

Abstract
KRAS is the most frequently mutated oncogene in cancer and KRAS mutation is commonly associated with poor prognosis and resistance to therapy. Since the KRAS oncoprotein is, as yet, not directly druggable, efforts to target KRAS mutant cancers focus on identifying vulnerabilities in downstream signaling pathway or in stress-response pathways that are permissive for strong oncogenic signaling. One aspect of KRAS biology that is not well appreciated is the potential biological differences between the many distinct KRAS activating mutations. This review draws upon insights from both clinical and experimental studies to explore similarities and differences among KRAS alleles. Historical and emerging evidence supports the notion that the specific biology related to each allele might be exploitable for allele-specific therapy.
Keywords: RAS, KRAS, oncogene, cancer genetics, alleles
K-RAS is an Oncogenic GTPase
The Rat sarcoma (RAS) oncogene family is comprised of three members (KRAS, HRAS, and NRAS) that play an important role in human cancer [1]. All RAS genes encode 21 kD monomeric GTPases that function to transduce extracellular signals to intracellular signal transduction cascades. The on/off state of RAS proteins is determined by nucleotide binding, with the GTP-bound form existing in an active signaling conformation. Missense mutations in RAS proteins alter the homeostatic balance of GDP and GTP binding toward the active state, either by reducing GTP hydrolysis or by increasing the rate of GTP loading. There has been considerable work over the past several decades comparing mutant and wild-type (WT) forms of RAS [1, 2], with relatively little attention paid to potential differences between the specific mutations that give rise to the activated oncoproteins. This review explores the broad array of evidence for functionally distinct allelic forms of RAS. Although the concepts that are discussed are likely relevant to HRAS and NRAS, the focus herein is largely on KRAS because the vast majority of contemporary data – from biochemical to cellular to clinical/epidemiological – pertain to this family member. Indeed, KRAS is unique among the RAS family members in that its mutation frequency in human cancer is considerably higher than HRAS and NRAS and the KRAS gene encodes for two distinct protein forms (KRAS4A and KRAS4B) through alternative splicing. All of the common KRAS activating mutations produce mutant forms of both KRAS4A and KRAS4B.
KRAS Allelic Variation
As the sequences of whole cancer genomes become available, we are getting an unparalleled look at frequency and variability of KRAS alleles across the spectrum of primary and metastatic cancers (Figure 1A,B) [3]. Among the cancers where KRAS mutations are most common – pancreatic ductal adenocarcinoma (PDAC), colorectal cancer (CRC), and non-small cell lung cancer (NSCLC) – codon 12 mutations predominate, accounting for nearly 90% of all KRAS mutations (Figure 1C). Interestingly, the likelihood of different missense changes at codon 12 is dependent on cancer type (Figure 1D). In NSCLC, the increased frequency of specific alleles can be explained by invoking a mutational mechanism; the G12C (GGT to TGT) and G12V (GGT to GTT) alleles result from a classical smoking-induced mutation (G:C to T:A transversion, [4]) and are the most frequent KRAS mutations in NSCLC. This pattern of enrichment for similar mutational events does not extend beyond NSCLC, however, and it seems highly unlikely that the preponderance of specific codon 12 alleles in different cancer contexts can be explained by mutational patterns (Figure 1D). The frequency of mutations in other codons is variable, but non-codon 12 mutations account for a significant proportion on KRAS activating alleles in some cancers. For example, mutations in codons 13, 146, and 117 are common in CRC relative to NSCLC and PDAC (Figure 1C). Interestingly, when looking across the entire spectrum of cancers, mutations at codons 146 (A146T or A146V) and 117 (K117N or K117R) are nearly selective for CRC relative to other cancer types [5, 6] (Box 1).
Figure 1. KRAS Mutations in Human Cancers.
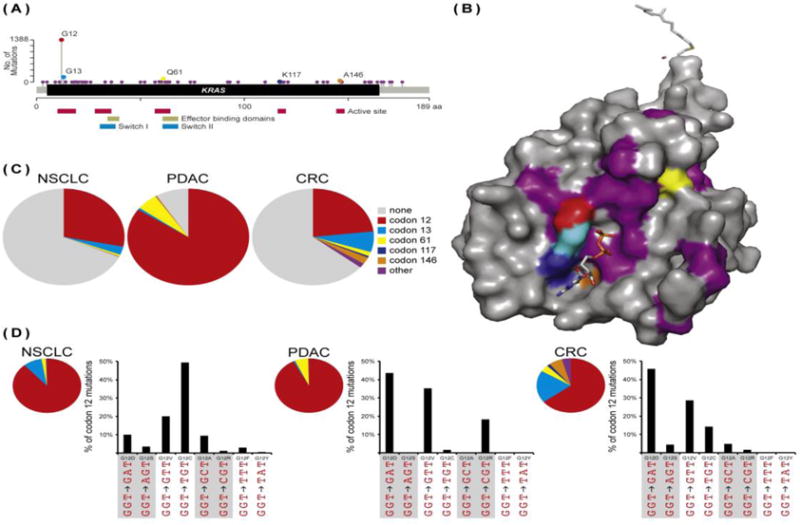
(A) The spectrum of cancer-associated KRAS mutations. Across all cancers, mutations in 51 different amino acids have been identified in at least one case. The functional significance of many of these mutations is unclear. Only 5 amino acids are mutated recurrently: codons 12, 13, 61, 117, and 146. Important functional regions of the protein are highlighted below the linear representation of the KRAS coding region. (B) Clustering of KRAS mutations. All of the common activating mutations alter the balance of KRAS•GDP to KRAS•GTP by affecting the active site of the enzyme. Commonly mutated residues are color-coded to correspond with panel A. This GDP-bound structure of full length KRAS (PDB code 5TAR) comes from [81]. (C) The diversity of KRAS alleles. Codon 12 mutations predominate in the “big 3” cancers: NSCLC, PDAC, and CRC. In NSCLC, codon 13 mutations also comprise a significant percentage of cancers, while in PDAC codon 61 mutations are more frequent. CRC stands out in the diversity of KRAS alleles. Here, codon 12 mutations account for only 65% of KRAS alleles. (D) Codon 12 allele choice. In NSCLC, the two most common alleles, G12C and G12V, result from the same type of mutation, a G to T transversion. In PDAC and CRC, codon 12 allele choice does not appear to be driven by mutational pattern. All data were collected from cBioportal [6].
