

Abstract
Background
The favorable decay properties of 43Sc and 44Sc for PET make them promising candidates for future applications in nuclear medicine. An advantage 43Sc (T1/2 = 3.89 h, Eβ+ av = 476 keV [88%]) exhibits over 44Sc, however, is the absence of co-emitted high energy γ-rays. While the production and application of 44Sc has been comprehensively discussed, research concerning 43Sc is still in its infancy. This study aimed at developing two different production routes for 43Sc, based on proton irradiation of enriched 46Ti and 43Ca target material.
Results
43Sc was produced via the 46Ti(p,α)43Sc and 43Ca(p,n)43Sc nuclear reactions, yielding activities of up to 225 MBq and 480 MBq, respectively. 43Sc was chemically separated from enriched metallic 46Ti (97.0%) and 43CaCO3 (57.9%) targets, using extraction chromatography. In both cases, ~90% of the final activity was eluted in a small volume of 700 μL, thereby, making it suitable for direct radiolabeling. The prepared products were of high radionuclidic purity, i.e. 98.2% 43Sc were achieved from the irradiation of 46Ti, whereas the product isolated from irradiated 43Ca consisted of 66.2% 43Sc and 33.3% 44Sc. A PET phantom study performed with 43Sc, via both nuclear reactions, revealed slightly improved resolution over 44Sc. In order to assess the chemical purity of the separated 43Sc, radiolabeling experiments were performed with DOTANOC, attaining specific activities of 5–8 MBq/nmol, respectively, with a radiochemical yield of >96%.
Conclusions
It was determined that higher 43Sc activities were accessible via the 43Ca production route, with a comparatively less complex target preparation and separation procedure. The product isolated from irradiated 46Ti, however, revealed purer 43Sc with minor radionuclidic impurities. Based on the results obtained herein, the 43Ca route features some advantages (such as higher yields and direct usage of the purchased target material) over the 46Ti path when aiming at 43Sc production on a routine basis.
Electronic supplementary material
The online version of this article (10.1186/s41181-017-0033-9) contains supplementary material, which is available to authorized users.
Keywords: Radionuclide production, Cyclotron, 43Sc, 43Ca, 46Ti, Radiolabeling, PET phantom, PET/CT imaging
Background
Nuclear imaging methods offer the possibility to follow disease processes in the body on a cellular and molecular level, thus, providing valuable information to oncology, cardiology and neurology (Bybel et al. 2008, Kitson et al. 2009). The two most widely-employed imaging techniques in nuclear medicine are Single Photon Emission Computed Tomography (SPECT) and Positron Emission Tomography (PET) (Ramogida and Orvig 2013). Traditionally, short-lived, non-metallic PET radionuclides such as 11C, 13N, 15O and, primarily, 18F are used as tracers by their incorporation into small organic molecules via covalent bonds. However, radiolabeling of peptide, antibody and other protein-based targeting agents is hampered by elaborated radiosynthetic processes necessary to introduce short-lived radionuclides into more complex molecular structures (Wadas et al. 2010). Metallic radionuclides usually feature prolonged decay times and, thus, they are considered to be better matches for the previously-mentioned targeting moieties, having long biological half-lives. The incorporation of such radiometals into a chelator, which itself is conjugated to a biomolecule, becomes possible by exploiting their vast coordination chemistry (Wadas et al. 2010, Ramogida and Orvig 2013).
The radiometal 68Ga achieved an important role in oncological PET (Banerjee and Pomper 2013, Velikyan 2014), as its decay characteristics (T1/2 = 68 min, Eβ+ av = 830 keV, [89%]) allow the acquisition of high quality images, for example, the visualization of neuroendocrine tumors and their metastases by 68Ga-labeled somatostatin analogues, as demonstrated in a number of clinical studies (Gabriel et al. 2007, Kwekkeboom et al. 2010). The commercial 68Ge/68Ga generator system ensures an easy and flexible availability of 68Ga, however, only a limited quantity of radioactivity (equivalent to two patient doses when using a new generator) can be obtained per elution (Eppard et al. 2013, Rösch 2013). The short half-life of 68Ga entails a close proximity of the production facility in question, which is obliged to follow the guidelines of good manufacturing practice (GMP) in most countries, to an operating PET scanner (Breeman, et al. 2011). The feasibility of centralizing the production and distribution of 68Ga-radiopharmaceuticals is compromised by the resulting high overall costs which, in turn, encouraged the quest for alternate options.
In this respect, 44Sc was proposed as a suitable alternative to 68Ga for clinical PET imaging (Pruszynski et al. 2010, Rösch 2012). Its decay is characterized by the emission of positrons with lower energy (Eβ+ av = 632 keV [94%]) compared to 68Ga, allowing for PET imaging with a potentially improved spatial resolution (Bunka et al. 2016, Domnanich et al. 2016). Considering its physical half-life of 4.04 h (Garcia-Torano et al. 2016), centralized production of radiopharmaceuticals and their transportation to remotely-located hospitals becomes attainable. Additionally, with the employment of 44Sc, radiolabeling of a broader variety of biomolecules with slower pharmacokinetic profiles comes within reach (Chakravarty et al. 2014, van der Meulen et al. 2015). The production of 44Sc in sufficient amounts for radiopharmaceutical purposes, as well as the in vitro and in vivo characterizations of 44Sc-labeled compounds, was the topic of a number of studies (Rösch and Baum 2011, Pruszynski et al. 2012, Müller et al. 2013, Chakravarty et al. 2014, Hernandez et al. 2014, Singh et al. 2017). Moreover, the chemical behavior of Sc(III) was shown to more closely resemble those of the other rare earth elements, which are commonly used as therapeutics (e.g., 90Y and 177Lu) (Reubi et al. 2000, Majkowska-Pilip and Bilewicz 2011, Müller et al. 2013, Umbricht et al. 2017). It is hypothesized, however, that the clinical application of 44Sc may be compromised by the high dose burden to the personnel caused by the co-emission of 1157 keV γ-rays with 99.9% intensity.
It was since proposed to introduce another positron-emitting scandium radionuclide – 43Sc – which encompasses similarly favorable decay characteristics as 44Sc, but comes with a main γ-line of much lower energy and intensity (T1/2 = 3.89 h, Eβ+ av = 476 keV [88%], Eγ = 372 keV [23%]) (Walczak et al. 2015). To date, successful production of 43Sc was described by α-particle irradiation of natural calcium and enriched 40Ca through the nuclear reactions 40Ca(α,p)43Sc and 40Ca(α,n)43Ti → 43Sc, respectively. The obtained product was of high radionuclidic purity, and after its separation from the target material, successful radiolabeling was demonstrated using a derivative of the macrocyclic polyaminocarboxylic chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) (Szkliniarz et al. 2015, Walczak et al. 2015, Szkliniarz et al. 2016). Deuteron irradiation of enriched 42Ca targets was suggested as another possible 43Sc production channel, via the 42Ca(d,n)43Sc nuclear reaction (Walczak et al. 2015), however, the number of cyclotrons providing α-particle or deuteron beams is limited. The nuclear reactions 43Ca(p,n)43Sc and 46Ti(p,α)43Sc (Fig. 1) require only protons with low energies (<50 MeV) (International Atomic Energy Agency 2008, Koning et al. 2015, Experimental Nuclear Reaction Data (EXFOR) 2017), which can be generated by most biomedical cyclotrons and renders large-scale 43Sc production possible.
Fig. 1.
