

Abstract
SCN5A encodes the voltage-gated Na+ channel NaV1.5 that is responsible for depolarization of the cardiac action potential and rapid intercellular conduction. Mutations disrupting the SCN5A coding sequence cause inherited arrhythmias and cardiomyopathy, and single-nucleotide polymorphisms (SNPs) linked to SCN5A splicing, localization, and function associate with heart failure–related sudden cardiac death. However, the clinical relevance of SNPs that modulate SCN5A expression levels remains understudied. We recently generated a transcriptome-wide map of microRNA (miR) binding sites in human heart, evaluated their overlap with common SNPs, and identified a synonymous SNP (rs1805126) adjacent to a miR-24 site within the SCN5A coding sequence. This SNP was previously shown to reproducibly associate with cardiac electrophysiological parameters, but was not considered to be causal. Here, we show that miR-24 potently suppresses SCN5A expression and that rs1805126 modulates this regulation. We found that the rs1805126 minor allele associates with decreased cardiac SCN5A expression and that heart failure subjects homozygous for the minor allele have decreased ejection fraction and increased mortality, but not increased ventricular tachyarrhythmias. In mice, we identified a potential basis for this in discovering that decreased Scn5a expression leads to accumulation of myocardial reactive oxygen species. Together, these data reiterate the importance of considering the mechanistic significance of synonymous SNPs as they relate to miRs and disease, and highlight a surprising link between SCN5A expression and nonarrhythmic death in heart failure.
Keywords: Cardiology, Genetics
Keywords: Genetic variation, Ion channels, Noncoding RNAs
Introduction
The onset and clinical course of heart failure is shaped by a complex interplay of environmental and genetic factors that influence a host of biological processes, including gene regulatory mechanisms. Optimal cardiac function relies on finely tuned expression of genes related to cardiac structure, energetics, conduction, and contraction. Tight regulation of cardiac ion channels governing heart rate and rhythm is vital, since slight changes in ion conductance can trigger arrhythmia, elevating one’s risk for sudden cardiac death. While prior research has established that heart failure invokes cardiac ion channel remodeling with arrhythmias and that some mutations in ion channels cause cardiomyopathy with heart failure (1, 2), it remains unclear how alterations in channels related to cardiac conduction may influence nonarrhythmic deaths in heart failure patients.
The cardiac action potential is initiated by sodium current through the heart’s primary voltage-gated sodium channel, NaV1.5, encoded by SCN5A. NaV1.5 is critical for normal cardiac function, and mutations in NaV1.5 and NaV1.5-interacting genes that modify its function and trafficking cause arrhythmic syndromes (Brugada, long QT, inherited conduction) (3–8) and cardiomyopathies with contractile dysfunction (9, 10). Moreover, genome-wide association studies (GWAS) have identified several common SNPs within the SCN5A locus linked to electrocardiographic measures (PR, QT, and QRS intervals) (11–16) and Brugada syndrome (17), an inherited arrhythmic disease with sudden cardiac death. The clear relevance of SCN5A to arrhythmias has fueled vigorous research of cellular mechanisms controlling NaV1.5 biosynthesis, posttranslational processing, localization, and function (18–20). Among these efforts, researchers have identified alternatively spliced SCN5A transcript isoforms that associate with fatal arrhythmias in heart failure subjects (21) and have begun characterizing miR-mediated regulation via the SCN5A 3′-untranslated region (3′-UTR) (22–24). However, given the complex nature of this sizable gene (nearly 30 exons spanning > 80 kb), continued efforts are needed to further define SCN5A transcript regulatory mechanisms; for example, no studies have yet assessed miR functions within the expansive SCN5A coding region.
MiRs have been established as key effectors in cardiovascular biology and disease (25–27). These short RNAs are incorporated into Argonaute (Ago) proteins, producing effector complexes capable of base-pairing with and repressing target transcripts via translational inhibition and mRNA destabilization (28–30). Canonically, miRs engage 3′-UTRs containing sequences complementary to their seed (miR nucleotide positions 2–8; ref. 31). This minimal degree of sequence recognition complicates research efforts to identify biologically relevant miR-target sites. To overcome this, we recently performed high-throughput sequencing of cross-linked immunoprecipitates (HITS-CLIP) to identify Ago2-associated miRs and their bound sequences in human myocardial tissues, yielding approximately 4,000 miR-target sites across more than 2,000 mRNAs (32), with approximately 50% of the sites overlapping coding regions. In exploring the interface of these sites with human genetic variations, we identified an intriguing interaction between miR-24 and rs1805126, a synonymous SNP in the SCN5A coding sequence, and postulated that this SNP would modulate SCN5A expression and outcomes in heart failure patients.
Results
Regulation of human SCN5A by miR-24 is modulated by the common SNP rs1805126.
The SCN5A coding SNP rs1805126 was previously found to associate with electrocardiographic measures across several independent GWAS cohorts (12, 13, 15, 16, 33), but is synonymous (i.e., does not alter the amino acid sequence), and thus has been overlooked as being a causal variant. However, our new cardiac Ago2 HITS-CLIP data point to the possibility that this polymorphism may modulate the function of an adjacent miR-24 site (Figure 1A); notably, this was the most robustly engaged region by Ago2 across the entire SCN5A mRNA (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI95710DS1). As this SNP does not interfere with miR-target base-pairing (Figure 1A), we speculated that a change in mRNA structure (supported by computational RNA-fold analysis; Supplemental Figure 2) might confer an allele-specific response of SCN5A mRNAs to miR-24 activity. Indeed, PITA miR target prediction analysis, which incorporates target-site accessibility parameters (34), indicated that the C allele represents the more thermodynamically favorable miR-24 target, compared with T (32). This was supported by our previous experiments testing traditional luciferase-based 3′-UTR reporters for each allele, revealing that miR-24 more strongly suppresses the C allele compared with the T allele (32). To move beyond this artificial system (i.e., coding region placed into reporter gene 3′-UTR), we assessed the impact of miR-24 on SCN5A gene expression derived from full-length cDNA expression plasmids engineered to harbor either the rs1805126 T or C allele. Cotransfection studies in both mouse N2a and human HEK293 cells demonstrated that miR-24 mimics strongly suppress NaV1.5 expression (Figure 1, B and C and Supplemental Figure 3), and consistent with the luciferase reporter data, the rs1805126 C allele was repressed to a greater degree than the T allele (relative to control miR mimics, P < 0.01). The enhanced response to miR-24 treatment was further supported by corresponding quantitative PCR (QPCR) data in N2a cell studies, showing a nearly 40% decrease in SCN5A C allele transcripts and virtually no change in T allele transcripts (Figure 1D, P< 0.01). To address the possibility that additional miR-24 sites and/or other mechanisms may contribute to the robust miR-24–mediated SCN5A silencing, we introduced synonymous mutations into the experimental miR-24 seed sequence within the full-length SCN5A expression construct, and this completely abolished the miR-24 inhibitory effect on SCN5A expression (Figure 1, B and D).
