

Abstract
Frogeye leaf spot, caused by Cercospora sojina Hara, is a common disease of soybean in most soybean-growing countries of the world. In this study, we report a high-quality genome sequence of C. sojina by Single Molecule Real-Time sequencing method. The 40.8-Mb genome encodes 11,655 predicated genes, and 8,474 genes are revealed by RNA sequencing. Cercospora sojina genome contains large numbers of gene clusters that are involved in synthesis of secondary metabolites, including mycotoxins and pigments. However, much less carbohydrate-binding module protein encoding genes are identified in C. sojina genome, when compared with other phytopathogenic fungi. Bioinformatics analysis reveals that C. sojina harbours about 752 secreted proteins, and 233 of them are effectors. During early infection, the genes for metabolite biosynthesis and effectors are significantly enriched, suggesting that they may play essential roles in pathogenicity. We further identify 13 effectors that can inhibit BAX-induced cell death. Taken together, our results provide insights into the infection mechanisms of C. sojina on soybean.
Keywords: Cercospora sojina, soybean, genome, pathogenicity
1. Introduction
The causal agent of frogeye leaf spot (FLS), Cercospora sojina Hara, is a worldwide destructive pathogen on soybean.1 It was first reported in Japan in 1915.2 After that, many soybean growing countries were reported the occurrence of this disease, such as the USA, China, and Argentina.3 The main measurement to control this disease is to grow resistant soybean varieties or to apply chemical fungicides, which usually lose the effects rapidly in fields due to race differentiation and gene mutations of the pathogen.4–6 FLS causes about 10–60% yield loss in soybean growing regions, such as Argentina and Nigeria, and it has been reported to be the most expensive disease in the history of soybean production in Argentina.3
Taxonomically, C. sojina belongs to the order Capnodiales in the class of Dothideomycetes. Currently, it is known that there are 22 races of C. sojina in Brazil and 12 races in the USA.1 Later, using 93 isolates of C. sojina and 38 putative soybean differentials, Mian et al. proposed a core set of 11 races that represent the major diversity of the 93 isolates in the USA.1 We previously reported the 14 races in north China7; of the identified races of C. sojina, the occurrence frequency of race 1 is 43.5%, emerging as the dominant race among others, which causes yield loss up to 38% in the field.7
Despite the importance of C. sojina, the infection mechanism and the genetic information are not known for this pathogen. For example, most of the species can produce a non-specific coloured mycotoxin cercosporin in the Cercospora genus, which is indispensable for their pathogenicity,8 but C. sojina may not produce cercosporin.9 Does the genome harbour the cercosporin biosynthesis genes? Nowadays, the main strategy to unravel the mystery of the pathogen infection mechanisms is to obtain their genome information. Magnaporthe grisea, one of the best studied fungi, was sequenced at genome level in 2005.10 The genome annotation shows that the pathogen may carry over 700 secreted proteins, and most of them are believed to be virulence effectors. Later, several effectors have been implicated in suppressing immune responses in rice.11,12 The other genes, such as MoEnd3, MoSwi6, and MoHYR1 have been demonstrated to be essential for appressorium formation, melanin accumulation, and reactive oxygen species (ROS) scavenging.13–15 Similarly, the genome sequence availability and gene annotation have helped to uncover the infection mechanism of Verticillium dahliae substantially, where this pathogen shows distinct infection structure compared with M. oryzae and other fungi.16,17
Although the breeding programme and fungicide application make great success in controlling FLS in last decades, their efficiencies are facing challenges recently.4,7 In fact, Cercospora species may undergo positive selections and rapid evolution. Soares et al. reported that more Cercospora species were able to infect soybean and caused similar disease symptom as C. kikuchii, the closest species of C. sojina.3 Importantly, they detected interlineage recombination among Cercospora species, along with a high frequency of mutations linked to fungicide resistance.3 Moreover, it has been observed that C. sojina populations are genetically diverse and likely undergoing sexual reproduction.18 The above-mentioned reports imply that C. sojina could adapt to the changing environment flexibly.
In this study, we report the 40.8 Mb complete genome sequence of C. sojina race 1. The genome annotation and whole genome transcriptome assays reveal that C. sojina encodes a different set of proteins that distinguishes it from other fungi species in terms of infection strategy and disease development. We demonstrate that the secondary metabolites and effectors play essential roles in the soybean–C. sojina pathosystem. Moreover, the genome information can be further used for comparative genomic studies with other Cercospora species to unravel their evolution and infection mechanism on soybean.
2. Materials and methods
2.1. Fungal growth conditions and DNA preparation
Cercospora sojina (C. sojina) race 1 was isolated from soybean fields in Heilong Jiang province of China. Briefly, the fungi single spore was isolated from the soybean seedling infested by C. sojina race 1. The isolated spores were grown on potato dextrose agar (PDA) medium at 28°C. Then, the mycelia were removed from the media and ground in liquid nitrogen. Genomic DNA was extracted using a modified cetyltrimethylammonium bromide (CTAB) method.19
2.2. Genome sequencing and assembly
Cercospora sojina race1 genome was sequenced by Single Molecule Real-Time (SMRT) method in Biomarker Technologies (Beijing, China).20 DNA libraries with 270 bp and 10 kb inserts were constructed. The 270-bp library was constructed following Illumina’s standard protocol, including fragmentation of genomic DNA, end repair, adaptor ligation and PCR amplification. The 270-bp library was quantified using 2100 Bioanalyzer (Agilent, USA) and subjected to paired-ended 150 bp sequencing by Illumina HiSeq4000. The sequencing data (filtered reads: 2.99G, sequencing depth: 90×) were used to estimate the genome size, repeat content, and heterozygosity. Then, the 10-kb library was constructed following PacBio’s standard methods, including fragmentation of genomic DNA, end repair, adaptor ligation, and templates purification. The 10-kb library was quantified by 2100 Bioanalyzer (Agilent, USA) and sequenced by SMRT, and the sequencing data (filtered reads: 4.92G, sequencing depth: 123×) was assembled by CANU (Version-1.2) with default parameters.21 Finally, Illumina reads were used for error correction and gap filling with SOAPdenovo GAPCLOSER v1.12.22
2.3. Genome annotations
Protein-encoding genes were annotated by a combination of three independent ab initio predicators GeneMark (Version 4.30),23 SNAP24, and Glimmer.25 Transcriptome data were incorporated into PASA26 to improve quality of C. sojina annotation. Briefly, the transcriptome assemblies were mapped to the genome using Trinity.27 Then PASA alignment assemblies based on overlapping transcript alignments from Trinity and use EVidenceModeler (EVM) to compute weighted consensus gene structure annotations. Finally, PASA was used to update the EVM consensus predictions.
Functional annotations for all predicted gene models were made using multiple databases, including Swiss-Prot, nr, KEGG28, and COG29 by blastP with E-values of ≤1e−5. Domain-calling analyses of protein-encoding genes were performed using the Pfam database30 and HMMER.31 Potential virulence-related proteins were identified by searching against the pathogen–host interaction database (PHI-base)32 by blastP with E-values of ≤1e−5. Blast2GO33 was used for Go enrichment analysis of genes that only belong to C. sojina, and do not have homologue in the non-plant pathogen Aspergillus nidulans and Neurospora crassa.
2.4. Repetitive sequences and whole genome DNA methylation analysis
Tandem repeat sequences were identified using Tandem Repeats Finder (TrF).34 Transposable elements (TEs) were excavated strictly using three softwares, including a de novo software Repeat Modeler (http://repeatmasker.org/RepeatModeler/) and two database-based softwares Repeat Protein Masker (www.repeatmasker.org/cgi-bin/RepeatProteinMaskRequest) and Repeat Masker (www.repeatmasker.org/). All the parameters are set as default. Whole-genome DNA modification detection and motif analysis were performed according to Blow et al.35 using the PacBio SMRT software (version = 2.2.3, www.pacb.com).
