

Abstract
Arousal from sleep in response to CO2 is a critical protective phenomenon. Dysregulation of CO2-induced arousal contributes to morbidity and mortality from prevalent diseases, such as obstructive sleep apnea and sudden infant death syndrome. Despite the critical nature of this protective reflex, the precise mechanism for CO2-induced arousal is unknown. Because CO2 is a major regulator of breathing, prevailing theories suggest that activation of respiratory chemo- and mechano-sensors is required for CO2-induced arousal. However, populations of neurons that are not involved in the regulation of breathing are also chemosensitive. Among these are serotonin (5-HT) neurons in the dorsal raphe nucleus (DRN) that comprise a component of the ascending arousal system. We hypothesized that direct stimulation of these neurons with CO2 could cause arousal from sleep independently of enhancing breathing. Dialysis of CO2-rich acidified solution into DRN, but not medullary raphe responsible for modulating breathing, caused arousal from sleep. Arousal was lost in mice with a genetic absence of 5-HT neurons, and with acute pharmacological or optogenetic inactivation of DRN 5-HT neurons. Here we demonstrate that CO2 can cause arousal from sleep directly, without requiring enhancement of breathing, and that chemosensitive 5-HT neurons in the DRN critically mediate this arousal. Better understanding mechanisms underlying this protective reflex may lead to interventions to reduce disease-associated morbidity and mortality.
SIGNIFICANCE STATEMENT Although CO2-induced arousal is critical to a number of diseases, the specific mechanism is not well understood. We previously demonstrated that serotonin (5-HT) neurons are important for CO2-induced arousal, as mice without 5-HT neurons do not arouse to CO2. Many have interpreted this to mean that medullary 5-HT neurons that regulate breathing are important in this arousal mechanism. Here we found that direct application of CO2-rich aCSF to the dorsal raphe nucleus, but not the medullary raphe, causes arousal from sleep, and that this arousal was lost with genetic ablation or acute inhibition of 5-HT neurons. We propose that 5-HT neurons in the dorsal raphe nucleus can be activated directly by CO2 to cause arousal independently of respiratory activation.
Keywords: 5-HT, arousal, chemosensation, CO2, serotonin, sleep
Introduction
Sleep is a necessary state of reduced consciousness that all mammals are subjected to on a daily basis (Zimmerman et al., 2008). Without sleep, cognitive function declines, memory consolidation is impaired, motor performance wanes (Killgore, 2010), and in some cases animals die (Rechtschaffen et al., 1983). Although this is a necessary state of unconsciousness, it can be dangerous to be completely insensitive to stimuli for prolonged periods. Therefore, the body retains the ability to detect and respond to, with arousal for instance, certain stimuli, especially life-threatening ones. One such arousal stimulus is CO2 (Berthon-Jones and Sullivan, 1984; Buchanan and Richerson, 2010). Elevations in CO2 commonly occur with disorders of breathing, such as acute airway obstruction. Impairment of CO2-induced arousal is thought to play an important role in the sudden infant death syndrome (SIDS) (Kinney et al., 2009) and may contribute to the pathophysiology of sudden unexpected death in epilepsy (SUDEP) (Richerson and Buchanan, 2011; Devinsky et al., 2016). In diseases, such as obstructive sleep apnea (OSA), arousal resulting from elevated CO2 is thought to restore tone to the airway musculature, thereby opening the airway and normalizing serum CO2. However, the accompanying repeated arousals disrupt sleep architecture and contribute to morbidity (Malhotra and White, 2002).
While CO2 is known to be a robust arousal stimulus (Berthon-Jones and Sullivan, 1984), the mechanism by which CO2 causes arousal is somewhat controversial (Buchanan and Richerson, 2010; Guyenet and Abbott, 2013; Kaur et al., 2017). Given that CO2 provides the major drive to breathe, many believe respiratory activation by CO2 leads to arousal. Indeed, there is ample evidence that arousal can occur due to activation of pulmonary and diaphragmatic stretch receptors when breathing is stimulated by CO2 (Gleeson et al., 1990). While this undoubtedly contributes to arousal from CO2, arousal may also occur through direct activation of chemosensitive neurons that comprise components of the ascending arousal system (Saper et al., 2001). Among these neurons are serotonin (5-hydroxytryptamine; 5-HT) neurons in the midbrain dorsal raphe nucleus (DRN). Like their counterparts in the medulla which modulate breathing in response to CO2 (Richerson, 2004), DRN 5-HT neurons are also chemosensitive (Severson et al., 2003). Mice with a genetic deletion of 5-HT neurons in the CNS (Lmx1bf/f/p mice; Zhao et al., 2006) lack a CO2-induced arousal response despite maintaining an intact arousal response to hypoxia, as well as to auditory and tactile stimulation (Buchanan and Richerson, 2010). These mice also display a reduced hypercapnic ventilatory response (Hodges et al., 2008). While data from these mice demonstrate that 5-HT neurons are important for CO2-induced arousal (Buchanan and Richerson, 2010), which specific neuronal subpopulation is primarily responsible remains unclear (Darnall et al., 2005; Kaur et al., 2013).
Here we investigated, in unanesthetized sleeping mice, whether 5-HT neurons in the DRN are sufficient to detect a rise in CO2 and cause arousal. For this, we used a reverse microdialysis protocol similar to that used to probe respiratory chemosensors (Li and Nattie, 1997; Hodges et al., 2004; da Silva et al., 2010) in the chronic 5-HT neuron-deficient Lmx1bf/f/p mice, mice with acute systemic or site-specific inactivation of 5-HT neurons, and following systemic or site-specific pharmacological manipulation of 5-HT receptors. Together, we found that 5-HT neurons in the DRN, but not medullary raphe, were critical for arousal to CO2; medullary 5-HT neurons were more important for modulating breathing. As deficits in CO2-induced arousal responses likely contribute to diseases with high morbidity, such as OSA, and mortality, such as SIDS (Kinney et al., 2009) and SUDEP (Richerson and Buchanan, 2011), these findings will lend insights into improving strategies to reduce disease-related morbidity and mortality.
Materials and Methods
Animals
All procedures on mice were conducted at the Yale School of Medicine or the University of Iowa Roy J. and Lucille M. Carver College of Medicine and were approved by the respective Institutional Animal Care and Use Committees. Care was taken to minimize animal numbers and reduce pain and distress from the procedures. Adult male mice from the following genetic lines were used in these studies: Lmx1bf/f and Lmx1bf/f/p, Htr1aKO and Htr1aRR, Pet1-Cre, and C57BL/6J mice. Lmx1bf/f, Lmx1bf/f/p, Htr1aKO, Htr1aRR, and Pet1-Cre mice were bred in our animal care facilities at the Yale and the University of Iowa. Lmx1bf/f and Lmx1bf/f/p mice were originally obtained from Zoufeng Chen at Washington University (St. Louis). Htr1aKO and Htr1aRR mice were originally obtained from Cornelius Gross at the European Molecular Biology Laboratory (Monterotondo, Italy). Pet1-Cre [B6.Cg-Tg(Fev-cre)1Esd/J] (Scott et al., 2005) and C57BL/6J mice were originally obtained from The Jackson Laboratory. All mice were housed in a room with a 12:12 light-dark cycle (7:00 AM to 7:00 PM) and food and water available ad libitum. All of the experiments were performed between 9:00 AM and 2:00 PM.
In Htr1aRR mice (Audero et al., 2013), all 5-HT1A receptors are first eliminated in the animal and then reoverexpressed onto only 5-HT neurons. Because the 5-HT1A receptor is an inhibitory autoreceptor (Barnes and Sharp, 1999), and in this model it is replaced only onto 5-HT neurons, systemic application of the 5-HT1A receptor agonist, (±)-2-dipropylamino-8-hydroxy-1,2,3,4-tetrahydronaphthalene (8-OH-DPAT), causes inactivation of 5-HT neurons (Audero et al., 2013). Htr1aKO littermates of the Htr1aRR mice do not have the 5-HT1A receptor reoverexpressed on 5-HT neurons and thus lack all 5-HT1A receptors. These animals were used as controls.
