2 SNPs per locus) using KASPAR technology. Of these 714 follow-up CeAD cases, 55 were excluded because of the unavailability of information on the dissection site, leaving us with 659 CeAD cases for analysis. To avoid population stratification bias, the association of the top SNPs with CeAD was analyzed separately by inclusion region (see Supplementary Table 4 for the matching algorithm), and meta-analysis (fixed effects, inverse variance weighted) was performed on the results.
Statistics
Genome-wide association analysis comparing CeAD cases to healthy controls
Analyses were based on an additive genetic model. We used logistic regression to estimate OR values with corresponding 95% CIs, adjusting for sex. The CADISP-1 and CADISP-2 cohorts were analyzed separately because genotyping was carried out on different platforms (Supplementary Table 1). Moreover, because Finnish participants had a distinct ancestral origin relative to other populations (Supplementary Fig. 4), CADISP-1 was divided into CADISP-1 non-Finnish and CADISP-1 Finnish cohorts. Hence, sample sizes (CeAD-cases/controls) were as follows: 772/8,972 for CADISP-1 non-Finnish, 170/287 for CADISP-1 Finnish and 451/5,157 for CADISP-2. The first ten principal component scores were used as covariates for the CADISP-1 non-Finnish and CADISP-2 cohorts, and the first principal component score was used for the CADISP-1 Finnish cohort (Supplementary Note). Genomic control was applied to each of the three GWAS results (CADISP-1 non-Finnish, CADISP-1 Finnish and CADISP-2). Thereafter, a combined analysis was performed by inverse variance–weighted meta-analysis with a fixed-effects model (Supplementary Note). P < 5 × 10−8 was considered genome-wide significant.
Follow-up study
Because of the limits on DNA availability, six loci (ten SNPs) were chosen for follow-up genotyping in the phase 3 samples uniquely on the basis of statistical rankings in the GWAS meta-analysis of phase 1 and 2 samples (CADISP-1 and CADISP-2). We examined 6 SNP markers at the 5 most significantly associated loci from the genotyped panel (P < 1 × 10−5 in the GWAS) in the 659 CeAD and 2,648 control samples available for follow-up. In addition, two SNPs from the imputed locus on chromosome 18q22.1 were selected for genotyping in follow-up samples (Supplementary Table 5). We also genotyped two proxies for two of the most significantly associated SNPs (P < 1 × 10−5) from the GWAS meta-analysis (rs12215208, R2 = 0.39 with rs9349379; rs6761601, R2 = 0.67 with rs6741522).
First, we calculated the association statistics for the follow-up samples, using a threshold of P = 5.00 × 10−3 to assign significance (with Bonferroni correction for ten SNPs, which is conservative because of positive LD between some markers); irrespective of significance, we calculated combined association test statistics using meta-analysis and evaluated evidence of heterogeneity between GWAS and follow-up data. Second, we applied a Bayesian approach to evaluate the significance of associations using Wakefield’s approximate Bayes factor12. ABF and BFDP values are provided in Supplementary Tables 6 and 7.
Bayesian approach
We followed the approach developed by Wakefield12 to consider the posterior odds on the null hypothesis (no association of a SNP with CeAD) for sequential studies from a Bayesian perspective. In summary, we obtained estimates of the log relative risk , standard error and Wald statistic for a SNP from the logistic regression model (based on an additive genetic model), which was performed as our primary analysis to generate frequentist P values (Table 3 and Supplementary Table 5). Thereafter, we calculated Wakefield’s ABF as follows:
where W is the variance on the prior for the effect size, R=W/(V1W+V2W+V1V2), V1 and V2 are squared standard errors, and z1 and z2 are Wald statistics, each from the discovery GWAS (stages 1 and 2 combined) and the follow-up study (stage 3). A smaller ABF value corresponds to more evidence of the alternative hypothesis, i.e., the association of a SNP with CeAD.
Wakefield proposed three different specifications of priors for the log relative risk. Because our aim in this study was to find common susceptibility variants for CeAD, we followed an effect-MAF independence prior as , with RRu = 1.5. RRu is an upper value above which we believe that relative risks will occur with low probability; q is the prior probability of a relative risk above RRu; Φ (.) is the distribution function of a standard normal random variable.