2.5. Phylogenetic analysis and synteny analysis
The sequences of fungi were downloaded from DOE Joint Genome Institute (JGI).36 A group of consistent phylogenetic ‘backbone’ genes (phylogeneticly conserved) in the fungi genome were used to construct phylogenetic tree.37 Putative ‘backbone’ genes of the other 11 fungi were identified using stand-alone blast with E-values of ≤1e−20. And the ‘backbone’ genes were concatenated into one sequence. Sequence alignment was done using MUSCLE38 and the phylogenetic tree was generated by MEGA 7.039 using a UPGMA method. Ustilago maydis was used as outgroup. Synteny of Cercospora zeae-maydis, C. sojina, and Pseudocercospora fijiensis was analysed using GATA.40
2.6. Comparison analysis of carbohydrate-active enzymes and secondary metabolism genes
The proteome of C. sojina and 14 other above-mentioned fungal species were downloaded from DOE Joint Genome Institute (JGI).36 HMMER 3.0 packages were used for homology search.41 Family-specific HMM profiles were downloaded from dbCAN database.42 The executable file hmmscan and the hmmscan-parser script provided by dbCAN were used to generate and extract the searching results, respectively.
Putative polyketide synthases (PKS) and non-ribosomal peptide synthases (NRPS) genes were identified using the web-based software SMURF with default settings.43 The modules of different domains in individual NRPS and PKS proteins were identified via searching the antiSMASH database (antibiotics and Secondary Metabolite Analysis Shell).44 The core genes were annotated using stand-alone BLAST (E-values ≤1e−10) against Swiss-Prot database.
2.7. Secondary metabolites extraction and quantification
Cercosporin extraction and quantification were performed according to the method described by Shim and Dunkle.45 The pigments of C. sojina were induced in the complete medium (CM) with 20 mmol/l cyclic adenosine monophosphate (cAMP) or starvation treatments for 4 days at 28°C. The supernatants were collected and purified by the C18-SPE cartridge. The elutes in 40% methanol fraction were further purified by a HPLC on C18 preparation column. The fractions with grey, light yellow, and dark grey were further identified by a reverse phase HPLC with a PDA detector (Shimadzu Corporation, Japan).
2.8. Transcriptome analysis and quantitative RT-PCR
For transcriptome analysis, the fungus was grown in minimal nutrient medium (3 g NaNO3, 1 g K2HPO4, 0.5 g MgSO4⋅7H2O, 0.5 g KCl, 0.01 g FeSO4, and 30 g Sucrose per litre) at 28°C for 6 days. For starvation treatment, the fungal mycelia was transferred to minimal nutrient medium lacking NaNO3, then was harvested at 24 and 48 h, respectively. Each treatment had three biological replicates.
Library preparation and bioinformatics analysis were performed according to the method of Yang et al.46 One microgram RNA per sample was subjected to RNA-seq library construction. The RNA-seq libraries were quantified using 2100 Bioanalyzer (Agilent, USA), and sequenced (paired-end, 100 bp each) by the Illumina genome analyzer (Hiseq 2000; Illumina, USA). Quantification of gene expression levels were estimated by fragments per kilobase of transcript per million fragments mapped. Differential expression analysis was performed using the DESeq R package.
For quantitative RT-PCR, infected leaves were collected at indicated time points. Total RNA was extracted by Trizol method (Invitrogen). The cDNAs were synthesized using HiScript II Q RT SuperMix kit with genomic DNA wiper (Vazyme Biotech). Reactions were performed on CFX96TM Real-time System (Bio-RAD) with the SYBR qPCR Mix (Vazyme Biotech). The 2−ΔΔCT method was used for calculating the relative gene expression levels. Actin gene of C. sojina was used as the internal control.
2.9. Functional study of putative effectors
The putative effector genes were cloned into binary vector pMD-1 (T7 tag) driven by 35S promoter using ClonExpress II One Step Cloning Kit (Vazyme Biotech). Then, these constructs were transformed into Agrobacterium tumefaciens strain EHA105 by electroporation. Leaves of 4-week-old Nicotiana benthamiana were infiltrated with EHA105 strains harbouring indicated effectors using needleless syringes. GFP and Phytophothora sojae effector Avr1b served as the negative control and positive control, respectively. Twenty-four hours later, plants were infiltrated with EHA105 harbouring pVX-BAX (OD600 = 0.4). Cell death symptoms were evaluated and photographed at 72 h after pVX-BAX infiltration. Results are representatives of six biological replicates.
3. Results
3.1. Assembly of C. sojina genome
Like most of the fungi, C. sojina showed similar infection cycle, but it also demonstrated some distinctions (Supplementary Fig. S1). It does not form appressorium, but infects the plants by branched hyphae through open stomata (Supplementary Fig. S1C and D). Compared with other hemibiotrophic fungi, FLS disease development is relatively slower (Supplementary Fig. S1C). In order to investigate the infection mechanism, we therefore extracted the genomic DNA from the mycelia and sequenced the pathogen at genome level.
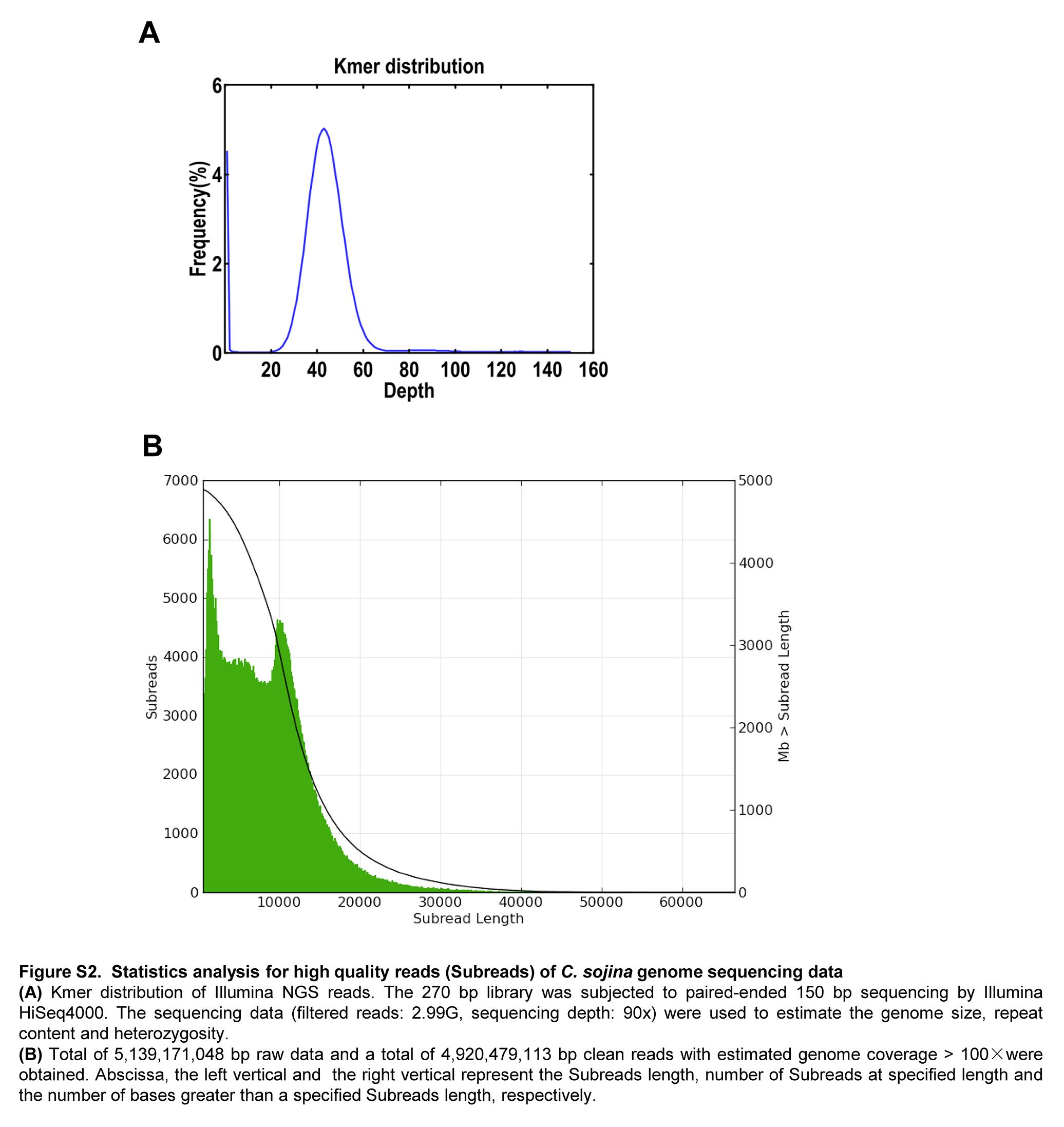
The genome of C. sojina race 1 was assembled from the data generated by a recently developed Single Molecule Real-Time (SMRT) sequencing technique, an effective method to decode the difficulty to detect but important regions, such as non-coding regions and repetitive elements, which can assist in obtaining gapless eukaryotic genome sequence.20
Subreads distribution analyses confirm the high quality of the 10-kb library (Supplementary Fig. S2 and Table S1). The sequencing data (4,920,479,113 bp clean reads) were de novo assembled using CANU,21 leading to the generation of 62 contigs, with an N50 length of 1.59 Mb and a total assembly size around ∼40.84 Mb (Table 1 and Supplementary Table S2). Twenty-four largest scaffolds were displayed by circos-plot (Fig. 1). A total of 11,655 protein-coding genes are predicted, in which the gene density is ∼285 genes per 1 Mb. However, 277 tRNA and 281 pseudogenes are predicted in the genome. Notably, 8,474 putative protein-coding genes were supported by the RNA-seq data. It is also worth noting that the genome size of C. sojina race 1 is much larger than C. sojina isolate S9, where the genome was assembled by 124 bp library and genome size was estimated around ∼30.8 M.47
Table 1.