Figure 1. The synonymous coding SCN5A SNP, rs1805126, modulates the miR-24–SCN5A interaction.

(A) Schematic of the conserved miR-24–SCN5A interaction identified by Ago2 HITS-CLIP (read coverage shown above). The site resides within the coding region of the SCN5A terminal exon and is adjacent to the synonymous SNP rs1805126 (*) that has previously been linked to heart rhythm abnormalities by GWAS. (B–D) The effect of miR-24 on SCN5A expression, and the potential impact of rs1805126 on this interaction, was tested in cell culture experiments. Mouse N2a cells were cotransfected in triplicate with synthetic pre-miRs (4 nM) and human full-length SCN5A expression plasmids harboring either the C or T allele for rs1805126, or synonymous mutations that disrupt the miR-24 seed site (mut*, base mutations are indicated in bold in panel A). At 48 hours after transfection, Western blot and QPCR analyses were used to measure protein and mRNA expression. (B) Representative Western blots show that miR-24 strongly suppresses NaV1.5 expression, having a more robust effect on the C allele, relative to T. Densitometry analysis of Western blot data (n = 9 biological replicates from 3 separate studies for C versus T, n = 4 for mutated) supports a significant allele-specific difference in miR-24 suppression of NaV1.5 levels (C), as does QPCR analyses measuring SCN5A mRNA levels (D; n = 6 biological replicates from 2 studies). Data are represented as the mean ± SEM, and P values were obtained using 2-tailed unpaired t test comparing the indicated groups.
miR-24 overexpression inhibits NaV1.5 expression and sodium current density.
To determine if miR-24 functionally inhibits the activity of the SCN5A-encoded NaV1.5 sodium channel in heart cells, we performed single-cell patch-clamp analyses in cultured neonatal rat cardiomyocytes (NRCMs); of note, the miR-24 SCN5A target seed sequence is conserved in rodents (Figure 1A), and NRCMs have been shown to express high levels of endogenous miR-24 (35). Consistent with this, we found that a luciferase-based miR-24 sensor transgene was suppressed in NRCMs and that this was blocked by treatment with miR-24 inhibitors (i.e., anti-miRs, Supplemental Figure 4A). We next tested whether miR-24 overexpression could provoke a functional interaction with rat Scn5a mRNA in NRCMs. NRCMs treated with miR-24 mimics displayed a marked decrease in sodium current density (>4-fold, P < 0.001), as compared with cells treated with control miR (Figure 2). Notably, the degree of miR-24–induced sodium current suppression was similar to that observed in NRCMs treated with Scn5a-targeted RNAi, and in complementary studies, NRCMs treated with miR-24 mimics showed an approximately 50% decrease in NaV1.5 protein expression (Figure 2, D and E, P < 0.01). However, anti–miR-24 treatment in NRCMs had no effect on NaV1.5 sodium current density or expression (Supplemental Figure 4, B–E). It is possible that miR-24 does not naturally or strongly engage Scn5a mRNA in cultured NRCMs, since proximal nucleotide differences make the site considerably less favorable compared with human SCN5A (Supplemental Figure 5).
Figure 2. Effect of miR-24 overexpression on SCN5A function and expression in neonatal rat cardiomyocytes.

NRCMs were cotransfected with synthetic pre-miRs (25 nM) and plasmids coexpressing miRs/siRNAs and EGFP. Na+ current in EGFP-positive cells was measured 2–4 days later using patch clamp. Shown are representative traces of cardiac Na+ currents and pulse protocol (A) and the current-voltage relationship of Na+ current (B) in NRCMs transfected with miR-Neg1 (miR-N, negative control), miR-24, or siSCN5A (positive control). (C) Sodium current density at –10 mV is also plotted. These patch-clamp data support the hypothesis that miR-24 potently suppresses sodium current density in NRCMs, to a similar degree as Scn5a-targeted siRNA. Data are represented as the mean ± SEM. Sample numbers combined from 3 separate experiments are denoted in the figure, and P values were calculated using 1-way ANOVA with Dunnett’s post hoc test (versus miR-Neg1). (D) NRCMs were transfected with 25 nM synthetic pre-miR mimics and endogenous NaV1.5 protein levels were measured by Western blot 48 hours later; a representative Western blot image is shown. (E) Combined Western blot (WB) densitometry analysis from 2 separate experiments (n = 6 total biological replicates per treatment; mean ± SEM) is plotted. Pre–miR-29 and -Neg1 serve as negative controls, and β-actin or GAPDH levels were used for normalization. *P value derived using 1-way ANOVA with Dunnett’s post hoc test (versus miR-Neg1).
The rs1805126 C allele associates with adverse outcomes in heart failure patient cohorts.
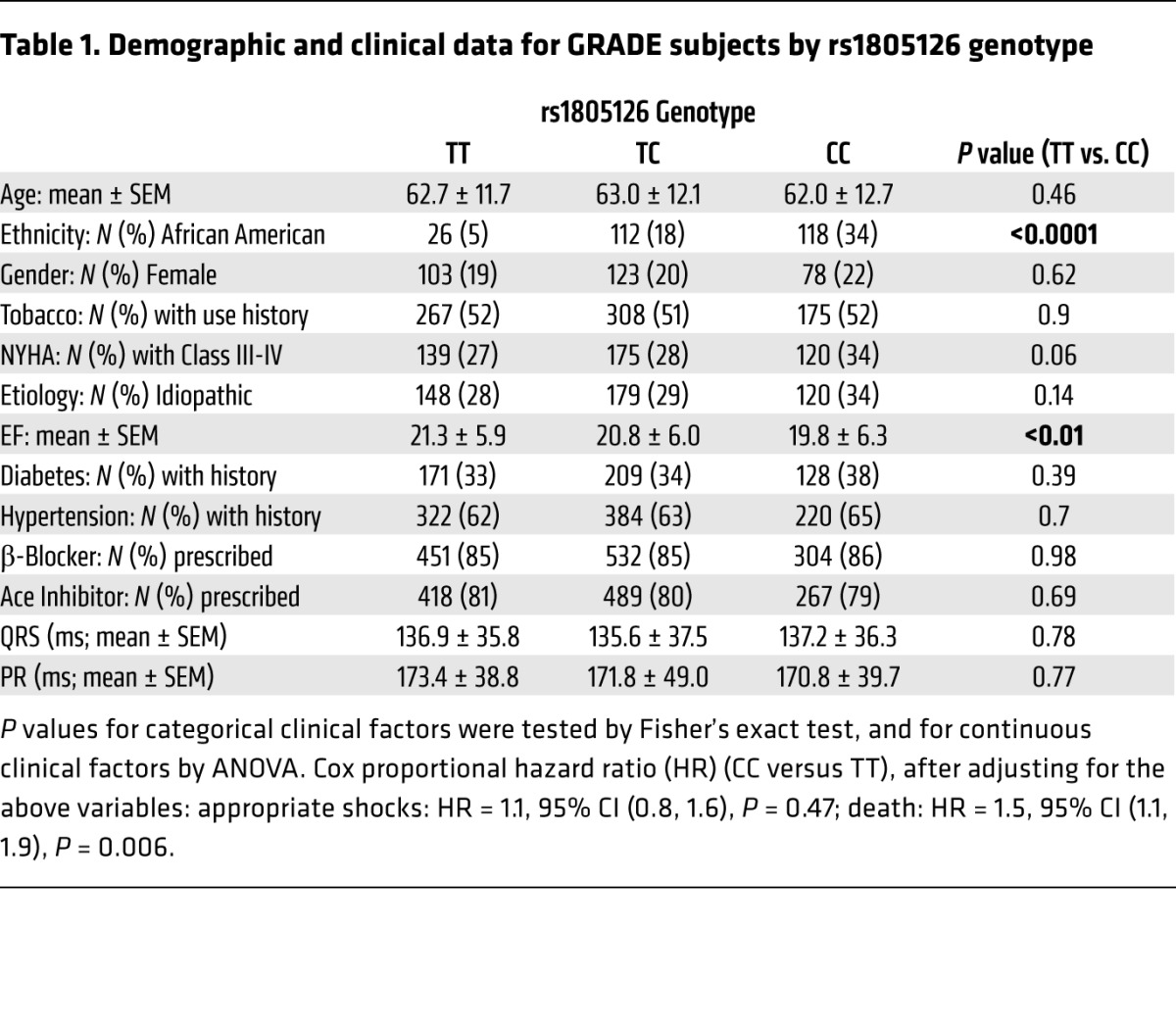
