

Abstract
Finger millet (Eleusine coracana (L.) Gaertn) is an important crop for food security because of its tolerance to drought, which is expected to be exacerbated by global climate changes. Nevertheless, it is often classified as an orphan/underutilized crop because of the paucity of scientific attention. Among several small millets, finger millet is considered as an excellent source of essential nutrient elements, such as iron and zinc; hence, it has potential as an alternate coarse cereal. However, high-quality genome sequence data of finger millet are currently not available. One of the major problems encountered in the genome assembly of this species was its polyploidy, which hampers genome assembly compared with a diploid genome. To overcome this problem, we sequenced its genome using diverse technologies with sufficient coverage and assembled it via a novel multiple hybrid assembly workflow that combines next-generation with single-molecule sequencing, followed by whole-genome optical mapping using the Bionano Irys® system. The total number of scaffolds was 1,897 with an N50 length >2.6 Mb and detection of 96% of the universal single-copy orthologs. The majority of the homeologs were assembled separately. This indicates that the proposed workflow is applicable to the assembly of other allotetraploid genomes.
Keywords: hybrid de novo assembly, finger millet, allotetraploid genome, whole-genome optical mapping
1. Introduction
After sorghum and pearl millet, finger millet (Eleusine coracana (L.) Gaertn) is the coarse cereal that is most widely cultivated in dry regions under rain-fed conditions. Finger millet is an allotetraploid (2n = 4× = 36) with an AABB genome that belongs to the family Poaceae and subfamily Chloridoideae. It was domesticated from the wild allotetraploid E. africana in Uganda to Ethiopia more than 5000 years ago. It is widely accepted that the A genome progenitor is the wild diploid species E. indica (2n = 2× = 18) or one of its close relatives, including E. tristachya, while the B genome progenitor is unknown or most likely extinct.1,2,3,4 Finger millet is one of the richest sources of mineral elements such as calcium, iron, manganese, and zinc, in addition to being a fairly good source of vitamins and several essential amino acids.5 Among all millets, finger millet is often considered as the best source of lysine.6 With its high satiety value and low glycemic index, finger millet has excellent potential as a nutraceutical cereal. It is grown widely, including in India, many African countries, China, and, historically until the present day, even in Japan.7
Despite its importance, very little research emphasis has been given to finger millet; hence, this species is classified as an orphan or underutilized crop.7,8 The major reasons for this classification include a distant relationship to any of the major cereals, a large genome size, and particularly, its tetraploidy. Thus, limited efforts have been made toward the generation of genomic resources for this species, including sequencing of its genome. The genome size of finger millet was estimated to be 3.34–3.87 pg (2C),7,9 which corresponds to 1.63–1.89 Gb in haploid (1C) cells assuming that 1 pg DNA corresponds to 978 Mbp with a GC content of 50%. Several attempts have also been made to prospect novel genes linked to the drought-stress acclimation response,10–13 and a small number of SSR markers for the construction of genetic linkage maps have been reported.14 The EcNAC1 gene of E. coracana, which encodes a NAC family transcriptional factor, was shown to be induced by drought and to confer resistance to drought and other stresses when expressed in tobacco.10 However, compared with the progress made in generating genomic information in sorghum and pearl millet, efforts in finger millet remain scarce. The absence of such genomic resources has been the major impediment in improving finger millet through the adoption of molecular breeding approaches.
The genome assembly and annotation of polyploid species has been a major challenge in genomics, because homeologous genes (i.e. duplicated genes derived from genome duplication) are by definition highly similar and their separate assembly is difficult. About 35% of vascular plants are estimated to be recent polyploids,15,16 and it has long been known that important crop species are polyploids.17 However, the genome assemblies of only a handful of polyploid species have been reported. More importantly, the quality of the polyploid assembly, in particular the scaffold length, tends to be low in such reports. The presence of duplicated genes and polyploidy split the assembly, thus leading to shorter scaffolds and contigs.18,19
Some polyploid species have, however, been assembled; e.g. Nicotiana tabacum,20Gossypium hirsutum,21Triticum aestivum,22 and Brassica juncea.23 The genome assembly of hexaploid wheat, Triticum aestivum (TGACv1),22 was performed via chromosome separation, albeit with an N50 of several kb.22 A combined approach of PacBio long reads and Bionano Genomics optical genome mapping succeeded in assembling the genome of Brassica juncea, which is an allotetraploid, with an N50 scaffold that was 1.5 Mb long.23 Subsequently, pseudochromosomes were constructed using the diploid genome and genetic linkage maps. However, in previous polyploid assemblies, little was reported on the validation of the separation of homeologs in cases with unknown parental species. Another potential assembly strategy that can be used when all the parental species are known is the assembly of the genome sequences based on the parental diploid species. This was employed for the allotetraploid Arabidopsis kamchatica using the two parental species A. halleri and A. lyrata;24,25 however, this cannot be simply applied to finger millet because one of the parental species is unknown. Recently, the rubber tree genome was assembled using Dovetail Chicago method26 (Dovetail Genomics, LLC, Santa Cruz, CA, USA) with a scaffold N50 of 96.8 kb, and they revealed the paleotetraploidy of the genome. The Chicago library is a longer range technique than mate-pair library, which is based on chromosome conformation capture sequencing (Hi-C) protocol.27 The barley genome was assembled in chromosome length scale with Hi-C library.28 NRGene Ltd (Ness-Ziona, Israel) has succeeded recently in assembling some crop genomes, e.g. the genome of maize with a very high scaffold N50 of 35.5 Mb, using the DeNovoMAGICTM software (http://nrgene.com/products-technology/denovomagic/).29 They require some PCR-free paired-end libraries and mate-pair libraries for the DeNovoMAGICTM, but the company has not reported its algorithm and strategy in detail.
Here, we report the whole-genome draft sequencing and assembly of a tetraploid finger millet, cultivar PR202 (IC: 479099), using a novel polyploid genome assembly workflow. This cultivar was chosen for this work considering the extent of its adaptability. A selection from the Godavari regions of Southern India was chosen for this study based on its resilience to drought and high-temperature stresses. This cultivar is being used as the national check in all of the multilocation yield-evaluation trials performed by the All India Coordinated Small Millets Improvement Project (AICSMIP) (http://millets.res.in/aicrp_small.php).30
2. Materials and methods
Seeds of finger millet (variety name, PR202; IC number, 479099; the IC number is the indigenous collection number issued by the National Bureau of Plant Genetic Resources, India) that were stocked at the Project Coordinating Unit (Small Millets), University of Agricultural Sciences, GKVK, Bangalore, India) were used for germination. DNA was extracted from young leaves of finger millet plants grown in a plant chamber at 20 °C to 22 °C, 3 weeks later for sequencing and 2 months later for genome scanning. In this section, the methods used for DNA and RNA isolation and preparation of libraries for sequencing are described.