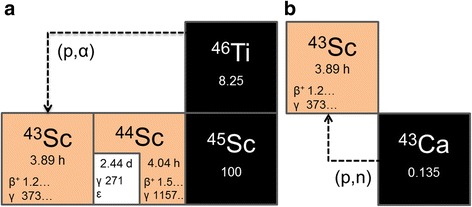
Production of 43Sc from 46Ti (a) and 43Ca (b) via the nuclear reactions 46Ti(p,α)43Sc and 43Ca(p,n)43Sc, respectively
In this work, the feasibility of proton-induced production of 43Sc using 43Ca and 46Ti as target materials has been demonstrated – to our knowledge – for the first time, with the final product quality tested by means of radiolabeling. The image resolution of 43Sc was investigated and compared to that of 44Sc using Derenzo phantoms and a preclinical PET scanner.
Methods
Chemicals
Enriched 46TiO2 (97.0 ± 0.2% 46Ti, 0.44% 47Ti, 2.28% 48Ti, 0.15% 49Ti, 0.13% 50Ti, Isoflex, USA) was reduced to metallic 46Ti powder with calcium hydride (CaH2, 98% metals basis, Mg <1%, Alfa Aesar, Germany; 99.9% trace metals basis, Sigma Aldrich, USA), argon (Ar, 99.9999%, Linde, Germany) and acetic acid (CH3COOH, 100% Suprapur, Merck, Germany) and then used as target material. Prior to irradiation, a preceding scan for trace metals (Ca, Cr, Cu, Fe, Ir, K, Mg, Mn, Mo, Na, Sb, Si, Sn, Sr, Ti, U, Y, Zn, Zr) was performed by ICP-OES (Perkin Elmer Optima 3000). Enriched 43CaCO3 (28.5% 40Ca, 1.05% 42Ca, 57.9 ± 1.8% 43Ca, 12.36% 44Ca, <0.003% 46Ca, 0.19% 48Ca, Trace Sciences International, USA) and graphite powder (99.9999%, Alfa Aesar, Germany) were used for the preparation of 43Ca targets. The chemical separation of Sc(III) from Ti(III) and Ca(II) was performed with N,N,N′,N′-tetra-n-octyldiglycolamide, non-branched resin (DGA, particle size 50–100 μm, TrisKem International, France). SCX cation exchange cartridges (100 mg Bond Elut SCX, particle size 40 μm, Agilent Technologies Inc., USA) served for the concentration of Sc(III). Furthermore, MilliQ water, hydrochloric acid (HCl, 30% Suprapur, Merck, Germany) and sodium chloride (NaCl, Trace Select, ≥99.999%, Fluka Analytical, Germany) were used for the chemical separation procedures. The application of oxalic acid dihydrate ((COOH)2·2H2O, Trace Select, ≥99.9999% metals basis, Fluka Analytical, Germany) and ammonia solution (NH3, 25%, Suprapur, Merck, Germany) enabled full recycling of the target material. DOTANOC acetate was obtained from ABX GmbH, Germany, and used for the radiolabeling of the final product as a means of quality control.
Reduction of enriched 46TiO2
The reduction of 46TiO2 to metallic 46Ti was performed with calcium hydride (Alfa Aesar) at Helmholtzzentrum für Schwerionenforschung (GSI) in Darmstadt, Germany. The detailed procedure has been outlined elsewhere (Lommel et al. 2014).
In order to increase the reduction yield, the reduction process for 46TiO2 was optimized at the Paul Scherrer Institute (PSI) with natural TiO2. Enriched 46TiO2 and natTiO2 (1.15 × 10−3 mol per tablet), respectively, were mixed with a 2–4 fold molar excess of CaH2 (2.3–4.6 × 10−3 mol per tablet) (Sigma Aldrich) and subsequently ground to a very fine powder, over a period of 25 min, with an agate mortar in a dry argon atmosphere. A tablet with a diameter of 10 mm was prepared by placing the finely-ground mixture in between two layers of ~80 mg CaH2 and pressing it with a pressure of 3 t for 30–40 s. This tablet was placed in a small tantalum boat inside a nickel tube, which was evacuated to pressures of 10−3–10−5 mbar. The temperature was gradually increased to 800–1000 °C over a period of 60–120 min and maintained at this level for about 30 min. After cooling to room temperature, the reduction products were retrieved and the metallic 46Ti isolated from the co-produced calcium oxide using dilute acetic acid. Further details on the isolation procedure can be found elsewhere (Lommel et al. 2014). The resultant reduced 46Ti metal was directly used for the preparation of the targets.
The reduction yield was determined by boiling an aliquot of the reduced product (approx. 5–10 mg) in 2–3 mL concentrated HCl for 10–15 min. Under these conditions, the reduced 46Ti metal was dissolved completely (Straumanis and Chen 1951) while the insoluble residue, consisting of TiO and TiO2 (Perry 2011, Rumble 2018), was collected and weighed. The ratio of soluble to insoluble species served as an indication for the degree of reduction. X-ray diffraction (XRD) analysis (Philips X’PertPro X-ray diffractometer, wavelength: Cu Kα = 1.541 Å) was additionally employed to identify the chemical speciation of the product.
Manufacturing and irradiation of 46Ti and 43CaCO3 targets
Targets were prepared by placing 9–28 mg reduced 46Ti metal powder or 8–12 mg enriched 43CaCO3 on top of ~150 mg graphite powder and pressed into pills with a pressure of 5–7 t. The resulting thickness of the target in question was between 0.4 and 0.5 mm with a diameter of 16 mm. After encapsulation in aluminum, the target was introduced into a target holder system. The Injector 2 cyclotron at PSI produces 72 MeV protons and was used for the irradiations presented herein. The required lower beam energies were achieved using niobium degrader discs of various thicknesses (3.2–3.5 mm). 46Ti targets were irradiated with proton energies of 15.1 ± 1.9 MeV at beam currents of 30 μA for 60–420 min, whereas, proton energies of 12.0 ± 2.3 MeV and 10.4 ± 2.6 MeV, respectively, at 50 μA were applied to the 43CaCO3 targets for 90–220 min. The impinging energies were calculated with SRIM-2010 (Ziegler et al. 2010). After the end of bombardment (EOB), the activated targets were detached from the target station and the aluminum encapsulation removed.
Separation of 43Sc from 46Ti
A chemical separation system (schematic shown in Fig. 2) was developed to separate 43Sc from 46Ti. The irradiated target was transferred into a conical glass vial (30 mL, Schmizo AG, Switzerland) with an integrated charcoal filter (2 mL chromatography column, BioRad, France, filled with charcoal, Merck, Germany)on top. The target was dissolved in 4–5 mL of boiling 8.0 M HCl for 15–20 min. The concentration of the obtained HCl solution was adjusted to ~4.0 M with the addition of MilliQ water. The insoluble graphite disc remained in the glass vial while the radioactive solution was pumped through a filter (1 mL cartridge fitted with a 10 μm frit, ISOLUTE SPE Accessories, UK) before loading onto a 1 mL column cartridge, containing ~85 mg DGA extraction chromatographic resin. Negligible sorption of Ti(III) and a simultaneous strong Sc(III) retention on DGA resin at HCl molarities below 6 M (Pourmand and Dauphas 2010) facilitated its application for the separation of these elements. The reaction vessel was rinsed with 2.0 mL 4.0 M HCl and the solution pumped through the DGA resin. Further 5.0 mL 4.0 M HCl were applied directly onto the DGA column to ensure complete removal of residual 46Ti. The 43Sc fraction was finally eluted with 4.0 mL 0.1 M HCl directly onto a second column containing SCX cation exchange resin to concentrate the 43Sc in a small volume. From this resin, 43Sc was eluted with 700 μL 4.8 M NaCl/0.13 M HCl (pH 0–0.5) and partitioned into three fractions. The volumes of the fractions were 100, 400 and 200 μL, respectively, with the second fraction directly used for radiolabeling.