Genome features of Cercospora sojina
| Features | C. sojina |
|---|---|
| Size (bp) | 40,835,411 |
| Coverage | 120× |
| (G+C) percentage (%) | 53.12 |
| N50 (bp) | 1,594,385 |
| Protein-coding genes | 11,655 |
| Average gene length (bp) | 1,441 |
| Gene density (no. gene per Mb) | 285 |
| tRNA genes | 277 |
| Pseudogene | 281 |
Figure 1.

Circos-plot of C. sojina. The largest 24 scaffolds of C. sojina are displayed by circos-plot (Mb scale). The circos from outside to inside are: (a) 24 largest scaffolds; (b) DNA methylations; (c) GC content; (d) carbohydrate enzymes; (e). putative effectors; (f) PHI-base genes; (g) duplicated genes; The DNA methylations and GC contents are statistical results of 20 kb non-overlapping windows. The inner lines link duplicated genes.
3.2. Repetitive elements and potential methylation sites
Repetitive DNA sequence and TEs play important roles in the evolution, the genome structure, and gene functions of fungi.48 A total of 11,138,239 bp (∼11 M) repeat sequences were identified in C. sojina genome, including DNA transposon, LTR retrotransposon, tandem repeat sequence and other unclassified transposons (Fig. 2A). The repeat sequence accounts for 25.56% of the genome. Interestingly, the majority of repetitive sequences (96.36%) are TEs, whereas the tandem repeat sequences just account for 0.93%. Notably, DNA transposon and LTR retrotransposons account for 28 and 25% of all TEs, respectively.
Figure 2.

Repeat elements and DNA methylation sites of C. sojina. (A) The percentage of different types of repetitive sequences in the C. sojina genome. (B) Statistic analysis of candidate DNA methylation sites from primary sequence of C. sojina genome. m4C, m6A, and the unidentified represent 4-methyl-cytosine, 6-methyl-adenosine, and the unidentified methylation sites, respectively. (C) Distribution of repetitive elements and different types of DNA methylations in scaffold 1 of C. sojina. Black histogram indicates the distribution of repetitive elements. All data are statistical results of 20 kb windows. Asterisks indicate regions with high and low frequency of DNA methylations, respectively. (D, E) The number of different type of methylations per 20 kb was calculated in total genome and repetitive elements of C. sojina.
DNA methylation is involved in many important cell processes, such as genomic imprinting and gene transcription regulation.49 Although DNA methylation has been found in higher plants and animals for years, it is just reported in some fungi recently.50 Using SMRT, we were able to detect m6A and m4C methylation in particular.51 In total, 1,015,733 m4C (4-methyl-cytosine) and 17,409 m6A (6-methyl-adenosine) were identified in C. sojina genome (Fig. 2B). However, majority of methylation sites (8,453,041) were uncategorized. Interestingly, most of the categorized DNA methylations are m4C, accounting for 98.3%, whereas m6A only accounts for 1.7%. In consistent with the identified methylation sites, we detect multiple motifs that may be recognized by transmethylase specifically (Supplementary Table S3). However, compared with m6A, m4C DNA methylations occur with low frequency in the regions of repetitive elements (Fig. 2C–E).
3.3. Comparative genomic analysis
The evolutionary relationship of C. sojina and other fungi species was analysed using a group of phylogenetic backbone genes of the fungi.37 Phylogenetic analysis reveals that C. sojina is evolutionally close to Cercospora zeae-maydis, a plant pathogen that can cause leaf spot disease on maize52 (Fig. 3A). In addition, C. sojina is also close to the other three Dothideomycetes pathogen Pseudocercospora fijiensis, Sphaerulina musiva, and Dothistroma septosporum (Fig. 3A). The C. sojina homologous proteins show an average identity of 79.9, 65.9, 67.5, and 65.8% with that of C. zeae-maydis, P. fijiensis, S. musiva, and D. septosporum, respectively.
Figure 3.

Phylogenetic and synteny analysis of C. sojina with other fungal species. (A) Phylogenetic tree of different fungal species. The UPGMA phylogenetic tree was constructed based on the consistent phylogenetic backbone genes of the fungi. The number represents branch lengths. (B) Synteny of Cercospora zeae-maydis, C. sojina, and Pseudocercospora fijiensis. Rectangle boxes represent order of gene models. Non-coding regions are not depicted.
Although C. sojina is evolutionarily distant from the non-plant pathogen Aspergillus nidulans and Neurospora crassa, 47.18 and 43.07% proteins of C. sojina have homologues in A. nidulans and N. crassa, respectively. Therefore, we examined the potential gene family expansions in C. sojina. The results show that 5,652 genes exclusively exist in C. sojina genome but not in A. nidulans and N. crassa. However, near 70% of these genes cannot be annotated from current databases (Supplementary Fig. S3A). Go enrichment analysis of the 1,675 annotated genes reveals that most of the genes are involved in metabolic process, biosynthetic process, and response to stresses or stimuli (Supplementary Fig. S3B). Notably, these genes are predicted to have binding function (ion binding or protein binding), hydrolase activity, transferase activity or oxidoreductase activity (Supplementary Fig. S3C). As C. sojina is evolutionarily distant from the non-plant pathogens, we speculate that the gene family expansion, a common event in the evolution of phytopathogenic fungi,52,53 might have occurred in C. sojina genome, which eventually makes C. sojina be a plant pathogen.
In addition, synteny analysis of C. sojina genome with the other three genomes of Dothideomycetes spp., the C. zeae-maydis, P. fijiensis, and Mycosphaerella graminicola, reveals that C. sojina genome displays different synteny with those fungi (Fig. 3B and Supplementary Fig. S4). Of all the sequenced genomes, C. zeae-maydis shows highest synteny with C. sojina. For example, scaffolds 2, 4, and 5 of C. zeae-maydis correspond well with the scaffold 1 of C. sojina, and Scaffold 3 and 10 show well syntney to the scaffold 3 of C. sojina (Supplementary Fig. S4A). Importantly, we observed that the 24 largest scaffolds, which accounts for 91.2% of C. sojina genome, show very high synteny with the 13 core chromosomes of M. graminicola,53 but not with the rest 8 dispersed chromosomes (Supplementary Fig. S4C), indicating that C. sojina shares the conserved and core genes of Dothideomycetes.