Box 1. Rare KRAS alleles.
Among the “big 3” cancers that exhibit a high frequency of KRAS mutation, CRC stands out in its diversity of alleles. Indeed, across all cancers mutation of K117 and A146 is exceedingly rare, although their significant frequencies in CRC make them among the top 5 mutated codons across all cancers. Multiple myeloma (MM) is an interesting exception. The overall frequency of KRAS mutation in MM is quite high (22% of cases) and mutations in codons 117 and 146 occur at frequencies similar to CRC, representing 4% and 6% percent of alleles. Unlike CRC (and NSCLC and PDAC), however, codon 12 is not the most common site of mutation. Instead, Q61 is the most commonly mutated residue in MM [6]. The underlying molecular mechanisms that drive the unique diversity of KRAS mutations in CRC and MM are unclear, but might reflect the unique biology of these tissues or genetic interactions between KRAS and other mutations that are common in these contexts. While the vast majority of cancer associated KRAS mutations occur in codons 12, 13, 61, 117, and 146, there are rarer mutations that confer unique biological properties upon KRAS. For example, A59T has a 5-fold reduction in GTPase activity and a 10-fold increase in exchange, and is also a site of auto-phosphorylation in v-RAS [72]. A59E, a phospho-mimetic mutation, is as common as A59T in human cancers. How the autophosphorylation of A59T, or the phospho-mimetic activity of A59E, affects hydrolysis and exchange is not known. D119N, which occurs in a single B cell lymphoma cell line (NU-DUL-1), is an oncogenic exchange allele, but it also increases affinity for xanthosine nucleotides, making it an XTPase [73]. Is it possible that other exchange alleles affect KRAS function by altering its nucleotide preference? Finally, some rare oncogenic alleles were predicted long before they were found in human cancers [74]. One example is D33E was predicted to be oncogenic based on early structural studies, was later found to occur in NSCLC and CRC, and was identified in a relatively recent screen for oncogenic KRAS alleles [75].
One issue that cannot be overlooked when thinking about oncogenic KRAS mutants is the function of the WT allele. Data from genetically engineered mouse models suggest that loss of the WT KRAS allele enhances tumorigenesis induced by the mutant [7, 8]. Whether the WT allele is a bona fide tumor suppressor, or whether the ratio of WT and mutant alleles is the more critical factor, allelic imbalance caused by loss of WT KRAS or amplification of mutant KRAS is frequently seen in tumors from mice and humans and can affect the response to tumors to therapy [9, 10]. It remains to be determined whether specific alleles are more or less likely to be associated with allelic imbalance, but it stands to reason that mutant proteins with different biochemical properties could be differentially sensitive to up-regulation at the level of expression.
Biochemistry of KRAS Alleles
Even before mutations in cellular RAS (c-RAS) genes were found in human cancers, much of our fundamental knowledge about the biochemistry and cell biology of oncogenic forms of RAS proteins had been established. That RAS proteins bind GTP [11], are associated with the plasma membrane [12], and are prenylated [13] was established through studies of viral RAS (v-RAS). Comparisons between v-RAS and c-RAS – which differ at codons 12 and 59 – revealed that v-RAS exhibits autokinase activity and has reduced GTPase activity relative to the cellular form [14, 15]. The discovery that v-RAS and c-RAS differ in their GTPase activities led to the identification of cellular co-factors – guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs) – that regulate the nucleotide binding state of RAS proteins [2]. While most of these biochemical studies were performed on the viral or cellular forms of H-RAS, the catalytic domains of the RAS family members are nearly identical. As such, historical and contemporary studies of RAS biochemistry can reveal much about potential differences between KRAS alleles.
The structural conformation of KRAS, and therefore its biological activity, is governed by its nucleotide binding state. The ratio of KRAS•GDP to KRAS•GTP is controlled by the relative rates of exchange and hydrolysis and the oncogenic activity of mutant KRAS results from an increase in the rate of intrinsic nucleotide dissociation and/or a decrease in the rate of intrinsic or GAP-induced hydrolysis. The most common KRAS activating mutations (those at codons 12, 13, 61, 117, and 146) cluster around the nucleotide binding pocket (Figure 1B). G12 and G13 are located on the P-loop, which is necessary to stabilize the nucleotide in the active site, but they have distinct effects on the biochemistry of KRAS. Most codon 12 mutants affect both intrinsic and GAP-induced hydrolysis without changing the rate of nucleotide exchange, while codon 13 mutants affect hydrolysis and also increase intrinsic exchange by approximately 10-fold [16, 17]. Q61 is located at the N-terminus of switch II, where it participates in the conformational changes associated with this region during the interconversion between structural states. The current hypothesis for catalysis of GTP is that Q61 helps to coordinate and stabilize the transition state of the hydrolysis reaction. As such, Q61 mutants have the lowest hydrolysis rates among all KRAS alleles [16, 18].
The hydrocarbon portion of the K117 side chain packs against the guanine ring of the nucleotide and the charged amine group of K117 forms a salt-bridge with the backbone carbonyl of G13 in the P-loop, creating a dual role in stabilizing the nucleotide and promoting hydrolysis [19]. K117N, the most common cancer associated mutation, enhances nucleotide exchange [20]. A146 likely plays a role in nucleotide specificity, as this residue is present in a tightly packed space near the guanine base of the nucleotide [21]. Mutations at A146 promote nucleotide exchange (1,000-fold increase over WT) in the absence of guanine nucleotide exchange factor, but do not affect intrinsic GTPase activity [22].
In summary, it is clear that KRAS can be activated by alleles that have very large effects on hydrolysis (G12, Q61) or nucleotide exchange (A146), or by mutations that subtly affect hydrolysis and nucleotide exchange (G13, K117). This concept is demonstrated even more effectively by the inherited mutations that arise in the RASopathies, a group of inherited disorders – including Noonan syndrome (NS), Costello syndrome (CS), Cardiofaciocutaneous (CFC) syndrome, and Neurofibromatosis type 1 (NF1) – that result from germ line mutations in genes of the MAPK pathway, such as KRAS (Box 2).
Box 2. KRAS alleles in RASopathies.