Next, we assessed the clinical significance of the miR-24–SCN5A–SNP interaction in heart failure patients. We determined rs1805126 genotypes in 1,658 cardiomyopathy patients (mean age = 62 years, mean ejection fraction [EF] = 21%; 79% male, 79% Caucasian and 20% African American, 70% ischemic) with implantable cardioverter defibrillators (ICDs) from the prospective, multicenter, NIH-funded Genetic Risk Assessment of Defibrillator Events (GRADE) study (36), identifying 565 TT (34%), 694 TC (42%), and 399 CC (24%) subjects. Kaplan-Meier analyses revealed that CC homozygotes had markedly higher mortality rates, as compared with T allele carriers (hazard ratio [HR] = 1.5, P = 0.002; Figure 3A). This risk appeared to be independent of other potential confounding variables, including heart failure etiology (ischemic or idiopathic) and ethnicity (Supplemental Figure 6), the latter being of notable significance given that the CC genotype is approximately 3 times more prevalent among African Americans. This was supported by multivariate analyses to estimate the effect of rs1805126 genotype on mortality, adjusting for other clinically relevant variables and comorbidities (listed in Table 1); this yielded a Cox proportional HR = 1.5, 95% CI (1.1, 1.9), P = 0.006 (CC versus TT). Interestingly, rs1805126 genotype did not significantly associate with appropriate ICD shocks for ventricular tachycardia or ventricular fibrillation, a surrogate for arrhythmic sudden cardiac death (Figure 3B), as might be expected given the well-established relationships among SCN5A, rs1805126, and heart rhythm. Moreover, whole-cohort analyses did not reveal significant genotype-based differences in PR or QRS intervals (Table 1); of note, the aforementioned GWAS links were established in very large cohorts without heart failure, whereas heart failure subjects in GRADE had significantly greater variability in PR and QRS intervals due to conduction disease. By contrast, the CC genotype in GRADE was associated with indications of worsening heart failure, with CC subjects having significantly reduced left ventricular ejection fractions (LVEFs) compared with TT subjects (P = 0.002) and trending towards poorer New York Heart Association (NYHA) heart failure classification scores (P = 0.06; Figure 3C and Table 1).
Figure 3. Association of rs1805126 genotype with heart failure outcomes.

(A–C) Patient samples from GRADE (a multicenter prospective study of heart failure outcomes) were genotyped for the rs1805126 SNP and Kaplan-Meier analyses were performed to compare overall survival rates from all-cause death (A), and freedom from appropriate ICD shocks (B), a surrogate for sudden cardiac death. Homozygous CC individuals had significantly poorer survival outcomes relative to the other genotypes (i.e., T allele carriers). No differences in shock outcomes were observed between the groups. P values were obtained using log-rank test. Cohort sizes are as follows: TT = 532, TC = 628, and CC = 358. (C) CC homozygous individuals had lower left ventricular ejection fractions (LVEF; mean ± SEM; n = 541 TT, 673 TC, and 385 CC). **P < 0.01 using 1-way ANOVA with Dunnett’s post hoc test, CC versus TT genotypes. (D) A replication study was performed in the GRAHF cohort (based on DNA sample availability); consistent with GRADE, rs1805126-CC homozygous GRAHF patients showed a near-significant trend towards worse heart failure outcomes (i.e., lower A-HeFT composite scores). Group mean (dark line) and 95% confidence intervals (light shading) are plotted; P value determined by 2-tailed unpaired t test. Cohort sizes are as follows: TT = 32, TC = 99, and CC = 139.
Table 1. Demographic and clinical data for GRADE subjects by rs1805126 genotype.
To determine if these observations extend beyond the GRADE cohort, we evaluated the association of rs1805126 genotypes with heart failure outcomes in 270 African American cardiomyopathy patients (mean age = 57 years, mean EF = 24%; 61% male, 26% ischemic) from the Genetic Risk of Heart Failure in African Americans (GRAHF) study, a subgroup of the African American Heart Failure Trial (A-HeFT) with available DNA samples (37). Genotyping of rs1805126 identified 32 TT (12%), 99 TC (37%), and 139 CC (51%) subjects, consistent with previous observations that the C allele is more common in African Americans. Association of these genotypes with accompanying clinical data revealed that CC patients showed a near-significant trend towards having worse A-HeFT composite scores (trial primary endpoint, which summarizes death, hospitalization, and change in quality of life; Figure 3D, P = 0.06 for CC versus TT). Of note, genotype-based subgroups showed no difference in other clinically relevant variables and comorbidities (Supplemental Table 1), and adjusting for sex, age, etiology and diabetes yielded a statistically significant association (P = 0.05), further reinforcing the link between the rs1805126-CC genotype and adverse outcomes in heart failure patients.
The rs1805126 C allele associates with decreased SCN5A expression in human hearts.