2.1. Preparation of DNA libraries for sequencing
Good-quality and high-molecular-weight genomic DNA (gDNA) was isolated using a combination of CTAB and Genomic tip 500/G column (Qiagen AG, Hombrechtikon, Switzerland) purification method, in accordance with the protocol ‘Preparing Arabidopsis Genomic DNA for Size-Selected ∼20 kb SMRTbell™ Libraries’ (http://www.pacb.com/wp-content/uploads/2015/09/Shared-Protocol-Preparing-Arabidopsis-DNA-for-20-kb-SMRTbell-Libraries.pdf). The DNA pellet obtained using the conventional CTAB method was dissolved in 1 ml of Dnase- and RNase-free distilled water (Life Technologies, Carlsbad, CA, USA) and purified by passing through Genomic tip 500/G columns, to obtain intact DNA with a size above 60 kb. For library preparation, the TruSeq DNA NanoSample Prep Kit v2 (Illumina Inc., San Diego, CA, USA) was used in the following steps. DNA samples (1 µg) were sonicated on a Covaris instrument (Covaris, Woburn, MA, USA) using settings specific for a fragment size of 550 bp. The fragmented DNA samples were size selected using AMPure beads (Beckman Coulter, Inc., Brea, CA, USA), end-repaired, and polyadenylated. TruSeq adapters containing the index for multiplexing were ligated to the fragmented DNA samples. Fragments containing TruSeq adapters at both ends were selectively enriched by PCR. The quality and quantity of the enriched libraries were validated using Qubit® 2.0 Fluorometer (Life Technologies) and TapeStation system (Agilent, Waldbronn, Germany). The products were a smear with an average fragment size of approximately 700 bp. The libraries were normalized to 10 nM in Tris-HCl 10 mM, pH 8.5 with 0.1% Tween 20. The 3–8 and 20 kb mate-pair libraries were prepared according to Lucigen’s NxSeq® Long Mate Pair Kit protocol and NxSeq® 20 kb Mate Pair Library kit protocol (Lucigen Corporation, Middleton, WI, USA), respectively. Briefly, genomic DNA was sheared using Covaris G-TUBES (Covaris, Woburn, MA, USA) to the desired size (3, 5, 8, and 20 kb), end-repaired, A-tailed, and ligated to adaptors. The insert was size selected and ligated to a unique coupler that contained encrypted Chimera Code™ sequences. The circularized inserts were treated with exonuclease for the removal of unwanted linear DNA and digested with a selection of endonucleases to produce correctly sized di-tags (400–900 bp). Biotin capture was used to remove unwanted DNA fragments prior to the addition of the Junction Code™ Reagent. The library was recircularized and PCR amplified and the quality was assessed using a Qubit® 2.0 Fluorometer (Life Technologies), and the TapeStation system (Agilent, Waldbronn, Germany). The final mate-pair libraries were normalized to 10 nM in Tris–HCl 10 mM, pH 8.5 with 0.1% Tween 20.
2.2. Preparation of RNA libraries for sequencing
RNA was extracted from young leaves of finger millet 1 month after germination. Freshly germinated finger millet plants were exposed to drought stress, in which the plants were not watered for 7 days. Tissue samples were taken from the young leaves before and after the exposure to drought conditions. RNA was extracted using an RNeasy plant kit (Qiagen AG, Hombrechtikon, Switzerland). The quantity and quality of the isolated RNA was determined using a Qubit® 2.0 Fluorometer (Life Technologies) and a Bioanalyzer 2100 (Agilent, Waldbronn). For library preparation, TruSeq Stranded mRNA Sample Prep Kit (Illumina) was used in the subsequent steps. Briefly, total RNA samples (1 µg) were ribosome depleted and then reverse transcribed into double-stranded cDNAs, with addition of actinomycin during first-strand synthesis. The cDNA samples were fragmented, end-repaired, and polyadenylated before the ligation of TruSeq adapters. The adapters contained the index for multiplexing. Fragments containing TruSeq adapters at both ends were selectively enriched with PCR. The quality and quantity of the enriched libraries were assessed using a Qubit® 2.0 Fluorometer and a Bioanalyzer 2100 (Agilent, Waldbronn). Products were a smear with an average fragment size of ∼360 bp. The libraries were normalized to 10 nM in Tris-HCl 10 mM, pH 8.5 with 0.1% Tween 20.
2.3. Library preparation and sequencing for PacBio RS II
The SMRT Bell library was produced using a DNA Template Prep Kit 1.0 (PacBio p/n 100-259-100). The concentration of the input gDNA was measured using a Qubit Fluorometer dsDNA Broad Range assay (Life Technologies p/n 32850). gDNA (10 µg) was mechanically sheared to an average size distribution of 10 kb using a Covaris gTube (Kbiosciences p/n 520079). A Bioanalyzer 2100 12K DNA Chip assay (Agilent p/n 5067-1508) was used to assess the fragment size distribution. Sheared gDNA (5 µg) was DNA-damage repaired and end-repaired using polishing enzymes. A blunt-end ligation reaction followed by exonuclease treatment was performed to create the SMRT Bell template. A Blue Pippin device (Sage Science, Inc., Beverly, MA, USA) was used to size select the SMRT Bell template and enrich for large fragments (>10 kbp). The size-selected library was quality inspected and quantified on Agilent Bioanalyzer 12 kb DNA Chip and Qubit Fluorimeter (Life Technologies). A ready-to-sequence SMRT Bell-Polymerase Complex was created using a P6 DNA/Polymerase Binding Kit 2.0 (PacBio p/n 100-236-500), according to the manufacturer’s instructions. The PacBio RS II instrument was programmed to load and sequence the sample on PacBio SMRT cells v3.0 (PacBio p/n 100-171-800), acquiring one movie of 360 min per SMRT cell. A MagBead loading (PacBio p/n 100-133-600) method was chosen to improve the enrichment of the longer fragments. After the run, a sequencing report was generated for each cell via the SMRT portal, to assess adapter dimer contamination, sample-loading efficiency, average read length, and number of filtered sub-reads.