The various RASopathy syndromes have overlapping, but distinct, phenotypic presentations, including cardiac defects, craniofacial features, developmental and growth delays, and neurocognitive impairment [76], and also have an increased risk of developing cancer [77]. One of the most common RASopathies, neurofibromatosis type I, is caused by mutation of the NF1 gene, with encodes for a RAS GAP. KRAS mutations are restricted to NS and CFC syndrome. Like cancer-associated KRAS alleles, most of the known RASopathy alleles increase the steady state levels of KRAS•GTP and they can do so in different ways [78]. Some alleles (V14I, Q22E, F28L, and F156L) enhance intrinsic and GEF-induced nucleotide exchange [78], while others (F28L, P34R, Q22E/R, G60R, D153V, and F156L) affect intrinsic and/or GAP-induced hydrolysis [20, 78]. K5N and D153V do not affect the GTPase cycle, but instead affect the orientation at the plasma membrane [79]. The mechanism of activation of T58I is very interesting. Like many of the germ-line and somatic KRAS mutants, it exhibits reduced intrinsic GTPase activity and loss of sensitivity to GAP-induced hydrolysis. Unlike most mutants, however, T58I shows selective sensitivity to GAP – it is resistant to NF1-induced hydrolysis, but retains sensitivity to p120 GAP-induced hydrolysis [80]. Although all of the RASopathy-associated KRAS alleles show some level of biochemical activation relative to WT protein, they are typically attenuated in their activation state relative to the common cancer-associated alleles. As such, there is likely to be a biochemical activation threshold that a given allele must cross in order to be oncogenic. Most RASopathy alleles fall below this threshold, but some – including mutations at V14, Q22, F28, T58, and G60 – have been identified in sporadic cancers.
Epidemiology of KRAS Alleles
One question that arises from somatic genetic analysis is whether KRAS allele choice influences the clinical aspects of a given cancer. The prognostic value of KRAS mutations has been studied extensive in many different cancer contexts and in some of these studies the role of different alleles has been examined. For example, in PDAC, G12D mutation, the most common allele, is associated with a lower probability of survival, while patients with G12R mutation fare better [23] (Figure 2A). In CRC, multiple independent analyses of large cohorts have identified a correlation between G12D and G12V mutations and worse overall survival [24– 26] (Figure 2B), while in patients with early stage NSCLC, although KRAS mutation is generally associated with shorter overall survival, patients with the frequent G12V or G12C mutations do better than those with rarer codon 12 mutations [27]. In short, the prognostic value of a codon 12 mutation depends upon (1) which allele is present and (2) which cancer is under investigation. If/how any of these observations might relate to the propensity for different codon 12 alleles to arise in different cancer contexts is unclear.
Figure 2. The Prognostic Significance of KRAS Alleles.
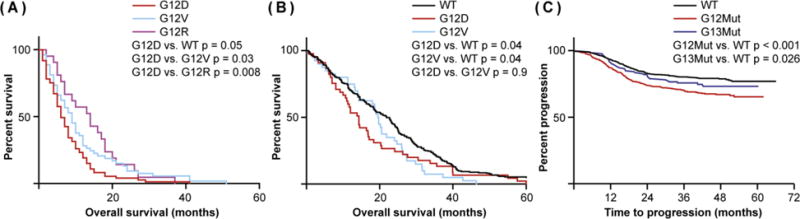
(A) Correlation between different codon 12 alleles and survival in pancreatic cancer. In PDAC, G12D mutations are associated with worse overall survival than are G12V mutations. G12R mutations, by contrast, correlate with better overall survival. Data are derived from [23]. (B) Correlation between different codon 12 alleles with survival in colorectal cancer. Unlike in PDAC, where G12V survival is significantly different from G12D, both mutations are associated with reduced overall survival in CRC. Data are derived from [26]. (C) Correlation between progression and mutations at different amino acids in colorectal cancer. Codon 12 mutations are associated with reduced progression-free survival in CRC, whereas progression is not statistically different in patients with WT or codon 13 mutant cancer. Data are derived from [29].
While alternate mutations at the same codon convey the subtleties of KRAS as a prognostic factor, comparing the relative prognostic value of mutations in different codons begins to highlight more clearly the apparent context dependence of KRAS oncogenicity. In CRC patients, codon 12 mutations are typically associated with worse overall survival, relative to patients with KRAS wild-type (WT) cancers, while codon 13 mutations are not [25, 28, 29] (Figure 2C), possibly because codon 12 mutations, but not codon 13 mutations, are associated with advanced disease and increased likelihood of lymph node metastasis [30]. Interestingly, the benefit of having a codon 13 mutation is restricted to cancers from the right side colon [29]. Moreover, CRC patients with codon 146 mutations appear to exhibit improved overall survival relative to patients with other mutations, while patients with codon 61 mutations exhibit poor overall and progression-free survival [31, 32]. Whether this difference in survival is a direct function of KRAS allele status, or whether it reflects a genotype-independent (i.e. physiologic) feature of CRC, is unclear; cancers with codon 12/13 mutations are enriched in right-side CRC, which have reduced survival relative to left-sided CRCs [33], while those with codon 61/146 mutations are equally distributed along the colon [34].
KRAS Mutational Status as a Predictive Factor
Chemotherapy remains the first-line treatment for most cancers. In patients with advanced CRC who are treated with folinic acid, fluorouracil, and oxaliplatin (FOLFOX), the standard first-line therapy for CRC, KRAS mutation is predictive of inferior response and this overall effect is driven largely by those cancers expressing G12D [35]. Consistent with the observation that patients with G12V mutations do better than those with other codon 12 mutations in terms of overall survival [27], late stage lung cancers expressing G12V appear to respond better to platinum-based chemotherapy than do cancers expressing other KRAS alleles [36]. This G12V-selective sensitivity to cisplatin was validated in isogenic cells lines ectopically expressing different codon 12 alleles, indicating that it is an intrinsic property of G12V-mutant lung cancers [37].