Surgical implantation of guide cannulae, EEG/EMG head mounts, and temperature telemeters
Mice were anesthetized with isoflurane (1%–5% induction; 0.5%–2% maintenance; inhaled). Once anesthetized (as determined by nonresponsiveness from a tail pinch), the animal's scalp was shaved and secured into a stereotaxic apparatus (Stoelting). The scalp was cleaned with betadine and a 1 cm longitudinal incision was made in the midline. Four small holes were made using a 23 gauge hypodermic needle 2 mm anterior to bregma and 2 mm anterior to λ ±2 mm from midline. Stainless-steel machine screws (0.1 in anterior and 0.125 in posterior; 000–120; Small Parts) were soldered to stainless-steel wires and then secured into the four holes to serve as EEG electrodes. Before attaching the screws to the EEG/EMG head mount, a guide cannula (CMA7; CMA) was implanted into the DRN (−4.6 anteroposterior, ±0 mediolateral, −1.8 dorsoventral) or medullary raphe (−5.2 anteroposterior, ±0 mediolateral, −5.0 dorsoventral), with the skull level. Anteroposterior and mediolateral measurements were from bregma, and dorsoventral measurements were from the dural surface. The guide cannula was then secured with dental acrylic glue (Lang Dental); and once firmly in place, the electrode wires were soldered to the six-pin EEG/EMG head socket (Pinnacle Technology or Bisco Industries). A pair of wire electrodes attached to the head socket was inserted in bilateral nuchal muscles to serve as EMG electrodes. All wires were then sealed with dental acrylic for stability and to decrease noise, and the skin was closed with 3-0 nylon suture (Ethicon). At the same time, a temperature telemeter was implanted either intraperitoneally (G2 E-mitter; Mini-Mitter) or subcutaneously over the scapulae (IPTT-300; Bio Medic Data Systems) as previously described (Buchanan et al., 2015; Hajek and Buchanan, 2016; Purnell et al., 2017). The animals received meloxicam (2.0 mg/kg) before surgery and for 2 d afterward and were allowed to recover for at least 7 d before being studied.
EEG/EMG data acquisition
Animals were fitted with a small preamplifier (8202-SL; Pinnacle Technology) directly attached to the implanted head socket and connected to a commutator (8204; Pinnacle Technology) that was linked by a USB cable to a desktop computer (Dell). EEG/EMG signals were simultaneously routed through a data conditioning amplifier (EX4-400 Quad Channel Differential Amplifier, Dagan; or Model 440 Instrumentation Amplifier, Brownlee Precision), BNC patch panel (BNC-2090-A; National Instruments), and to a data acquisition board (PCI-6221; National Instruments) in the same computer and collected using Sirenia Acquisition software (Pinnacle Technology) and software custom written in MATLAB (The MathWorks).
Plethysmography data acquisition
The recording chamber was fitted with a low-volume pressure transducer (DC002NDR5; Honeywell International) and the signal transferred to the A-D converter and ultimately displayed within MATLAB. Ambient temperature and relative humidity were recorded with an electronic thermometer (Physiotemp Instruments) and a relative humidity/temperature sensor (HIH-4602-A; Honeywell International) and the same custom software. Breath volumes were calibrated by measuring the voltage output produced by metered breaths delivered with a mechanical ventilator (300 μl; 150 breaths/min; Mini-Vent; Harvard Apparatus). Body temperature data were acquired from intraperitoneal telemeters by a telemetry receiver (ER-4000, Energizer Receiver; Mini-Mitter) placed under the recording chamber, transmitted to the computer via a serial port, and sampled every 10 s with custom MATLAB software, as previously described (Buchanan et al., 2015). Body temperature signals from subcutaneous telemeters were sampled every 1–2 min with a telemetry wand reader (DAS-7007S; Bio Medic Data Systems), as previously described (Purnell et al., 2017). Relative humidity, ambient temperature, body temperature, and atmospheric pressure (obtained from www.wunderground.com) were used to calculate VT using standard methods (Drorbaugh and Fenn, 1955; Buchanan et al., 2014).
Hypercapnic and hypoxic gas challenges
Animals were placed into a custom 350 cm3 cast acrylic recording chamber and allowed to acclimate for at least 1 h on at least 2 consecutive days before the experimental trial. Room air (RA; 21% O2, 79% N2) was piped into the chamber at baseline and was changed to hypercapnia (7% CO2 with 21% O2 and 72% N2; 10 min) or hypoxia (10% O2, 90% N2; 2 min) as needed (Buchanan and Richerson, 2010). After a baseline recording period in RA for at least 60 min, animals received an injection (i.p. or s.c.) of test substance or vehicle, underwent microdialysis of normal or acidified aCSF, or test substance, or were subjected to optogenetic inactivation of 5-HT neurons (described below). At least 30 min after drug administration and with the animal asleep, the gas in the chamber was changed to either hypercapnia or hypoxia. For animals receiving multiple gas challenges, 20 min after the end of the first gas challenge, the animals were challenged with the other test gas. Half the animals were randomly exposed to hypercapnia first, and half were exposed to hypoxia first. The gas was changed back to RA between test gas exposures. Gas exposures were initiated after mice were asleep as determined by real-time observation of EEG/EMG data in accordance with amplitude and frequency parameters described in Vigilance state determination and analysis. Flow rates were maintained at 400–450 ml/min with a flow meter (WU-32446–33; Cole-Parmer). All compressed gas containers were obtained from Airgas East or Praxair.
Vigilance state determination and analysis
Data were scored in 10 s epochs as wake (W), nonrapid eye movement sleep (NREM), or rapid eye movement sleep (REM) using Sirenia SleepPro software (Pinnacle Technology) and custom software written in MATLAB. Vigilance state was determined based on the EEG/EMG frequency characteristics as previously described (Buchanan and Richerson, 2010) and as follows: W characterized by low-amplitude, high-frequency (7–13 Hz) EEG with high EMG power and activity; NREM characterized by high-amplitude, low-frequency (0.5–4 Hz) EEG with moderate to low EMG power and no activity; REM characterized by moderate amplitude, moderate frequency (4.5–8 Hz) EEG with minimal EMG power, except for brief bursts and minimal activity correlating with EMG bursts. In the wake state, the EEG showed a low-amplitude, high-frequency signal with a fast Fourier transform between 4 and 9 Hz, in addition to substantial EMG activity. In NREM sleep, the EEG showed a high-amplitude lower frequency signal, with a fast Fourier transform between 1 and 3 Hz, in addition to an absent or low EMG activity. In REM sleep, the EEG signal showed low amplitude and higher frequency in addition to low EMG activity. The fast Fourier transform power spectra was created for each 10 s epoch of data using software from MATLAB and used to verify scoring.
Reverse microdialysis for application of test substances
The aCSF was equilibrated with either 5% or 25% CO2 (with 21% O2 and balance N2). The composition of the aCSF was (in mm) as follows: 152 Na+, 3.0 K+, 2.1 Mg2+, 2.2 Ca2+, 131 Cl−, and 26 HCO3−. The Ca2+ was added after the aCSF was warmed to 37°C and equilibrated with CO2. The pH of each solution was checked to confirm that the equilibration process produced an appropriate pH level (control/normal = 7.4, acidosis = 6.8).
All animals were allowed to acclimate to the chamber and EEG tether/dialysis tubing tether for at least 1 h per day on 2 d before experimental study. Approximately 1 h before the beginning of the experiment, animals were removed from their cage, the dummy cannula was removed, and the dialysis probe (CMA7; CMA; pores <6 kDa; tip length, 1 mm; tip diameter 240 μm) was inserted. Animals were then placed into the recording chamber, attached to EEG and dialysis tether through a small aperture in the chamber lid. Throughout the experiment, RA was piped through the chamber.
Normal aCSF equilibrated with 5% CO2 was dialyzed using syringe pumps (PHD 2000; Harvard Apparatus; rate 45 μl/min) from the time the mouse was placed into the chamber until the dialysate was changed to 25% CO2 equilibrated aCSF (acidosis) or to an equivalent 5% CO2 equilibrated aCSF (control) solution. Measurements were based on the entire 20 min test period. Each dialysis test was initiated after mice were noted to be asleep as determined by real-time observation of the EEG/EMG recording, eye closure, and absence of motor activity using frequency and amplitude parameters as described above. After the 20 min trial, dialysis solution was returned to the original solution.
Virus infection and optical fiber placement
Under isoflurane anesthesia, an adeno-associated virus (AAV) with archaerhodopsin and yellow fluorescent protein (rAAV5-EF1a-DIO-eArch3.0-EYFP, 0.5 μl of ∼3.4 × 1012 particles/ml; Gene Therapy Center Vector Core, Chapel Hill, NC) was stereotactically injected into the DRN over a period of 10 min (coordinates relative to bregma: −4.6 anteroposterior, ±0.0 mediolateral, −1.8 dorsoventral) (Franklin and Paxinos, 1997). The syringe was left in place for 10 min to allow virus spread and then slowly retracted to prevent reflux. As the virus was Cre-dependent and administered to Pet1-Cre mice, Cre recombination led to expression selectively in 5-HT neurons. During the same surgery, a fiberoptic cannula (200 μm core; 0.22 nA; Doric Lenses) was placed with the tip directly above the DRN. Control animals were injected with a virus not expressing a functional opsin (rAAV5-EF1a-DIO-EYFP, 0.5 μl of ∼6.5 × 1012 particles/ml). Animals received meloxicam (2.0 mg/kg, s.c.) before the surgery and for 2 d afterward. Experiments commenced at least 30 d after viral injection to allow full viral transfection.
Optogenetic inhibition of DRN 5-HT neurons
On trial days, the animal was connected for EEG/EMG recording as described, and an optical patch cable coupled to the light source (561 nm laser; Opto Engine) was attached to the implanted optic fiber using a zirconia ferrule (Doric Lenses). Trials were conducted with the laser on for 5 min, and with the laser off. The laser output was calibrated to 14 mW before each experiment and verified afterward using a digital power meter (PM100D; Thorlabs).