2.4. Next-generation optical mapping
A Bionano Prep™ Plant Tissue DNA Isolation Kit was used according to the Bionano Prep™ Plant Tissue DNA Isolation Base Protocol (Document number: 30068, Document revision: B, Bionano Genomics, Inc., San Diego, CA, USA). Briefly, 1 g of young leaves collected 2 months after germination was fixed in formaldehyde, to protect the nuclei and DNA against mechanical shearing. The leaves were then homogenized with a rotor-stator apparatus, to break the tough plant cell wall, followed by a density gradient purification for the recovery of intact nuclei. These samples were embedded in low-melt agarose plugs for proteinase K (Qiagen AG, Hombrechtikon, Switzerland) and RNase A (Qiagen AG, Hombrechtikon) digestion and subsequent purification of high-molecular-weight (HMW) DNA. The resulting HMW DNA was processed further according to the IrysPrep™ Labeling-NLRS User Guide (Document number: 30024, Document revision: G, Bionano Genomics) using an NLRS Kit (Bionano Genomics). In brief, 300 ng of HMW DNA was nicked by Nt.BspQI (New England Biolabs, Ipswich, MA, USA) and labeled. These reactions provided sequence specificity for an optimal label density of 8–15 labels per 100 kbp. The nick enzyme Nt.BspQI was selected by predicting the label density as 11.9 labels per 100 kbp using Bionano Genomics Knickers (ver. 1.5.5.0, http://www.bnxinstall.com/knickers/Knickers.htm) beforehand. The labeled nicks were repaired to restore strand integrity, and the labeled DNA was stained for backbone visualization. Labeled nicks were subsequently detected as dots on a string when run on a Bionano Irys® system instrument.
2.5. Sequencing and optical mapping
Sequencing was performed on an Illumina NextSeq500, MiSeq instrument (Illumina) and a PacBio RS II system (Pacific Bioscience of California, Inc., CA, USA). Genome optical mapping was carried out on a Bionano Irys® system (Bionano Genomics). The three paired-end libraries with insertion lengths of 250, 550, and 700 bases were sequenced on an Illumina NextSeq500 apparatus (reagent version 2 kits 2 × 150 cycles). In contrast, the four mate-pair libraries with insert sizes of 3, 5, 8, and 20 kb were sequenced on an Illumina MiSeq instrument (reagent version 3 kits 2 × 300 cycles). Long reads were obtained from PacBio RS II using a total of 23 SMRT cells. The nicked-labeled HMW DNA was run on a Bionano IrysChip® flowcell that scanned 6 times for 96.5× coverage.
2.6. Genome size estimation
Approximately 0.5 cm2 of fresh leaf tissue of finger millet, together with an equal amount of leaf tissue of tomato (Lycopersicon esculentum; used as an internal standard [diploid genome size, 1.96 pg31]), was chopped with a sharp razor blade in extraction buffer (CyStain PI Absolut P; Sysmex Europe GmbH, Norderstedt, Germany). The suspension of nuclei was filtrated and incubated in staining solution containing propidium iodide (PI) and RNase A (CyStain PI Absolut P; Sysmex). The suspension of stained nuclei was analyzed using a flow cytometer (CyFlow Space, Sysmex).
2.7. Hybrid de novo assembly workflow and gene annotation
The hybrid de novo assembly workflow is shown in Fig. 1. Adapters that contaminated the sequenced paired-end reads and low-quality bases were trimmed using Trimmomatic (ver. 0.33)32 on the SUSHI framework33 after sequencing. Regarding the mate-pair libraries, first, chimera reads were filtered out, and chimera code and linker sequences from the 5′ end were trimmed using the script IlluminaChimera-Clean5.py provided by Lucigen Co., Wisconsin, USA, followed by another script, IlluminaNxSeqJunction-Split9.py, to segregate the reads into proper mate-pair reads and unsplit reads via the detection of junction sequences. Subsequently, Platanus (ver. 1.2.4)34 was executed using the preprocessed paired-end reads to assemble contigs (Fig. 1, step 1), which were then used as anchors to connect the PacBio long reads on DBG2OLC (commit id: 7f1712ae76b015b1f4efee81a5d4e1305701197a)35 (Fig. 1, step 2). Contigs from the Platanus assembly that were longer than 1 kb were used for the input of the DBG2OLC hybrid assembly. Note that many short contigs were filtered out to adopt a conservative approach aimed at avoiding misassembly, as short and fragmented contigs stemmed most probably from complex regions, such as repeats and duplicates. The contigs from the DBG2OLC hybrid assembly were polished using Pilon (ver. 1.21)36 with the accurate Illumina short reads, to correct misassemblies (NGS contigs) (Fig. 1, step 3). We used only half amount of the paired-end reads (250 base insert size) for the polishing. Subsequently, the polished contigs were scaffolded with the mate-pairs using SSPACE standard (ver. 3.0)37 (NGS scaffolds) (Fig. 1, step 4-1). We only used the proper mate-pair reads for scaffolding. Independently, optical genome mapping was performed on a Bionano Irys® system (Fig. 1, step 4-2), followed by de novo assembly with the detected molecules using the IrysView® and Bionano IrysSolve® software (ver. 2.1) (BNG contigs) (Fig. 1, step 5). The de novo assembly was performed twice: the first de novo assembly used the estimated nose parameters by mapping the detected molecules to the NGS contigs, and the second assembly used autonoise (autodetection of appropriate noise parameters). The hybrid scaffolding was performed using Bionano IrysSolve® (ver. 2.1) with both the NGS scaffolds and the BNG contigs. Finally, GMclose (ver. 1.6)38 filled the gaps in the super-scaffolds using all the contigs assembled by Platanus. The actual executed commands as a shell script with configuration files and parameters are provided in Supplementary File S1.
Figure 1.