Fig. 2.
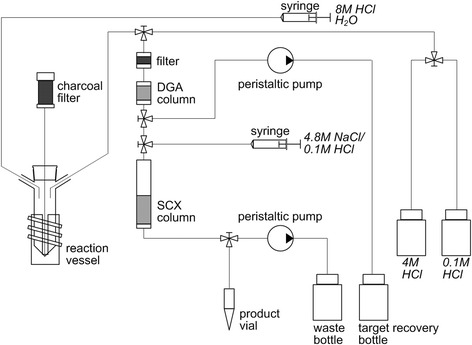
Schematic diagram of the 46Ti/43Sc separation system
Separation of 43Sc from 43Ca
The chemical separation of 43Sc from 43Ca was performed as for the previously reported procedure for 44Sc (van der Meulen et al. 2015). In brief, the irradiated target was dissolved in HCl and the separation of 43Sc performed on a DGA extraction chromatographic column. 43Sc was directly eluted onto a SCX cation exchange resin cartridge, from where it was eluted in a small volume of NaCl/HCl and directly used for radiolabeling.
Target recycling
The Ti-containing eluate collected from the DGA column was heated to boiling and the pH adjusted to 8.0 with 25% ammonia solution. As a consequence, a black, flaky precipitate formed immediately and transformed into white TiO2 over a waiting period of 40 min. The precipitate was filtered through a glass filter crucible (30 mL, pore size 10–16 μm, Duran Group GmbH, Germany), heated to 400 °C, in air, and kept at this temperature for 1 h to ensure complete oxidation. XRD-measurements were employed for the specification analysis of the recovered target material.
Recycling of the valuable enriched 43Ca target material from the collected waste fraction was performed as described previously for 44Ca (van der Meulen et al. 2015).
Radionuclidic purity
A N-type high-purity germanium (HPGe) coaxial detector (Canberra, France) in combination with the InterWinner software package (version 7.1, Itech Instruments, France) was employed for analyzing the radionuclidic purity of the final products. The measurements were performed with an aliquot of 2.5–5.0 MBq 43Sc and the counting time adjusted to ensure a statistical measurement error of <5%. Further γ-spectroscopy measurements of the same samples with long counting times were performed several days post-separation, aiming to determine low-level activities originating from long-lived radionuclidic impurities.
PET phantom study using 43Sc
An aliquot was withdrawn from the second fraction of the respective eluate and diluted with 70% ethanol to a volume ratio of 3:1 (aliquot of 43Sc eluate: ethanol). The holes (diameter ranging from 0.8–1.3 mm in 0.1 mm steps) of a polycarbonate Derenzo phantom (D = 19.5 mm, H = 15.0 mm) were filled with 600 μL of the diluted solution. Particular care was taken to avoid the inclusion of air bubbles (Bunka et al. 2016). The phantom was closed with a screw cap and aligned on a small-animal PET/CT scanner (eXplore VISTA PET/CT, GE Healthcare, Spain). The determined total radioactivity in the phantom was ~4–8 MBq at the beginning of the PET scan. The energy window was set to 400–700 keV and each phantom scanned for 30 min. Using the post-processing software VivoQuant™ (version 2.00, inviCRO Imaging Service and Software, Boston USA), one representative single transversal section was selected and analyzed at three different phantoms depths. The resulting intensity plots of the profiles were transferred to Origin® 2016 (OriginLab). The full-width at half-maximum (FWHM) was determined for each slice and used for calculating the arithmetic mean and standard deviation. A detailed description of the quantification of the relative resolution is described by Bunka et al. (Bunka et al. 2016).
Radiolabeling for quality control of the produced 43Sc
Radiolabeling of a DOTA-functionalized peptide at pre-defined specific activities was employed for quality control of the product. As non-radioactive metal impurities would compete with the radionuclide for complexation by a DOTA chelator, this method serves as a reasonable benchmark to evaluate the success of a chemical separation (Severin et al. 2012, Valdovinos et al. 2014). After separation of 43Sc from the 46Ti and 43CaCO3 target material, the activity of the obtained eluate was quantitatively determined with a dose calibrator (ISOMED 2010, Nuclear-Medizintechnik Dresden, GmbH, Germany – calibrated on a fortnightly basis). The quality of the 43Sc was investigated by means of its radiolabeling capability with DOTANOC. The required activity (20–50 MBq) was withdrawn from the vial and mixed with sodium acetate solution (0.5 M, pH 8), in order to obtain a pH of 3.5–4.5, followed by the successive addition of DOTANOC (3.5–14.3 μL of a 0.7 mM solution in MilliQ water, ABX GmbH, Advanced Biochemical Compounds, Germany). The reaction mixture was incubated at 95 °C for 15 min. High performance liquid chromatography (HPLC) with a C-18 reversed-phase column (Xterra™ MS, C18, 5 μm, 150 × 4.6 mm, Waters, USA) was employed in order to determine the radiolabeling yield. Before analysis, the addition of 10 μL 2 mM Na-DTPA solution ensured the complexation of free radiometals. A UV (LaChrom L-7400) and a radiodetector (Berthold, HPLC Radioactivity Monitor, LB 506B) were used for detection. The analysis sequence comprised the gradual change of the mobile phase from 95% A (MilliQ water containing 0.1% trifluoracetic acid) and 5% B (acetonitrile) to 20% A and 80% B, over a period of 15 min and at a flow rate of 1.0 mL/min.
Results
Reduction of enriched 46TiO2
The yield of the reduction procedure of 46TiO2 to metallic 46Ti, performed at GSI, was determined by dissolution of the metallic product in conc. HCl as 95.7%. Comparable yields of 90–98% were verified by the authors of the study when reducing 50TiO2 and employing energy-dispersive X-ray spectroscopy (EDX) for analysis (Lommel et al. 2014).
While the method using the vacuum-based system at PSI was successful, several parameters were optimized using natTiO2, aiming to enhance the yield of the reduction process. The use of four-fold surplus of reducing agent in a finer granulated form with 46TiO2 and natTiO2 was found to be a suitable starting mixture. The mixed powder was embedded in between two layers of CaH2. Reduction yields of 96–99%, determined by the dissolution test in boiling HCl and verified by XRD analysis, were achieved at pressures <10−4 mbar and by increasing the temperature to 950–1000 °C. The XRD spectra are given in the Additional file 1: Figure S2 a-b.
Production of 43Sc from 46Ti via the (p,α) nuclear reaction
The activities of the 46Ti-targets at the end of bombardment (EOB) ranged between 60 and 225 MBq 43Sc, however, theoretically achievable activities (A(43Sc)calc) were estimated to be between 590 and 2340 MBq (Table 1). The calculations were performed by taking into account the mass of 46Ti, the irradiation time (tirr), the proton flux (Φ) and the cross section for the reaction 46Ti(p,α)43Sc amounting to 36 ± 2 mbarn (Carzaniga et al. 2017). Differences between the experimental and the calculated activity are expressed by the factor f(43Sc), which were typically in the range between 5.6 and 12.3. The values of f(43Sc) characterize how many times larger the theoretical activity is than that experimentally obtained. In one particular production run, an exceptionally high activity of ~1.0 GBq 43Sc was generated under identical irradiation conditions. The resulting, rather low value of f(43Sc) = 1.9, clearly demonstrates the potential of this approach. Formulae used for the calculations of A(43Sc)calc and f(43Sc) are given in the Additional file 1: Figure S1 a-b.