RA was infused into the recording chamber. Arousal trials were initiated after the animal was asleep, determined by EEG/EMG recordings and behavioral observation, as described above. The latency to arousal was scored in real time and verified post hoc. The following conditions were tested in a randomized order on different days: arousal to CO2 (7% CO2, 21% O2, balanced in N2) or RA with laser on and laser off. Three trials were performed in each condition, with at least 10 min between trials.
Tissue fixation and anatomical confirmation
Animals were anesthetized with ketamine (50–75 mg/kg i.p.)/xylazine (5–7.5 mg/kg i.p.). Once anesthetized, a small horizontal incision was made in the abdominal skin and muscle. The incision was then extended anteriorly to cut through the sternum, and the ribs were raised to expose the heart. A small incision was made to cut the right atrium while a 25 gauge needle was placed into the left ventricle. PBS (30 ml) was perfused until all blood was flushed from the circulation and then 4% PFA (50 ml) was perfused. Solutions were maintained at 4°C for perfusion. Brains were then immediately extracted and placed in vials containing 4% PFA for 24 h. They were either stored at 4°C until sectioned (40–80 μm) with a vibratome (Leica Biosystems) or cryo-protected with 30% sucrose for at least 24 h, embedded in Tissue-Tek optimum cutting temperature compound (VWR International), and stored at −80°C until sectioned (20–60 μm) with a cryostat (Leica Biosystems). The sections were counterstained with cresyl violet. We identified anatomic landmarks and verified the site of dialysis probe placement by using a mouse brain atlas for reference (Franklin and Paxinos, 1997).
Virus infection sites were verified by the presence of the fluorescent marker in the DRN. The signal was amplified by washing the sections in 0.1 m PBS with 0.3% Triton X-100, blocking in 10% goat serum (in 0.3% Triton X-100/PBS) for 1 h, and overnight incubation with a mouse anti-GFP antibody at 4°C (1:200 in 10% normal goat serum in 0.3% Triton X-100/PBS; DSHB-GFP-12A6; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, Iowa). The next day, sections were washed in 0.3% Triton X-100/PBS and incubated with the secondary antibody goat anti-mouse 488 for 1 h (1:200 in 0.3% Triton X-100/PBS; A11001; Invitrogen, Thermo Fisher Scientific).
Drugs/test substances
8-OH-DPAT, α-phenyl-1-(2-phenylethyl)-4-piperidinemethanol (MDL 11,939), and 2,5-dimethoxy-4-iodoamphetamine (DOI) were obtained from Tocris Biosciences. All drugs were dissolved in aCSF for intracranial application and saline for intraperitoneal or subcutaneous application.
Experimental design and statistical analyses
Experiment 1.
Determine whether stimulation of DRN 5-HT neurons with acidified aCSF causes arousal from sleep. Arousal latency, sleep profiles, and ventilatory responses were determined for 10 Lmx1bf/f and 10 Lmx1bf/f/p mice following perfusion of normal or acidified aCSF into the DRN. Arousal latency was also determined in 6 C57BL/6J mice following perfusion of normal or acidified aCSF in to the DRN. Each animal received both test substances during wake and sleep at least 2 d apart allowing within-subjects comparisons using paired t test with false discovery rate (FDR) correction for multiple comparisons. Comparisons were also made between genotypes using t test.
Experiment 2.
Determine whether stimulation of medullary 5-HT neurons with acidified aCSF causes arousal from sleep. Arousal latency, sleep profiles, and ventilatory responses were determined for 6 Lmx1bf/f and 6 Lmx1bf/f/p mice following perfusion of normal or acidified aCSF in to the medullary raphe. Each animal received both test substances during wake and sleep at least 2 d apart allowing within-subjects comparisons using paired t test with FDR correction for multiple comparisons. Comparisons were also made between genotypes using t test.
Experiment 3.
Determine whether acute inactivation of all 5-HT neurons in the CNS prevents arousal to inspired CO2. Arousal latency to inspired CO2 or hypoxia challenge was determined for 6 Htr1aKO and 6 Htr1aRR mice following systemic injection of 8-OH-DPAT or vehicle. Each animal was exposed to both gas challenges following treatment with each test substance at least 2 d apart allowing within-subjects comparisons using paired t test with FDR correction for multiple comparisons. Comparisons were also made between genotypes using t test.
Experiment 4.
Determine whether acute inactivation of DRN 5-HT neurons in Htr1aRR mice prevents arousal to inspired CO2. Arousal latency to inspired CO2 or hypoxia challenge was determined for 6 Htr1aKO and 6 Htr1aRR mice following perfusion of 8-OH-DPAT or vehicle into the DRN. Each animal was exposed to both gas challenges following treatment with each test substance at least 2 d apart allowing within-subjects comparisons using paired t test with FDR correction for multiple comparisons. Comparisons were also made between genotypes using t test.
Experiment 5.
Determine whether acute optogenetic inactivation of DRN 5-HT neurons prevents arousal to inspired CO2. Arousal latency to inspired CO2 or RA challenge was determined for 6 Pet1-Cre mice transfected with rAAV5-EF1a-DIO-eArch3.0-EYFP and 6 Pet1-Cre mice transfected with rAAV5-EF1a-DIO-EYFP into the DRN with laser off or on. Each animal received both gas challenges on different days and was tested with laser off and laser on, allowing within-subjects comparisons using paired t test with FDR correction for multiple comparisons.
Experiment 6.
Determine whether restoration of 5-HT1A receptors onto 5-HT neurons normalizes sleep architecture. Sleep states were scored for 24 h in 5 Htr1aKO and 5 Htr1aRR mice house in a 12:12 light-dark cycle. Comparisons were made between genotypes and between light-dark conditions using t test.
Experiment 7.
Determine whether systemic blockade of 5-HT2A receptors prevents arousal following application of acidified aCSF in the DRN. Arousal latency to perfusion of normal or acidified aCSF was determined for 6 Lmx1bf/f mice following systemic application of MDL 11,939 or vehicle. Each mouse received all four combinations of intra-DRN and systemic test substances at least 2 d apart, allowing for within and between-subjects comparisons using paired t test with FDR correction for multiple comparisons.
Experiment 8.
Determine whether local application of 5-HT2A receptor agonist restores arousal following application of acidified aCSF in the DRN in Lmx1bf/f/p mice. Arousal latency to perfusion of normal or acidified aCSF was determined for 6 Lmx1bf//f/p mice following perfusion of DOI or vehicle into the DRN. Each mouse received all four test substance combinations at least 2 d apart, allowing for within and between-subjects comparisons using paired t test with FDR correction for multiple comparisons.
Experiment 9.
Determine whether local application of 5-HT2A receptor antagonist prevents arousal following application of acidified aCSF in the DRN. Arousal latency to perfusion of normal or acidified aCSF was determined for 6 Lmx1bf/f mice following perfusion of MDL 11,939 or vehicle into the DRN. Each mouse received all four test substance combinations of at least 2 d apart, allowing for within- and between-subjects comparisons using paired t test with FDR correction for multiple comparisons.
Statistical analyses.
Interactions between genotype, gas composition, test substance, and vigilance state were analyzed for relevant physiologic variables as indicated above. The significance threshold was p < 0.05 for all conditions. Analyses were accomplished using Microsoft Excel, OriginPro 9.1 (OriginLab), and Systat, version 11.0.
Results
Stimulation of DRN with acidosis-induced arousal from sleep in Lmx1bf/f, but not Lmx1bf/f/p mice
To determine whether stimulation of the DRN with acidosis was sufficient to induce arousal from sleep, Lmx1bf/f and Lmx1bf/f/p mice were implanted with microdialysis cannulae directed toward the DRN (Fig. 1A,F) and received selectively targeted exchanges of normal (control) or acidified aCSF into the DRN during NREM sleep. Application of normal aCSF to the DRN did not induce arousal from sleep in Lmx1bf/f or 5-HT neuron deficient Lmx1bf/f/p mice (mean latency 3.36 ± 0.67 min and 3.21 ± 0.89 min, respectively; n = 10; p = 0.7736; Fig. 1B–D). This latency is similar to that which would be expected by chance in these mice (Buchanan and Richerson, 2010). Application of acidified aCSF to the DRN of Lmx1bf/f mice caused arousal from sleep (mean latency 0.55 ± 0.39 min; n = 10; Fig. 1B,D); however, similar stimulation did not cause arousal in Lmx1bf/f/p mice (3.29 ± 0.59 min; n = 10; Fig. 1C,D). In addition to reducing the latency to arousal, continued exposure of the DRN region to acidosis resulted in sustained wakefulness in Lmx1bf/f, but not Lmx1bf/f/p, mice (Fig. 1E).
Figure 1.