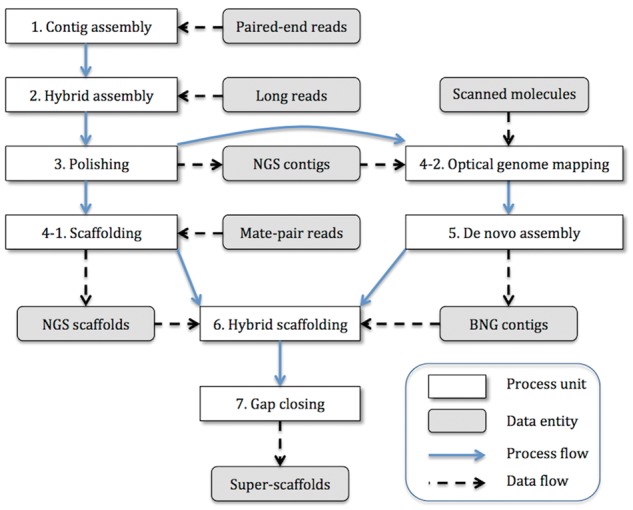
Hybrid de novo assembly workflow. In the figure, the white box represents a process, the gray box represents a data entity, the blue solid arrow shows the process flow, and the black dashed arrow shows the data flow. The hybrid de novo assembly was executed as follows: (1) contig assembly using only paired-end reads (Platanus ver. 1.2.4), (2) hybrid contig assembly using contigs >1 kb from the first step and long reads (DBG2OLC commit id: 7f1712ae76b015b1f4efee81a5d4e1305701197a), (3) polishing of the assembled contig with paired-end reads (Pilon ver. 1.21) (NGS contigs), (4) using the polished contigs in the two subsequent independent steps, (4-1) scaffolding of the polished contigs with mate-pair reads (NGS scaffolds), (4-2) after converting scanned molecules into a CMAP file and optical genome mapping to the NGS contigs, (5) de novo assembly of scanned molecules based on the parameters of the molecular-quality report and autonoise (autodetection of appropriate noise parameters) with the NGS contigs (BNG contigs), (6) hybrid scaffolding with both NGS scaffolds (step 4-1 output) and BNG contigs (step 5 output), and (7) gap closing (GMclose ver. 1.6) of the scaffolds with all the contigs generated in the first step (step 1 output). Finally, the workflow produced the super scaffolds. The raw data preprocesses, such as adapter trimming and removing chimera code from mate-pair reads, are not shown in this workflow.
The genes were predicted and annotated using MAKER (ver. 2.31.8)39 based on the rice genome (Oryza sativa ssp. japonica IRGSP-1.0, ENA Assembly: GCA_001433935.1) and the RNAseq data that were obtained from young leaves. The rice cDNAs and RNAseq data for the EST evidence and the rice proteins for the protein homology evidence were used for the MAKER job. To render the GFF file, the input of MAKER, the RNAseq data were mapped to the assembled reference sequences using STAR aligner (ver. 2.5.2b)40 and the GFF annotation file was constructed via transcriptome construction using StringTie (ver. 1.3.3b).41 The rice-specific repeat-making library obtained from Repbase database42 was used for RepeatMasker (ver. 4.0.5).43 The rice model was used for Augustus (ver. 3.2.0)44 gene prediction in the MAKER pipeline. The detail options are written in the Supplementary File S1. For assessing the quality of the genome assembly and annotation, we executed BUSCO (ver. 2.0.1)45 using the published plant universal single-copy orthologs. We also performed an RNAseq data alignment to the final assembled genome using STAR (ver. 2.5.2b)40 and checked the mapping statistics.
3. Results and discussion
In this section, we report the genome assembly and discuss the results of its validation.
3.1. Sequencing and assembly statistics
Using the flow cytometry experiment, we estimated that the size of the finger millet genome is 1.5 Gb. In total, short reads amounting to 140× coverage and long reads amounting to 16× coverage were sequenced. We scanned the DNA molecules on a Bionano Irys® system and detected 144,761 Mb molecules in a total of six runs (Supplementary Table S1). Based on the estimated genome size, the sequenced coverage depth for each library is reported in Table 1 (PacBio subreads length distribution and calculated insert size of distribution are shown in Supplementary Figs S1 and S2, respectively).
Table 1.
Raw sequence data
| Library type | DNA|RNA | Sequencer | Insert length (bases) | Number of bases | Coverage depth |
|---|---|---|---|---|---|
| Paired-end | DNA | Illumina NextSeq | 250 | 93,783,186,606 | 63× |
| Paired-end | DNA | Illumina NextSeq | 550 | 63,519,282,198 | 42× |
| Paired-end | DNA | Illumina NextSeq | 700 | 50,551,135,834 | 34× |
| Mate-pair | DNA | Illumina MiSeq | 3,000 | 789,059,46 | 0.053× |
| Mate-pair | DNA | Illumina MiSeq | 5,000 | 364,070,088 | 0.24× |
| Mate-pair | DNA | Illumina MiSeq | 8,000 | 1,138,183,671 | 0.76× |
| Mate-pair | DNA | Illumina MiSeq | 20,000 | 1,366,910,280 | 0.91× |
| PacBio CLR | DNA | PacBio RS II | – | 25,086,179,201 | 16.7× |
| Paired-end | RNA | Illumina HiSeq4000 | – | 148,856,244,082 | 99.2× |
The number of sequenced raw data in each library and the coverage depth based on the genome size, 1.5 Gb as estimated by flow cytometry, are shown.
The statistics of each step in the hybrid assembly are shown in Table 2. The total assembled size was 1,188,784,944 bases, which is 78.2% of the genome size estimated by flow cytometry. The hybrid contig assembly by DBG2OLC and the scaffolding by SSPACE enhanced the continuity from the Platanus contig assembly, although the mate-pair sequenced coverage was less than 10×, and the total assembly size decreased compared with the Platanus contig assembly. This could be explained by the low sequencing coverage of the PacBio long reads, rather than the filtering out of the shorter Platanus contigs. At the final hybrid scaffolding and gap closing, the N50 of the final super-scaffolds increased to 2.68 Mb, which is approximately twice as long as the NGS scaffolds. The hybrid scaffolding generated super-scaffolds that concatenated the NGS scaffolds based on the BNG contigs that were assembled from the optical genome mapping. Some conflicts were detected between NGS scaffolds and BNG contigs, which split the conflicting NGS scaffolds into smaller pieces. This had the major advantage of correcting the polyploid genome assembly, as scaffolding with only mate-pair short reads may fail to solve the long-range homeologous complexity and may sometimes lead to mis-scaffolding.46,47
Table 2.
Assembly statistics at each assembly step
| Statistics | Contig assembly | Hybrid assembly | Scaffold | Hybrid scaffold |
|---|---|---|---|---|
| Total sequences | 2,812,919 | 6,374 | 2,387 | 1,897 |
| Total bases | 1,307,217,455 | 1,067,045,564 | 1,069,478,541 | 1,188,784,944 |
| Min sequence length | 115 | 3,408 | 3,911 | 1,244 |
| Max sequence length | 27,802 | 2,072,336 | 5,237,708 | 13,553,037 |
| Average sequence length | 464.72 | 167,405.96 | 448,042.96 | 626,665.76 |
| Median sequence length | 154.00 | 102,084.50 | 237,045.00 | 92,178.00 |
| N25 length | 3,419 | 520,874 | 1,581,824 | 5,029,714 |
| N50 length | 1,410 | 285,549 | 905,318 | 2,683,090 |
| N75 length | 311 | 145,770 | 457,768 | 1,232,573 |
| N90 length | 133 | 76,519 | 216,067 | 419,121 |
| (G + C)s | 43.15% | 43.83% | 43.57% | 40.98% |
| Ns | 0.00% | 0.00% | 0.73% | 6.63% |
Assembly statistics at each step are summarized. Contig assembly represents the contigs that were assembled using Platanus with preprocessed paired-end reads. Hybrid assembly represents the contigs that were generated using DBG2OLC with the Platanus contigs (> 1 kb long) and PacBio long reads. Scaffold is the result of the SSPACE standard scaffolding with mate-pair reads, and Hybrid scaffold is the hybrid scaffolding result with both Bionano Genomics de novo assembled contigs and the SSPACE scaffolds, followed by GMclose gap closing.