Table 1.
Comparison between the experimental A(43Sc)exp and the calculated activities A(43Sc)calc, obtained from proton irradiations of enriched 46Ti targets, measured at EOB
| Number of irradiations | m(46Ti) [mg] | tirr [min] | A(43Sc)exp at EOB [MBq] | A(43Sc)calc [MBq] | f(43Sc) |
|---|---|---|---|---|---|
| 2 | 10 | 180–240 | 110–140 | 930–990 | 7.2–8.4 |
| 5 | 11 | 110–240 | 60–180 | 590–1080 | 6.0–12.2 |
| 3 | 12 | 120–240 | 130–150 | 700–1180 | 5.6–8.7 |
| 1 | 15 | 390 | 1030 | 1990 | 1.9 |
| 1 | 16 | 420 | 225 | 2210 | 9.9 |
| 1 | 17 | 420 | 190 | 2340 | 12.3 |
Chemical behavior of 43Sc and 46Ti on DGA resin and their separation
The chemical behavior of Sc(III) and Ti(III) on DGA resin was investigated using ~30 mg naturalTi metal, spiked with trace amounts of radioactive 46Sc (1.7 kBq) and 44Ti (2.3 kBq). γ-Spectroscopy was employed in order to quantify the 46Sc and 44Ti radioactivity in each fraction (the resulting elution profile is shown in Fig. 3). Using 9.2 mL 4.0 M HCl solution, Sc(III) was quantitatively sorbed on DGA resin, while Ti(III) was not retained. Before the final elution of Sc(III) with 4.0 mL 0.1 M HCl, the resin was rinsed with additional 5.0 mL 4.0 M HCl to ensure complete removal of Ti(III).
Fig. 3.
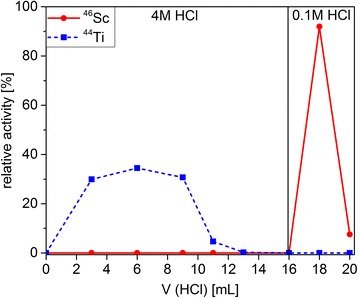
Elution profile of 44Ti/nat.Ti (blue squares) and 46Sc (red dots) on DGA extraction chromatographic resin. Each fraction was measured by γ-spectroscopy until the statistical measurement error was <5%
With the developed separation system (Fig. 2), 89.7 ± 3.1% of the total 43Sc activity could be eluted in a small volume using 4.8 M NaCl/0.13 M HCl as eluent. Fractionized collection revealed that ~90% of the eluted 43Sc activity were obtained in the second fraction (400 μL), the remaining 10% were divided between the first (100 μL) and third fraction (200 μL). The residual overall 43Sc activity (~10%) was left on the graphite disc, the DGA and SCX resin columns, respectively.
Production of 43Sc from 43Ca via the (p,n) nuclear reaction and separation
Total activities of 380–720 MBq were generated by the irradiation of 43CaCO3 targets, which consisted of 66.2% 43Sc and 33.3% co-produced 44Sc (Table 3), consequently, resulting in final yields of 250–480 MBq pure 43Sc radioactivity at EOB (Table 2). Theoretically accessible activities of 1270–3200 MBq 43Sc were calculated based on beam energies of 12 MeV at a corresponding cross section of 275 mbarn (Carzaniga et al. 2017). The discrepancies between the experimental and the calculated activities, once more expressed by the factor f(43Sc), were within the range between 3.9 and 6.8, which is slightly lower than those determined for the 46Ti(p,α)43Sc route.
Table 3.
The radionuclide inventory of the 43Sc eluate, isolated from irradiated 46Ti and 43CaCO3 targets is shown, together with the calculated values at EOB. Cross section data for the nuclear reactions 46Ti(p,α)43Sc, 43Ca(p,n)43Sc and 44Ca(p,n)44Sc were taken from Carzaniga et al. (Carzaniga et al. 2017), while the data for all other nuclear reactions was retrieved from the TENDL 2015 database (Koning et al. 2015)
| 43Sc eluate isolated from irradiated 46Ti | ||||
| Radionuclide inventory at EOB [%] | ||||
| Isotope | Nuclear reaction | Calculated prediction | Experimental results | |
| 14.6/15 MeV | 15.1 ± 1.9 MeV | |||
| 43Sc | 46Ti(p,α)43Sc | 99.1 | 98.2 ± 0.3 | |
| 44gSc | 47Ti(p,α)44gSc | 0.9 | 1.5 ± 0.2 | |
| 44mSc | 47Ti(p,α)44mSc | 1.3 × 10−2 | 4.2 × 10−2 ± 1.6 × 10−2 | |
| 46Sc | 47Ti(p,2p)46Sc49Ti(p,α)46Sc | 2.2 × 10−4 | 1.1 × 10−2 ± 5.7 × 10−3 | |
| 47Sc | 50Ti(p,α)47Sc48Ti(p,2p)47Sc | 3.0 × 10−3 | 9.6 × 10−3 ± 4.7 × 10−3 | |
| 48Sc | 49Ti(p,2p)48Sc | 1.6 × 10−7 | 1.7 × 10−2 ± 7.0 × 10−3 | |
| 86Y, 87Y, 87mY, 88Y | – | 0.16, 2.7 × 10−2, 9.5 × 10−2, 1.0 × 10−2 | ||
| 43Sc eluate isolated from irradiated 43CaCO3 | ||||
| Radionuclide inventory at EOB [%] | ||||
| Isotope | Nuclear reaction | Calculated prediction | Experimental results | |
| 9.9/10.0 MeV | 12.4/12.0 MeV | 10.4 ± 2.6/12.1 ± 2.3 MeV | ||
| 43Sc | 43Ca(p,n)43Sc | 67.4 | 65.5 | 66.2 ± 1.5 |
| 44gSc | 44Ca(p,n)44gSc | 32.4 | 34.3 | 33.3 ± 1.5 |
| 44mSc | 44Ca(p,n)44mSc | 0.1 | 0.2 | 0.2 ± 5.3 × 10−2 |
| 46Sc | 46Ca(p,n)46Sc | 1.5 × 10−5 | 1.2 × 10−5 | – |
| 47Sc | 48Ca(p,2n)47Sc | 2.0 × 10−2 | 2.6 × 10−2 | 2.2 × 10−2 ± 1.0 × 10−2 |
| 48Sc | 48Ca(p,n)48Sc | 5.7 × 10−2 | 2.3 × 10−2 | 0.1 ± 2.9 × 10−2 |
| 86Y, 87Y | – | – | 0.2, 1.0 × 10−2 | |
Table 2.
Comparison of the measured total activities A(43/44Sc)exp, the experimental 43Sc activities A(43Sc)exp as well as the calculated activities A(43Sc)calc, obtained from proton irradiations of enriched 43CaCO3 targets, measured at EOB
| Number of irradiations | m(43CaCO3) [mg] | tirr [min] | A(43/44Sc)exp at EOB [MBq] | A(43Sc)exp at EOB [MBq] | A(43Sc)calc [MBq] | f(43Sc) |
|---|---|---|---|---|---|---|
| 1 | 8 | 90 | 380 | 250 | 1270 | 5.0 |
| 4 | 9 | 90–220 | 440–670 | 290–440 | 1420–2910 | 3.9–6.6 |
| 1 | 10 | 220 | 720 | 480 | 3200 | 6.8 |
The isolation of 90.4 ± 5.5% of the total 43Sc/44Sc activity was possible in a small volume (700 μL) of 4.8 M NaCl/0.13 M HCl eluent by using the previously-developed separation system (van der Meulen et al. 2015). The residual ~10% of 43Sc/44Sc activity were left on the remaining components of the setup, e.g. graphite disc, DGA and SCX resin columns, respectively.