Considering PO = π0/(1− π0) to be the prior odds on the null hypothesis, where π0 is the prior probability of the null (Pr[H0]), the posterior probability of H0 is given by the BFDP, with BFDP = ABF × PO/(1 + ABF × PO).
Specificity analysis
We tested whether the SNPs yielding the most significant associations with CeAD were also associated with non-CeAD ischemic stroke. We ran a logistic regression adjusted for sex and principal components, as described above, for non-Finnish Europeans (421 cases and 8,972 controls) and Finns (162 cases and 287 controls), followed by a fixed-effects inverse variance–weighted meta-analysis. Statistical significance was defined by false discovery rate (FDR)-corrected P < 0.05.
Sensitivity analyses
For the most significant associations, we stratified on and tested for interaction with sex, and we examined whether the most significant SNPs predicted an earlier age of onset of CeAD. We then evaluated the stability of the associations according to dissection site (carotid or vertebral) and the presence of cerebral ischemia (ischemic stroke or transient ischemic attack). Finally, we performed analyses stratified on the presence of two CeAD risk factors, migraine and cervical trauma in cases; case-only association analyses of the top CeAD SNPs with dissection site, the presence of cerebral ischemia, migraine and trauma were used as surrogate tests of heterogeneity or interaction, with P < 0.05 being considered significant51.
Candidate gene approach
We examined the associations of CeAD with published CeAD susceptibility SNPs and with SNPs in COL3A1, the gene harboring causal mutations for vascular Ehlers-Danlos syndrome, a rare etiology of CeAD16. We also tested for pleiotropic effects of susceptibility SNPs for intracranial aneurysms, thoracic aortic aneurysms and dissections, other subtypes of ischemic stroke and migraine (Supplementary Note). An FDR-corrected P value of <0.05 was considered to be statistically significant. We performed a sign test to evaluate whether associations of CeAD with alleles previously associated with migraine and ischemic stroke subtypes involved the same risk alleles more often than predicted by chance. A right-tailed P value of 0.05 was considered to be statistically significant.
Ethics
The study protocol was approved by local authorities in all participating centers and conducted according to national rules concerning ethics committee approval and informed consent.
Supplementary Material
Acknowledgments
The authors thank the staff and participants of all CADISP centers for their important contributions. The CADISP study has been supported by INSERM, Lille 2 University, Institut Pasteur de Lille and Lille University Hospital and received funding from the European Regional Development Fund (FEDER funds) and Région Nord-Pas-de-Calais in the framework of Contrat de Projets Etat-Region 2007–2013 Région Nord-Pas-de-Calais (grant 09120030), Centre National de Génotypage, the Emil Aaltonen Foundation, the Paavo Ilmari Ahvenainen Foundation, the Helsinki University Central Hospital Research Fund, the Helsinki University Medical Foundation, the Päivikki and Sakari Sohlberg Foundation, the Aarne Koskelo Foundation, the Maire Taponen Foundation, the Aarne and Aili Turunen Foundation, the Lilly Foundation, the Alfred Kordelin Foundation, the Finnish Medical Foundation, the Orion Farmos Research Foundation, the Maud Kuistila Foundation, the Finnish Brain Foundation, the Biomedicum Helsinki Foundation, Projet Hospitalier de Recherche Clinique Régional, Fondation de France, Génopôle de Lille, Adrinord, the Basel Stroke Funds, the Käthe-Zingg-Schwichtenberg-Fonds of the Swiss Academy of Medical Sciences and the Swiss Heart Foundation.