3.4. The secretome and potential effectors
The genome of C. sojina contains 11,655 protein-coding genes, covering approximately 41% sequence of the genome (Table 1). Among them, a total of 9,506 genes were annotated using multiple databases (Supplementary Fig. S5 and Table S4). Domain calling analysis reveals that more than 7,500 types of domains exist in C. sojina proteome. The arsenal of potentially secreted proteins were predicted, and proteins containing a signal peptide, but lacking transmembrane domain and glycosylphosphatidylinositol (GPI) modification site were considered as secreted proteins. A combination of software tools for the prediction of transmembrane domain,54 signal peptide motifs,55 and GPI modification site indicate that C. sojina has similar number of potential secreted proteins (∼750) when compared with other fungal species.56
Pathogen-secreted effectors play critical roles in facilitating the proliferation of pathogens, often by suppressing plant immune system.57 In total, 233 proteins are predicted as the putative small (≤400 amino acids) cysteine-rich (≥4 cysteine residues) proteins. Through domain calling analysis, 205 functional motifs and domains were found in 141 putative effectors, including 60 effectors with multiple domains. Notably, the most abundant domain is PF14295.4 (n = 6), which mediates protein−protein interactions. Other common domains include abhydrolase domain (PF12697.5, n = 5), Hydrolase domain (PF12146.6, n = 5), and PAN domain (PF00024.24, n = 5).
3.5. Up-regulation of pathogenicity-related genes by whole genome transcription assays
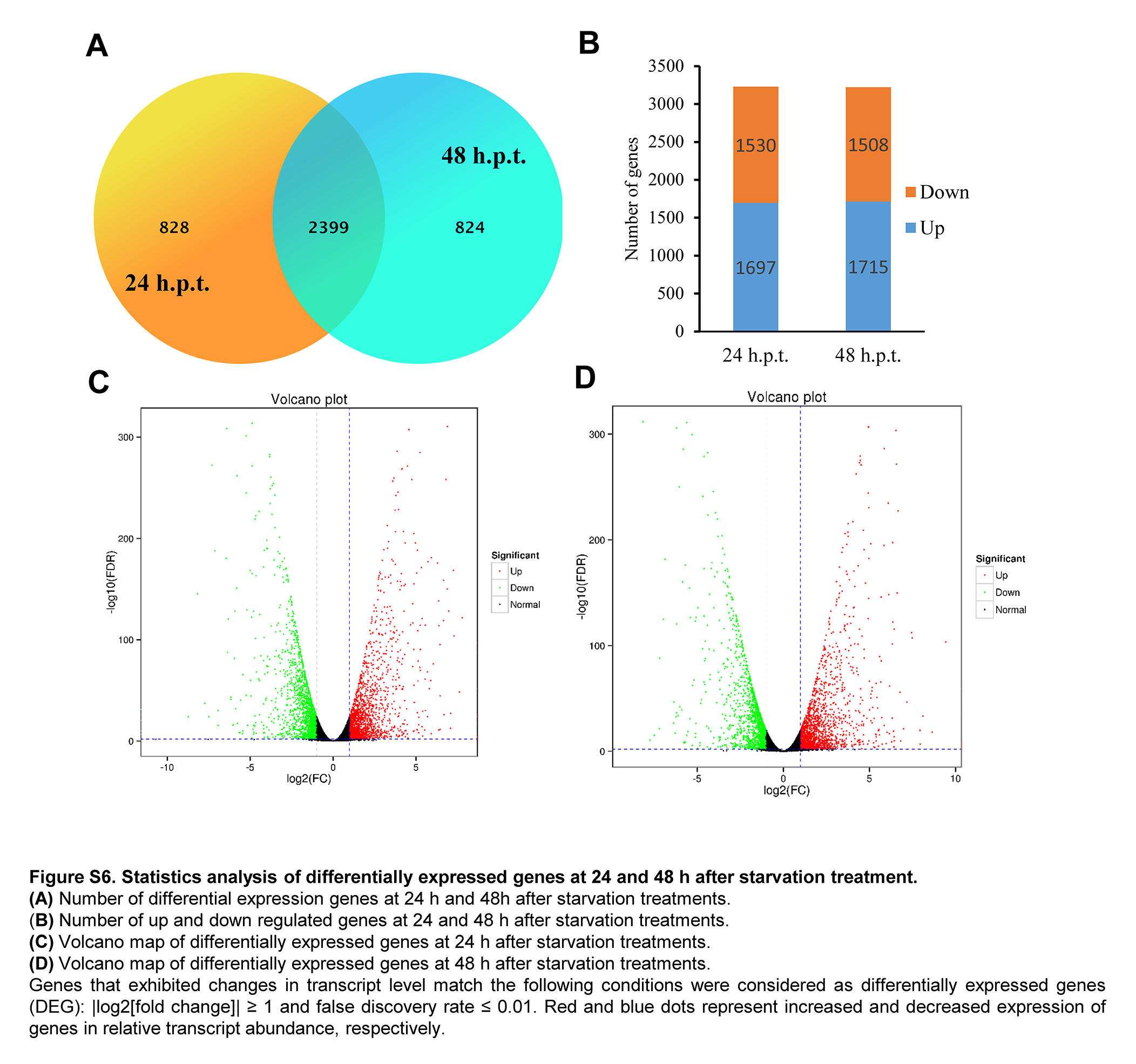
Because the infection progress of C. sojina on soybean is very slow, it is difficult to collect enough samples to examine the gene expression of the in planta hyphae. However, starvation treatments could mimic the physiology of pathogen during infection.58 Therefore, we used the mycelia that were grown in nutrient-limited culture for 24 and 48 h, and performed transcriptome analysis by RNA sequencing; 3,227 and 3,223 differentially expressed genes (DEGs) were identified at 24 and 48 h post-starvation treatment (hpt), respectively (Supplementary Fig. S6). A total of 4,051 DEGs were identified during starvation treatment, and 2,399 genes were differentially expressed at both 24 and 48 hpt. Of all the DEGs, 1,530 and 1,508 genes were upregulated, while 1,697 and 1,715 genes were downregulated at 24 and 48 hpt, respectively (Supplementary Fig. S6).
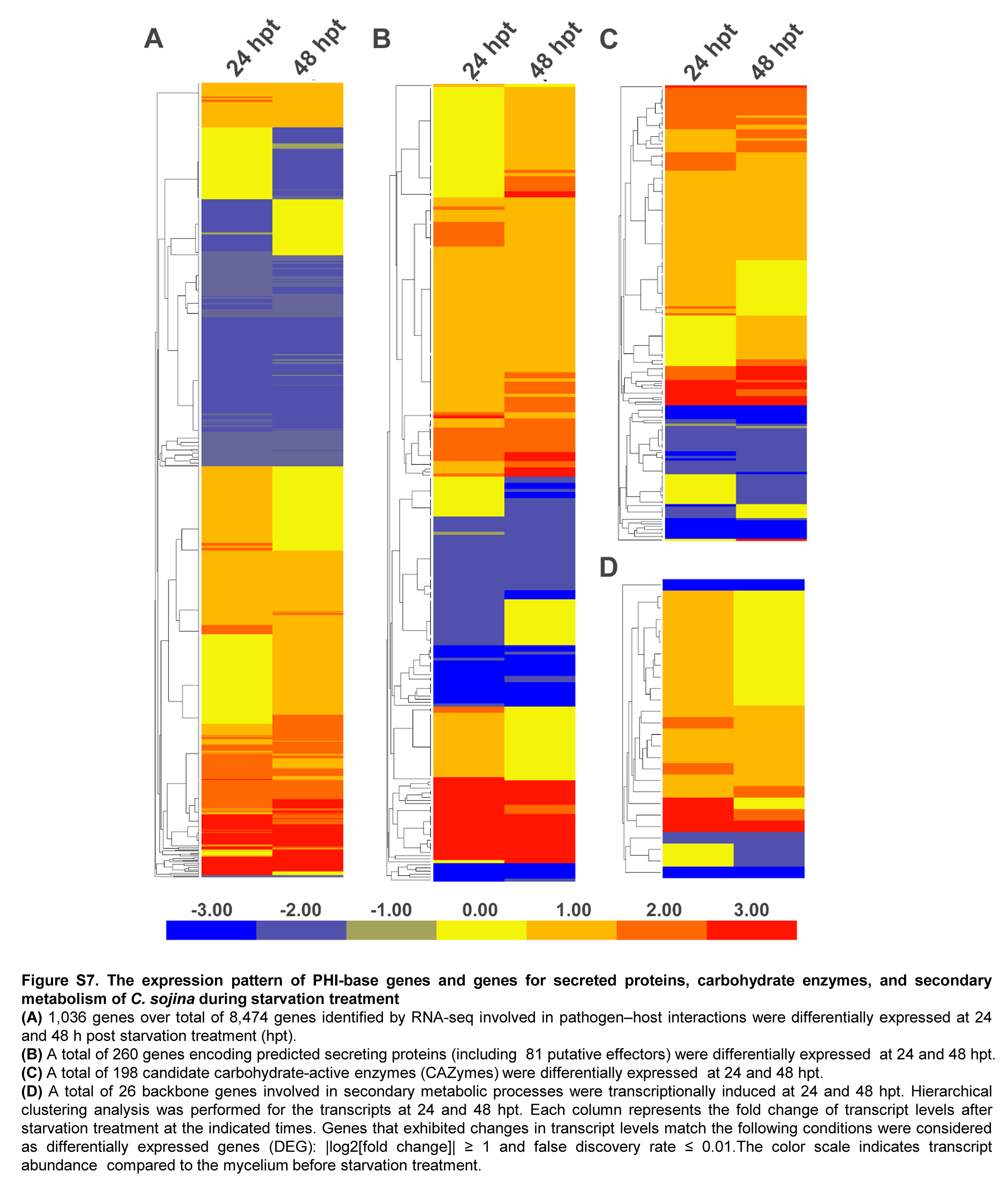
Notably, four classes of DEGs caused our attention. These genes are annotated to be involved in PHI, secretome, putative carbohydrate-active enzymes (CAZymes), and secondary metabolic processes. First, 1,036 PHI genes are differentially expressed after starvation treatment (Supplementary Fig. S7A). A total of 591 PHI genes are significantly upregulated, demonstrating the important roles of these genes in responding to stimulus. Second, 260 secreted protein-coding genes, including 81 effectors, are differentially expressed (Supplementary Fig. S7B). There is 62.5% (50/80) effector-coding gene expression being significantly upregulated, and some effectors with conserved domains, such as Wall Stress-responsive Component domain, glycoside hydrolase, fungal hydrophobin, cutinase, leucine rich repeat or peptidase domains, may play critical roles in fungal pathogenicity (Supplementary Table S5). Third, 198 CAZymes were differentially expressed. Interestingly, almost half of them are glycoside hydrolases (87/198), implying their essential roles in early infection (Supplementary Fig. S7C). Further, the secondary metabolism-related genes, including 5 PKS and 16 NRPS/NRPS-like genes are significantly upregulated (Supplementary Fig. S7D). These genes are likely involved in mycotoxin biosynthesis in fungi. Therefore, the increased expression of PKS and NRPS/NRPS-like genes suggests that these genes may play essential roles in mycotoxins biosynthesis (Supplementary Table S6).
3.6. Gene clusters for secondary metabolites
Cercospora sojina genome encodes 16 non-ribosomal peptide synthetases (NRPS), 20 PKS, 18 fatty acid synthases, 3 terpene synthases, 2 geranylgeranyl diphosphate synthases, and 1 terpenoid cyclases (Supplementary Tables S7 and S8). These enzymes are involved in synthesis of secondary metabolites, including mycotoxins, pigments, and alkaloids. Annotation of secondary metabolite biosynthesis genes shows that C. sojina lacks PKS-NRPS hybrids, PKS-like proteins, and dimethylallyl tryptophan synthases (Supplementary Tables S7 and S8).