Target recycling
A γ-spectroscopic measurement of the 46Ti-containing fraction indicated the presence of 48V (T1/2 = 16 days), presumably being formed in the nuclear reaction 48Ti(p,n)48V. In order to avoid any co-precipitation, the liquid was set aside until 48V was completely decayed to stable 48Ti. Consequently, the target recycling process was developed with natural titanium. The achieved overall recovery yield for the precipitation of natTiO2 was 97.6%, with XRD measurements confirming the chemical identity of the product. The XRD spectrum is given in the Additional file 1: Figure S3.
The recovery of enriched 43Ca target material was performed according to the previously-described method used for 44Ca (van der Meulen et al. 2015) at an equivalent efficiency. The 43Sc obtained by irradiation of recovered material, proved to be of the same quality as with targets from newly-purchased 43CaCO3. An unchanged radionuclidic purity of the obtained 43Sc eluate confirmed the absence of trace element impurities in the recycled 43CaCO3.
Radionuclidic purity of 43Sc produced from 46Ti and 43Ca target material
Irradiation of 46Ti targets (97.0% enriched) with protons yielded a product of high radionuclidic purity, containing 98.2% 43Sc and only 1.5% 44Sc. In comparison, the 43Sc eluate isolated from proton irradiated 43CaCO3 (57.9% enriched) contained 66.2% 43Sc and 33.3% 44Sc. Long-term γ-spectroscopic measurements determined low activity levels of 0.079% 44mSc, 46Sc, 47Sc, 48Sc and 0.34% 44mSc, 47Sc, 48Sc in the final products of irradiated 46Ti and 43Ca targets, respectively (Table 3). All radionuclides of Sc were formed in (p,n), (p,2n), (p,α) and (p,2p) nuclear reactions, with stable isotopes of titanium (46Ti, 47Ti, 48Ti, 49Ti, 50Ti) and calcium (40Ca, 42Ca, 43Ca, 44Ca, 46Ca, 48Ca), being present in the respective target material. In the case of the 46Ti/43Sc production route, the amount of 46Sc was found to exceed the prediction by a factor of ~50, while no long-lived 46Sc, even if predicted, could be determined in the final eluate available from the 43Ca/43Sc route. Low levels of various Y radionuclides (86Y, 87Y, 87mY and 88Y) were present in products isolated from 46Ti and 43CaCO3 target material, in total, amounts of 0.29% and 0.19%, respectively. No Y isotopes were observed in the 43Sc eluate obtained from 46Ti target material which was reduced at PSI, however.
PET phantom study
PET images of Derenzo phantoms were acquired on a small-animal PET/CT scanner using 44Sc as well as 43Sc, produced via the 46Ti and 43Ca routes (radionuclidic purity: 98.2% and 66.2% 43Sc), respectively. A simple visual comparison already suggests a favorable image quality and an improved resolution for 43Sc in comparison to 44Sc (Fig. 4). The numerical expression of these differences was derived by means of the FWHM, which was determined for a hole diameter of 1.3 mm. The resolution was found to be the best for 43Sc obtained from 46Ti, followed by 43Sc from 43Ca and, finally, by 44Sc (Table 4), hence, the calculated FWHM values corroborate the visual evaluation. The observed sequence is in agreement with the expectations according to the average positron energies of the respective radionuclides.
Fig. 4.
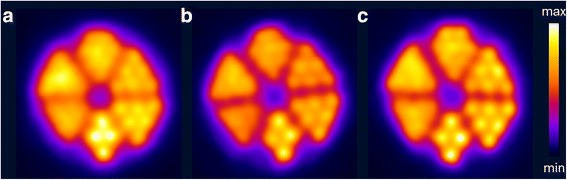
Transversal slices of PET scans of Derenzo phantoms (hole diameter ranging from 0.8–1.3 mm in 0.1 mm steps) filled with >99% 44Sc (a), 66.2% 43Sc (b) and 98.2% 43Sc (c). The acquisition of the PET scans was performed in an energy window of 400–700 keV for 30 min, in order to obtain a total number of ~6 × 107 coincidences
Table 4.
FWHM determined for phantom hole-diameters of 1.3 mm for 44Sc and 43Sc in three different sections of the PET scan
| Radionuclide | Radionuclidic purity [%] | Eβ+ average [keV] | FWHM [mm] |
|---|---|---|---|
| 44Sc | >99 (<1% 44mSc) | 632 | 2.12 ± 0.04 |
| 43Sc from 43Ca | 66.2 (33.3% 44Sc) | 476 | 2.04 ± 0.06 |
| 43Sc from 46Ti | 98.2 (1.5% 44Sc) | 476 | 1.87 ± 0.14 |
Quality control of 43Sc
Quality control of the 43Sc (Fig. 5) was performed by radiolabeling of DOTANOC, isolated from irradiated 43CaCO3 targets. Reproducibly, >96% radiochemical purity were achieved at a specific activity of 8 MBq 43Sc per nmol DOTANOC. Lower activity concentrations of the 43Sc eluate, obtained from irradiated 46Ti targets, rendered radiolabeling at high specific activities more challenging. The radiolabeling reaction could be reproducibly performed at 5 MBq/nmol.
Fig. 5.
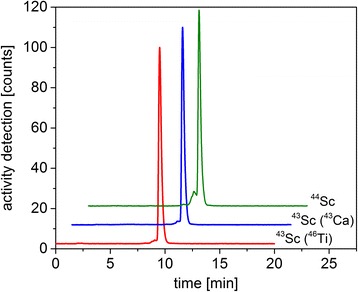
HPLC chromatograms of 43Sc isolated from 46Ti and 43CaCO3 target material and 44Sc, directly after the radiolabeling reaction with DOTANOC (the chromatograms of 43Sc- (43Ca) and 44Sc-DOTANOC are shifted up and sideways for better visibility). The retention times of free 43Sc and 44Sc were determined to be 2.2 ± 0.2 min and 9.7 ± 0.3 min for 43Sc/44Sc-DOTANOC
Discussion
Enriched 46Ti is only commercially available as 46TiO2. As titanium dioxide requires hot sulfuric or hydrofluoric acid for dissolution, the reduction prior to target manufacturing was necessary. After conversion to the metallic state, the dissolution of 46Ti under less stringent conditions becomes possible. The initial reduction process described elsewhere (Lommel et al. 2014) was changed to a vacuum-based system. A four-fold molar excess of reducing agent in a fine granulated formulation with 46TiO2, pressures <5 × 10−3 mbar and temperatures of 950–1000 °C were identified to be crucial conditions to achieve maximum yields. The optimized procedure allowed for reduction yields between 96% and 99%, determined by the dissolution test in concentrated HCl and verified by XRD analysis (Additional file 1: Figure S2 a-b).