The effect of KRAS alleles on therapeutic response extends to targeted therapies as well. For example, NSCLC patients with G12C and G12V mutations exhibit shorter progression free survival in response to sorafinib, a RAF and vascular endothelial growth factor receptor (VEGFR) inhibitor, relative to those with other KRAS alleles [38]. The most significant association between KRAS and targeted therapies relates to response to inhibition of the epidermal growth factor receptor (EGFR) and there are significant and compelling data on this relationship. Antibody-based inhibition of EGFR was initially demonstrated to benefit patients who’s CRCs were refractory to other available therapies [39]. This effect was found to be specific to patients who did not have an exon 2 mutation (codons 12, 13) in KRAS [40]. Subsequent studies demonstrated that mutations in exons 3 and 4 of KRAS (codons 61, 146) could underlie the lack of response in patients that do not have codon 12 or 13 mutations [41, 42]. As a result, CRC patients with mutations in codon 12, 13, 59, 61, 117, and 146 in KRAS (or NRAS) are excluded from receiving antibody-based EGFR inhibitor therapy [43]. Given the extensive data on clinical responses to EGFR inhibition, it is possible to ask whether all KRAS alleles are equal in their ability to confer resistance to anti-EGFR therapy. Retrospective analyses suggest that CRCs with G13D mutations appear to respond to EGFR inhibition, while cancers with codon 12 mutations are resistant [44–46]. In contrast, codon 13 mutations are correlated with worse progression-free survival (PFS) in NSCLC patients treated with EGFR tyrosine kinase inhibitors (TKIs) [47, 48], highlighting potential alternate behavior of this allele in CRC and NSCLC.
Altogether, although clinical/epidemiological data are highly variable from study to study, there is some provocative evidence for clinically important allelic variation in KRAS. Nevertheless, it is important to understand the limitations of genetic/genomic and clinical/epidemiological studies of human cancers. To start, comparing allele frequencies is a useful way to think about potential differences between alleles, but it should not be used as a singular way to measure the importance of any given allele in cancer, especially given that some rare alleles (e.g. G12R in CRC) are associated more several clinical behaviors [25]. Moreover, like all clinical and epidemiological studies, those focusing specifically on KRAS alleles are complicated by issues with cohort size, tumor subtyping, tumor staging, treatments, endpoints, genetic background, and even the methods used to collect mutational data, making interpretation and validation difficult. Finally, given that the relative effects of different alleles are not consistent between cancers, the differences in prognostic and predictive power might not be linked directly or solely to KRAS function, but instead to more complicated issues of interaction with context or genetic background. For example, in CRC, PIK3CA mutations are more common in a KRAS mutant setting than in a KRAS wild-type setting [49], whereas KRAS and PIK3CA mutations are mutually exclusive in NSCLC [6].
Ectopic Expression of KRAS Alleles in Experimental Systems
While studies of human cancer patients are not yet able to provide a definitive link between KRAS and its unique alleles with particular clinical behaviors, they provide statistical correlations that are useful for generating experimentally testable hypotheses. The RAS field was keen to the concept of allelic variation very early on, although at that time, because isoform-specific functions were not as well appreciated, most studies focused on HRAS. For example, in cells ectopically expressing mutant forms of H-RAS, all amino acid substitutions at codon 12, with the exception of G12P, were found to induce transformation [50]. Many of these alleles are exceedingly rare in human cancers, however, because they require more than one nucleotide substitution. One of the first papers to relate differences in both biological and biochemical activities between alleles, and to highlight quantitative differences, focused on codon 61 mutations in H-RAS. In this study, 15 of 17 amino acid substitutions at codon 61 had transforming activity in mouse fibroblasts, with Q61V, Q61L, Q61K, Q61A having the greatest activity [51]. All mutants that were capable of transformation had a significant effect on intrinsic GTPase activity and GAP-induced hydrolysis [18, 51]. Although these studies focused on HRAS, rather than KRAS, it provides a means to compare in vitro transforming activity to in vivo mutation frequency. If one considers all of the single nucleotide changes that can change the amino acid at codon 61 (Figure 3A,B), the transforming activity of HRAS alleles in fibroblasts correlates extremely well with HRAS allele frequency in human cancers (Figure 3C), indicating that allele selection for HRAS is likely driven by biological differences between alleles.
Figure 3. In vitro Prediction of HRAS and KRAS Allele Frequencies.
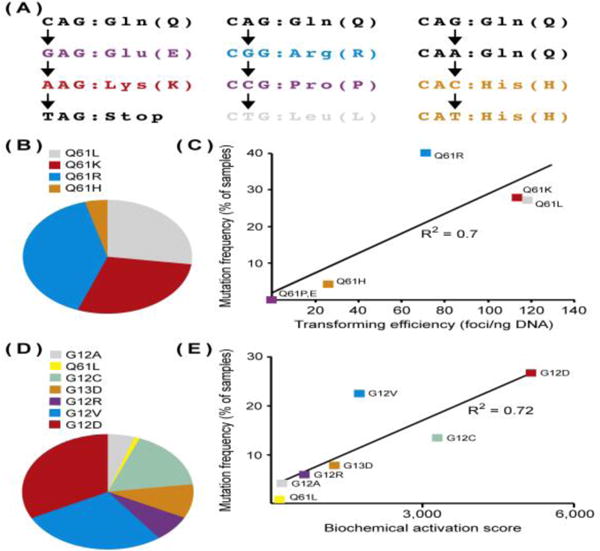
(A) HRAS codon 61 mutations resulting from single nucleotide changes. Q61 can be changed to six different amino acids. (B) Frequency of HRAS codon 61 alleles across human cancers. Of the six codon 61 alleles that can result from a single nucleotide change, only 4 are naturally occurring in human cancers. Data were collected from cBioportal [6]. (C) Correlation between in vitro transforming activity and allele frequency for HRAS codon 61 mutants. Alleles that do not occur in human cancers are not transforming in mouse fibroblasts. Data for transforming activity (defined as the number of NIH3t3 foci that develop per ng of transfected HRAS cDNA expression vector) were taken from [51]. (D) Relative frequencies of different KRAS alleles across all cancers. Note that this graph includes data only for the alleles listed. Data were collected from cBioportal [6]. (E) Correlation between in vitro biochemical activity and allele frequency for KRAS mutants. The biochemical activation score was calculated by multiplying the rate of intrinsic hydrolysis [Khydrolysis 10−5 (sec−1)] by the affinity for the RAS binding domain (RBD) of RAF-1 [nmol/L]. Data for biochemical activity were taken from [16].