Currently, the so-called de novo assembly method based on next-generation and third-generation sequencing technologies is classified mainly into two approaches: (i) de Bruijn graph (DBG) assembly and (ii) overlap-layout consensus (OLC) assembly.48,49 Although the DBG method is more efficient computationally regarding both memory and calculation time, the fragment size parameter, usually called k-mer length, critically influences the results and often causes a misassembly because of the small fragment k-mers on homologous or repetitive genome regions. Some assemblers, such as Platanus,34 solve this problem by using multiple k-mer sizes for the construction of the DBG. As another solution for the homeologous complexity, longer reads produced from a single-molecule sequencer combined with the OLC method may be beneficial. However, high-coverage sequencing of long reads may solve and correct the wrongly sequenced bases, albeit at a high cost. The hybrid de novo assembly described in this article, which uses both accurate short reads and inaccurate long reads with a low coverage depth, represents another solution regarding assembly cost and calculation efficiency; moreover, the beneficial feature of long reads can be applied to solve the homeologous complexity encountered during polyploid genome assembly. The classification of reads via homeolog phasing50 based on long reads or the parental diploid genome may improve the polyploid assembly further. In addition, further longer molecule information from such as Bionano Irys® system is very promising, not only for super-scaffolding, but also for solving the long-range homeologous complexity in polyploid genome assembly, as demonstrated by the results reported in this article.
3.2. Gene annotation and assembly validation
After the hybrid assembly, we conducted gene annotation prediction using MAKER and 62,348 genes were predicted. The distribution of Annotation Edit Distance (AED)39 tagged by MAKER is shown in Fig. 2A, in which more than 91% of the annotated genes (57,066 genes among a total of 62,348 genes) show an AED <0.5. This result indicates that the annotation is well supported by the evidence. For the assessment of the quality of the assembly and annotation, the assembled scaffolds and predicted genes were analyzed on BUSCO (Fig. 2B). In the assembled scaffolds (shown in Fig. 2B, ‘genome’), 96.5% of the universal single-copy genes (1,389 genes among a total of 1,440 genes) were identified, supporting the quality of the assembly. Among the 1,389 single-copy genes detected, 606 genes were detected as being complete single-copy genes, 783 genes were found to be duplicates, and 766 of the 783 duplicates were exactly twice found from two different scaffolds. An NCBI BLAST search of the complete single-copy genes (606) that were detected only once in the assembled genome found an additional 556 homologous partial genes with a shorter alignment length (40% of the 1,389 complete single-copy genes detected). This indicates that the majority of the homeologous genes were assembled separately (see section 3.3). In tetraploid species, two copies may be found for each ‘single-copy gene’, although duplicated genes may be gradually lost because of redundancy (called nonfunctionalization or homeolog loss).51,52 Similar results were obtained from the BUSCO analysis of proteins and transcripts (Fig. 2, ‘proteins’ and ‘transcripts’), but the missing and fragmented proportion was increased compared with the ‘genome’ analysis. It is well known that one of the homeologs may not be expressed (or silenced) after polyploidization, because of redundancy; thus, some genes were not annotated and the missing and fragmented proportion increased among transcripts and proteins because of the lack of evidence. Moreover, we searched the predicted proteins in the Pfam database53 and found that 74% of predicted proteins were present in this database (Supplementary Fig. S3). RNAseq data obtained using other tissues and conditions may improve the gene annotation process.
Figure 2.

Annotation edit distance (AED) and benchmark of universal single-copy orthologs. (A) The distribution of AED tagged as a gene by MAKER. (B) Benchmark result of the plant set of universal single-copy orthologs produced by BUSCO. In each bar, the pale-blue color indicates the number of genes that were detected completely as single-copy genes in the genome, the dark-blue color indicates the number of genes that were detected as single-copy genes but were counted more than twice, the yellow color indicates the number of genes that were detected as single-copy genes but not completely, and the red color indicates the number of single-copy genes that were not detected among the plant universal single-copy orthologs. The ‘genome’ is the BUSCO result that was obtained by searching the assembled genome FASTA file, and the ‘transcripts’ and ‘proteins’ constitute the BUSCO result that was obtained by searching the transcript FASTA file and amino acid FASTA file produced by MAKER.
Furthermore, we aligned the data from six RNAseq sequenced libraries to the assembled genome for validation: 95.9% of the reads were mapped, which indicates that the assembled genome may cover more than 95% of the coding region (STAR alignment statistics is shown in Supplementary Table S2).
3.3. Polyploid assembly validation and phylogenetic analysis
To validate the quality of the polyploid assembly, i.e. to determine whether the homeologs were assembled in different scaffolds, we analyzed our assembly data of three genes for which Sanger sequencing data of E. coracana had been already reported. First, we performed an NCBI BLAST (ver. 2.2.31+)54 search of the published low-copy-number marker phosphoenolpyruvate carboxylase 4 (Pepc4)1 and granule-bound starch synthase I-like (waxy)2 in the assembled scaffolds. The reciprocal BLAST best-hit search with a very low E-value threshold identified exactly two scaffolds with two different completely matching sequences from different clades (the NCBI BLAST search result is shown in Supplementary File S2). Second, we conducted a molecular phylogenetic analysis using MEGA4 (ver. 7.0.26)55 based on the detected low-copy-number homeologs with the published sequences of close relatives1 (Supplementary Fig. S4). As shown in these figures, the homeologs detected were clearly separated in different clusters. This result indicates that, in these gene regions, the homeologs were successfully assembled, regardless of the presence of highly homologous base sequences. Moreover, the trees supported the contention that E. indica is the wild diploid progenitor of the A genome, because one of the homeologs clustered with the sequences of E. indica.