Activities of 60–225 MBq 43Sc could be regularly obtained by the irradiation of the enriched 46Ti targets, while 250–480 MBq 43Sc (equivalent to total activities of 380–720 MBq 43Sc/44Sc) was produced via the 43Ca(p,n)43Sc nuclear reaction. The 2–4-fold higher 43Sc activity available from 43Ca irradiations can be attributed to the higher nuclear cross section of the 43Ca(p,n)43Sc reaction in comparison to the 46Ti(p,α)43Sc nuclear reaction (Additional file 1: Figure S4). A theoretical 43Sc yield obtainable via the 46Ti route, was calculated to be 0.6–2.3 GBq, whereas 1.3–3.2 GBq 43Sc were calculated for the 43Ca route. The discrepancies to the experiment results in this work can be mainly explained by the rather large energy degradation of the proton beam (using niobium), from the initial 72 MeV to ~10–15 MeV, resulting in a beam with a broad spread of energies at diminished intensities. Another addition to the discrepancy of results is the lack of beam diagnostics closer than 80 cm from the target. The exceptionally high yield of ~1 GBq became attainable when the proton beam was precisely positioned, at a specific energy, on the target material.
A product of high radionuclidic purity was isolated from irradiated 46Ti samples, containing only 1.5% 44Sc and 0.079% of all other Sc radioisotopes, which is comparable to the calculations determined from the cross section measurements (Koning et al. 2015, Carzaniga et al. 2017). The quantity of 46Sc (0.011%, T1/2 = 83.8 d) was about 50 times higher than expected, however, the percentage can be still related to the threshold value for the long-lived 68Ge (T1/2 = 270.8 d) impurity in the 68Ga generator eluate (0.001%) set by the European Pharmacopoeia (Council of Europe 2013). γ-Spectroscopic measurements of the 43Sc eluate, isolated from irradiated 43CaCO3 targets, revealed the presence of 66.2% 43Sc and 33.3% 44Sc. The co-formation of 44Sc originated from 44Ca (12.36% in the target material) via the (p,n) nuclear reaction, as the cross section maxima of both reactions, 44Ca(p,n)44Sc and 43Ca(p,n)43Sc, are centered around 9–10 MeV (Additional file 1: Figure S4). Performing the irradiation with proton energies of 10 or 12 MeV did not influence the ratio of 43Sc and 44Sc, which is in compliance with the calculations performed (Koning et al. 2015, Carzaniga et al. 2017), hence, the isolated product can be rather considered as a combination of two PET nuclides, with comparable decay properties. Trace activities of various Y radioisotopes were identified in both products, conceivably produced via (p,n) and (p,α) nuclear reactions with natSr and natZr impurities, both being present as low-level contaminants in the target materials (Additional file 1: Table S5). A scan for impurities of trace metals by ICP-OES in enriched 46TiO2 and 46Ti metal suggests that they were probably introduced in the course of the reduction process performed at GSI. These impurities were later eliminated with the use of calcium hydride of higher chemical purity, hence, the introduction of trace metals was avoided, confirmed by the absence of any Y isotopes in the 43Sc eluate. The limited quantity and high costs of 43CaCO3 did not allow for a similar analysis.
The separation of 43Sc from 43Ca target material was performed at a comparable yield (90.4 ± 5.5%) to the previous separations of 44Sc and 47Sc from irradiated 44Ca and 46Ca targets (98.0 ± 0.3% and 94.8 ± 2.1%) (van der Meulen et al. 2015, Domnanich et al. 2017). Using the developed 46Ti/43Sc separation system, about 90% of the initial 43Sc activity could be isolated within 45 min, which is about 10 times faster compared to all previously-reported radiochemical separation methods in this regard (Kolsky et al. 1998).
Radiolabeling of 43Sc with the model compound DOTANOC was utilized to assess the chemical purity of both eluates, obtained by irradiation from 46Ti and 43CaCO3 target material, respectively (Severin et al. 2012, Valdovinos et al. 2014). Reproducible radiosynthesis of 43Sc-DOTANOC was demonstrated at specific activities of 5–8 MBq/nmol. These results are slightly lower than previously achieved with eluates of 44Sc and 47Sc (van der Meulen et al. 2015, Domnanich et al. 2017), but still comparable when taking the obtained low 43Sc activity concentrations into consideration.
The establishment of a recovery process for the enriched 46Ti target material was an important part of the present study. A simple, fast and efficient method was employed to precipitate TiO2 with a high overall yield, however, it was only performed with natural titanium thus far. The established method for 44Ca was successfully employed, for the recycling of the enriched 43Ca target material. Both recycling methods have great potential to significantly reduce the production cost as the current prices for the target materials amount to about 30 USD and 260 USD for 1 mg of enriched 46Ti and 43Ca, respectively.
The resolution of a PET image is, among other factors, influenced by the radionuclide’s positron range. The discriminability between two different radioactive sources in an image is described by the spatial resolution which is, on a particular PET system, only dependent on the radionuclide’s positron energy (Palmer et al. 2005). Herein it was shown, that scanning at slightly improved resolution is demonstrated when using 43Sc instead of 44Sc. An intermediate resolution is shown with 43Sc obtained from 43Ca (66.2% radionuclidic purity) target material. These findings are in line with the expectations, based on the correlation between lower positron energy and enhanced image quality. To date, no FWHM values have been published for 43Sc, but the results in this work are comparable with previously published data for 11C and 89Zr (Palmer et al. 2005, Bunka et al. 2016). This modest gain in resolution, demonstrated using a small animal PET scanner, is unlikely to play a significant role in clinical practice. Speculation whether the co-emitted γ-radiation of 43Sc (Eγ = 372 keV [23%]) will interfere with the image resolution of a clinical PET scanner should not emerge, as modern PET systems based on scintillation crystals use energy windows in between 430 and 650 keV for the detection of annihilation photons (the γ-energy lies outside this range and should be cut off by the standard energy window) (Gnesin 2017). The absence of co-emitted high energetic gamma rays as they are emitted by 44Sc at high intensity (Eγ = 1157 keV [100%]), would be clearly advantageous regarding the dose burden to patients and medical staff.
Conclusion
The production of several hundred MBq 43Sc was demonstrated for the first time by proton irradiation of enriched 46Ti and 43Ca. Only moderate proton energies of 10–15 MeV are required for the respective nuclear reactions, 46Ti(p,α)43Sc and 43Ca(p,n)43Sc, which are available from most commercial biomedical cyclotrons. It was shown that higher 43Sc radioactivities were produced via the 43Ca route. At the same time, this production pathway is accompanied by a comparatively less complex target preparation and separation procedure. Even though the irradiation of 46Ti yielded a product of higher radionuclidic purity, the eluate obtained from irradiated 43CaCO3 can be rather considered as combination of two PET nuclides. The cost of enriched 43Ca is significantly higher than that of 46Ti, however, the expenses are kept within limits by the implementation of recycling procedures. Based on the results obtained, it can be concluded that the 43Ca route features several advantages over the 46Ti path when aiming at a production of 43Sc on a routine basis. In future, the production will be optimized to reproducibly obtain high quantities of 43Sc in order to assess the gain in resolution in a clinical setting. Further studies in this regard are necessary for the upscale of production by means of increasing target material quantities. This is currently underway, utilizing a biomedical cyclotron.
Additional file
Supplementary experimental data. (DOCX 445 kb)
Acknowledgments
The authors thank Dr. Bettina Lommel (from GSI); Dr. Martina Benešová, Walter Hirzel, Alexander Sommerhalder, Muhamet Djelili and Raffaella M. Schmid (from PSI); and Claudia Keller (from ETH Zürich) for technical assistance.