To understand the molecular basis of the association between the rs1805126 SNP and heart failure mortality, we first evaluated genotype-related alterations in myocardial gene expression, given the effects of rs1805126 on miR-24–mediated SCN5A silencing and the absence of nonsynonymous SNPs in linkage disequilibrium with it (r2 ≥ 0.4; Supplemental Table 2). For this, we assessed human cardiac tissue samples from nonfailing hearts (i.e., unused heart transplant donors, Supplemental Tables 2 and 3). Considering our in vitro data showing that the rs1805126 C allele promotes stronger SCN5A suppression by miR-24, we hypothesized that myocardial SCN5A expression would be lower in CC, relative to TT, homozygous individuals. Western blot and QPCR analyses showed that CC hearts have approximately 50%–60% less NaV1.5 protein (Figure 4, A and B; P < 0.01) and SCN5A mRNA (Supplemental Figure 7A) compared with TT hearts, while miR-24 levels were similar (Supplemental Figure 7B). Genotype-related associations with SCN5A mRNA levels were further evaluated in larger cohorts using available data derived from cardiac eQTL studies (one headed by Kenneth B. Margulies and the other by Connie Bezzina [University of Amsterdam, The Netherlands]) (38, 39), each offering transcriptional profiling and genome-wide SNP-typing data on hundreds of nonfailing myocardial tissue samples. Notably, reanalysis and query of these datasets revealed that rs1805126-CC hearts exhibit approximately 15%–20% lower SCN5A mRNA levels, relative to TT hearts (Figure 4C, P ≤ 0.05; sample information in Supplemental Table 4), while heterozygotes show intermediate expression (Supplemental Figure 8). In follow-up work with the Margulies lab, we further interrogated this association by allele-specific expression analysis using myocardial RNA sequencing (RNA-seq) data derived from 29 individuals heterozygous for rs1805126 (Supplemental Table 5). In the majority of these heterozygous samples, C allele SCN5A transcripts were expressed at lower levels, as compared with those harboring the T allele (~12% mean difference, P < 0.001; Figure 4D), providing strong evidence to further support this reproducible association.
Figure 4. Association of rs1805126 genotype with cardiac SCN5A expression.

(A and B) Western blot (WB) using antibodies against both N- and C-terminal ends of SCN5A and accompanying densitometry analysis shows that NaV1.5 protein expression is decreased in nonfailing CC homozygous human cardiac tissue samples (n = 7), relative to TT samples (n = 6). β-Catenin serves as loading control. **P < 0.01 by 2-tailed unpaired t test. (C) Reanalysis of available genome-wide human cardiac eQTL data from nonfailing hearts reveals that CC samples express less SCN5A mRNA, compared with TT samples; cohorts from the University of Pennsylvania (left) and The Netherlands (right). Normalized microarray probe intensity (log2) is plotted; further analysis indicates approximately 10%–20% lower SCN5A mRNA in CC versus TT. Sample numbers are denoted in the figure. *P ≤ 0.05 by 2-tailed unpaired t test. (D) Allele-specific expression analysis in heterozygous individuals was done using RNA-seq data obtained from nonfailing and failing human hearts (n = 29), offering further support that the C allele is expressed at lower levels, relative to T. ***P < 0.001 by 1-sample t test.
Decreased Scn5a expression leads to accumulation of cardiac reactive oxygen species in mice.
As an initial step towards understanding how decreased SCN5A expression may compromise myocardial health and function, we focused on the established Scn5a heterozygous knockout (Scn5a+/–) mouse strain. These mice are known to express Scn5a mRNA and NaV1.5 protein at 50% levels, relative to wild-type littermates, and develop aging-related signs of adverse myocardial remodeling (e.g., increased fibrosis and conduction slowing), which manifest after 12 months of age. Considering that oxidative stress is often at the root of profibrotic signaling cascades in the heart and is increased in heart failure, we hypothesized that Scn5a+/– mice may exhibit increased accumulation of reactive oxygen species (ROS). To address this, we measured oxidation of dihydroethidium (DHE, a surrogate marker of steady-state levels of superoxide) in fresh-frozen cardiac tissues harvested from 7- to 8-month-old male wild-type and Scn5a+/– mice, prior to the appearance of overt myocardial remodeling. Relative to wild-type littermate hearts, Scn5a+/– hearts showed a striking and highly significant increase in DHE oxidation (~2.5-fold, P < 1 × 10–20; Figure 5), and Western blot analysis done in parallel confirmed a 50% decrease in cardiac NaV1.5 expression in Scn5a+/– mice (Supplemental Figure 9). Together, these data support a potentially novel link between reduced expression of the cardiac sodium channel and downstream escalation in oxidative stress, which represents an established pathway towards adverse myocardial remodeling and dysfunction and could explain the worsened LV function and increased mortality in rs1805126-CC heart failure patients.
Figure 5. Evaluation of oxidative stress in Scn5a heterozygous knockout mouse hearts.

Oxidation of dihydroethidium (DHE, a surrogate marker of steady-state levels of superoxide) was measured in fresh-frozen cardiac tissue sections collected from 7- to 8-month-old male Scn5a+/– mice and wild-type littermates (n = 3 each). Positive controls included sections treated with antimycin A (AntA, a mitochondrial electron transport chain blocker that is known increase ROS generation). Images were captured at ×40 magnification. Scale bars: 100 μm. (A) Representative photomicrographs of DHE-stained cardiac sections are shown. (B) Signal intensity quantified in 90 cells per group (i.e., 30 per mouse) is plotted (mean ± SEM). ****P < 0.0001 by 2-tailed t test.
Discussion
Previous studies have demonstrated that SCN5A is under regulatory control by miRs targeting the 3′-UTR (22–24); however, our studies are the first to our knowledge to evaluate miR binding within the coding sequence, an often overlooked region in miR-related investigations. Indeed, our previous HITS-CLIP data revealed that the most prominent human cardiac Ago2 binding site within SCN5A lay within the terminal coding exon. Here, our follow-up studies indicate that this site is functionally engaged by miR-24, a miR that is commonly upregulated in failing human and rodent hearts (40, 41), revealing a potentially novel and potent disease-relevant suppressor of SCN5A expression. Furthermore, we demonstrate that the synonymous SNP rs1805126 modulates this regulation, providing a potential molecular mechanism to account for established associations between this variant and heart rhythm changes in human subjects.
Specifically, our data link lower myocardial SCN5A expression to the rs1805126-CC genotype, which was previously associated with highly significant increases in PR and QRS intervals (13); this is consistent with the prolonged PR that coincides with decreased NaV1.5 expression in patients with loss-of-function SCN5A mutations and in Scn5a heterozygous knockout mice (42, 43). In our current work, we did not find a significant association between rs1805126 genotypes and PR and QRS intervals in GRADE subjects; our study was underpowered for this, especially considering that heart failure causes these measures to become highly variable across patients with frequent bundle branch block and need for pacing and biventricular pacing. In addition, the prior related GWAS were done on very large populations of normal individuals and found only relatively small alterations in PR and QRS intervals (2.5 and 0.7 ms, respectively).
Beyond cardiac conduction, we uncovered an unanticipated and significant association between rs1805126 and all-cause mortality in the GRADE cohort, signifying the first observed link to our knowledge between a common SCN5A SNP and nonarrhythmic death in heart failure. We did not see evidence of an association between rs1805126 genotype and ventricular tachyarrhythmias, a surrogate of sudden cardiac death, and the presence of an ICD precludes asystole and severe bradyarrhythmias as contributors to mortality in this population. Notably, we found that the association with mortality was present in the African American subgroup of GRADE, and provided additional supportive evidence for this association in the African American GRAHF cohort, with rs1805126-CC subjects having worse heart failure outcomes as evidenced by lower A-HeFT composite score. Together, these results justify follow-up studies in other heart failure patient cohorts to further examine the robustness of this association.