Stimulation of DRN with acidosis-induced arousal from sleep in Lmx1bf/f but not Lmx1bf/f/p mice. A, Sagittal schematic view of mouse brain demonstrating cannula placement into DRN (green). Medullary raphe depicted in purple for reference. B, EEG and EMG traces depicting lack of arousal from sleep with perfusion of normal aCSF into DRN (left) and arousal from sleep with perfusion of acidified aCSF into DRN (right) in an Lmx1bf/f mouse. Calibration: vertical, 20 μV; horizontal, 20 s. C, EEG and EMG traces depicting lack of arousal from sleep with perfusion of normal (left) or acidified (right) aCSF into DRN of an Lmx1bf/f/p mouse. Calibration as in B. D, Mean arousal latencies for Lmx1bf/f (black) and Lmx1bf/f/p (red) mice treated with normal (control; solid bars) or acidified (acidosis; open bars) aCSF. Data are mean ± SEM. n = 10 per group. *p < 0.001. E, Percentage change in time spent in Wake (left) and NREM (right) for Lmx1bf/f (black) and Lmx1bf/f/p (red) mice while DRN perfused with acidified aCSF (acidosis; open bars) compared with normal aCSF (control; solid bars). n = 10 per group. *p < 0.001. F, Nissl-stained section to verify cannula placement toward DRN. Arrow indicates cannula tract. Scale bar, 100 μm. Aq, Aqueduct of Sylvius. G–I, Bar graphs represent mean effects of perfusion of normal or acidified aCSF into DRN on respiratory frequency (G), tidal volume (H), and minute ventilation (I) for Lmx1bf/f (black) and Lmx1bf/f/p (red) mice during Wake (left) and NREM (right). n = 10 per group.
The phenotypically wild-type mice (Lmx1bf/f) tested are littermates of our Lmx1bf/f/p mice and therefore carry two copies of the loxP-flanked Lmx1b gene (Zhao et al., 2006). To control for possible effects of these transgenes, C57BL/6J mice were tested for their ability to arouse from sleep in response to hypercapnia. Latency to arousal was reliably reduced in these mice with acidified aCSF compared with normal aCSF mice (0.48 ± 0.41 min vs 3.17 ± 0.63 min; n = 6; p < 0.001). These latencies were comparable with those for Lmx1bf/f (Fig. 1B,D).
Stimulation of medullary raphe, but not the DRN, with acidosis increased breathing in Lmx1bf/f, but not in Lmx1bf/f/p mice
To determine whether stimulation of the DRN with acidosis was sufficient to increase breathing, Lmx1bf/f and Lmx1bf/f/p mice underwent simultaneous breathing assessment via whole-body plethysmography while normal or acidified aCSF was applied to the DRN during wakefulness or NREM sleep. Application of acidified aCSF to the DRN in Lmx1bf/f or Lmx1bf/f/p mice had no effect on breathing in either genotype when stimuli were delivered during waking (Fig. 1G–I). There was an increase in breathing frequency and subsequently minute ventilation in Lmx1bf/f mice, which coincided with arousal when acidified aCSF was applied during sleep (Fig. 1G–I); however, this was thought to be due to the vigilance state change and not due to stimulation of the DRN per se, as spontaneous arousal and arousal induced by other means (e.g., tactile stimulation) caused similar changes in breathing (Buchanan and Richerson, 2010). There was no change in breathing when the DRN of Lmx1bf/f/p mice were stimulated with acidosis during sleep (Fig. 1G–I), likely because there was no arousal.
To determine whether stimulation of the medullary raphe with acidosis was sufficient to increase breathing, Lmx1bf/f and Lmx1bf/f/p mice were implanted with microdialysis cannulae directed into the medullary raphe (Fig. 2A,I), and neurons in this region were stimulated with acidosis during wakefulness or NREM sleep. Application of acidified aCSF to the medullary raphe did significantly increase breathing compared with treatment with normal aCSF (Fig. 2). Application of acidosis to the medullary raphe during wakefulness in Lmx1bf/f mice increased breathing to a greater degree than during sleep (Fig. 2B,D–F), consistent with the well-established reduction in respiratory chemosensitivity during sleep (Bulow, 1963). Stimulation of the medullary raphe with acidified aCSF during wakefulness or sleep in Lmx1bf/f/p mice had a less robust effect on breathing (Fig. 2C–F) consistent with the known reduction in chemosensitivity in these animals (Hodges et al., 2008).
Figure 2.

Stimulation of medullary raphe with acidosis increased ventilation during wakefulness and sleep. A, Sagittal schematic view of mouse brain demonstrating cannula placement into medullary raphe (purple). DRN depicted in green for reference. Representative plethysmography traces from an Lmx1bf/f (B) and an Lmx1bf/f/p (C) mouse while the medullary raphe was perfused with normal aCSF (control; top traces) or acidified aCSF (acidosis; bottom traces) during Wake (left) or NREM (right). Calibration: vertical, 0.5 μV; horizontal, 0.5 s. Bar graphs represent mean effects of perfusion of normal or acidified aCSF into medullary raphe on respiratory frequency (D), tidal volume (E), and minute ventilation (F) for Lmx1bf/f (black) and Lmx1bf/f/p (red) mice during wake (left) and NREM (right). n = 6 per group. *p < 0.05. G, Mean latency to arousal following application of normal (control; solid bars) and acidified (control; open bars) aCSF into the medullary raphe in Lmx1bf/f (black) and Lmx1bf/f/p (red) mice. n = 6 per group. H, Percentage change in time spent in Wake (left) or NREM (right) for Lmx1bf/f (black) and Lmx1bf/f/p (red) mice while DRN perfused with acidified (acidosis; open bars) or normal aCSF (control; solid bars). n = 6 per group. I, Nissl-stained section to verify cannula placement toward the raphe. Arrow indicates cannula tract. Scale bar, 1000 μm. Aq, Aqueduct of Sylvius.
Stimulation of medullary raphe with acidosis did not induce arousal from sleep in mice of either genotype
To determine whether the medullary raphe plays a direct role in acidosis-induced arousal, Lmx1bf/f and Lmx1bf/f/p underwent simultaneous assessment of arousal while medullary raphe neurons were stimulated with acidosis during NREM sleep. Delivery of acidified aCSF did not change the latency to arousal from sleep compared with delivery of normal aCSF in Lmx1bf/f (3.74 ± 0.48 vs 3.89 ± 0.57 min; n = 6; p = 0.836) or Lmx1bf/f/p (3.86 ± 0.52 vs 3.68 ± 0.71 min; n = 6; p = 0.793; Fig. 2G) mice. Similarly, delivery of normal or acidified aCSF did not change the percentage of time spent in any vigilance state in Lmx1bf/f or Lmx1bf/f/p mice (Fig. 2H).
Acute inactivation of 5-HT neurons via systemic or DRN-specific application of 8-OH-DPAT to Htr1aRR mice blocked CO2-induced arousal from sleep
To further verify the role of 5-HT neurons in CO2-induced arousal, we determined whether acute inactivation of 5-HT neurons could prevent arousal to inspired CO2 by using the Htr1aRR mouse (Audero et al., 2013).
Systemic application of 8-OH-DPAT (0.5 mg/kg) to Htr1aRR mice before inspired CO2 challenge during NREM sleep increased the latency to arousal compared with pretreatment with vehicle (4.54 ± 0.64 min vs 1.01 ± 0.44 min; n = 6; p < 0.001; Fig. 3A,B). As a control, CO2-induced arousal was examined in Htr1aKO. Htr1aKO mice had slightly longer arousal latency to CO2 following systemic application of saline compared with Htr1aRR mice (1.78 ± 0.25 min vs 1.01 ± 0.44 min; n = 6; p < 0.05; Fig. 3B). Systemic application of 8-OH-DPAT had no additional effect on latency to arousal in response to inspired CO2 in Htr1aKO mice (1.92 ± 0.35 min; n = 6; p = 0.618; Fig. 3B). To ensure that the blockade was specific to CO2-induced arousal, the arousal response to inspired hypoxia (10% O2, balance N2; 2 min) was also tested following systemic application of saline or 8-OH-DPAT. 8-OH-DPAT application had no effect on hypoxia-induced arousal compared with saline treatment (0.73 ± 0.38 vs 0.85 ± 0.47 min; n = 6; p = 0.823). These arousal latencies were comparable with those previously seen for hypoxia (Buchanan and Richerson, 2010; Buchanan et al., 2015).
Figure 3.