L.H.B., S.T.E. and P.A.L. were supported, in part, by a grant from the Swiss National Science Foundation (33CM30-124119). S.D. is supported by a Chair of Excellence from the French National Research Agency (ANR). S.D. and M.D. are supported by a grant from the Leducq Foundation. M.D. is supported by the Vascular Dementia Research Foundation. I.F.-C. is supported by the Miguel Servet programme (CP12/03298) from the Spanish Ministry of Health (Instituto de Salud Carlos III). G.K. is a member of the Deutsche Forschungsgemeinschaft Cluster of Excellence ‘Inflammation at Interfaces’. P.S. is supported by a Department of Health (UK) senior fellowship. A.M.S. is supported by the American Heart Association/American Stroke Association National Clinical Research Program (AHA 3CRP14140001). V.T. is supported by Fonds Wetenschappelijk Onderzoek Flanders.
More detailed acknowledgments can be found in the Supplementary Note.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
Study conception and design were performed by S.D., Y.K., M.L., D.L., P.A. and J.D. Acquisition of data was carried out by S.D., T.M.M., M.K., S.T.E., A. Pezzini, V.T., H.S.M., M.D., R.D., E.T., A.M.S., Y.S., S.A., Y.B., V.C., A.B., A.G., M.S., J.C., C. Lamy, E.M., S.B., L.H.B., A.J.G., P.M., J.J.M., P.S., E.B., B.G., E.G.v.d.H., I.F.-C., K.J., I.W., M.A.N., F.-E.D.L., C.J., Y.-C.C., A.J.M., C. Lichy, L.D., L.K., M.N., P.A.L., T. Brandt, G.B.B., H.-E.W., C.G., T. Böttcher, M.C., D.A., M.A.I., M.M.B.B., A. Padovani, J.F.M., G.K., A.R., B.B.W., E.-B.R., D.Z., T.T., M.L., D.L., P.A. and J.D. Statistical analysis and interpretation of the data were performed by S.D., Y.K., C.W., Y.-C.C., G.C., M.L., P.A. and J.D. The manuscript was drafted by S.D., Y.K., M.L., P.A. and J.D. Critical revision of the manuscript was performed by S.D., Y.K., T.M.M., S.T.E., C.W., M.L., P.A., J.D., A. Pezzini, V.T., H.S.M., E.T., A.M.S., J.C., J.J.M., P.S., I.F.-C., A.J.M., P.A.L., M.A.I., D.Z., T.T., M.L., D.L., P.A. and J.D. Annotation for expression quantitative trait loci was performed by A.D.J. Funding was obtained by S.D., S.T.E., A. Pezzini, V.T., H.S.M., M.D., S.B., A.J.G., P.M., J.J.M., P.S., B.G., F.-E.D.L., C.J., P.A.L., G.B.B., H.-E.W., M.C., D.A., M.M.B.B., J.F.M., A.R., B.B.W., E.-B.R., D.Z., T.T., M.L., D.L., P.A. and J.D.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Debette S, Leys D. Cervical-artery dissections: predisposing factors, diagnosis, and outcome. Lancet Neurol. 2009;8:668–678. doi: 10.1016/S1474-4422(09)70084-5. [DOI] [PubMed] [Google Scholar]
- 2.Rubinstein SM, Peerdeman SM, van Tulder MW, Riphagen I, Haldeman S. A systematic review of the risk factors for cervical artery dissection. Stroke. 2005;36:1575–1580. doi: 10.1161/01.STR.0000169919.73219.30. [DOI] [PubMed] [Google Scholar]
- 3.Debette S, et al. Association of vascular risk factors with cervical artery dissection and ischemic stroke in young adults. Circulation. 2011;123:1537–1544. doi: 10.1161/CIRCULATIONAHA.110.000125. [DOI] [PubMed] [Google Scholar]
- 4.Arnold M, et al. Vascular risk factors and morphometric data in cervical artery dissection: a case-control study. J Neurol Neurosurg Psychiatry. 2009;80:232–234. doi: 10.1136/jnnp.2008.151324. [DOI] [PubMed] [Google Scholar]
- 5.Debette S, Markus HS. The genetics of cervical artery dissection: a systematic review. Stroke. 2009;40:e459–e466. doi: 10.1161/STROKEAHA.108.534669. [DOI] [PubMed] [Google Scholar]
- 6.Kathiresan S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deloukas P, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freilinger T, et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat Genet. 2012;44:777–782. doi: 10.1038/ng.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anttila V, et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat Genet. 2013;45:912–917. doi: 10.1038/ng.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Debette S, et al. Differential features of carotid and vertebral artery dissections: the CADISP study. Neurology. 2011;77:1174–1181. doi: 10.1212/WNL.0b013e31822f03fc. [DOI] [PubMed] [Google Scholar]