As an initial step towards interrogating the mechanistic basis of the association between decreased SCN5A expression and worse heart failure outcomes, we considered previous observations that Scn5a+/– mice develop aging-related signs of adverse myocardial remodeling (43), and we speculated that elevated ROS may be at the root of this pathology. Interestingly, previous reports have shown that mitochondrial ROS can alter NaV1.5 channel activity (44); however, our new data showing elevated ROS in hearts from Scn5a+/– mice provide the first evidence to our knowledge that suggest a bidirectional relationship between NaV1.5 and ROS. Along these lines, one recent study demonstrated that increasing persistent late Na+ current in mouse hearts leads to structural derangements, mitochondrial injury, and fibrosis (45).
Overall, the results presented herein support future studies to look beyond 3′-UTRs for clinically significant miR-SNP interactions. To our knowledge, we have discovered only the second known example of a disease-associated miR-SNP interaction found within a coding region (46), likely due to related studies having a restricted focus on 3′-UTRs (47, 48). Beyond this, our data highlight the need for follow-up investigations to further define the extensive regulatory controls that influence SCN5A expression, assessing if rs1805126-CC genotype risk is compounded by environmental factors or other common SCN5A SNPs (49, 50). In addition, our data reveal a need to characterize noncanonical functions of NaV1.5 in cardiomyocytes, focusing on whether its mechanistic influence on provoking ROS accumulation occurs secondarily through aberrations in sodium-calcium balance, rates of oxidative phosphorylation versus glycolysis, endosomal/lysosomal acidification, or altering NADPH oxidase activities by weakening dystrophin multiprotein scaffolding complex integrity (18, 51–55).
Methods
Human cardiac tissue samples.
LV cardiac tissues were obtained from the Myocardial Applied Genomics Network (MAGNet; www.med.upenn.edu/magnet). All subjects donating tissue provided consent under an approved IRB protocol, and provided clinical information that is confidentially linked to the specimens by a study number. LV free-wall tissue was harvested at the time of cardiac surgery from subjects with heart failure undergoing transplantation and from unused donor hearts. The heart was perfused with cold cardioplegia prior to cardiectomy to arrest contraction and prevent ischemic damage. Tissue specimens were then obtained and frozen in liquid nitrogen and stored at –80°C until used. For Western blot analysis, total protein of human left ventricles was harvested by homogenizing tissues in lysis buffer (50 mM Tris at pH 7.4, 150 mM NaCl, 10 mM NaF, 1 mM Na3VO4, 1 mM EGTA, 1 mM EDTA, 0.5% Triton X-100, 0.5% Na deoxycholate, 0.1% SDS) containing protease inhibitors (Roche). Protein concentrations were determined using the Pierce BCA Assay (Thermo Fisher Scientific).
Plasmids.
The rs1805126 C allele expression plasmid was generated by site-directed mutagenesis performed on a plasmid containing full-length human SCN5A cDNA (rs1805126 T allele) under control of the CMV promoter. For this, the Phusion site-directed mutagenesis protocol (New England BioLabs) was used with the following 5′-phosphorylated primers: 5′-/Phos/GCCCTGTCTGAGCCACTCCGTATC-3′ and 5′-/Phos/GTCGGCAAAGTCAGACAGGACCGAATAC-3′ (Integrated DNA Technologies). For the miR-24 dual luciferase sensor plasmid, DNA oligos were used to insert the following sequence: 5′-GACTGAGCCACCTTGAAATGCTCCAACCACTGAGCCAGGTTTGCCGTACTGAGCCA-3′, to introduce 3 mIR-24 8mer binding sites downstream of the Renilla luciferase expression cassette (using XhoI and NotI restriction enzyme sites) in the psiCheck2 plasmid (Promega).
Full-length Scn5a knockdown studies.
Mouse N2a and human HEK293 cells (which do not express endogenous SCN5A at detectable levels by Western blot, data not shown; ATCC cell lines) were seeded in 24-well plates (80,000 and 200,000 cells/well for N2a and HEK293, respectively), and the next day, cells were cotransfected with synthetic pre-miRs (4 nM) along with human full-length SCN5A expression plasmids (100 ng) using Lipofectamine 2000 (Life Technologies). After 48 hours, cells were harvested in 200 μl of RIPA buffer, and half of the lysate was immediately added to 900 μl of TRIzol reagent (Invitrogen) for RNA isolation. Western blot and QPCR analyses were performed as described below.
Western blot.
Protein samples were resolved by standard SDS-PAGE before being transferred to a 0.45-μm PVDF membrane (Millipore). The membrane was blocked with 2%–5% milk in 1× PBST (0.05% Tween 20), after which rabbit anti-NaV1.5 antibody (1:200; Alomone Labs, ASC-005), anti–β-catenin (1:200; Abcam, ab2365), or anti–β-actin (1:2,000; Sigma-Aldrich, A5441) diluted in blocking buffer was added and incubated overnight at 4°C. Membranes were washed with 1× PBST, incubated for 1 hour with HRP-conjugated goat anti-mouse or anti-rabbit antibody (Jackson ImmunoResearch, 115-035-146 and 111-035-144) diluted 1:10,000 in blocking buffer, and then washed again. Chemiluminescence detection was performed using ECL-Plus substrate (Amersham Biosciences) followed by either film exposure or imaging on a Bio-Rad GelDoc (used for densitometry analyses).
QPCR.
Total RNA was isolated from N2a cells or 50–100 mg of human cardiac tissues using TRIzol reagent, and 250–500 ng was subjected to DNase treatment and first-strand cDNA synthesis using standard methods. QPCR analyses for human SCN5A and normalizer gene (β-actin or GAPDH) transcripts were performed using custom-designed SYBR Green assays, done in triplicate and detected on a Viia 7 QPCR machine (Life Technologies). Primers pairs were as follows: human SCN5A (forward: 5′-GCCATCTTCACAGGCGAGTGTATTG-3′, reverse: 5′-GGGGAGAAGAAGTACTTCTGGATGATG-3′), mouse β-actin (forward: 5′-CTGAACCCTAAGGCCAACCGTG-3′, reverse: 5′-GTGGTACGACCAGAGGCATACAG-3′), and human GAPDH (forward: 5′-GAAGGTGAAGGTCGGAGTC-3′, reverse: 5′-GCAACAATATCCACTTTACCAGAG-3′). Small RNA QPCR analyses were performed using commercially available stem-loop reverse trancription primers and accompanying TaqMan assays for miR-24, U6, and RNU48 (Life Technologies). Standard curve and melt curves were evaluated for quality control as needed, and relative mRNA expression was determined by the ddCt method.
NRCM preparation and transfection.