Abbreviations
- CT
computed tomography
- DOTA
1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid
- EOB
end of bombardment
- EOS
end of separation
- FWHM
full-width at half-maximum
- GMP
good manufacturing practice
- HPLC
high performance liquid chromatography
- p.i.
post injection
- PET
positron emission tomography
- SPECT
single photon emission computed tomography
Authors’ contributions
KAD developed and performed the production and separation process of 43Sc, did the reduction and radiolabeling experiments, analyzed the data and drafted the manuscript. RE supported and supervised the 46TiO2 reduction process and reviewed the manuscript. SJ assisted with the separation, as well as the reduction processes. CM performed the PET phantom study and revised the manuscript. AT assisted with calculations and reviewed the manuscript. MB, SB and RS reviewed the manuscript. NPvdM oversaw the development of the production and separation process of 43Sc, supervised the whole study and revised the manuscript. All authors read and approved the final manuscript.
Funding
The research was financially supported by the Swiss National Science Foundation (CR23I2 156,852).
Ethics approval and consent to participate
This article does not contain any studies with human participants performed by any of the authors. All applicable international, national, and/or institutional guidelines for the care and use of test animals were followed.
Competing interest
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Katharina A. Domnanich, Email: katharina.domnanich@psi.ch
Robert Eichler, Email: robert.eichler@psi.ch.
Cristina Müller, Email: cristina.mueller@psi.ch.
Sara Jordi, Email: sara.jordi@psi.ch.
Vera Yakusheva, Email: v.yakusheva@gsi.de.
Saverio Braccini, Email: saverio.braccini@lhep.unibe.ch.
Martin Behe, Email: martin.behe@psi.ch.
Roger Schibli, Email: roger.schibli@psi.ch.
Andreas Türler, Email: andreas.tuerler@dcb.unibe.ch.
Nicholas P. van der Meulen, Email: nick.vandermeulen@psi.ch
References
- Banerjee SR, Pomper MG. Clinical applications of Gallium-68. Appl Radiat Isot. 2013;76:2–13. doi: 10.1016/j.apradiso.2013.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breeman WAP, de Blois E, Chan HS, Konijnenberg M, Kwekkeboom DJ, Krenning EP. 68Ga-labeled DOTA-peptides and 68Ga-labeled radiopharmaceuticals for positron emission tomography: current status of research, clinical applications, and future perspectives. Semin Nucl Med. 2011;41(4):314–321. doi: 10.1053/j.semnuclmed.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Bunka M, Müller C, Vermeulen C, Haller S, Türler A, Schibli R, van der Meulen NP. Imaging quality of 44Sc in comparison with five other PET radionuclides using Derenzo phantoms and preclinical PET. Appl Radiat Isot. 2016;110:129–133. doi: 10.1016/j.apradiso.2016.01.006. [DOI] [PubMed] [Google Scholar]
- Bybel B, Brunken RC, DiFilippo FP, Neumann DR, Wu G, Cerqueira MD. SPECT/CT imaging: clinical utility of an emerging technology. Radiographics. 2008;28(4):1097–1113. doi: 10.1148/rg.284075203. [DOI] [PubMed] [Google Scholar]
- Carzaniga, T. S., M. Auger, S. Braccini, M. Bunka, A. Ereditato, K. P. Nesteruk, P. Scampoli, A. Türler and N. Van der Meulen (2017). Measurement of 43Sc and 44Sc production cross-section with an 18 MeV medical PET cyclotron. Appl Radiat Isot. [DOI] [PubMed]
- Chakravarty R, Goel S, Valdovinos HF, Hernandez R, Hong H, Nickles RJ, Cai W. Matching the decay half-life with the biological half-life: ImmunoPET imaging with 44Sc-labeled Cetuximab fab fragment. Bioconjug Chem. 2014;25(12):2197–2204. doi: 10.1021/bc500415x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Council of Europe (2013). Gallium (68Ga) Edotreotide injection. 01/2013:2482. Council of Europe. European pharmacopeia 9.0, European Directorate for the Quality of Medicines and Healthcare: 2.
- Domnanich, K. A., C. Müller, M. Benešová, R. Dressler, S. Haller, U. Köster, B. Ponsard, R. Schibli, A. Türler and N. Van der Meulen (2017). 47Sc as useful β−-emitter for the radiotheragnostic paradigm: a comparative study of feasible production routes. EJNMMI Radiopharmacy and Chemistry 2(5). [DOI] [PMC free article] [PubMed]
- Domnanich, K. A., C. Müller, R. Farkas, R. M. Schmid, B. Ponsard, R. Schibli, A. Türler and N. Van der Meulen (2016). 44Sc for labeling of DOTA- and NODAGA- functionalized peptides: preclinical in vitro and in vivo investigations. EJNMMI Radiopharmacy and Chemistry 8(1): 19. [DOI] [PMC free article] [PubMed]
- Eppard E, Loktionova NS, Rosch F. Quantitative online isolation of 68Ge from 68Ge/68Ga generator eluates for purification and immediate quality control of breakthrough. Appl Radiat Isot. 2013;82:45–48. doi: 10.1016/j.apradiso.2013.07.020. [DOI] [PubMed] [Google Scholar]
- Experimental Nuclear Reaction Data (EXFOR). (2017). Retrieved 04.09.2017, From https://www-nds.iaea.org/exfor/exfor.htm.
- Gabriel M, Decristoforo C, Kendler D, Dobrozemsky G, Heute D, Uprimny C, Kovacs P, Von Guggenberg E, Bale R, Virgolini IJ. 68Ga-DOTA-Tyr3-octreotide PET in neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and CT. J Nucl Med. 2007;48(4):508–518. doi: 10.2967/jnumed.106.035667. [DOI] [PubMed] [Google Scholar]
- Garcia-Torano E, Peyres V, Roteta M, Sanchez-Cabezudo AI, Romero E, Martinez Ortega A. Standardisation and precise determination of the half-life of 44Sc. Appl Radiat Isot. 2016;109:314–318. doi: 10.1016/j.apradiso.2015.12.007. [DOI] [PubMed] [Google Scholar]
- Gnesin, S. (2017). Personal discussion on the standard energy window for clinical PET scanners.
- Hernandez R, Valdovinos HF, Yang Y, Chakravarty R, Hong H, Barnhart TE, Cai W. 44Sc: an attractive isotope for peptide-based PET imaging. Mol Pharm. 2014;11(8):2954–2961. doi: 10.1021/mp500343j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Atomic Energy Agency (2008). Cyclotron Produced Radionuclides: Principles and Practice Technical Report Series No 465.