When mutant forms of KRAS were expressed in murine fibroblasts, codon 12 alleles exhibited the strongest transforming activity, but only just slightly greater than those at codons 13, 61, 146 and 117 [50, 52, 53], possibly because codon 12 mutants were unique in their ability to promote PI3K signaling in this context [54]. Global transcriptional analysis of cells expressing mutant forms of KRAS provided an interesting observation; cells expressing different codon 12 alleles had similar gene expression patterns, but they were significantly different from those expressing G13D, Q61H, A146T, and K117N, even though the transforming activity of codon 12 mutants was only slightly greater than the others [53]. Indeed, it is likely that in vitro transformation assays read out only a portion of the oncogenic properties of KRAS, thus providing an incomplete view of the differences between alleles.
More recently, Park and colleagues ectopically expressed different oncogenic alleles of KRAS in the zebrafish pancreas [55]. Using tumor incidence as a measure of transforming activity, the strongest KRAS allele was G12R, followed by G12D, Q61L, G12V, G21C, A146T, Q61R, and G13D. Some alleles (G12A, G12F, G12S, G13C) did not induce any pancreatic tumors. The authors interpreted their results as evidence that PDAC selects for particular mutants. How do these data compare to human genetic data? In the zebrafish system, G12A and G12S alleles did not induce tumors and these alleles are extremely rare in human PDAC (Figure 1D). Nevertheless, G12R was a stronger allele than G12D in zebrafish, but human PDAC has much greater frequency of G12D than G12R (Figure 1D). Moreover, A146T induced tumors in zebrafish pancreas, but this allele is exceedingly rare in the human disease. It is likely that the ectopic expression of mutant alleles in the zebrafish system can overcome the weak transforming activity of some, but not all, KRAS alleles. This fact highlights the inherent limitations of using ectopic expression systems, which commonly cause super physiological expression of KRAS that can lead to mis-localization of KRAS to regions of the cell where it is not normally present and produce artifactual levels of biochemical activation.
Endogenous Mutant KRAS Alleles in Experimental Systems
One approach for studying the biology of endogenous alleles is to compare primary human cancers or human cancer cell lines with different KRAS mutations. For example, in a gene expression analysis of primary NSCLCs, those expressing G12C or G12V had distinct gene expression profiles relative to cell lines expressing other alleles [38]. The problem with these types of studies is that it is impossible to separate any given KRAS allele from the genetic background with which it is associated. This is, of course, a major problem with interpreting epidemiological studies as well.
An alternative to studying different alleles in different cell lines is to use isogenic cells engineered to express different alleles. For example SW48 CRC cells, which normally have WT KRAS, were engineered to express G12V, G12D, or G13D and then profiled for protein and phospho-protein expression via mass spectrometry [56]. The codon 12 mutants had a greater effect on the proteome of SW48 cells than did G13D, for example in the up-regulation of proteins involved in gluconeogenesis. DCLK1, which plays a role in colon cancer stem cells [57], was up-regulated in codon 12 mutants relative to WT and G13D, consistent with prior work demonstrating that G12D locks colon cancer cells into a stem-like state [58]. Using similar knock-in cells, G12V was found to promote resistance to anti-EGFR therapy, while cells expressing G13D and WT were sensitive [46]. This result is consistent with the clinical observation that patients with G13D CRCs benefit from anti-EGFR therapy [44–46]. Although there are caveats to using engineered isogenic cells (i.e. clonal selection, genetic drift), they do provide a more genetically controlled for directly comparing alleles than do cell line panels.
The laboratory mouse provides a genetically controlled in vivo experimental system in which to study allelic variation for KRAS. While the allelic diversity of genetically engineered mouse models (GEMMs) does not reflect human cancers – the commonly used GEMMs have G12D and G12V alleles [59, 60] – multiple studies have taken advantage of natural allelic selection that occurs following chemical mutagenesis. For example, WT mice treated with 3-methylcholanthrene develop late stage lung tumors harboring G12V, G12R, G12D, or G13D alleles [61]. These mice also develop benign lesions that have G12C mutations, suggesting that G12C is insufficient (on its own, and in this context) to promote cancer. Similarly, WT mice treated with urethane develop KRAS mutant lung adenomas and adenocarcinomas. The adenomas typically have Q61L mutations, while the adenocarcinomas typically have Q61R mutations [62], suggesting that these alleles have different in vivo activities. Interestingly, urethane induces Q61R mutations in animals carrying a single knockout allele of KRAS [63], suggesting an allele-specific interaction with the WT form of the protein.
Translating KRAS Biochemistry into Cellular Phenotypes
It is likely that the core biochemical properties (hydrolysis and exchange) of mutant KRAS proteins only partially account for their oncogenic activities. Mutant forms of KRAS differ from WT and from other mutants, for example, in their affinity for RAF and other effectors [16, 64]. And, although no single biochemical quality of the different alleles predicts KRAS allele frequency, a metric that accounts for GTPase activity and effector binding, a “biochemical activation score”, can predict allele frequency in primary human cancers (Figure 3D,E). Clearly, the oncogenic activity of different alleles at the cellular level is a function how much the mutation affects nucleotide binding state and, independently, the interaction with downstream effectors.
KRAS is commonly perceived as a “molecular switch” (Figure 4A). This is a useful conceptualization for a single molecule, but it is less relevant when thinking about the entire population of KRAS molecules in a cell. No single molecule can be partially “ON”, but the population of KRAS molecules is probably never strictly “ON” or “OFF”. By extension, it may be more appropriate to think of KRAS as a dimmer rather than a switch (Figure 4B). Changes to hydrolysis or exchange turn the dimmer right or left and the overall KRAS activation state, at the population level, is graded rather than binary. When this graded level of GTP binding state is combined with differing effector affinity of KRAS alleles, there is potential to produce widely varying quantitative levels of functional activation. This leads to the question of whether cancers with different alleles actually have different KRAS activation states, or whether compensatory mutations that co-occur with weak alleles serve to increase the overall level of KRAS activation.
Figure 4. A Non-switch Model for KRAS Activation.