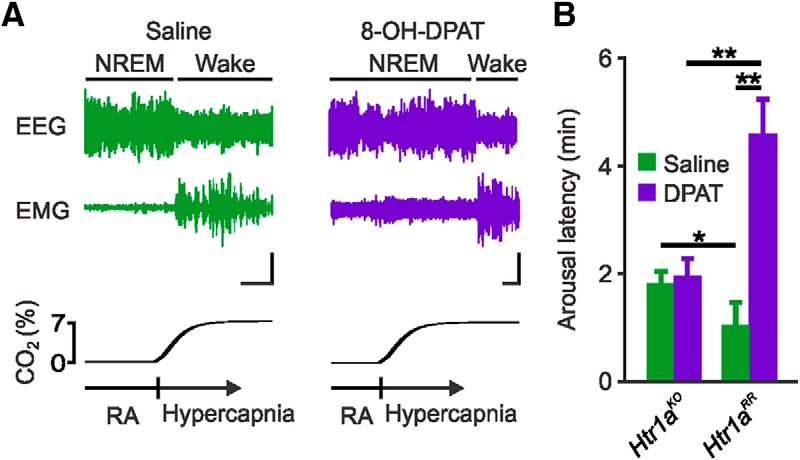
Inactivation of 5-HT neurons through systemic application of 8-OH-DPAT to Htr1aRR mice prevented arousal to CO2. A, EEG and EMG traces depicting arousal response following 7% CO2 challenges 60 min after saline (left) or 8-OH-DPAT (0.5 mg/kg; right) injection (100 μl, s.c.) in a Htr1aRR mouse. Calibration: vertical, 5 μV; horizontal, 30 s. B, Mean latency to arousal following CO2 challenge 60 min after saline (green) or 8-OH-DPAT (purple) injections in Htr1aKO and Htr1aRR mice as indicated. n = 6 per group. *p < 0.05. **p < 0.001.
Given that systemic application of 8-OH-DPAT would silence 5-HT neurons in both the DRN and medullary raphe, to verify that the arousal blockade occurred via action of 8-OH-DPAT in the DRN and not in the medullary raphe, separate groups of Htr1aRR and Htr1aKO mice were implanted with microdialysis cannulae directed at the DRN. Application of normal aCSF into the DRN during inspired CO2 challenge in NREM sleep had no effect on CO2-induced arousal. Application of 8-OH-DPAT into the DRN during CO2 challenge in NREM sleep increased the latency to arousal compared with application of normal aCSF in Htr1aRR (3.213 ± 0.43 min vs 0.43 ± 0.17 min; n = 6; p < 0.05; Fig. 4A,B). Application of normal aCSF or 8-OH-DPAT to the DRN of Htr1aKO mice had no effect on CO2-induced arousal (0.38 ± 0.23 min vs 0.46 ± 0.09 min; n = 6; p = 0.783; Fig. 4B). It should be noted that pharmacological blockade (Monti and Jantos, 2004) or activation (Monti et al., 1990) of 5-HT1A receptors can alter sleep architecture; however, effects are usually reported >2 h after application. This may explain why changes were not seen after our acute treatments. Genetic deletion of 5-HT1A receptors is also known to alter sleep architecture primarily by reducing the amount of REM sleep during the early daytime (Boutrel et al., 2002). To assess sleep architecture in Htr1aRR and Htr1aKO mice, animals were implanted with EEG/EMG head mounts, 24 h recordings with video were performed, and data were assigned to a vigilance state in 10 s epochs. We observed a small but statistically significant reduction in REM sleep during the daytime in Htr1aKO mice that was corrected by reoverexpression of the 5-HT1A receptor onto 5-HT neurons in Htr1aRR mice (light: 75.50 ± 7.69 min Htr1aKO vs 55.33 ± 10.19 min Htr1aRR, n = 5, p < 0.05; dark: 23.70 ± 8.76 min Htr1aKO vs 26.17 ± 6.38 min Htr1aRR, n = 5, p = 0.364). There was no significant difference between the genotypes in the amount of time spent in wake (light: 199.70 ± 16.76 min Htr1aKO vs 209.40 ± 11.77 min Htr1aRR, n = 5, p = 0.238; dark: 438.13 ± 31.66 min Htr1aKO vs 409.93 ± 48.06 min Htr1aRR, n = 5, p = 0.231) or NREM (light: 444.40 ± 21.14 min Htr1aKO vs 454.83 ± 10.90 min Htr1aRR, n = 5, p = 0.254; dark: 257.73 ± 24.11 min Htr1aKO vs 283.50 ± 45.42 min Htr1aRR, n = 5, p = 0.226).
Figure 4.
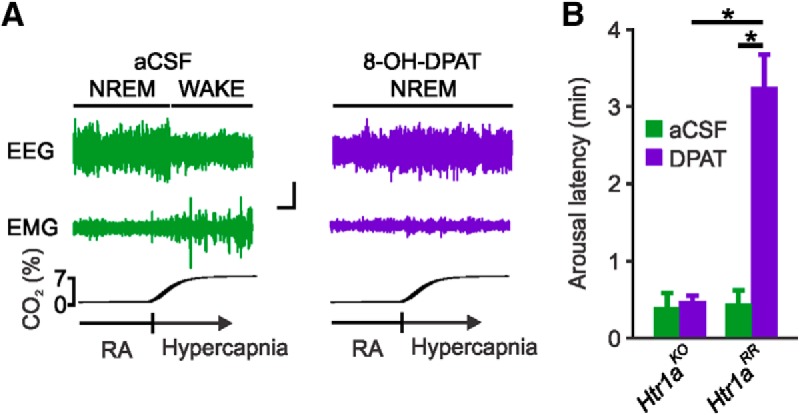
Inactivation of DRN 5-HT neurons through direct application of 8-OH-DPAT to DRN of Htr1aRR mice prevented arousal to CO2. A, EEG and EMG traces depicting arousal response to 7% CO2 during perfusion of DRN with normal aCSF (left) or aCSF containing 8-OH-DPAT (1 mm; right) in an Htr1aRR mouse. Calibration: vertical, 20 μV; horizontal, 20 s. B, Latency to arousal to CO2 challenge during perfusion of DRN with normal aCSF (green) or 8-OH-DPAT (purple) in Htr1aKO and Htr1aRR mice as indicated. n = 6 per group. *p < 0.001.
Acute optogenetic inactivation of 5-HT neurons in the DRN blocked CO2-induced arousal from sleep
To further verify that inhibition of DRN 5-HT neurons is sufficient to block CO2-induced arousal from sleep, we selectively expressed the fluorescently labeled inhibitory opsin archaerhodopsin (Arch) in DRN 5-HT neurons by injecting a Cre-inducible AVV vector in the DRN in Pet1-Cre mice (Fig. 5A,B). Without light stimulation, exposure to 7% CO2 was associated with a shorter arousal latency compared with RA exposure (mean latency 0.67 ± 0.10 min and 2.72 ± 0.29 min, respectively; n = 6; p < 0.001; Fig. 5C,D). During optogenetic inhibition of DRN 5-HT neurons, the latency to arousal to CO2 significantly increased compared with when the light source was off (1.68 ± 0.41 min and 0.67 ± 0.10 min; n = 6; p = 0.049; Fig. 5C,D). There was no change in the mean arousal latency within the RA condition when neurons were optogenetically inhibited (2.72 ± 0.29 min and 2.88 ± 0.58 min; n = 6; p = 0.712; Fig. 5D). As additional control, a separate group of Pet1-Cre mice received an AAV vector without opsin. These mice also aroused quickly to CO2 (0.88 ± 0.30 min compared with RA laser off 2.71 ± 0.08 min; n = 6; p = 0.012). Importantly, there was no effect of optogenetic inhibition in the RA (2.71 ± 0.08 min and 2.82 ± 0.56 min; n = 6; p = 0.828) or CO2 condition (0.88 ± 0.30 min and 1.03 ± 0.43 min; n = 6; p = 0.385; Fig. 5D) when animals received the AAV without opsin. No fluorescent staining was detectable in wild-type mice injected with the virus (Fig. 5B).
Figure 5.

Optogenetic inhibition of DRN 5-HT neurons prevented arousal to CO2. A, Schematic of virus transfection and placement of fiberoptic cannula into DRN (green). Medullary raphe depicted in purple for reference. B, Coronal sections showing virus transfection in the DRN of a Pet1-Cre mouse (top) and lack of expression in a wild-type mouse (bottom). C, EEG and EMG traces depicting arousal from sleep to CO2 without (left) and with (right) laser stimulation in a Pet1-Cre mouse transfected with Arch. Green traces represent time of laser on. Calibration: vertical, 20 μV; horizontal, 60 s. D, Mean arousal latencies in mice expressing Arch (bottom) and in control mice not expressing a functional opsin (top) to room air (open bars) and CO2 (closed bars) exposure without (black) and with (green) laser stimulation. Data are mean ± SEM. n = 6 per group. *p < 0.05. **p < 0.001.