NRCMs were isolated from 1- to 2-day-old Sprague-Dawley rats (Harlan Laboratories). The hearts were surgically extracted and the ventricles digested overnight using a standardized trypsin and collagenase-based protocol (Worthington Biochemical). The isolated cells were plated in a mixture of Dulbecco’s modified Eagle’s medium (DMEM) with F12 supplemented with HEPES, glutamine, insulin-transferrin-selenium (ITS), and bromodeoxyuridine (to inhibit fibroblast replication), at a field density of 10,000 cells/cm2 on glass coverslips coated with laminin, and stored at 37°C in a 5% CO2 incubator. Culture reagents were purchased from Gibco-BRL. After 2 hours, cells were cotransfected with synthetic pre-miRs (25 nM) along with miR or siRNA expression plasmids coexpressing EGFP (100 ng), using Lipofectamine 2000 (Life Technologies). For anti-miR studies, LNA-based or 2′-O-methyl–based miR inhibitors (Exiqon and Integrated DNA Technologies) were transfected into NRCMs at 10–50 nM along with 100 ng of an EGFP expression plasmid. Media were then changed 3 hours later to cardiomyocyte medium with gentamycin (50 μg/ml, Life Technologies), and 5% stripped horse serum.
Whole-cell patch clamp.
After 2–4 days of transfection, a conventional whole-cell patch-clamp technique was used to record Na+ currents from EGFP-positive NRCMs. Patch pipettes with a resistance of 1.5 ± 0.2 MΩ when filled with pipette solution contained (in mmol/l) 10 NaF, 110 CsF, 20 CsCl, 10 EGTA, 10 HEPES (titrated to pH 7.35 with CsOH) and the bath solution contained (in mmol/l) 145 NaCl, 4.5 KCl, 1 MgCl2, 1.5 CaCl2, 10 HEPES (pH 7.35 with CsOH). In all recordings, the potentials were corrected for the liquid junction and 80% of the series resistance was compensated, yielding a maximum voltage error of ±1 mV. Low-pass filtered signals (5 kHz) from a patch clamp amplifier (Axopatch 200B, Molecular Devices) were digitized in an AD/DA converter (Digidata 1200, Molecular Devices) at 20 kHz and stored in a PC for later analysis. Na+ channel data were analyzed with Origin software (Microcal). Cell capacitance was recorded directly from the patch amplifier after nullifying the transients following patch rupture. Data were accepted only when access resistance in voltage-clamp mode was below 7 MΩ. To minimize time-dependent drift in gating parameters, all protocols were initiated 5 minutes after whole-cell configuration was obtained. A 200-ms prepulse to –120 mV was used to eliminate inactivation, and 200-ms test pulses between –80 and +30 mV were used to activate the channel. All electrophysiological experiments were performed at room temperature (23°C–25°C).
GRADE and GRAHF samples and genotyping.
The GRADE and GRAHF patient cohorts have been previously reported (36, 37). In brief, subjects included in this study are from the National Heart, Lung, and Blood Institute–sponsored (NHLBI-sponsored), prospective, observational, multicenter GRADE study, designed to identify genetic modifiers of arrhythmic risk and heart failure outcomes. Inclusion criteria were patients who were 18 years of age or older with a diagnosis of at least moderate-to-severe systolic LV dysfunction (EF ≤ 30%) and who had an ICD. Subjects were excluded if they had intractable class IV heart failure and conditions (other than heart failure) that were expected to limit survival to less than 6 months. The primary endpoint in this study was time to first appropriate ICD shock, and secondary endpoints included all-cause death. The GRAHF cohort derives from a subgroup of the A-HeFT study; inclusion criteria include self-designation as African Americans, heart failure due to systolic dysfunction, and standard background therapy for heart failure with neurohormonal blockade, including angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists, and β-blockers. Based on available DNA and successful genotyping, a total of 1,658 GRADE patients and 270 GRAHF patients were considered for the current analyses. Genotyping was performed using a commercially available TaqMan SNP assay (rs1805126) and TaqMan Genotyping Universal Master Mix (Life Technologies). Detection was done on a Viia 7 QPCR machine and genotyping calls were obtained using the analysis software’s default settings.
LD analysis.
The rs1805126 SNP id was input into the HaploReg v4.1 web-based tool (Broad Institute), and the options were adjusted to set the linkage disequilibrium (LD) threshold to 0.4. The analysis was run using the 1000G Phase 1 population data for EUR (European) and AFR (African) populations, which are most relevant to the GRADE cohort. Data are provided in Supplemental Table 2.
Microarray-based SCN5A eQTL analyses.
Previously published human cardiac transcriptomic data were obtained from the NCBI Gene Expression Omnibus (GEO) database (University of Pennsylvania, GSE57338 and The Netherlands, GSE55231), and accompanying sample genome-wide genotyping data were either downloaded (GSE55230) or made available through collaboration with Kenneth B. Margulies. For the latter, DNA samples were genotyped using Affymetrix Genome-Wide SNP Array 6.0, and quality control (QC) filters were applied to exclude unreliable samples, samples with cryptic relatedness, and samples that were not genetically inferred Caucasian; in addition, SNPs were eliminated if there was significant departure from Hardy-Weinberg equilibrium (P < 10–6). Subject SNP genotypes for rs1805126 and normalized SCN5A mRNA probe intensities were retrieved from the data for subsequent genotype-based expression quantitative trait loci (eQTL) analyses.
Allele-specific expression analyses.
Total RNA was extracted from human cardiac tissue samples (n = 60) using the miRNeasy Kit (Qiagen) including DNAse treatment. For RNA-seq, library prep was conducted using Illumina truSeq stranded mRNA kit followed by the Nugen Ovation amplification kit. Resultant fastq files were assessed for quality control using the FastQC program. Fastq files were aligned against the human reference genome (hg19/hGRC37) using the GSNAP aligner in SNP tolerant mode and converted to BAM files using SAMtools (56). Duplicate reads in the BAM files were flagged using the MarkDuplicates program from Picard tools. The R package asSEQ and custom R scripts were used to generate per-allele specific counts of SCN5A from RNA-seq and genotype data (57). Briefly, per-sample haplotype information was estimated from Affymetrix SNP genotyping data using the software package SHAPEIT with the 1000 Genomes reference panel (58). Then for each sample a list of heterozygous SNPs was used to create a new BAM file for each haplotype per sample. Reads mapping to the SCN5A exons and UTRs were counted from each haplotype-specific BAM file using the htseq-count application. Allele-specific expression was plotted as C allele counts/T allele counts (Figure 4D), and 1-sample 2-sided t test was performed on the allele-specific expression values for C allele counts/sum (T and C allele counts) against the hypothetical mean value of 0.5.
DHE oxidation levels in wild-type and Scn5a+/– mouse heart tissue sections.
Scn5a+/– mice (backcrossed more than 10 times to the C56BL6/J strain) were a gift from Dan Roden (Vanderbilt University, Nashville, Tennessee, USA). Mice were housed in a controlled temperature environment on a 12-hour light/dark cycle, and food and water were provided ad libitum. Fresh frozen hearts were collected from 7- to 8-month-old male Scn5a+/– and wild-type littermate mice (n = 3 per group) and then cut into 10-μm sections for DHE analysis. Briefly, the tissue sections were labeled with 10 μM DHE in Dulbecco’s PBS containing 5 mM sodium pyruvate for approximately 20–25 minutes at 37°C, after which they were imaged by confocal microscopy. The red fluorescence intensity of each section was quantified using ImageJ software (NIH) (30 cells per mouse, i.e., 90 cells per group) and all groups were normalized to the wild-type control group to obtain the normalized mean fluorescence intensity (NMFI). Antimycin A (a mitochondrial electron transport chain blocker that is known to increase DHE fluorescence) was used as a positive control at 10 μM.