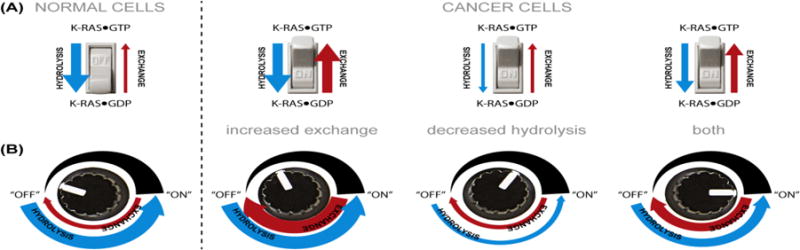
(A) The classical switch analogy. KRAS proteins are typically described as being “OFF” or “ON” as a function of nucleotide binding state. In a normal cell, the homeostatic balance of hydrolysis and exchange keeps KRAS in a GDP-bound state and the switch is OFF. Cancer cells can flip the switch by (1) increasing exchange, (2) decreasing hydrolysis, or (3) doing both. This model views the entire cellular population of KRAS molecules as a binary switch. (B) A dimmer analogy. This model does not consider the nucleotide binding state of individual proteins, but rather the “OFF” or “ON” state of the entire population of KRAS molecules in a cell. This model allows for different quantitative levels of KRAS activation, which is more consistent with the distinct mechanisms of activation of different activating mutations.
Prospects for Allele-Specific Therapies
The ultimately goal of studying individual KRAS mutations is to identify allele-specific therapeutic strategies. Thus far, this has only been achieved for G12C, where inhibitors have been developed that can covalently bind to the cysteine and inhibit the activated oncoprotein [65, 66]. One such inhibitor (ARS853) interacts selectively with GDP-bound KRAS and prevents the nucleotide exchange that is required for full activation of codon 12 alleles [67]. This is the same mechanism proposed for the inhibition of KRAS-induced transformation by lysine 104 acetylation [68]. While direct inhibition of other alleles has not been achieved, inhibition of exchange has been explored previously as a therapeutic strategy. For example, a synthetic peptide that binds to RAS and interferes with its SOS interaction inhibits GEF-induced nucleotide exchange [69]. Perhaps it would be possible to make a similar peptide that prevents SOS-independent nucleotide exchange in mutants that have increased intrinsic exchange, thereby locking these mutant KRAS molecules into a GDP-bound state.
An alternative approach is to identify allele-specific effector utilization. Because each allele has slightly different biochemical properties, the downstream pathways engaged are likely to be both quantitatively and qualitatively different [64], as highlighted in the prior studies that have compared gene expression and proteomic profiles of cells expressing different alleles [38, 56]. The best existing example of this strategy is the differential sensitivity of CRC cell lines expressing different KRAS alleles to inhibition of MEK1/2. Cell lines expressing A146T are sensitive to MEK1/2 inhibition as a single agent, while cell lines expressing other KRAS activating mutations are not [31]. The molecular mechanism underlying this differential response is not known, and whether this observation will extend to mouse models and/or human patients remains to be seen, but it provides a clue that allele-specific targeting of downstream effectors has potential as a therapeutic strategy.
Concluding Remarks
There is compelling evidence that passive mutational mechanisms and active biological selection drive allele choice in cancer [70]. For example, smoking-related mutation patterns clearly drive codon 12 allele selection in lung cancer (Figure 1D). In contrast, biochemical properties of different KRAS alleles likely contribute to allele selection over a broad spectrum of cancer types (Figure 3E). Other aspects of allele selection are less well characterized (see Outstanding Questions), for example whether KRAS expression level matters, as suggested recently in studies of codon usage [71]. In the end, the “how?” and “why?” of allele choice are less important than the “so what?”. The most important question of all is whether differences between alleles can be exploited to identify allele-specific therapeutic opportunities.
Outstanding Questions.
Are the epidemiological correlations with different alleles due to underlying biological differences between mutant forms of KRAS, for example in their abilities to promote metastasis, or are they due to something else, like allele-specific co-segregating mutations in other genes?
Do specific mutations preferentially affect the function of KRAS4A or KRAS4B?
Are different mutant forms of KRAS substrates for different GEFs and GAPs and do they preferentially engage distinct sets of effectors?
Studies of mouse models suggest that genetic background plays a role in a tumor’s choice of allele. Does the human genome carry modifiers of KRAS allele choice, for example single nucleotide polymorphisms that directly or indirectly affect KRAS expression?
Why are colorectal cancer and multiple myeloma distinct in the spectrum of KRAS alleles that arise? Is the diverse allelic spectrum somehow linked to the underlying biology of the tissue of origin?
Is the signaling downstream from KRAS alleles qualitative different or quantitative different? Are differences in downstream signaling sufficient to form the basis for allele-specific therapies?
Trends.
KRAS is the most commonly mutated oncogene in human cancer, with particularly high frequency in cancers of the pancreas, colon, and lung.
KRAS mutation is associated with poor prognosis, yet there are no effective therapies to specifically treat cancers expressing mutant forms of the KRAS oncoprotein.
The lack of detailed understanding of the biological properties of oncogenic KRAS has been an impediment to the identification of therapeutic targets.
Recent clinical, epidemiological, and experimental studies have provided clarity into the complexities of KRAS genetics in cancer.