Next, we analyzed EcNAC1, which is the best-studied gene of E. coracana related to the drought response.10 NAC is one of the largest families of plant-specific transcription factors and has been shown to be involved in diverse processes, such as growth, development, and abiotic stresses. The EcNAC1 of the E. coracana GPU28 variety was induced by drought stresses and conferred tolerance to various abiotic stresses, such as simulated osmotic stress by polyethylene glycol (PEG) and mannitol and salinity stresses in the overexpression lines of tobacco.10 Based on our assembly of PR202, we found a sequence that was similar to, and clustered with, the EcNAC1 of GPU28, together with another sequence from the Trichy1 variety (GenBank accession ID: KU500625.1) (Supplementary Fig. S5). The assembly also had another copy, which is likely to be a homeolog in a different scaffold. It would be valuable to study this copy, which may well have drought-related functions.
In addition, we searched homologous genes as ‘duplicated genes’ in the predicted transcripts using NCBI BLAST and found 57,913 genes that had more than two copies in the genome (Supplementary File S3). Among these duplicates, we found 34,678 genes as reciprocal BLAST best-hit genes located in different scaffolds. Assuming that these genes are homeologs, we found that 56% of all 62,348 genes were considered homeologs, which is consistent with the ratio (55% (766/1389)) of duplicated single-copy genes detected exactly twice in BUSCO (the analytical process and results are shown as a shell script in Supplementary File S3). To check the location of these homeologs, we calculated the gene density (the number of genes per Mb) and the homeolog density (the number of homeologs per Mb) in each scaffold (Supplementary Fig. S6 A1–2, B1–2). Panels A1 and B1 of Supplementary Fig. S6 show the density against the scaffold length of all genes and homeologs, respectively, and panels A2 and B2 show the density distribution of all genes and homeologs, respectively, as well. The genome-wide gene density was 52.4 genes per Mb and the genome-wide homeolog density was 29.2 homeologs per Mb. As shown in these figures, all genes and homeologs were distributed similarly over the genome. Nonetheless, there was a small number of scaffolds (>1 kb long) with high homeolog density (Supplementary Fig. S6, B1), which indicates that there are areas that are dense in homeologs compared with the genome-wide homeolog distribution.
3.4. Summary of the assembly of the genome of polyploid finger millet
Over half of the world’s freshwater resources are currently being used for irrigating agricultural crops. The domestic and industrial demands of the burgeoning population have led to an unprecedented withdrawal of freshwater.56 Combined with the erratic pattern of rainfall caused by the ensuing global climate change, agriculture production is expected to be severely affected in the years to come.57 This scenario urgently necessitates the cultivation of more water-productive crops and/or the development of improved cultivars with a superior water-use efficiency. Millets, with their C4 photosynthetic metabolism, are generally considered to possess higher levels of drought tolerance and are generally cultivated under rain-fed conditions. Because of their higher resilience to harsh climatic conditions, millets exhibit a wider adaptation, and hence can be cultivated in a wider range of agro-ecosystems. Therefore, millets currently represent an integral component of human diets in most impoverished areas, especially in the tropical and subtropical regions of Africa and Southeast Asia.
We assembled the genome of the allotetraploid finger millet (Eleusine coracana (L.) Gaertn, variety: PR202, IC: 479099) via a novel pipeline using various types of sequencers. The size of the assembled genome was about 1.2 Gb, while the genome size measured by flow cytometry was 1.5 Gb. Considering that the majority of the grass genomes are likely to be composed of repetitive elements, the coverage of the genome is sufficiently high. MAKER predicted 62,348 genes, which is roughly the double of the 35,679 genes identified in diploid rice (Oryza sativa Japonica IRGSP-1.0); this is also consistent with a good coverage of the genome. Three lines of evidence supported the good coverage. First, we provided a polyploid assembly with a long scaffold size (N50 length >2.5 Mb), while the assembly of polyploid genomes tends to be fragmented. It is true that the N50 may not mean correct assembly and scaffolding, but we avoided too-greedy scaffolding. Second, 96.5% of the 1,440 universal single-copy orthologs were identified, and 95.9% of RNAseq reads were mapped to the assembled genome, which supported the adequateness of the assembly. Third, the majority of these universal sets had two copies, suggesting the separation of homeologs was adequate in our assembly strategy. Concomitantly, however, we found a significant number of genes that had a single copy. There are several likely explanations for this finding. (i) One of the duplicated genes tends to be lost because of redundancy. Polyploidization was estimated to have occurred well before the domestication of millet (0.50–2.71 mya), in contrast with other crops, in which polyploidization occurred through domestication during the last thousand years.58,59 Thus, it is possible that many duplicated genes were lost. (ii) The published plant universal single-copy gene set is not collected by random sampling from the entire plant genes in terms of the rate of gene loss. Despite the frequent occurrence of genome duplication events throughout angiosperms, these genes tend to be lost and become single copy, potentially because of dosage balance or redundancy. (iii) The assembly size is less than the genome size estimated by flow cytometry. Most of the lacking sequences are likely to be repetitive sequences, but it is also likely that parts of the genic regions were not assembled. (iv) The assembler could not deal with two homeologs; thus, one copy was missing or two copies were combined. We verified that the two copies of the Pepc4 gene, waxy and NAC, were assembled separately. Currently, not many genome sequences from diploid E. indica are available, preventing the systematic validation of homeologs. Further improvement of assembly algorithms and validation would be valuable in this field of research. It is still possible to improve the assembly and annotation of this genome. Nevertheless, the allotetraploid finger millet genome information reported here is expected to be of much value for further genomic research of this millet at the molecular level, as well as for studies aimed at improving finger millet productivity and bioavailability. We have successfully shown the applicability of the multiple hybrid de novo assembly workflow to a complex allotetraploid genome.
Recently, the genome assembly of the E. coracana ML-365 variety was elucidated using Illumina and SOLiD sequencing data.60 We emphasize that our data and analyses were targeted at the analysis of complex polyploid genomes. The N50 of our assembly (>2.5 Mb) was two orders of magnitude higher than that of ML-365 (about 24 kb) and would allow RNAseq analysis of each homeolog separately, as well as resequencing analyses. Our results illustrate the strength of single-molecule sequencing and optical genome mapping in assembling polyploid genomes.