In the Cercospora genus, most of the species can produce a non-specific mycotoxin cercosporin. However, it has been disputed that if C. sojina produces cercosporin.9 Nevertheless, we identified a similar gene cluster with eight cercosporin biosynthesis genes in C. sojina genome (Fig. 4A). These eight genes display high amino acid sequence similarity to C. nicotianae cercosporin biosynthesis genes in the same tandem order (Fig. 4A). Furthermore, we observed the increased transcription of the eight genes during infection (Fig. 4B). These data imply that C. sojina may produce cercosporin during infection. However, we were unable to detect the cercosporin in either cultured mycelium or infected plant tissue according to the method that was used in other Cercospora species (Supplementary Fig. S8).
Figure 4.

Putative gene clusters for cercosporin biosynthesis in C. sojina. (A) The eight cercosporin toxin biosynthetic genes in C. sojina genome. The genes involved in cercosporin biosynthesis of Cercospora nicotianae (C. nicotianae) were blasted against C. sojina using BlastP. The black arrow represents the direction of sense strand. (B) Expression of candidate genes involved in cercosporin biosynthesis at 48 h after C. sojina infection. The mRNA levels were detected using qRT-PCR. Values are means ± SD (n = 3 biological replicates).
Pigments are the other important group of secondary metabolites for successful invasion of pathogens. Generally, pathogen-produced pigment is able to protect pathogen from host oxidative stress during infection.59 We found that C. sojina genome encodes multiple putative PKS that are responsible for pigment production (Fig. 5A and Supplementary Table S7). We also found that C. sojina can produce some grey pigments, and the pigment was significantly induced by both starvation and cAMP treatments (Fig. 5B), suggesting that the pigments may be related to pathogen virulence. Therefore, we further isolated and partially purified the pigments. Three major components, the grey, the light yellow, and the dark grey pigments were obtained (Supplementary Fig. S9), and the dark grey pigment is the most abundant one.
Figure 5.

Putative PKS gene clusters for pigment production in C. sojina. (A) Genes encoding methyltransferases, cytochrome P450s, oxidoreductases, dehydrogenases, acyltransferases, MFS transporters, and transcriptional factors were clustered with PKS genes. These clusters are responsible for the pigment synthesis. The black arrows represent the direction of sense chain. The orange arrows highlight PKS genes. The blue and grey arrows represent putative pigment biosynthesis genes and other genes that are not involved in pigment biosynthesis. (B) Pigments produced by C. sojina in different media. The supernatants were collected at 4 days after treatments on mycelia. Czapek medium served as a negative control. Results are representatives of three biological replicates. PDB and CM represent potato-dextrose broth medium and complete medium, respectively.
3.7. Carbohydrate-active enzymes
Successful phytopathogenic fungi can break down and utilize the plant cell wall polysaccharides by CAZymes. Cercosporasojina harbours 596 predicted CAZymes (Supplementary Table S9). Compared with other fungi in Dothideomycetes, C. sojina has a larger group of potential carbohydrate esterases, which can catalyze the O-de- or N-deacylation of substituted saccharides60,61 (Supplementary Table S9). In C. sojina genome, there are around 23.5% potential secreted proteins (177/752) that were predicted as CAZyme, demonstrating that C. sojina may employ a large group of CAZymes to digest host cell walls during invasion.
Interestingly, one of the families of CAZymes, the glycoside hydrolase GH109 family, is highly enriched (Fig. 6). The biochemical function of GH109 family is proved to be α-N-acetylgalactosaminidase (αNAGAL), which can cleave the terminal alpha-linked N-acetylgalactosamine epitope of blood group A.62 However, it has been shown that soybean lectin can specifically bind N-acetyl galactosamine, a component of fungal cell wall.63 These data suggest that αNAGAL may be able to compete with lectin to bind N-acetylgalactosamine. We find that C. sojina encodes 14 putative GH109 genes, which is more than most of fungi, such as M. oryzae, B. cinerea, and N. crassa (Supplementary Table S10). These data suggest that the expansion of GH109 family in C. sojina may contribute to overcome lectin-mediated resistance in soybean.
Figure 6.

Comparison of carbohydrate enzymes between C. sojina and nine other fungal species. Nine species were selected to compare with C. sojina. Pi, Phytophothora infestans; Ps, Phytophothora sojae; Nc, Neurospora crassa; Bc, Botrytis cinerea; Cm, Cercospora zeae-maydis; Cs, Cercospora sojina; An, Aspergillus nidulans; Vd, Verticillium dahliae; Mo, Magnaporthe oryzae; Fg, Fusarium graminearum. GH, glycoside hydrolase; GT, glycosyltransferase; PL, polysaccharide lyase; CE, carbohydrate esterase; AA, auxiliary activity family; CBM, carbohydrate-binding module family. The numbers of gene families were normalized by Z score.