Systemic application of the 5-HT2A receptor antagonist MDL 11,939 prevented arousal from sleep following direct stimulation of the DRN with acidosis
We previously found that CO2-induced arousal could be blocked with systemic application of the 5-HT2A receptor antagonist MDL 11,939, and that arousal to inspired CO2 could be recovered in Lmx1bf/f/p mice with systemic application of the 5-HT2A agonist DOI or (4-bromo-3,6-dimethoxybenxocyclobuten-1-yl)methylamine hydrobromide (Buchanan et al., 2015). This raised the question of where the 5-HT2A receptors might be activated and whether 5-HT neurons are necessary for CO2-induced arousal. To determine whether blockade of 5-HT2A receptors could prevent arousal from direct acidification of the DRN, we dialyzed normal or acidified aCSF into the DRN in Lmx1bf/f mice at least 30 min after systemic application of MDL 11,939 (10 mg/kg; i.p.) while the animals were asleep. Compared with vehicle treatment, systemically applied MDL 11,939 increased the latency to arousal in response to acidified aCSF (0.552 ± 0.346 min vs 4.438 ± 0.605 min; n = 6; p < 0.05; Fig. 6A). Vehicle or MDL 11,939 treatment had no effect on latency to arousal from perfusion of normal aCSF (4.162 ± 0.518 min vs 3.919 ± 0.696 min; n = 6; p = 0.673). Similarly, we systemically applied the 5-HT2A receptor agonist DOI to Lmx1bf/f/p mice before simulation of the DRN region with acidified aCSF. This was insufficient to restore the arousal response to acidosis in the DRN (3.936 ± 0.806 min for normal aCSF vs 4.438 ± 0.605 min for acidified aCSF; n = 6; p = 0.338; Fig. 6B), suggesting that, although 5-HT2A receptors are expressed in the DRN region (Monti, 2010), activation of 5-HT2A receptors in some other downstream target region is required for arousal. To further examine this hypothesis, MDL 11,939 was dialyzed into the DRN along with normal or acidified aCSF in Lmx1bf/f mice. Application of MDL 11,939 directly into the DRN before perfusion of acidified aCSF into the DRN had no effect on the latency to arousal to the acidified aCSF compared with coadministration of vehicle and acidified aCSF (0.766 ± 0.427 min vs 0.506 ± 0.173 min; n = 6; p = 0.419; Fig. 6A), supporting that 5-HT2A receptor activation outside of the DRN is required for arousal to CO2.
Figure 6.
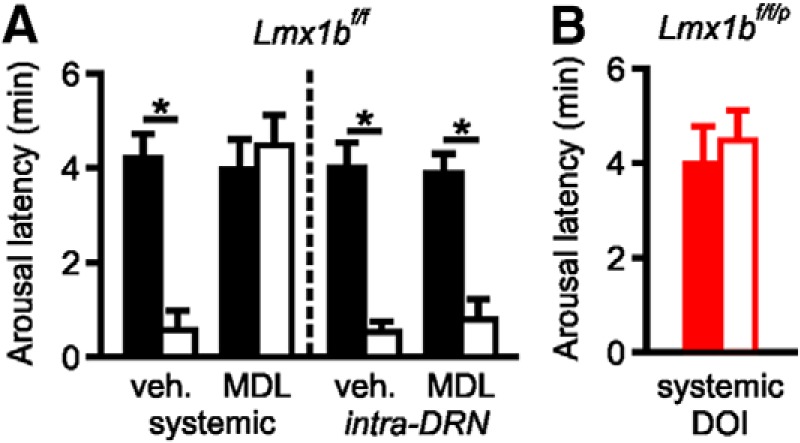
Systemic inhibition of 5-HT2A receptors prevents acidosis-induced arousal in Lmx1bf/f mice, but systemic activation of 5-HT2A receptors does not restore acidosis-induced arousal in Lmx1bf/f/p mice. A, Latency to arousal induced by perfusion of DRN with acidified aCSF following systemic (10 mg/kg; left) or intra-DRN (1 mm; right) application of vehicle or MDL 11,939 in Lmx1bf/f mice. n = 6 per group. *p < 0.05. B, Latency to arousal induced by perfusion of DRN with acidified aCSF following systemic application of DOI (1 mg/kg) in Lmx1bf/f/p mice. n = 6 per group.
Discussion
Arousal from sleep in response to hypercapnia is an important defense mechanism that is relevant to diseases, such as SIDS (Kinney et al., 2009), OSA (Chamberlin, 2013; Kaur et al., 2013), and SUDEP (Richerson and Buchanan, 2011). The data presented here indicate that 5-HT neurons in the DRN, and not those in the medullary raphe, contribute to a direct pathway for CO2-induced arousal that does not require ventilatory activation.
In diseases, such as OSA, it is well established that airway obstruction causes apnea that leads to arousal (Malhotra and White, 2002). It is further established that this arousal is critical to restore airway tone and patency, allow resumption of breathing, and prevent death (Malhotra and White, 2002). It is thought that arousal occurs as a result of derangements in blood gases consequent to apnea (i.e., reduced airflow leads to accumulation of CO2 and reduction in O2). Aberrant blood gas conditions, namely, hypoxia and hypercapnia, are known to potently activate breathing: the former through primarily peripheral oxygen chemosensors in the carotid bodies and aortic arch, and the latter through activation of CO2 chemosensors within the CNS, such as 5-HT neurons in the medullary raphe (Buchanan and Richerson, 2009). While the mechanisms underlying CO2-induced arousal are not well understood, a body of evidence suggests that arousal occurs via enhancement of respiratory effort against a closed airway and consequent activation of mechano-, stretch-, and/or pressure sensors in the chest wall, diaphragm, airway, and lung (Gleeson et al., 1990). Activation of sensors in these locations leads to activation of ascending pathways that most likely involve the nucleus tractus solitarius and parabrachial nucleus (PBN) (Buchanan and Richerson, 2009; Chamberlin, 2013; Kaur et al., 2013; McSharry et al., 2014). However, it is also known that brain regions involved in regulation of arousal can be activated by blood gas changes in the absence of increased tidal volume and ventilation (Ayas et al., 2000). Indeed, many of the brain loci that constitute the sleep–wake regulating ascending arousal system can be activated by CO2. These include the noradrenergic neurons of the locus ceruleus (Pineda and Aghajanian, 1997), histaminergic neurons in the tuberomammillary nucleus (Johnson et al., 2005), orexinergic neurons in the lateral hypothalamus (Sunanaga et al., 2009; Johnson et al., 2012), and 5-HT neurons in the DRN (Severson et al., 2003). There is also evidence that regions, such as the retrotrapezoid nucleus (Guyenet and Abbott, 2013) and PBN, are activated by CO2 (Kaur et al., 2013; Yokota et al., 2015; Kaur et al., 2017) to mediate CO2-induced arousal. Although there is no evidence that the PBN is itself chemosensitive, it is heavily interconnected with other chemosensitive regions and may play a role in mediating CO2-induced arousal (Kaur et al., 2013; Yokota et al., 2015), by in turn activating downstream forebrain structures involved in maintenance of arousal (Kaur et al., 2017).
Therefore, it stands to reason that a rise in CO2 and the resulting acidosis could directly activate one or more of these components to cause arousal independently of increases in breathing. A leading candidate for such activation is 5-HT neurons of the DRN (Buchanan and Richerson, 2010). 5-HT neurons in the DRN robustly increase their firing in response to a physiologic decrease in pH in vitro (Severson et al., 2003) and to increases in inspired CO2 in vivo (Veasey et al., 1997). Lmx1bf/f/p mice do not arouse to CO2, whereas their phenotypically wild-type littermates arouse readily (Buchanan and Richerson, 2010). Lmx1bf/f/p mice also arouse easily to hypoxia and other nonspecific arousal stimuli (Buchanan and Richerson, 2010), demonstrating that there is no overall arousal deficit. Here we demonstrate that direct stimulation of DRN with acidosis causes arousal from sleep in Lmx1bf/f but not Lmx1bf/f/p mice, indicating both that DRN stimulation is sufficient to cause arousal and that 5-HT neurons are required. When arousal occurred, there was an associated increase in ventilation, but this was thought to be due to the arousal state change and not due to a direct effect of stimulation of DRN 5-HT neurons with acidosis as stimulation of these neurons during wakefulness did not affect ventilation. Furthermore, stimulation of the medullary raphe with acidosis during NREM sleep or wakefulness increased ventilation. However, this increase in breathing did not cause arousal from NREM sleep, suggesting that, although respiratory activation can cause arousal, the increase in ventilation induced by stimulation of the medullary raphe with acidosis was insufficient to cause arousal.
There are at least 16 5-HT receptors subtypes, grouped into seven numbered receptor subtype families (Barnes and Sharp, 1999). Many of these are expressed in components of the ascending arousal system that could be activated downstream of DRN activation by CO2 (Monti, 2010). We previously determined that systemic 5-HT2A receptor blockade is sufficient to prevent CO2-induced arousal (Buchanan et al., 2015). Here we demonstrated that systemic 5-HT2A receptor blockade is also sufficient to prevent arousal to direct acidification of the DRN. We further demonstrated that inactivation of 5-HT2A receptors within the DRN itself is insufficient to block arousal following direct acidification of the DRN. This indicates that, although there are 5-HT2A receptors within the DRN, activation of 5-HT2A receptors elsewhere, for instance, in some yet to be identified downstream effector site, is required for arousal to CO2.