- 11.Debette S, et al. CADISP-genetics: an international project searching for genetic risk factors of cervical artery dissections. Int J Stroke. 2009;4:224–230. doi: 10.1111/j.1747-4949.2009.00281.x. [DOI] [PubMed] [Google Scholar]
- 12.Wakefield J. Bayes factors for genome-wide association studies: comparison with P-values. Genet Epidemiol. 2009;33:79–86. doi: 10.1002/gepi.20359. [DOI] [PubMed] [Google Scholar]
- 13.Pezzini A, et al. Plasma homocysteine concentration, C677T MTHFR genotype, and 844ins68bp CBS genotype in young adults with spontaneous cervical artery dissection and atherothrombotic stroke. Stroke. 2002;33:664–669. doi: 10.1161/hs0302.103625. [DOI] [PubMed] [Google Scholar]
- 14.Pezzini A, et al. Migraine mediates the influence of C677T MTHFR genotypes on ischemic stroke risk with a stroke-subtype effect. Stroke. 2007;38:3145–3151. doi: 10.1161/STROKEAHA.107.491506. [DOI] [PubMed] [Google Scholar]
- 15.Longoni M, et al. The ICAM-1 E469K gene polymorphism is a risk factor for spontaneous cervical artery dissection. Neurology. 2006;66:1273–1275. doi: 10.1212/01.wnl.0000208411.01172.0b. [DOI] [PubMed] [Google Scholar]
- 16.Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673–680. doi: 10.1056/NEJM200003093421001. [DOI] [PubMed] [Google Scholar]
- 17.Low SK, et al. Genome-wide association study for intracranial aneurysm in the Japanese population identifies three candidate susceptible loci and a functional genetic variant at EDNRA. Hum Mol Genet. 2012;21:2102–2110. doi: 10.1093/hmg/dds020. [DOI] [PubMed] [Google Scholar]
- 18.Bilguvar K, et al. Susceptibility loci for intracranial aneurysm in European and Japanese populations. Nat Genet. 2008;40:1472–1477. doi: 10.1038/ng.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yasuno K, et al. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat Genet. 2010;42:420–425. doi: 10.1038/ng.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lemaire SA, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet. 2011;43:996–1000. doi: 10.1038/ng.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Donnell CJ, et al. Genome-wide association study for coronary artery calcification with follow-up in myocardial infarction. Circulation. 2011;124:2855–2864. doi: 10.1161/CIRCULATIONAHA.110.974899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu X, et al. Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet. 2012;44:890–894. doi: 10.1038/ng.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hager J, et al. Genome-wide association study in a Lebanese cohort confirms PHACTR1 as a major determinant of coronary artery stenosis. PLoS ONE. 2012;7:e38663. doi: 10.1371/journal.pone.0038663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehta NN. A genome-wide association study in Europeans and South Asians identifies 5 new loci for coronary artery disease. Circ Cardiovasc Genet. 2011;4:465–466. doi: 10.1161/CIRCGENETICS.111.960989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schunkert H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 27.Bevan S, et al. Genetic heritability of ischemic stroke and the contribution of previously reported candidate gene and genomewide associations. Stroke. 2012;43:3161–3167. doi: 10.1161/STROKEAHA.112.665760. [DOI] [PubMed] [Google Scholar]
- 28.Bellenguez C, et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet. 2012;44:328–333. doi: 10.1038/ng.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Traylor M, et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE Collaboration): a meta-analysis of genome-wide association studies. Lancet Neurol. 2012;11:951–962. doi: 10.1016/S1474-4422(12)70234-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smyth DJ, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. 2008;359:2767–2777. doi: 10.1056/NEJMoa0807917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calvet D, et al. Increased stiffness of the carotid wall material in patients with spontaneous cervical artery dissection. Stroke. 2004;35:2078–2082. doi: 10.1161/01.STR.0000136721.95301.8d. [DOI] [PubMed] [Google Scholar]