Of the annotated carbohydrate esterase genes, family CE1 and CE10 are two major subfamilies (Fig. 6). Both families encode proteins with the common activities of carboxylesterase and endo-1,4-β-xylanase.64 Besides, 34 potential carbohydrate-binding module (CBM) proteins were identified in C. sojina genome (Supplementary Table S9), which can digest carbohydrate complex extracellularly.64 Unexpectedly, only one CBM protein CBM1 was found in C. sojina genome (Supplementary Table S9). However, many phytopathogenic fungi carry plenty of CBM1. For example, Verticillium dahlia has an expansion of CBM1 containing protein family (∼30 genes).65
3.8. Functional analysis of putative effectors
Effectors are low molecular weight proteins that are secreted by bacteria, oomycetes or fungi to impair the host immune defence and to adapt to specific environment. In the maize pathogen U. maydis, it was observed that most of the genes in secreted protein-coding gene clusters were induced simultaneously in infected tissue.66 Phylogenetic analysis for 233 putative effectors reveals that 21 predicted effectors are grouped into clusters that contain 2 or 3 high sequence similarity genes in C. sojina (Fig. 7A). Clusters of putative effectors also suggest that local duplications might be involved in expansion of effectors in C. sojina. In addition, 40 putative effectors can be annotated by PHI database (Supplementary Table S11), and most of the annotated effectors have been implicated in fungal pathogenesis.
Figure 7.

Functional analysis of putative C. sojina effectors. (A) Phylogenetic analysis of putative effectors. The phylogenetic tree of 233 putative effectors is categorized into six super clades. (B) Selected C. sojina effectors suppress BAX-induced programmed cell death in N. benthamiana. N. benthamiana leaves were infiltrated with agrobacterium carrying C. sojina effector genes (OD600 = 0.4). GFP and Phytophothora sojae effector Avr1b were used as the negative and the positive controls, respectively. Twenty-four hours later, plants were infiltrated with pVX-BAX (OD600 = 0.4), then the photos were taken 3 days later. Results are representatives of six biological replicates. (C) The effectors that can suppress BAX-induced PCD are significantly up-regulated at 48 hpi. Soybean leaves were inoculated with C. sojina. Samples were collected at 0 and 48 hpi, respectively. Quantitative RT-PCR (qRT–PCR) were used to determine gene expression levels. Values are mean ± SD (n = 3 biological replicates).
Next, we attempt to investigate the effector function. Pro-apoptotic mouse protein BAX-induced programmed cell death (PCD) on N. benthamiana could physiologically resemble defence-associated hypersensitive response caused by pathogens, providing a valuable screening approach for effectors that can suppress defence-related PCD.67 We randomly selected 50 effectors and transiently expressed them in N. benthamiana to screen the potential effectors that can suppress BAX-triggered PCD (BT-PCD). Our results show that about 1/4 (13/50) selected effectors strongly suppress BT-PCD (Fig. 7B). Moreover, qRT-PCR results show that most of these putative effectors are transcriptionally induced at 48 h after C. sojina infection (Fig. 7C), implying they probably contribute to early infection on soybean.
4. Discussion
Cercospora species cause severe leaf spot and blight diseases on many crops worldwide.1 In this study, we sequenced the genome of the economically important fungi, C. sojina race 1. The genome size is ∼40.84 Mb, and 25.56% of the genome is composed of repeat sequences. However, compared with the genome size of sequenced but not annotated C. sojina isolate using NGS method (IMG genome id: 2506520004, https://img.jgi.doe.gov/cgi-bin/m/main.cgi), C. sojina race 1 has a larger genome size (Table 1 and Supplementary Table S12). In particular, the assembled C. sojina genome by SMRT generates ∼10 Mb repetitive sequences that are not detected by other sequencing techniques.
DNA methylation is an important research area of epigenetics in both eukaryotic and prokaryotic. The most studied type of DNA methylation in fungi is m5C, while in prokaryotic it is m4C and m6A.68 Until recently, the methylase and demethylase for m6A in eukaryotic were identified. However, in fungi, this type of DNA modification is poorly studied. Using SMRT, we identified m6A and m4C in C. sojina for the first time, in which we found 17,407 m6A and 1,015,733 m4C in C. sojina genome (Fig. 2B). The m6A frequency in this fungus is 435.2/Mb, which is slightly more than 341.3/Mb in yeast.69 In bacteria, m6A is regarded as an epigenetic signal for DNA-protein interactions.70 Surprisingly, we also observe that C. sojina contains large numbers of m4C, which is only reported in bacteria to our knowledge.68 Interestingly, the m4C modification occurs with lower frequency in the region of repetitive elements in C. sojina genome (Fig. 2C and D), indicating that m4C may be involved in the transposition of the transposons. Our results also demonstrate that SMRT is a powerful tool in studying fungal genome epigenetic modification.
It is worth noting that one of the enriched family is glycoside hydroxylate GH109 family (Fig. 6). This gene family encodes α-N-acetylgalactosaminidase (αNAGAL), an enzyme that can cleave N-acetyl galactosamine from the conjugated proteins.62 Interestingly, it has been found that lectin can specifically bind N-acetyl galactosamine.62 It is also found that soybean lectin can bind the N-acetyl galactosamine, a component of fungal cell wall.63 Therefore, the αNAGAL may compete with soybean lectin to bind N-acetyl galactosamine. It is known that soybean lectin and other plant lectins can inhibit hyphae growth and spore germination by binding their cell wall components in several fungi, such as Penicillia and Aspergilli species.71,72 We hypothesize that the expansion of GH109 family in C. sojina genome may contribute to overcome the lectin-mediated disease resistance in soybean.
CBMs are the most common non-catalytic modules associated with enzymes active in plant cell-wall hydrolysis.73 They can increase enzyme efficiency by anchoring the enzyme’s catalytic region to insoluble cellulose.74 However, only one CBM1 protein was found in C. sojina genome (Supplementary Table S9). In light of their relative slower infection process, the deficiency of CBMs in the genome may undermine C. sojina infection in terms of digesting plant cell walls.
It is believed that mycotoxin plays a critical role during pathogen infection.75 Mycotoxin cercosporin produced by Cercospora spp. is considered to be one of the key factors that can enhance their virulence, as their pathogenicity was remarkably impaired in the cercosporin-deficient mutants.75 However, C. sojina is one of the few Cercospora spp. that was reported not able to produce cercosporin, although there is a dispute.9 We made an effort to examine the cercosporin in either cultured mycelium or infected plant tissue. However, we are unable to detect cercosporin in any of the samples although the complete gene cluster for cercosporin biosynthesis exists in C. sojina genome (Fig. 4 and Supplementary Fig. S8). Therefore, our data imply that C. sojina may not employ cercosporin but other mycotoxin to enhance virulence.
Fungi-derived pigments can act as virulence factors to facilitate infection in plants, and are required for pathogen fitness by serving as UV protectants and ROS scavengers.59 The well-studied fungal pigment is melanin, which essentially contributes to fungal pathogenesis by altering cytokine responses, decreasing phagocytosis and scavenging ROS, as well as playing an important role in reinforcing fungal cell wall.76 Similar as melanin, we observed some pigment production was induced by cAMP or starvation treatments (Fig. 5B). We also identified the key gene clusters that encode PKS in C. sojina genome (Fig. 5A and Table S7). Moreover, the whole genome transcriptome assays also demonstrate that the key genes that are involved in pigment biosynthesis are significantly up-regulated. These data further support the assumption that the pigments produced by C. sojina are involved in its virulence.
In addition to secondary metabolites, pathogens usually harbour various virulence effectors. These effectors interfere with host immune responses to enhance virulence. For example, bacterial pathogen Pseudomonas syringae delivers over 30 effectors by type III secretion system during infection.77 Our work showed that more than one third of the effectors were upregulated during starvation (Supplementary Fig. S7B), and many of them can suppress BAX-induced cell death (Fig. 7B), similar to the finding in Phytophothora sojae.67 These data demonstrate that C. sojina can probably deploy effectors to promote infection.
In summary, we report a complete genome sequence of C. sojina by SMRT sequencing method. This sequencing method not only assists us to find the repetitive elements, but also to discover the DNA methylations in fungus. By the genome assembly and annotation, we hypothesize that the specific CAZymes, secondary metabolites, and effectors can help C. sojina to adapt to soybean successfully. Our work also lays the groundwork for future discoveries on this important soybean disease.