This raises the question of what happens downstream of DRN 5-HT neuron activation to ultimately lead to arousal from sleep. The DRN sends diffuse projections to cortex, thalamus, hypothalamus, brainstem, and other regions (Monti, 2010). Activation of many of these could be reasoned to affect arousal. Optogenetic activation of DRN 5-HT neurons causes an increase in wakefulness, a decrease in NREM sleep, and is correlated with cortical activity (Ito et al., 2013). Certainly, full arousal requires diffuse cortical activation, but whether this is brought about directly through projections from DRN to cortex, or requires initial activation of an intermediary site, such as within the brainstem or diencephalon is unknown. Recent evidence suggests that the PBN is a good candidate to be the intermediary site (Kaur et al., 2017). Further, it is unknown whether activation of any one of the six DRN subnuclei is sufficient to incite this arousal or whether only activation of specific subnuclei is capable of causing arousal. It is also unclear how local interconnections between dorsal and median raphe might be involved.
CO2-induced arousal in human disease
CO2-induced arousal is implicated in a number of diseases, including OSA, SIDS, SUDEP, and panic disorder. In OSA, apnea-induced arousal is crucial to reestablish airway tone, restore breathing, and prevent death (Malhotra and White, 2002). However, the repeated arousals from sleep leads to fragmented and reduced sleep, which is associated with high morbidity and can be associated with mortality, especially from associated accidents (Malhotra and White, 2002). Strategies are being developed to attempt to restore airway patency without arousal from sleep. However, these do not address the residual arousal that could occur from residual small increases in CO2 resulting from the initial airway occlusion. Such arousal would still fragment and reduce sleep and contribute to morbidity. Thus, it is important to recognize and account for these mechanisms in management strategies.
SIDS is thought to result, at least in part, from dysfunctional activation of respiratory and arousal systems by CO2 (Kinney et al., 2009). Brains from four separate cohorts of babies who fell victim to SIDS were found to have abnormalities in the brainstem 5-HT system (Kinney et al., 2009). While systematic examination of brains from SUDEP victims has not yet been undertaken, a preponderance of data implicates serotonergic dysregulation in SUDEP. SUDEP is thought to result in seizure-induced cardiac and respiratory dysfunction (Buchanan et al., 2014; Zhan et al., 2016). SUDEP propensity is increased by reduced arousal and consciousness following seizure (Richerson and Buchanan, 2011; Sowers et al., 2013), reduced monitoring (Nashef et al., 1995), and prone sleeping position (Tomson et al., 2008; Liebenthal et al., 2015). The conundrum in SUDEP is that CO2 levels typically rise following a seizure. Indeed, this rise in CO2 likely contributes to the mechanisms of seizure cessation (Lee et al., 1996). Thus, understanding mechanisms of CO2-induced arousal can aid in understanding of how a seizure could reduce effectiveness of these mechanisms and inform development of prophylactic strategies.
Mechanisms of arousal to CO2 are likely also relevant to panic disorder, which is thought to be triggered by a false suffocation alarm, or a hypersensitivity to CO2 (Klein, 1993; Leibold et al., 2015). The mechanisms of this false alarm are not well understood. Among efferents from the DRN are projections to limbic structures that are involved in panic. Better understanding these pathways may also aid in reducing morbidity associated with this disease.
Here we identify a novel pathway by which direct stimulation of 5-HT neurons in the DRN results in arousal from sleep and involve downstream activation of 5-HT2A receptors outside of the DRN. This is an important physiologic mechanism that needs to be further explored, as it may hold the key for reducing morbidity and mortality in several all too common diseases.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke K08 NS069667 and R01 NS095842, and the Beth & Nate Tross Research Fund to G.F.B., Niels Stensen Fellowship and Limburg University Fund/SWOL to N.K.L., and National Institutes of Health/National Institute of Neurological Disorders and Stroke T32 NS007421 to B.S.P.
The authors declare no competing financial interests.
References
- Audero E, Mlinar B, Baccini G, Skachokova ZK, Corradetti R, Gross C (2013) Suppression of serotonin neuron firing increases aggression in mice. J Neurosci 33:8678–8688. 10.1523/JNEUROSCI.2067-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayas NT, Brown R, Shea SA (2000) Hypercapnia can induce arousal from sleep in the absence of altered respiratory mechanoreception. Am J Respir Crit Care Med 162:1004–1008. 10.1164/ajrccm.162.3.9908040 [DOI] [PubMed] [Google Scholar]
- Barnes NM, Sharp T (1999) A review of central 5-HT receptors and their function. Neuropharmacology 38:1083–1152. 10.1016/S0028-3908(99)00010-6 [DOI] [PubMed] [Google Scholar]
- Berthon-Jones M, Sullivan CE (1984) Ventilation and arousal responses to hypercapnia in normal sleeping humans. J Appl Physiol 57:59–67. 10.1152/jappl.1984.57.1.59 [DOI] [PubMed] [Google Scholar]
- Boutrel B, Monaca C, Hen R, Hamon M, Adrien J (2002) Involvement of 5-HT1A receptors in homeostatic and stress-induced adaptive regulations of paradoxical sleep: studies in 5-HT1A knock-out mice. J Neurosci 22:4686–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan GF, Richerson GB (2009) Role of chemoreceptors in mediating dyspnea. Respir Physiol Neurobiol 167:9–19. 10.1016/j.resp.2008.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan GF, Richerson GB (2010) Central serotonin neurons are required for arousal to CO2. Proc Natl Acad Sci U S A 107:16354–16359. 10.1073/pnas.1004587107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan GF, Murray NM, Hajek MA, Richerson GB (2014) Serotonin neurones have anti-convulsant effects and reduce seizure-induced mortality. J Physiol 592:4395–4410. 10.1113/jphysiol.2014.277574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan GF, Smith HR, MacAskill A, Richerson GB (2015) 5-HT2A receptor activation is necessary for CO2-induced arousal. J Neurophysiol 114:233–243. 10.1152/jn.00213.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulow K. (1963) Respiration and wakefulness in man. Acta Physiol Scand Suppl 59:1–110. 10.1111/j.1748-1716.1963.tb02717.x [DOI] [PubMed] [Google Scholar]
- Chamberlin NL. (2013) Brain circuitry mediating arousal from obstructive sleep apnea. Curr Opin Neurobiol 23:774–779. 10.1016/j.conb.2013.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnall RA, Harris MB, Gill WH, Hoffman JM, Brown JW, Niblock MM (2005) Inhibition of serotonergic neurons in the nucleus paragigantocellularis lateralis fragments sleep and decreases rapid eye movement sleep in the piglet: implications for sudden infant death syndrome. J Neurosci 25:8322–8332. 10.1523/JNEUROSCI.1770-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva GS, Li A, Nattie E (2010) High CO(2)/H(+) dialysis in the caudal ventrolateral medulla (Loeschcke's area) increases ventilation in wakefulness. Respir Physiol Neurobiol 171:46–53. 10.1016/j.resp.2010.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Hesdorffer DC, Thurman DJ, Lhatoo S, Richerson G (2016) Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol 15:1075–1088. 10.1016/S1474-4422(16)30158-2 [DOI] [PubMed] [Google Scholar]
- Drorbaugh JE, Fenn WO (1955) A barometric method for measuring ventilation in newborn infants. Pediatrics 16:81–87. [PubMed] [Google Scholar]
- Franklin KB, Paxinos G (1997) The mouse brain in stereotactic coordinates. San Diego, CA: Academic. [Google Scholar]
- Gleeson K, Zwillich CW, White DP (1990) The influence of increasing ventilatory effort on arousal from sleep. Am Rev Respir Dis 142:295–300. 10.1164/ajrccm/142.2.295 [DOI] [PubMed] [Google Scholar]
- Guyenet PG, Abbott SB (2013) Chemoreception and asphyxia-induced arousal. Respir Physiol Neurobiol 188:333–343. 10.1016/j.resp.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajek MA, Buchanan GF (2016) Influence of vigilance state on physiologic consequences of seizures and seizure-induced death in mice. J Neurophysiol 115:2286–2293. 10.1152/jn.00011.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR, Martino P, Davis S, Opansky C, Pan LG, Forster HV (2004) Effects on breathing of focal acidosis at multiple medullary raphe sites in awake goats. J Appl Physiol 97:2303–2309. 10.1152/japplphysiol.00645.2004 [DOI] [PubMed] [Google Scholar]