Acknowledgments
I apologize to authors whose work was not cited due to space limitations, in particular those individuals who performed detailed early work on the biochemistry and structural biology of RAS proteins. A special thank you to Louis Buscail, Robert Jones, Pierre Laurent-Puig, Julien Taieb and Karine Le Malicot for sharing the clinical data that were used to create the curves in Figure 2. Christian Johnson generated the KRAS structure in Figure 1. This work was supported by NIH/NCI grants CA178017, CA199252, and CA195744.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Simanshu DK, et al. RAS Proteins and Their Regulators in Human Disease. Cell. 2017;170:17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zehir A, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edkins S, et al. Recurrent KRAS codon 146 mutations in human colorectal cancer. Cancer Biol Ther. 2006;5:928–32. doi: 10.4161/cbt.5.8.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z, et al. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat Genet. 2001;29:25–33. doi: 10.1038/ng721. [DOI] [PubMed] [Google Scholar]
- 8.To MD, et al. Interactions between wild-type and mutant Ras genes in lung and skin carcinogenesis. Oncogene. 2013;32:4028–33. doi: 10.1038/onc.2012.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Junttila MR, et al. Selective activation of p53-mediated tumour suppression in highgrade tumours. Nature. 2010;468:567–71. doi: 10.1038/nature09526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burgess MR, et al. KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell. 2017;168:817–829 e15. doi: 10.1016/j.cell.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scolnick EM, et al. Guanine nucleotide-binding activity as an assay for src protein of rat-derived murine sarcoma viruses. Proc Natl Acad Sci USA. 1979;76:5355–9. doi: 10.1073/pnas.76.10.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willingham MC, et al. Localization of the src gene product of the Harvey strain of MSV to plasma membrane of transformed cells by electron microscopic immunocytochemistry. Cell. 1980;19:1005–14. doi: 10.1016/0092-8674(80)90091-4. [DOI] [PubMed] [Google Scholar]
- 13.Sefton BM, et al. The transforming proteins of Rous sarcoma virus, Harvey sarcoma virus and Abelson virus contain tightly bound lipid. Cell. 1982;31:465–74. doi: 10.1016/0092-8674(82)90139-8. [DOI] [PubMed] [Google Scholar]
- 14.Shih TY, et al. Guanine nucleotide-binding and autophosphorylating activities associated with the p21src protein of Harvey murine sarcoma virus. Nature. 1980;287:686–91. doi: 10.1038/287686a0. [DOI] [PubMed] [Google Scholar]
- 15.Gibbs JB, et al. Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc Natl Acad Sci USA. 1984;81:5704–8. doi: 10.1073/pnas.81.18.5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunter JC, et al. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res. 2015;13:1325–35. doi: 10.1158/1541-7786.MCR-15-0203. [DOI] [PubMed] [Google Scholar]
- 17.Smith MJ, et al. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc Natl Acad Sci USA. 2013;110:4574–9. doi: 10.1073/pnas.1218173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donovan S, et al. GTPase activating proteins: critical regulators of intracellular signaling. Biochim Biophys Acta. 2002;1602:23–45. doi: 10.1016/s0304-419x(01)00041-5. [DOI] [PubMed] [Google Scholar]
- 19.Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 20.Schubbert S, et al. Biochemical and functional characterization of germ line KRAS mutations. Mol Cell Biol. 2007;27:7765–70. doi: 10.1128/MCB.00965-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pai EF, et al. Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 A resolution: implications for the mechanism of GTP hydrolysis. EMBO J. 1990;9:2351–9. doi: 10.1002/j.1460-2075.1990.tb07409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feig LA, Cooper GM. Relationship among guanine nucleotide exchange, GTP hydrolysis, and transforming potential of mutated ras proteins. Mol Cell Biol. 1988;8:2472–8. doi: 10.1128/mcb.8.6.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bournet B, et al. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin Transl Gastroenterol. 2016;7:e157. doi: 10.1038/ctg.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andreyev HJ, et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer. 2001;85:692–6. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imamura Y, et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin Cancer Res. 2012;18:4753–63. doi: 10.1158/1078-0432.CCR-11-3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones RP, et al. Specific mutations in KRAS codon 12 are associated with worse overall survival in patients with advanced and recurrent colorectal cancer. Br J Cancer. 2017;116:923–929. doi: 10.1038/bjc.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Izar B, et al. The prognostic impact of KRAS, its codon and amino acid specific mutations, on survival in resected stage I lung adenocarcinoma. J Thorac Oncol. 2014;9:1363–9. doi: 10.1097/JTO.0000000000000266. [DOI] [PubMed] [Google Scholar]
- 28.Margonis GA, et al. Association Between Specific Mutations in KRAS Codon 12 and Colorectal Liver Metastasis. JAMA Surg. 2015;150:722–9. doi: 10.1001/jamasurg.2015.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blons H, et al. Prognostic value of KRAS mutations in stage III colon cancer: post hoc analysis of the PETACC8 phase III trial dataset. Ann Oncol. 2014;25:2378–85. doi: 10.1093/annonc/mdu464. [DOI] [PubMed] [Google Scholar]
- 30.Li W, et al. Colorectal carcinomas with KRAS codon 12 mutation are associated with more advanced tumor stages. BMC Cancer. 2015;15:340. doi: 10.1186/s12885-015-1345-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janakiraman M, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 2010;70:5901–11. doi: 10.1158/0008-5472.CAN-10-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taieb J, et al. Adjuvant FOLFOX +/− cetuximab in full RAS and BRAF wildtype stage III colon cancer patients. Ann Oncol. 2017;28:824–830. doi: 10.1093/annonc/mdw687. [DOI] [PubMed] [Google Scholar]
- 33.Petrelli F, et al. Prognostic Survival Associated With Left-Sided vs Right-Sided Colon Cancer: A Systematic Review and Meta-analysis. JAMA Oncol. 2016;3:211–219. doi: 10.1001/jamaoncol.2016.4227. [DOI] [PubMed] [Google Scholar]
- 34.Morris VK, et al. Clinicopathologic characteristics and gene expression analyses of non-KRAS 12/13, RAS-mutated metastatic colorectal cancer. Ann Oncol. 2014;25:2008–14. doi: 10.1093/annonc/mdu252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zocche DM, et al. Global impact of KRAS mutation patterns in FOLFOX treated metastatic colorectal cancer. Front Genet. 2015;6:116. doi: 10.3389/fgene.2015.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cserepes M, et al. Subtype-specific KRAS mutations in advanced lung adenocarcinoma: a retrospective study of patients treated with platinum-based chemotherapy. Eur J Cancer. 2014;50:1819–28. doi: 10.1016/j.ejca.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Garassino MC, et al. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol. 2011;22:235–7. doi: 10.1093/annonc/mdq680. [DOI] [PubMed] [Google Scholar]