Supplementary data
Supplementary data are available at DNARES online.
Authors’ contributions
J.L. and S.M. conceived the research plans; X.L., J.C., and J.H. performed the experiments; Z.W., Z.G., and Y.C. provided technical assistance; X.L., J.C., and J.L. wrote the article.
Funding
The study was supported by Chinese Academy of Sciences (Strategic Priority Research Program Grant NO. XDB11020300), the National Natural Science Foundation of China (Grant 31570252 and Grant 31500220), and by the grant from the State Key Laboratory of Plant Genomics (Grant No. O8KF021011).
Conflict of interest
None declared.
Accession number
Data availability
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession NFUF00000000. The version described in this paper is version NFUF01000000.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1. Mian M.A.R., Missaoui A.M., Walker D.R., Phillips D.V., Boerma H.R.. 2008, Frogeye leaf spot of soybean: a review and proposed race designations for isolates of Cercospora sojina Hara, Crop Sci., 48, 14–24. [Google Scholar]
- 2. Gupta D.K., Singh M., Singh G., Srivastava L.S.. 1994, Sources of resistance in soybean (Glycine-max) to frog-eye leaf-spot caused by Cercosporidium-sojinum, Indian J. Agric. Sci., 64, 886–7. [Google Scholar]
- 3. Soares A.P.G., Guillin E.A., Borges L.L., et al. 2015, More Cercospora species infect soybeans across the Americas than meets the eye, PloS One, 10, e0133495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mian R., Bond J., Joobeur T., et al. 2009, Identification of soybean genotypes resistant to Cercospora sojina by field screening and molecular markers, Plant Dis., 93, 408–11. [DOI] [PubMed] [Google Scholar]
- 5. Galloway J. 2008, Effective management of soybean rust and frogeye leaf spot using a mixture of flusilazole and carbendazim, Crop Prot., 27, 566–71. [Google Scholar]
- 6. Zhang G.R., Pedersen D.K., Phillips D.V., Bradley C.A.. 2012, Sensitivity of Cercospora sojina isolates to quinone outside inhibitor fungicides, Crop Prot., 40, 63–8. [Google Scholar]
- 7. Ma S.M., Li B.Y.. 1997, Primary report on the identification for physiological races of Cercospora sojina Hara in northeast China, Acta Phytopathol. Sin., 27, 180. [Google Scholar]
- 8. Daub M.E., Ehrenshaft M.. 2000, The photoactivated Cercospora toxin cercosporin: contributions to plant disease and fundamental biology, Annu. Rev. Phytopathol., 38, 461–90. [DOI] [PubMed] [Google Scholar]
- 9. Goodwin S.B., Dunkle L.D., Zismann V.L.. 2001, Phylogenetic analysis of cercospora and mycosphaerella based on the internal transcribed spacer region of ribosomal DNA, Phytopathology, 91, 648–58. [DOI] [PubMed] [Google Scholar]
- 10. Dean R.A., Talbot N.J., Ebbole D.J., et al. 2005, The genome sequence of the rice blast fungus Magnaporthe grisea, Nature, 434, 980–6. [DOI] [PubMed] [Google Scholar]
- 11. Wang R., Ning Y., Shi X., et al. 2016, Immunity to rice blast disease by suppression of effector-triggered necrosis. Curr. Biol., 26, 2399–411. [DOI] [PubMed] [Google Scholar]
- 12. Park C.H., Chen S., Shirsekar G., et al. 2012, The Magnaporthe oryzae effector AvrPiz-t targets the RING E3 Ubiquitin Ligase APIP6 to suppress pathogen-associated molecular pattern–triggered immunity in rice. Plant Cell, 24, 4748–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li X., Gao C., Li L., et al. 2017, MoEnd3 regulates appressorium formation and virulence through mediating endocytosis in rice blast fungus Magnaporthe oryzae. PLoS Pathogens, 13, e1006449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qi Z., Wang Q., Dou X., et al. 2012, MoSwi6, an APSES family transcription factor, interacts with MoMps1 and is required for hyphal and conidial morphogenesis, appressorial function and pathogenicity of Magnaporthe oryzae. Mol. Plant Pathol., 13, 677–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang K., Czymmek K.J., Caplan J.L., et al. 2011, HYR1-mediated detoxification of reactive oxygen species is required for full virulence in the rice blast fungus. PloS Pathogens, 7, e1001335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou T.T., Zhao Y.L., Guo H.S.. 2017, Secretory proteins are delivered to the septin-organized penetration interface during root infection by Verticillium dahliae. PloS Pathol., 13, e1006275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao Y.L., Zhou T.T., Guo H.S.. 2016, Hyphopodium-specific VdNoxB/VdPls1-dependent ROS-Ca2+ signaling is required for plant infection by Verticillium dahliae. PloS Pathol., 12, e1005793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim H., Newell A.D., Cota-Sieckmeyer R.G., Rupe J.C., Fakhoury A.M., Bluhm B.H.. 2013, Mating-type distribution and genetic diversity of Cercospora sojina populations on soybean from Arkansas: evidence for potential sexual reproduction, Phytopathology, 103, 1045–51. [DOI] [PubMed] [Google Scholar]
- 19. Kim J.S., Seo S.G., Jun B.K., Kim J.W., Kim S.H.. 2010, Simple and reliable DNA extraction method for the dark pigmented fungus, Cercospora sojina, Plant Pathol. J., 26, 289–92. [Google Scholar]
- 20. Chin C.S., Alexander D.H., Marks P., et al. 2013, Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data, Nat. Methods, 10, 563–9. [DOI] [PubMed] [Google Scholar]
- 21. Berlin K., Koren S., Chin C.S., Drake J.P., Landolin J.M., Phillippy A.M.. 2015, Assembling large genomes with single-molecule sequencing and locality-sensitive hashing, Nat. Biotechnol., 33, 623–30. [DOI] [PubMed] [Google Scholar]
- 22. Luo R., Liu B., Xie Y., et al. 2012, SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler, Gigascience, 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ter-Hovhannisyan V., Lomsadze A., Chernoff Y.O., Borodovsky M.. 2008, Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training, Genome Res., 18, 1979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salamov A.A., Solovyev V.V.. 2000, Ab initio gene finding in Drosophila genomic DNA, Genome Res., 10, 516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Delcher A.L., Harmon D., Kasif S., White O., Salzberg S.L.. 1999, Improved microbial gene identification with GLIMMER. Nucleic Acids Res, 27, 4636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haas B.J., Zeng Q., Pearson M.D., Cuomo C.A., Wortman J.R.. 2011, Approaches to fungal genome annotation, Mycology, 2, 118–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haas B.J., Papanicolaou A., Yassour M., et al. 2013, De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis, Nat. Protoc., 8, 1494–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanehisa M., Goto S., Kawashima S., Okuno Y., Hattori M.. 2004, The KEGG resource for deciphering the genome, Nucleic Acids Res., 32, D277–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tatusov R.L., Galperin M.Y., Natale D.A., Koonin E.V.. 2000, The COG database: a tool for genome-scale analysis of protein functions and evolution, Nucleic Acids Res., 28, 33–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Finn R.D., Bateman A., Clements J., et al. 2014, Pfam: the protein families database, Nucleic Acids Res., 42, D222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eddy S.R. 1998, Profile hidden Markov models, Bioinformatics, 14, 755–63. [DOI] [PubMed] [Google Scholar]