- Hodges MR, Tattersall GJ, Harris MB, McEvoy SD, Richerson DN, Deneris ES, Johnson RL, Chen ZF, Richerson GB (2008) Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci 28:2495–2505. 10.1523/JNEUROSCI.4729-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Yanase M, Yamashita A, Kitabatake C, Hamada A, Suhara Y, Narita M, Ikegami D, Sakai H, Yamazaki M, Narita M (2013) Analysis of sleep disorders under pain using an optogenetic tool: possible involvement of the activation of dorsal raphe nucleus-serotonergic neurons. Mol Brain 6:59. 10.1186/1756-6606-6-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PL, Moratalla R, Lightman SL, Lowry CA (2005) Are tuberomammillary histaminergic neurons involved in CO2-mediated arousal? Exp Neurol 193:228–233. 10.1016/j.expneurol.2004.11.022 [DOI] [PubMed] [Google Scholar]
- Johnson PL, Samuels BC, Fitz SD, Lightman SL, Lowry CA, Shekhar A (2012) Activation of the orexin 1 receptor is a critical component of CO2-mediated anxiety and hypertension but not bradycardia. Neuropsychopharmacology 37:1911–1922. 10.1038/npp.2012.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Pedersen NP, Yokota S, Hur EE, Fuller PM, Lazarus M, Chamberlin NL, Saper CB (2013) Glutamatergic signaling from the parabrachial nucleus plays a critical role in hypercapnic arousal. J Neurosci 33:7627–7640. 10.1523/JNEUROSCI.0173-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Wang JL, Ferrari L, Thankachan S, Kroeger D, Venner A, Lazarus M, Wellman A, Arrigoni E, Fuller PM, Saper CB (2017) A genetically defined circuit for arousal from sleep during hypercapnia. Neuron 96:1153–1167.e5. 10.1016/j.neuron.2017.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killgore WD. (2010) Effects of sleep deprivation on cognition. Prog Brain Res 185:105–129. 10.1016/B978-0-444-53702-7.00007-5 [DOI] [PubMed] [Google Scholar]
- Kinney HC, Richerson GB, Dymecki SM, Darnall RA, Nattie EE (2009) The brainstem and serotonin in the sudden infant death syndrome. Annu Rev Pathol 4:517–550. 10.1146/annurev.pathol.4.110807.092322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DF. (1993) False suffocation alarms, spontaneous panics, and related conditions: an integrative hypothesis. Arch Gen Psychol 50:306–317. 10.1001/archpsyc.1993.01820160076009 [DOI] [PubMed] [Google Scholar]
- Lee J, Taira T, Pihlaja P, Ransom BR, Kaila K (1996) Effects of CO2 on excitatory transmission apparently caused by changes in intracellular pH in the rat hippocampal slice. Brain Res 706:210–216. 10.1016/0006-8993(95)01214-1 [DOI] [PubMed] [Google Scholar]
- Leibold NK, van den Hove DL, Esquivel G, De Cort K, Goossens L, Strackx E, Buchanan GF, Steinbusch HW, Lesch KP, Schruers KR (2015) The brain acid-base homeostasis and serotonin: a perspective on the use of carbon dioxide as human and rodent experimental model of panic. Prog Neurobiol 129:58–78. 10.1016/j.pneurobio.2015.04.001 [DOI] [PubMed] [Google Scholar]
- Li A, Nattie EE (1997) Focal central chemoreceptor sensitivity in the RTN studied with a CO2 diffusion pipette in vivo. J Appl Physiol 83:420–428. 10.1152/jappl.1997.83.2.420 [DOI] [PubMed] [Google Scholar]
- Liebenthal JA, Wu S, Rose S, Ebersole JS, Tao JX (2015) Association of prone position with sudden unexpected death in epilepsy. Neurology 84:703–709. 10.1212/WNL.0000000000001260 [DOI] [PubMed] [Google Scholar]
- Malhotra A, White DP (2002) Obstructive sleep apnoea. Lancet 360:237–245. 10.1016/S0140-6736(02)09464-3 [DOI] [PubMed] [Google Scholar]
- McSharry DG, Saboisky JP, Deyoung P, Jordan AS, Trinder J, Smales E, Hess L, Chamberlin NL, Malhotra A (2014) Physiological mechanisms of upper airway hypotonia during REM sleep. Sleep 37:561–569. 10.5665/sleep.3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti JM. (2010) The structure of the dorsal raphe nucleus and its relevance to the regulation of sleep and wakefulness. Sleep Med Rev 14:307–317. 10.1016/j.smrv.2009.11.004 [DOI] [PubMed] [Google Scholar]
- Monti JM, Jantos H (2004) Effects of the 5-HT1A receptor ligands flesinoxan and WAY 100635 given systemically or microinjected into the laterodorsal tegmental nucleus on REM sleep in the rat. Behav Brain Res 151:159–166. 10.1016/j.bbr.2003.08.023 [DOI] [PubMed] [Google Scholar]
- Monti JM, Pineyro G, Orellana C, Boussard M, Jantos H, Labraga P, Olivera S, Alvarino F (1990) 5-HT receptor agonists 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI) and 8-OH-DPAT increase wakefulness in the rat. Biogen Amine 7:145–151. 10.1016/0014-2999(90)94513-w [DOI] [Google Scholar]
- Nashef L, Fish DR, Garner S, Sander JW, Shorvon SD (1995) Sudden death in epilepsy: a study of incidence in a young cohort with epilepsy and learning difficulty. Epilepsia 36:1187–1194. 10.1111/j.1528-1157.1995.tb01061.x [DOI] [PubMed] [Google Scholar]
- Pineda J, Aghajanian GK (1997) Carbon dioxide regulates the tonic activity of locus coeruleus neurons by modulating a proton- and polyamine-sensitive inward rectifier potassium current. Neuroscience 77:723–743. 10.1016/S0306-4522(96)00485-X [DOI] [PubMed] [Google Scholar]
- Purnell BS, Hajek MA, Buchanan GF (2017) Time-of-day influences on respiratory sequelae following maximal electroshock-induced seizures in mice. J Neurophysiol 118:2592–2600. 10.1152/jn.00039.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechtschaffen A, Gilliland MA, Bergmann BM, Winter JB (1983) Physiological correlates of prolonged sleep deprivation in rats. Science 221:182–184. 10.1126/science.6857280 [DOI] [PubMed] [Google Scholar]
- Richerson GB. (2004) Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci 5:449–461. 10.1038/nrn1409 [DOI] [PubMed] [Google Scholar]
- Richerson GB, Buchanan GF (2011) The serotonin axis: shared mechanisms in seizures, depression, and SUDEP. Epilepsia 52 [Suppl. 1]:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Scammell T (2001) The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci 28:152–157. [DOI] [PubMed] [Google Scholar]
- Scott MM, Wylie CJ, Lerch JK, Murphy R, Lobur K, Herlitze S, Jiang W, Conlon RA, Strowbridge BW, Deneris ES (2005) A genetic approach to access serotonin neurons for in vivo and in vitro studies. Proc Natl Acad Sci U S A 102:16472–16477. 10.1073/pnas.0504510102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson CA, Wang W, Pieribone VA, Dohle CI, Richerson GB (2003) Midbrain serotonergic neurons are central pH chemoreceptors. Nat Neurosci 6:1139–1140. 10.1038/nn1130 [DOI] [PubMed] [Google Scholar]
- Sowers LP, Massey CA, Gehlbach BK, Granner MA, Richerson GB (2013) Sudden unexpected death in epilepsy: fatal post-ictal respiratory and arousal mechanisms. Respir Physiol Neurobiol 189:315–323. 10.1016/j.resp.2013.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunanaga J, Deng BS, Zhang W, Kanmura Y, Kuwaki T (2009) CO2 activates orexin-containing neurons in mice. Respir Physiol Neurobiol 166:184–186. 10.1016/j.resp.2009.03.006 [DOI] [PubMed] [Google Scholar]
- Tomson T, Nashef L, Ryvlin P (2008) Sudden unexpected death in epilepsy: current knowledge and future directions. Lancet Neurol 7:1021–1031. 10.1016/S1474-4422(08)70202-3 [DOI] [PubMed] [Google Scholar]
- Veasey SC, Fornal CA, Metzler CW, Jacobs BL (1997) Single-unit responses of serotonergic dorsal raphe neurons to specific motor challenges in freely moving cats. Neuroscience 79:161–169. 10.1016/S0306-4522(96)00673-2 [DOI] [PubMed] [Google Scholar]
- Yokota S, Kaur S, VanderHorst VG, Saper CB, Chamberlin NL (2015) Respiratory-related outputs of glutamatergic, hypercapnia-responsive parabrachial neurons in mice. J Comp Neurol 523:907–920. 10.1002/cne.23720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Q, Buchanan GF, Motelow JE, Andrews J, Vitkovskiy P, Chen WC, Serout F,Gummadavelli A, Kundishora A, Furman M, Li W, Bo X, Richerson GB, Blumenfeld H (2016) Impaired serotonergic brainstem function during and following seizures. J Neurosci 36:2711–2722. 10.1523/JNEUROSCI.4331-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZQ, Scott M, Chiechio S, Wang JS, Renner KJ, Gereau RW 4th, Johnson RL, Deneris ES, Chen ZF (2006) Lmx1b is required for maintenance of central serotonergic neurons and mice lacking central serotonergic system exhibit normal locomotor activity. J Neurosci 26:12781–12788. 10.1523/JNEUROSCI.4143-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman JE, Naidoo N, Raizen DM, Pack AI (2008) Conservation of sleep: insights from nonmammalian model systems. Trends Neurosci 31:371–376. 10.1016/j.tins.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]