- 38.Ihle NT, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104:228–39. doi: 10.1093/jnci/djr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jonker DJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 40.Karapetis CS, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 41.Loupakis F, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101:715–21. doi: 10.1038/sj.bjc.6605177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Douillard JY, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 43.Allegra CJ, et al. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J Clin Oncol. 2015;34:179–185. doi: 10.1200/JCO.2015.63.9674. [DOI] [PubMed] [Google Scholar]
- 44.Tejpar S, et al. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol. 2012;30:3570–7. doi: 10.1200/JCO.2012.42.2592. [DOI] [PubMed] [Google Scholar]
- 45.Mao C, et al. KRAS p.G13D mutation and codon 12 mutations are not created equal in predicting clinical outcomes of cetuximab in metastatic colorectal cancer: a systematic review and meta-analysis. Cancer. 2013;119:714–21. doi: 10.1002/cncr.27804. [DOI] [PubMed] [Google Scholar]
- 46.De Roock W, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304:1812–20. doi: 10.1001/jama.2010.1535. [DOI] [PubMed] [Google Scholar]
- 47.Metro G, et al. Impact of specific mutant KRAS on clinical outcome of EGFR-TKI-treated advanced non-small cell lung cancer patients with an EGFR wild type genotype. Lung Cancer. 2012;78:81–6. doi: 10.1016/j.lungcan.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 48.Eberhard DA, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900–9. doi: 10.1200/JCO.2005.02.857. [DOI] [PubMed] [Google Scholar]
- 49.Rosty C, et al. PIK3CA activating mutation in colorectal carcinoma: associations with molecular features and survival. PLoS One. 2013;8:e65479. doi: 10.1371/journal.pone.0065479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seeburg PH, et al. Biological properties of human c-Ha-ras1 genes mutated at codon 12. Nature. 1984;312:71–5. doi: 10.1038/312071a0. [DOI] [PubMed] [Google Scholar]
- 51.Der CJ, et al. Biological and biochemical properties of human rasH genes mutated at codon 61. Cell. 1986;44:167–76. doi: 10.1016/0092-8674(86)90495-2. [DOI] [PubMed] [Google Scholar]
- 52.Cespedes MV, et al. K-ras Asp12 mutant neither interacts with Raf, nor signals through Erk and is less tumorigenic than K-ras Val12. Carcinogenesis. 2006;27:2190–200. doi: 10.1093/carcin/bgl063. [DOI] [PubMed] [Google Scholar]
- 53.Smith G, et al. Activating K-Ras mutations outwith ‘hotspot’ codons in sporadic colorectal tumours – implications for personalised cancer medicine. Br J Cancer. 2010;102:693–703. doi: 10.1038/sj.bjc.6605534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guerrero S, et al. K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res. 2000;60:6750–6. [PubMed] [Google Scholar]
- 55.Park JT, et al. Differential in vivo tumorigenicity of diverse KRAS mutations in vertebrate pancreas: A comprehensive survey. Oncogene. 2015;34:2801–6. doi: 10.1038/onc.2014.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hammond DE, et al. Differential reprogramming of isogenic colorectal cancer cells by distinct activating KRAS mutations. J Proteome Res. 2015;14:1535–46. doi: 10.1021/pr501191a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Westphalen CB, et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J Clin Invest. 2014;124:1283–95. doi: 10.1172/JCI73434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haigis KM, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40:600–8. doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tuveson DA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–87. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 60.Guerra C, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–20. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 61.Leone-Kabler S, et al. Ki-ras mutations are an early event and correlate with tumor stage in transplacentally-induced murine lung tumors. Carcinogenesis. 1997;18:1163–8. doi: 10.1093/carcin/18.6.1163. [DOI] [PubMed] [Google Scholar]
- 62.Nuzum EO, et al. Specific Ki-ras codon 61 mutations may determine the development of urethan-induced mouse lung adenomas or adenocarcinomas. Mol Carcinog. 1990;3:287–95. doi: 10.1002/mc.2940030509. [DOI] [PubMed] [Google Scholar]
- 63.Westcott PM, et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature. 2015;517:489–92. doi: 10.1038/nature13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith MJ, Ikura M. Integrated RAS signaling defined by parallel NMR detection of effectors and regulators. Nat Chem Biol. 2014;10:223–30. doi: 10.1038/nchembio.1435. [DOI] [PubMed] [Google Scholar]
- 65.Ostrem JM, et al. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–51. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lim SM, et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed Engl. 2014;53:199–204. doi: 10.1002/anie.201307387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lito P, et al. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351:604–8. doi: 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang MH, et al. Regulation of RAS oncogenicity by acetylation. Proc Natl Acad Sci USA. 2012;109:10843–8. doi: 10.1073/pnas.1201487109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patgiri A, et al. An orthosteric inhibitor of the Ras-Sos interaction. Nat Chem Biol. 2011;7:585–7. doi: 10.1038/nchembio.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ostrow SL, et al. Variation in KRAS driver substitution distributions between tumor types is determined by both mutation and natural selection. Sci Rep. 2016;6:21927. doi: 10.1038/srep21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ali M, et al. Codon bias imposes a targetable limitation on KRAS-driven therapeutic resistance. Nat Commun. 2017;8:15617. doi: 10.1038/ncomms15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.John J, et al. Biochemical properties of Ha-ras encoded p21 mutants and mechanism of the autophosphorylation reaction. J Biol Chem. 1988;263:11792–9. [PubMed] [Google Scholar]
- 73.Schmidt G, et al. Biochemical and biological consequences of changing the specificity of p21ras from guanosine to xanthosine nucleotides. Oncogene. 1996;12:87–96. [PubMed] [Google Scholar]
- 74.Grand RJ, Owen D. The biochemistry of ras p21. Biochem J. 1991;279:609–31. doi: 10.1042/bj2790609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim E, et al. Systematic Functional Interrogation of Rare Cancer Variants Identifies Oncogenic Alleles. Cancer Discov. 2016;6:714–26. doi: 10.1158/2159-8290.CD-16-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tidyman WE, Rauen KA. Pathogenetics of the RASopathies. Hum Mol Genet. 2016;25:R123–R132. doi: 10.1093/hmg/ddw191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013;14:355–69. doi: 10.1146/annurev-genom-091212-153523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gremer L, et al. Germline KRAS mutations cause aberrant biochemical and physical properties leading to developmental disorders. Hum Mutat. 2011;32:33–43. doi: 10.1002/humu.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mazhab-Jafari MT, et al. Oncogenic and RASopathy-associated K-RAS mutations relieve membrane-dependent occlusion of the effector-binding site. Proc Natl Acad Sci USA. 2015;112:6625–30. doi: 10.1073/pnas.1419895112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schubbert S, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331–6. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- 81.Dharmaiah S, et al. Structural basis of recognition of farnesylated and methylated KRAS4b by PDEdelta. Proc Natl Acad Sci USA. 2016;113:E6766–E6775. doi: 10.1073/pnas.1615316113. [DOI] [PMC free article] [PubMed] [Google Scholar]