Statistics.
Statistical analyses were performed using available software and tools (e.g., GraphPad Prism and R package, with collaborative assistance from the University of Iowa Department of Biostatistics [P. Breheny]). GraphPad was used to generate Kaplan-Meier curves based on genotype for appropriate shocks and death outcomes, and log-rank tests were performed to obtain P values. For other demographic and clinical characteristics (Table 1 and Supplemental Table 1), statistical differences across genotypes were tested by Fisher’s exact test (for categorical factors), and by ANOVA (for continuous measures). Cox proportional hazards models were used to estimate HRs for comparing rs1805126 genotypes, both unadjusted and adjusted for the potentially confounding effects of relevant clinical variables. The other specific statistical tests are indicated throughout the manuscript, and for each, all data met the test assumptions and groups showed similar variance. Results were considered to be significant if P values were less than or equal to 0.05. Data presented as box-and-whisker plots show Tukey whiskers. All data points were included in statistical analyses with the exception of one overt outlier for SCN5A expression in the Netherlands cohort (CC sample with probe intensity = 6.48; Figure 4C and Supplemental Figure 7), as detected by ROUT (definitive outlier stringency, Q = 0.1%). Power analyses considered event rates for death and shock and SNP allele frequencies; based on the minor-allele frequency of rs1805126, the study was powered to greater than 90% to detect an HR of 1.5.
Study approval.
All animal studies were approved by the Institutional Animal Care and Use Committees (IACUC) at the University of Iowa. Deidentified human samples and accompanying clinical and genetic data were used for this study. Sample and data collection were previously approved by Institutional Review Boards at GRADE and A-HeFT (GRAHF) study sites and at the University of Pennsylvania. All human subjects provided written informed consent.
Author contributions
RLB and BL designed and conceived the project, supervised the research, and interpreted the data. XZ, JYY, JMM, and KAM carried out experimental work, analyzed data, and participated in data interpretation. RLB, MM, and PB carried out computational and statistical analyses. RG, HLB, SCD, PTE, AAS, RW, MS, HM, and BL played key roles in overseeing GRADE patient data acquisition, organization, and interpretation. ALT, CWY, AMF, and DMM played key roles in overseeing GRAHF patient data acquisition, organization, and interpretation. KBM, WHWT, and CSM provided critical human cardiac tissue samples and accompanying clinical and genotyping data. KI and DRS guided experimental design and helped with data interpretation. RLB, BL, and XZ wrote the manuscript.
Supplementary Material
Acknowledgments
This work was supported by the American Heart Association (14SDG18590008 to R.L. Boudreau), Roy J. Carver Trust (University of Iowa to R.L. Boudreau), NIH NCI (CA182804 to D.R. Spitz), NIH NHLBI (HL77398 to B. London, HL115955 to K. Irani and B. London, HL105993 to K.B. Margulies and W.H.W. Tang, and HL007121 to J.M. McLendon). We also acknowledge members of the Boudreau and London labs for scientific discussion and manuscript feedback.
Version 1. 02/19/2018
Electronic publication
Version 2. 02/20/2018
Correction made in figure 1.
Version 3. 03/01/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(3):1154–1163.https://doi.org/10.1172/JCI95710.
See the related Commentary at SCN5A: the greatest HITS collection.
Contributor Information
Xiaoming Zhang, Email: xiaoming-zhang@uiowa.edu.
Jin-Young Yoon, Email: jinyoung-yoon@uiowa.edu.
Michael Morley, Email: mmorley@mail.med.upenn.edu.
Rebecca Gutmann, Email: rebecca-gutmann@uiowa.edu.
Haider Mehdi, Email: haider-mehdi@uiowa.edu.
Samuel C. Dudley, Email: sdudley@umn.edu.
Patrick T. Ellinor, Email: ellinor@mgh.harvard.edu.
Raul Weiss, Email: raul.weiss@osumc.edu.
W.H. Wilson Tang, Email: tangw@ccf.org.
Madhurmeet Singh, Email: madhurmeetsingh@gmail.com.
Anne L. Taylor, Email: alt2127@cumc.columbia.edu.
Arthur M. Feldman, Email: Arthur.Feldman@tuhs.temple.edu.
Kaikobad Irani, Email: kaikobad-irani@uiowa.edu.
Douglas R. Spitz, Email: douglas-spitz@uiowa.edu.
Patrick Breheny, Email: patrick-breheny@uiowa.edu.
Barry London, Email: barry-london@uiowa.edu.
Ryan L. Boudreau, Email: ryan-boudreau@uiowa.edu.