- 32. Urban M., Cuzick A., Rutherford K., et al. 2017, PHI-base: a new interface and further additions for the multi-species pathogen–host interactions database, Nucleic Acids Res., 45, D604–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gotz S., Arnold R., Sebastian-Leon P., et al. 2011, B2G-FAR, a species-centered GO annotation repository, Bioinformatics, 27, 919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Benson G. 1999, Tandem repeats finder: a program to analyze DNA sequences, Nucleic Acids Res., 27, 573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blow M.J., Clark T.A., Daum C.G., et al. 2016, The epigenomic landscape of prokaryotes, PloS Genet., 12, e1005854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nordberg H., Cantor M., Dusheyko S., et al. 2014, The genome portal of the Department of Energy Joint Genome Institute: 2014 updates, Nucleic Acids Res., 42, D26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ebersberger I., Simoes R.D., Kupczok A., et al. 2012, A consistent phylogenetic backbone for the fungi, Mol. Biol. Evol., 29, 1319–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Edgar R.C. 2004, MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Res., 32, 1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kumar S., Stecher G., Tamura K.. 2016, MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets, Mol. Biol. Evol., 33, 1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nix D.A., Eisen M.B.. 2005, GATA: a graphic alignment tool for comparative sequence analysis, BMC Bioinformatics, 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mistry J., Finn R.D., Eddy S.R., Bateman A., Punta M.. 2013, Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions, Nucleic Acids Res., 41, e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yin Y., Mao X., Yang J., Chen X., Mao F., Xu Y.. 2012, dbCAN: a web resource for automated carbohydrate-active enzyme annotation, Nucleic Acids Res., 40, W445–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Khaldi N., Seifuddin F.T., Turner G., et al. 2010, SMURF: genomic mapping of fungal secondary metabolite clusters, Fungal Genet. Biol., 47, 736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Medema M.H., Blin K., Cimermancic P., et al. 2011, antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences, Nucleic Acids Res., 39, W339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shim W.B., Dunkle L.D.. 2002, Identification of genes expressed during cercosporin biosynthesis in Cercospora zeae-maydis, Physiol. Mol. Plant P., 61, 237–48. [Google Scholar]
- 46. Yang C., Li W., Cao J., et al. 2017, Activation of ethylene signaling pathways enhances disease resistance by regulating ROS and phytoalexin production in rice, Plant J., 89, 338–53. [DOI] [PubMed] [Google Scholar]
- 47. Zeng F., Wang C., Zhang G., Wei J., Bradley C.A., Ming R.. 2017, Draft genome sequence of Cercospora sojina isolate S9, a fungus causing frogeye leaf spot (FLS) disease of soybean. Genomics Data, 12, 79–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bodega B., Orlando V.. 2014, Repetitive elements dynamics in cell identity programming, maintenance and disease, Curr. Opin. Cell Biol., 31, 67–73. [DOI] [PubMed] [Google Scholar]
- 49. Jaenisch R., Bird A.. 2003, Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals, Nat. Genet., 33, 245–54. [DOI] [PubMed] [Google Scholar]
- 50. Ohm R.A., Feau N., Henrissat B., et al. 2012, Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi, PloS Pathog., 8, e1003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Flusberg B.A., Webster D.R., Lee J.H., et al. 2010, Direct detection of DNA methylation during single-molecule, real-time sequencing. Nature Methods, 7, 461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Benson J.M., Poland J.A., Benson B.M., Stromberg E.L., Nelson R.J.. 2015, Resistance to gray leaf spot of maize: genetic architecture and mechanisms elucidated through nested association mapping and near-isogenic line analysis, PloS Genet., 11, e1005045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Goodwin S.B., Ben M'Barek S., Dhillon B., et al. 2011, Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis, PloS Genet., 7, e1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krogh A., Larsson B., von Heijne G., Sonnhammer E.L.L.. 2001, Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes, J. Mol. Biol., 305, 567–80. [DOI] [PubMed] [Google Scholar]
- 55. Petersen T.N., Brunak S., von Heijne G., Nielsen H.. 2011, SignalP 4.0: discriminating signal peptides from transmembrane regions, Nat. Methods, 8, 785–6. [DOI] [PubMed] [Google Scholar]
- 56. Soanes D.M., Alam I., Cornell M., et al. 2008, Comparative genome analysis of filamentous fungi reveals gene family expansions associated with fungal pathogenesis, PloS One, 3, e2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Koeck M., Hardham A.R., Dodds P.N.. 2011, The role of effectors of biotrophic and hemibiotrophic fungi in infection, Cell Microbiol., 13, 1849–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang Y., Wu J., Park Z.Y., et al. 2011, Comparative secretome investigation of magnaporthe oryzae proteins responsive to nitrogen starvation, J. Proteome Res., 10, 3136–48. [DOI] [PubMed] [Google Scholar]
- 59. Liu G.Y., Nizet V.. 2009, Color me bad: microbial pigments as virulence factors, Trends Microbiol., 17, 406–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Biely P. 2012. Microbial carbohydrate esterases deacetylating plant polysaccharides. Biotechnol. Adv., 30, 1575–88. [DOI] [PubMed] [Google Scholar]
- 61. Aurilia V., Parracino A., D'Auria S.. 2008, Microbial carbohydrate esterases in cold adapted environments, Gene, 410, 234–40. [DOI] [PubMed] [Google Scholar]
- 62. Liu Q.P., Sulzenbacher G., Yuan H., et al. 2007, Bacterial glycosidases for the production of universal red blood cells, Nat. Biotechnol., 25, 454–64. [DOI] [PubMed] [Google Scholar]
- 63. Benhamou N., Ouellette G.B.. 1986, Ultrastructural-localization of glycoconjugates in the fungus ascocalyx-abietina, the scleroderris canker agent of conifers, using lectin gold complexes, J. Histochem. Cytochem., 34, 855–67. [DOI] [PubMed] [Google Scholar]
- 64. Zhao Z.T., Liu H.Q., Wang C.F., Xu J.R.. 2013, Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi, BMC Genomics, 14, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Klosterman S.J., Subbarao K.V., Kang S.C., et al. 2011, Comparative genomics yields insights into niche adaptation of plant vascular wilt pathogens, PloS Pathog., 7, e1002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kamper J., Kahmann R., Bolker M., et al. 2006, Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis, Nature, 444, 97–101. [DOI] [PubMed] [Google Scholar]
- 67. Wang Q.Q., Han C.Z., Ferreira A.O., et al. 2011, Transcriptional programming and functional interactions within the Phytophthora sojae RXLR effector repertoire, Plant Cell, 23, 2064–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dubey A., Jeon J.. 2016, Epigenetic regulation of development and pathogenesis in fungal plant pathogens, Mol. Plant Pathol. doi:10.1111/mpp.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ye P.H., Luan Y.Z., Chen K.N., Liu Y.Z., Xiao C.L., Xie Z.. 2017, MethSMRT: an integrative database for DNA N6-methyladenine and N4-methylcytosine generated by single-molecular real-time sequencing, Nucleic Acids Res., 45, D85–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wion D., Casadesus J.. 2006, N6-methyl-adenine: an epigenetic signal for DNA-protein interactions, Nat. Rev. Microbiol., 4, 183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Barkai-golan R., Mirelman D., Sharon N.. 1978, Studies on growth inhibition by lectins of Penicillia and Aspergilli. Arch. Microbiol., 116, 119–24. [DOI] [PubMed] [Google Scholar]
- 72. Guo P., Wang Y., Zhou X., et al. 2013, Expression of soybean lectin in transgenic tobacco results in enhanced resistance to pathogens and pests. Plant Science, 211, 17–22. [DOI] [PubMed] [Google Scholar]
- 73. Boraston A.B., Bolam D.N., Gilbert H.J., Davies G.J.. 2004, Carbohydrate-binding modules: fine-tuning polysaccharide recognition, Biochem. J., 382, 769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Receveur V., Czjzek M., Schulein M., Panine P., Henrissat B.. 2002, Dimension, shape, and conformational flexibility of a two domain fungal cellulase in solution probed by small angle X-ray scattering, J. Biol. Chem., 277, 40887–92. [DOI] [PubMed] [Google Scholar]
- 75. Yu J.H., Keller N.. 2005, Regulation of secondary metabolism in filamentous fungi, Annu. Rev. Phytopathol., 43, 437–58. [DOI] [PubMed] [Google Scholar]
- 76. Nosanchuk J.D., Stark R.E., Casadevall A.. 2015, Fungal melanin: what do we know about structure? Front Microbiol., 6, 1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Xu N., Luo X., Li W., Wang Z., Liu J.. 2017, The bacterial effector AvrB-induced RIN4 hyperphosphorylation is mediated by a receptor-like cytoplasmic kinase complex in Arabidopsis. Mol. Plant-Microbe In., 30, 502–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession NFUF00000000. The version described in this paper is version NFUF01000000.