Supplementary Material
Acknowledgements
We wish to thank Dr. Scott Monsma at Lucigen Co., Wisconsin, USA, for mate-pair library preparation, Carmen Näf at the University of Zurich for RNA extraction and library preparation, Aki Morishima for plant growth and genome size estimation by flow cytometry, Dr. Misako Yamazaki and Dr. Lucas Mohn at the University of Zurich for sample handling, and Dr. Dario Copetti at the University of Arizona for advice on gene annotation, and Dr. Jun Sese and Dr. Tony Kuo at the National Institute of Advanced Industrial Science and Technology for discussion about polyploid genome assembly. We also thank Dr. Lilian Gilgen and Damaris O’Brien at the Indo-Swiss Collaboration in Biotechnology (ISCB) office at the Federal Institute of Technology in Lausanne (EPFL), Nangsa Kamtzi and Isavel Schöchli at the University of Zurich, and Lydia Imhof and Jana Ploszaj at the Functional Genomics Center Zurich for the administrative support. Moreover, we would like to thank the members of the Evolutionary and Ecological Genomics Group at the University of Zurich and the members of Genomics/Transcriptomics Group at the Functional Genomics Center Zurich for useful discussions and suggestions.
Availability
All sequenced raw data in FASTQ format, scanned molecular data in BNX format, de novo assembly results by Bionano IrysSolve® in CMAP format, and the final scaffolds and annotation in FASTA and GFF format are available in the DDBJ Sequence Read Archive (DRA) database with the BioProject accession number: PRJDB5606 and BioSample number: SAMD00076255.
Supplementary data
Supplementary data are available at DNARES online.
Conflict of interest
John Baeten and Kees-Jan Francoijs are employees of Bionano Genomics.
Accession numbers
BioProject accession number: PRJDB5606
BioSample number: SAMD00076255, DRA accession number: DRA005897
Funding
This project is funded by the Indo-Swiss Collaboration in Biotechnology (ISCB), a JST CREST Grant (number JPMJCR16O3), and a KAKENHI Grant (number 16H06469, 16K21727), Japan.
References
- 1. Liu Q., Triplett J. K., Wen J., Peterson P. M.. 2011, Allotetraploid origin and divergence in Eleusine (Chloridoideae, Poaceae): evidence from low-copy nuclear gene phylogenies and a plastid gene chronogram. Ann. Bot., 108, 1287–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu Q., Jiang B., Wen J., Peterson P. M.. 2014, Low-copy nuclear gene and McGISH resolves polyploid history of Eleusine coracana and morphological character evolution in Eleusine. Turkish J. Bot., 38, 1–12. [Google Scholar]
- 3. Bisht M. S., Mukai Y.. 2001, Genomic in situ hybridization identifies genome donor of finger millet (Eleusine coracana). Theor. Appl. Genet., 102, 825–32. [Google Scholar]
- 4. Hilu K. W. 1988, Identification of the “A” genome of finger millet using chloroplast DNA. Genetics, 118, 163–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chandra D., Chandra S., Arora P., Sharma A. K.. 2016, Review of finger millet (Eleusine coracana (L.) Gaertn): a power house of health benefiting nutrients. Food Sci. Human Wellness, 5, 149–55. [Google Scholar]
- 6. Saleh A. S., Zhang Q., Chen J., Shen Q.. 2013, Millet grains: nutritional quality, processing, and potential health benefits. Compr. Rev. Food Sci. Food Safety, 12, 281–95. [Google Scholar]
- 7. Goron T. L., Raizada M. N.. 2015, Genetic diversity and genomic resources available for the small millet crops to accelerate a New Green Revolution. Front. Plant Sci., 6, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Armstead I., Huang L., Ravagnani A., Robson P., Ougham H.. 2009, Bioinformatics in the orphan crops. Brief. Bioinform., 10, 645–53. [DOI] [PubMed] [Google Scholar]
- 9. Mysore K. S., Baird V.. 1997, Nuclear DNA content in species of Eleusine (Gramineae): a critical re-evaluation using laser flow cytometry. Plant Syst. Evol., 207, 1–11. [Google Scholar]
- 10. Ramegowda V., Senthil-Kumar M., Nataraja K. N., Reddy M. K., Mysore K. S., Udayakumar M.. 2012, Expression of a finger millet transcription factor, EcNAC1, in tobacco confers abiotic stress-tolerance. PLoS One, 7, e40397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parvathi M. S., Nataraja K. N., Yashoda B. K., Ramegowda H. V., Mamrutha H. M., Rama N.. 2013, Expression analysis of stress responsive pathway genes linked to drought hardiness in an adapted crop, finger millet (Eleusine coracana). J. Plant Biochem. Biotechnol., 22, 193–201. [Google Scholar]
- 12. Nagarjuna K. N., Parvathi M. S., Sajeevan R. S., Pruthvi V., Mamrutha H. M., Nataraja K. N.. 2016, Full-length cloning and characterization of abiotic stress responsive CIPK31-like gene from finger millet, a drought-tolerant crop. Curr. Sci., 111, 890. [Google Scholar]
- 13. Rahman H., Jagadeeshselvam N., Valarmathi R., et al. 2014, Transcriptome analysis of salinity responsiveness in contrasting genotypes of finger millet (Eleusine coracana L.) through RNA-sequencing. Plant Mol. Biol., 85, 485–503. [DOI] [PubMed] [Google Scholar]
- 14. Dida M. M., Srinivasachary, Ramakrishnan S., Bennetzen J. L., Gale M. D., Devos K. M.. 2006, The genetic map of finger millet, Eleusine coracana. Theor. Appl. Genet., 114, 321–32. [DOI] [PubMed] [Google Scholar]
- 15. Wood T. E., Takebayashi N., Barker M. S., Mayrose I., Greenspoon P. B., Rieseberg L. H.. 2009, The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci., 106, 13875–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mayrose I., Zhan S. H., Rothfels C. J., et al. 2011, Recently formed polyploid plants diversify at lower rates. Science, 333, 1257. [DOI] [PubMed] [Google Scholar]
- 17. Leitch A. R., Leitch I. J.. 2008, Genomic plasticity and the diversity of polyploid plants. Science, 320, 481–3. [DOI] [PubMed] [Google Scholar]
- 18. Bodily P. M., Fujimoto M., Ortega C., et al. 2015, Heterozygous genome assembly via binary classification of homologous sequence. BMC Bioinform., 16, S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pryszcz L. P., Gabaldón T.. 2016, Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res., 44, e113–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sierro N., Battey J. N. D., Ouadi S., et al. 2014, The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun., 5, 3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang T., Hu Y., Jiang W., et al. 2015, Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol., 33, 531–7. [DOI] [PubMed] [Google Scholar]
- 22. Mayer K. F. X., Rogers J., Dole el J., et al. 2014, A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science, 345, 1251788–8. [DOI] [PubMed] [Google Scholar]
- 23. Yang J., Liu D., Wang X., et al. 2016, The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet., 48, 1225–32. [DOI] [PubMed] [Google Scholar]