References
- 1.Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87(2):425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- 2.Laitinen-Forsblom PJ, et al. SCN5A mutation associated with cardiac conduction defect and atrial arrhythmias. J Cardiovasc Electrophysiol. 2006;17(5):480–485. doi: 10.1111/j.1540-8167.2006.00411.x. [DOI] [PubMed] [Google Scholar]
- 3.Chen Q, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392(6673):293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 4.Bennett PB, Yazawa K, Makita N, George AL. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376(6542):683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 5.Bezzina C, et al. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85(12):1206–1213. doi: 10.1161/01.RES.85.12.1206. [DOI] [PubMed] [Google Scholar]
- 6.London B, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116(20):2260–2268. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watanabe H, et al. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. 2008;118(6):2260–2268. doi: 10.1172/JCI33891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makita N, et al. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J Clin Invest. 2008;118(6):2219–2229. doi: 10.1172/JCI34057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olson TM, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293(4):447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beckermann TM, McLeod K, Murday V, Potet F, George AL. Novel SCN5A mutation in amiodarone-responsive multifocal ventricular ectopy-associated cardiomyopathy. Heart Rhythm. 2014;11(8):1446–1453. doi: 10.1016/j.hrthm.2014.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfeufer A, et al. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet. 2009;41(4):407–414. doi: 10.1038/ng.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holm H, et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42(2):117–122. doi: 10.1038/ng.511. [DOI] [PubMed] [Google Scholar]
- 13.Magnani JW, et al. Sequencing of SCN5A identifies rare and common variants associated with cardiac conduction: Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium. Circ Cardiovasc Genet. 2014;7(3):365–373. doi: 10.1161/CIRCGENETICS.113.000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeff JM, et al. SCN5A variation is associated with electrocardiographic traits in the Jackson Heart Study. Circ Cardiovasc Genet. 2011;4(2):139–144. doi: 10.1161/CIRCGENETICS.110.958124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ritchie MD, et al. Genome- and phenome-wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation. 2013;127(13):1377–1385. doi: 10.1161/CIRCULATIONAHA.112.000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaunt TR, et al. Integration of genetics into a systems model of electrocardiographic traits using HumanCVD BeadChip. Circ Cardiovasc Genet. 2012;5(6):630–638. doi: 10.1161/CIRCGENETICS.112.962852. [DOI] [PubMed] [Google Scholar]
- 17.Bezzina CR, et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45(9):1044–1049. doi: 10.1038/ng.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abriel H. Cardiac sodium channel Nav1.5 and its associated proteins. Arch Mal Coeur Vaiss. 2007;100(9):787–793. [PubMed] [Google Scholar]
- 19.Mohler PJ, et al. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004;101(50):17533–17538. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atack TC, et al. Informatic and functional approaches to identifying a regulatory region for the cardiac sodium channel. Circ Res. 2011;109(1):38–46. doi: 10.1161/CIRCRESAHA.110.235630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao G, Dudley SC. SCN5A splicing variants and the possibility of predicting heart failure-associated arrhythmia. Expert Rev Cardiovasc Ther. 2013;11(2):117–119. doi: 10.1586/erc.12.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daimi H, et al. Regulation of SCN5A by microRNAs: miR-219 modulates SCN5A transcript expression and the effects of flecainide intoxication in mice. Heart Rhythm. 2015;12(6):1333–1342. doi: 10.1016/j.hrthm.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Y, et al. Post-transcriptional regulation of cardiac sodium channel gene SCN5A expression and function by miR-192-5p. Biochim Biophys Acta. 2015;1852(10 Pt A):2024–2034. doi: 10.1016/j.bbadis.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nassal DM, et al. KChIP2 is a core transcriptional regulator of cardiac excitability. Elife. 2017;6:e17304. doi: 10.7554/eLife.17304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boettger T, Braun T. A new level of complexity: the role of microRNAs in cardiovascular development. Circ Res. 2012;110(7):1000–1013. doi: 10.1161/CIRCRESAHA.111.247742. [DOI] [PubMed] [Google Scholar]
- 26.Quiat D, Olson EN. MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment. J Clin Invest. 2013;123(1):11–18. doi: 10.1172/JCI62876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olson EN. MicroRNAs as therapeutic targets and biomarkers of cardiovascular disease. Sci Transl Med. 2014;6(239):239ps3. doi: 10.1126/scitranslmed.3009008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bazzini AA, Lee MT, Giraldez AJ. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science. 2012;336(6078):233–237. doi: 10.1126/science.1215704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Djuranovic S, Nahvi A, Green R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science. 2012;336(6078):237–240. doi: 10.1126/science.1215691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eichhorn SW, et al. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol Cell. 2014;56(1):104–115. doi: 10.1016/j.molcel.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 32.Spengler RM, et al. Elucidation of transcriptome-wide microRNA binding sites in human cardiac tissues by Ago2 HITS-CLIP. Nucleic Acids Res. 2016;44(15):7120–7131. doi: 10.1093/nar/gkw640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith JG, et al. Genome-wide association study of electrocardiographic conduction measures in an isolated founder population: Kosrae. Heart Rhythm. 2009;6(5):634–641. doi: 10.1016/j.hrthm.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39(10):1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 35.Eulalio A, et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492(7429):376–381. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 36.AlJaroudi WA, et al. Effect of angiotensin-converting enzyme inhibitors and receptor blockers on appropriate implantable cardiac defibrillator shock in patients with severe systolic heart failure (from the GRADE Multicenter Study) Am J Cardiol. 2015;115(7):924–931. doi: 10.1016/j.amjcard.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McNamara DM, et al. Aldosterone synthase promoter polymorphism predicts outcome in African Americans with heart failure: results from the A-HeFT Trial. J Am Coll Cardiol. 2006;48(6):1277–1282. doi: 10.1016/j.jacc.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics. 2015;105(2):83–89. doi: 10.1016/j.ygeno.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koopmann TT, et al. Genome-wide identification of expression quantitative trait loci (eQTLs) in human heart. PLoS One. 2014;9(5):e97380. doi: 10.1371/journal.pone.0097380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Rooij E, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103(48):18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li RC, et al. In vivo suppression of microRNA-24 prevents the transition toward decompensated hypertrophy in aortic-constricted mice. Circ Res. 2013;112(4):601–605. doi: 10.1161/CIRCRESAHA.112.300806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papadatos GA, et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci U S A. 2002;99(9):6210–6215. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Veen TA, et al. Impaired impulse propagation in Scn5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation. 2005;112(13):1927–1935. doi: 10.1161/CIRCULATIONAHA.105.539072. [DOI] [PubMed] [Google Scholar]
- 44.Liu M, Liu H, Dudley SC. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res. 2010;107(8):967–974. doi: 10.1161/CIRCRESAHA.110.220673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan E, et al. Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J Clin Invest. 2016;126(1):112–122. doi: 10.1172/JCI84669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brest P, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43(3):242–245. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 47.Bhattacharya A, Ziebarth JD, Cui Y. PolymiRTS Database 3.0: linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014;42(Database issue):D86–D91. doi: 10.1093/nar/gkt1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bruno AE, Li L, Kalabus JL, Pan Y, Yu A, Hu Z. miRdSNP: a database of disease-associated SNPs and microRNA target sites on 3′UTRs of human genes. BMC Genomics. 2012;13:44. doi: 10.1186/1471-2164-13-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van den Boogaard M, et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J Clin Invest. 2014;124(4):1844–1852. doi: 10.1172/JCI73140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Viswanathan PC, Benson DW, Balser JR. A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. J Clin Invest. 2003;111(3):341–346. doi: 10.1172/JCI16879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koltai T. Cancer: fundamentals behind pH targeting and the double-edged approach. Onco Targets Ther. 2016;9:6343–6360. doi: 10.2147/OTT.S115438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carrithers MD, et al. Expression of the voltage-gated sodium channel NaV1.5 in the macrophage late endosome regulates endosomal acidification. J Immunol. 2007;178(12):7822–7832. doi: 10.4049/jimmunol.178.12.7822. [DOI] [PubMed] [Google Scholar]
- 53.Gavillet B, et al. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006;99(4):407–414. doi: 10.1161/01.RES.0000237466.13252.5e. [DOI] [PubMed] [Google Scholar]
- 54.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107(35):15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williams IA, Allen DG. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am J Physiol Heart Circ Physiol. 2007;293(3):H1969–H1977. doi: 10.1152/ajpheart.00489.2007. [DOI] [PubMed] [Google Scholar]
- 56.Wu TD, Reeder J, Lawrence M, Becker G, Brauer MJ. GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. Methods Mol Biol. 2016;1418:283–334. doi: 10.1007/978-1-4939-3578-9_15. [DOI] [PubMed] [Google Scholar]
- 57.Romanel A, Lago S, Prandi D, Sboner A, Demichelis F. ASEQ: fast allele-specific studies from next-generation sequencing data. BMC Med Genomics. 2015;8:9. doi: 10.1186/s12920-015-0084-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2011;9(2):179–181. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.