- Kitson SL, Cuccurullo V, Ciarmiello A, Salvo D, Mansi L. Clinical applications of positron emission tomography (PET) imaging in medicine: oncology, brain diseases and cardiology. Curr Radiopharm. 2009;2(4):10. doi: 10.2174/1874471010902040224. [DOI] [Google Scholar]
- Kolsky KL, Joshi V, Mausner LF, Srivastava SC. Radiochemical purification of no-carrier-added Scandium-47 for radioimmunotherapy. Appl Radiat Isot. 1998;49(12):1541–1549. doi: 10.1016/S0969-8043(98)00016-5. [DOI] [PubMed] [Google Scholar]
- Koning AJ, Rochman D, Kopecky J, Sublet C, Bauge E, Hilaire S, Romain P, Morillon B, Duarte H, van der Marck S, Pomp S, Sjostrand H, Forrest R, Henriksson H, Cabellos O, Goriely S, Leppanen J, Leeb H, Plompen A, Mills R. TENDL-2015: TALYS-based evaluated nuclear data library. Retrieved. 2015;05(03):2017. [Google Scholar]
- Kwekkeboom DJ, Kam BL, van Essen M, Teunissen JJ, van Eijck CH, Valkema R, de Jong M, de Herder WW, Krenning EP. Somatostatin-receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr Relat Cancer. 2010;17(1):R53–R73. doi: 10.1677/ERC-09-0078. [DOI] [PubMed] [Google Scholar]
- Lommel B, Beusch A, Hartmann W, Hubner A, Kindler B, Steiner J, Yakusheva V. Reduction of isotopically enriched 50Ti-dioxide for the production of high-intensity heavy-ion beam. J Radioanal Nucl Chem. 2014;299(2):977–980. doi: 10.1007/s10967-013-2615-7. [DOI] [Google Scholar]
- Majkowska-Pilip A, Bilewicz A. Macrocyclic complexes of scandium radionuclides as precursors for diagnostic and therapeutic radiopharmaceuticals. J Inorg Biochem. 2011;105(2):313–320. doi: 10.1016/j.jinorgbio.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Müller C, Bunka M, Reber J, Fischer C, Zhernosekov K, Turler A, Schibli R. Promises of cyclotron-produced 44Sc as a diagnostic match for trivalent β−-emitters: in vitro and in vivo study of a 44Sc-DOTA-folate conjugate. J Nucl Med. 2013;54(12):2168–2174. doi: 10.2967/jnumed.113.123810. [DOI] [PubMed] [Google Scholar]
- Palmer MR, Zhu XP, Parker JA. Modeling and simulation of positron range effects for high resolution PET imaging. IEEE Trans Nucl Sci. 2005;52(5):1391–1395. doi: 10.1109/TNS.2005.858264. [DOI] [Google Scholar]
- Perry, D. L. (2011). Handbook of inorganic compounds, CRC Press.
- Pourmand A, Dauphas N. Distribution coefficients of 60 elements on TODGA resin: application to ca, Lu, Hf, U and Th isotope geochemistry. Talanta. 2010;81(3):741–753. doi: 10.1016/j.talanta.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Pruszynski M, Loktionova NS, Filosofov DV, Rosch F. Post-elution processing of 44Ti/44Sc generator-derived 44Sc for clinical application. Appl Radiat Isot. 2010;68(9):1636–1641. doi: 10.1016/j.apradiso.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Pruszynski M, Majkowska-Pilip A, Loktionova NS, Eppard E, Roesch F. Radiolabeling of DOTATOC with the long-lived positron emitter 44Sc. Appl Radiat Isot. 2012;70(6):974–979. doi: 10.1016/j.apradiso.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Ramogida CF, Orvig C. Tumour targeting with radiometals for diagnosis and therapy. Chem Commun (Camb) 2013;49(42):4720–4739. doi: 10.1039/c3cc41554f. [DOI] [PubMed] [Google Scholar]
- Reubi JC, Schar JC, Waser B, Wenger S, Heppeler A, Schmitt JS, Macke HR. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur J Nucl Med. 2000;27(3):273–282. doi: 10.1007/s002590050034. [DOI] [PubMed] [Google Scholar]
- Rösch F. Scandium-44: benefits of a long-lived PET radionuclide available from the 44Ti/44Sc generator system. Curr Radiopharm. 2012;5(3):187–201. doi: 10.2174/1874471011205030187. [DOI] [PubMed] [Google Scholar]
- Rösch F. 68Ge/68Ga generators and 68Ga radiopharmaceutical chemistry on their way into a new century. J Postgrad Med Educ Res. 2013;47(1):7. [Google Scholar]
- Rösch F, Baum RP. Generator-based PET radiopharmaceuticals for molecular imaging of tumours: on the way to THERANOSTICS. Dalton Trans. 2011;40(23):6104–6111. doi: 10.1039/c0dt01504k. [DOI] [PubMed] [Google Scholar]
- Rumble, J. R. e. (2018). CRC handbook of chemistry and physics. Physical constants of organic compounds. B. R. CRC Press/Taylor & Francis, FL. Internet Version 2018.
- Severin GW, Engle JW, Valdovinos HF, Barnhart TE, Nickles RJ. Cyclotron produced 44gSc from natural calcium. Appl Radiat Isot. 2012;70(8):1526–1530. doi: 10.1016/j.apradiso.2012.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, van der Meulen NP, Müller C, Klette I, Kulkarni HR, Turler A, Schibli R, Baum RP. First-in-human PET/CT imaging of metastatic neuroendocrine neoplasms with cyclotron-produced 44Sc-DOTATOC: a proof-of-concept study. Cancer Biother Radiopharm. 2017;32(4):124–132. doi: 10.1089/cbr.2016.2173. [DOI] [PubMed] [Google Scholar]
- Straumanis ME, Chen PC. The corrosion of titanium in acids - the rate of dissolution in sulfuric, hydrochloric, Hydrobromic and Hydroiodic acids. Corrosion. 1951;7(7):9. doi: 10.5006/0010-9312-7.7.229. [DOI] [Google Scholar]
- Szkliniarz K, Jastrzebski J, Bilewicz A, Chajduk E, Choinski J, Jakubowski A, Janiszewska L, Leszczuk E, Lyczko M, Sitarz M, Stolarz A, Trzcinska A, Was B, Zipper W. Medical radioisotopes produced using the alpha particle beam from the Warsaw heavy ion cyclotron. Acta Phys Pol A. 2015;127(5):1471–1474. doi: 10.12693/APhysPolA.127.1471. [DOI] [Google Scholar]
- Szkliniarz K, Sitarz M, Walczak R, Jastrzebski J, Bilewicz A, Choinski J, Jakubowski A, Majkowska A, Stolarz A, Trzcinska A, Zipper W. Production of medical Sc radioisotopes with an alpha particle beam. Appl Radiat Isot. 2016;118:182–189. doi: 10.1016/j.apradiso.2016.07.001. [DOI] [PubMed] [Google Scholar]
- Umbricht CA, Benešová M, Schmid RM, Türler A, Schibli R, van der Meulen NP, Müller C. 44Sc-PSMA-617 for radiotheragnostics in tandem with 177Lu-PSMA-617 - preclinical investigations in comparison with 68Ga-PSMA-11 and 68Ga-PSMA-617. EJNMMI Res. 2017;7(1):9. doi: 10.1186/s13550-017-0257-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdovinos HF, Hernandez R, Barnhart TE, Graves S, Cai W, Nickles RJ. Separation of cyclotron-produced 44Sc from a natural calcium target using a dipentyl pentylphosphonate functionalized extraction resin. Appl Radiat Isot. 2014;95C:23–29. doi: 10.1016/j.apradiso.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen NP, Bunka M, Domnanich KA, Müller C, Haller S, Vermeulen C, Türler A, Schibli R. Cyclotron production of 44Sc: from bench to bedside. Nucl Med Biol. 2015;42(9):745–751. doi: 10.1016/j.nucmedbio.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Velikyan I. Prospective of 68Ga-radiopharmaceutical development. Theranostics. 2014;4(1):47–80. doi: 10.7150/thno.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Coordinating Radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Chem Rev. 2010;110(5):2858–2902. doi: 10.1021/cr900325h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak R, Krajewski S, Szkliniarz K, Sitarz M, Abbas K, Choinski J, Jakubowski A, Jastrzebski J, Majkowska A, Simonelli F, Stolarz A, Trzcinska A, Zipper W, Bilewicz A. Cyclotron production of 43Sc for PET imaging. EJNMMI Physics. 2015;2(1):10. doi: 10.1186/s40658-015-0136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler JF, Ziegler MD, Biersack JP. SRIM - the stopping and range of ions in matter. Nucl Instrum Methods Phys Res, Sect B. 2010;268(11–12):6. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary experimental data. (DOCX 445 kb)