- 24. Paape T., Hatakeyama M., Shimizu-Inatsugi R., et al. 2016, Conserved but attenuated parental gene expression in allopolyploids: constitutive zinc hyperaccumulation in the allotetraploid Arabidopsis kamchatica. Mol. Biol. Evol., 33, 2781–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Akama S., Shimizu-Inatsugi R., Shimizu K. K., Sese J.. 2014, Genome-wide quantification of homeolog expression ratio revealed nonstochastic gene regulation in synthetic allopolyploid Arabidopsis. Nucleic Acids Res., 42, e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pootakham W., Sonthirod C., Naktang C., et al. 2017, De novo hybrid assembly of the rubber tree genome reveals evidence of paleotetraploidy in Hevea species. Sci. Reports, 7, 41457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lieberman-Aiden E., van Berkum N. L., Williams L., et al. 2009, Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (New York, N.Y.), 326, 289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mascher M., Gundlach H., Himmelbach A., et al. 2017, A chromosome conformation capture ordered sequence of the barley genome. Nature, 544, 427–33. [DOI] [PubMed] [Google Scholar]
- 29. Yuan Y., Bayer P. E., Batley J., Edwards D.. 2017, Improvements in genomic technologies: application to crop genomics. Trends Biotechnol., 35, 547–58. [DOI] [PubMed] [Google Scholar]
- 30. Geetha K., Mani A. K., Sur M.. 2012, Identification of finger millet (Eleusine coracana Gaertn.) variety suitable to rainfed areas of north western zone of Tamil Nadu. Indian J. Agric. Res., 46, 60–4. [Google Scholar]
- 31. Dolezel J., Sgorbati S., Lucretti S.. 1992, Comparison of three DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiologia Plantarum, 85, 625–31. [Google Scholar]
- 32. Bolger A. M., Lohse M., Usadel B.. 2014, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxford, England), 30, 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hatakeyama M., Opitz L., Russo G., Qi W., Schlapbach R., Rehrauer H.. 2016, SUSHI: an exquisite recipe for fully documented, reproducible and reusable NGS data analysis. BMC Bioinform., 17, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kajitani R., Toshimoto K., Noguchi H., et al. 2014, Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res., 24, 1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ye C., Hill C. M., Wu S., Ruan J., Ma Z.. 2016, DBG2OLC: efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Reports, 6, 31900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walker B. J., Abeel T., Shea T., et al. 2014, Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One, 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boetzer M., Henkel C. V., Jansen H. J., Butler D., Pirovano W.. 2011, Scaffolding pre-assembled contigs using SSPACE. Bioinformatics, 27, 578–9. [DOI] [PubMed] [Google Scholar]
- 38. Kosugi S., Hirakawa H., Tabata S.. 2015, GMcloser: closing gaps in assemblies accurately with a likelihood-based selection of contig or long-read alignments. Bioinformatics, 31, btv465. [DOI] [PubMed] [Google Scholar]
- 39. Cantarel B. L., Korf I., Robb S. M., et al. 2007, MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res., 18, 188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dobin A., Davis C. A., Schlesinger F., et al. 2013, STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pertea M., Pertea G. M., Antonescu C. M., Chang T. C., Mendell J. T., Salzberg S. L.. 2015, StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol., 33, 290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jurka J., Kapitonov V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J.. 2005, Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res., 110, 462–7. [DOI] [PubMed] [Google Scholar]
- 43. Saha S., Bridges S., Magbanua Z. V., Peterson D. G.. 2008, Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res., 36, 2284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stanke M., Diekhans M., Baertsch R., Haussler D.. 2008, Using native and synthetically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, 24, 637–44. [DOI] [PubMed] [Google Scholar]
- 45. Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M.. 2015, BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31, 3210–2. [DOI] [PubMed] [Google Scholar]
- 46. Staňková H., Hastie A. R., Chan S., et al. 2016, BioNano genome mapping of individual chromosomes supports physical mapping and sequence assembly in complex plant genomes. Plant Biotechnol. J., 14, 1523–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fierst J. L. 2015, Using linkage maps to correct and scaffold de novo genome assemblies: methods, challenges, and computational tools. Front. Genet., 6, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bruijn de N. G. 1946, A combinatorial problem. Proc. Sect. Sci., 49, 758–64. [Google Scholar]
- 49. Compeau P. E. C., Pevzner P. A., Tesler G.. 2011, How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol., 29, 987–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krasileva K. V., Buffalo V., Bailey P., et al. 2013, Separating homeologs by phasing in the tetraploid wheat transcriptome. Genome Biol., 14, R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Innan H., Kondrashov F.. 2010, The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet., 11, 4. [DOI] [PubMed] [Google Scholar]
- 52. Koh J., Soltis P. S., Soltis D. E.. 2010, Homeolog loss and expression changes in natural populations of the recently and repeatedly formed allotetraploid Tragopogon mirus (Asteraceae). BMC Genomics, 11, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Finn R. D., Bateman A., Clements J., et al. 2014, Pfam: the protein families database. Nucleic Acids Res., 42, D222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J.. 1990, Basic local alignment search tool. J. Mol. Biol., 215, 403–10. [DOI] [PubMed] [Google Scholar]
- 55. Tamura K., Dudley J., Nei M., Kumar S.. 2007, MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol. Biol. Evol., 24, 1596–9. [DOI] [PubMed] [Google Scholar]
- 56. Elliott J., Deryng D., Müller C., et al. 2014, Constraints and potentials of future irrigation water availability on agricultural production under climate change. Proc. Natl. Acad. Sci., 111, 3239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wheeler T., Braun J. von.. 2013, Climate change impacts on global food security. Science, 341, 508–13. [DOI] [PubMed] [Google Scholar]
- 58. Brown A. H. D. 2010, Variation under domestication in plants: 1859 and today. Philos. Trans. Roy. Soc. B Biol. Sci., 365, 2523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Meyer R. S., DuVal A. E., Jensen H. R.. 2012, Patterns and processes in crop domestication: an historical review and quantitative analysis of 203 global food crops. New Phytol., 196, 29–48. [DOI] [PubMed] [Google Scholar]
- 60. Hittalmani S., Mahesh H. B., Shirke M. D., et al. 2017, Genome and transcriptome sequence of finger millet (Eleusine coracana (L.) Gaertn.) provides insights into drought tolerance and nutraceutical properties. BMC Genomics, 18, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.