- 32.Golledge J, Eagle KA. Acute aortic dissection. Lancet. 2008;372:55–66. doi: 10.1016/S0140-6736(08)60994-0. [DOI] [PubMed] [Google Scholar]
- 33.Norman PE, Davis TM, Le MT, Golledge J. Matrix biology of abdominal aortic aneurysms in diabetes: mechanisms underlying the negative association. Connect Tissue Res. 2007;48:125–131. doi: 10.1080/03008200701331524. [DOI] [PubMed] [Google Scholar]
- 34.Shantikumar S, Ajjan R, Porter KE, Scott DJ. Diabetes and the abdominal aortic aneurysm. Eur J Vasc Endovasc Surg. 2010;39:200–207. doi: 10.1016/j.ejvs.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 35.Callaghan FM, et al. Wall stress of the cervical carotid artery in patients with carotid dissection: a case-control study. Am J Physiol Heart Circ Physiol. 2011;300:H1451–H1458. doi: 10.1152/ajpheart.00871.2010. [DOI] [PubMed] [Google Scholar]
- 36.Kurth T, et al. Headache, migraine, and structural brain lesions and function: population based Epidemiology of Vascular Ageing-MRI study. Br Med J. 2011;342:c7357. doi: 10.1136/bmj.c7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chasman DI, et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet. 2011;43:695–698. doi: 10.1038/ng.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Metso TM, et al. Migraine in cervical artery dissection and ischemic stroke patients. Neurology. 2012;78:1221–1228. doi: 10.1212/WNL.0b013e318251595f. [DOI] [PubMed] [Google Scholar]
- 39.Allen PB, Greenfield AT, Svenningsson P, Haspeslagh DC, Greengard P. Phactrs 1–4: a family of protein phosphatase 1 and actin regulatory proteins. Proc Natl Acad Sci USA. 2004;101:7187–7192. doi: 10.1073/pnas.0401673101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allain B, et al. Neuropilin-1 regulates a new VEGF-induced gene, Phactr-1, which controls tubulogenesis and modulates lamellipodial dynamics in human endothelial cells. Cell Signal. 2012;24:214–223. doi: 10.1016/j.cellsig.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 41.Jarray R, et al. Depletion of the novel protein PHACTR-1 from human endothelial cells abolishes tube formation and induces cell death receptor apoptosis. Biochimie. 2011;93:1668–1675. doi: 10.1016/j.biochi.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 42.Fils-Aimé N, et al. MicroRNA-584 and the protein phosphatase and actin regulator 1 (PHACTR1), a new signaling route through which transforming growth factor-β mediates the migration and actin dynamics of breast cancer cells. J Biol Chem. 2013;288:11807–11823. doi: 10.1074/jbc.M112.430934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goumans MJ, Liu Z, ten Dijke P. TGF-β signaling in vascular biology and dysfunction. Cell Res. 2009;19:116–127. doi: 10.1038/cr.2008.326. [DOI] [PubMed] [Google Scholar]
- 44.Loeys BL, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, et al. Genetic associations with expression for genes implicated in GWAS studies for atherosclerotic cardiovascular disease and blood phenotypes. Hum Mol Genet. 2014;23:782–795. doi: 10.1093/hmg/ddt461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Westra HJ, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bown MJ, et al. Abdominal aortic aneurysm is associated with a variant in low-density lipoprotein receptor–related protein 1. Am J Hum Genet. 2011;89:619–627. doi: 10.1016/j.ajhg.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee VH, Brown RD, Jr, Mandrekar JN, Mokri B. Incidence and outcome of cervical artery dissection: a population-based study. Neurology. 2006;67:1809–1812. doi: 10.1212/01.wnl.0000244486.30455.71. [DOI] [PubMed] [Google Scholar]
- 49.Pruim RJ, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 51.Thomas D. Gene—environment-wide association studies: emerging approaches. Nat Rev Genet. 2010;11:259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.