

Abstract
Congenital long QT syndrome (LQTS) is an inherited channelopathy associated with life-threatening arrhythmias. LQTS type 2 (LQT2) is caused by mutations in KCNH2, which encodes the potassium channel hERG. We hypothesized that modifier genes are partly responsible for the variable phenotype severity observed in some LQT2 families. Here, we identified contributors to variable expressivity in an LQT2 family by using induced pluripotent stem cell–derived cardiomyocytes (iPSC-CMs) and whole exome sequencing in a synergistic manner. We found that iPSC-CMs recapitulated the clinical genotype-phenotype discordance in vitro. Importantly, iPSC-CMs derived from the severely affected LQT2 patients displayed prolonged action potentials compared with cells from mildly affected first-degree relatives. The iPSC-CMs derived from all patients with hERG R752W mutation displayed lower IKr amplitude. Interestingly, iPSC-CMs from severely affected mutation-positive individuals exhibited greater L-type Ca2+ current. Whole exome sequencing identified variants of KCNK17 and the GTP-binding protein REM2, providing biologically plausible explanations for this variable expressivity. Genome editing to correct a REM2 variant reversed the enhanced L-type Ca2+ current and prolonged action potential observed in iPSC-CMs from severely affected individuals. Thus, our findings showcase the power of combining complementary physiological and genomic analyses to identify genetic modifiers and potential therapeutic targets of a monogenic disorder. Furthermore, we propose that this strategy can be deployed to unravel myriad confounding pathologies displaying variable expressivity.
Keywords: Cardiology, Genetics
Keywords: Arrhythmias, Ion channels, iPS cells
Introduction
Long QT syndrome (LQTS) is an inherited arrhythmogenic disorder attributable to mutations in genes contributing to the major cardiac ionic currents, with an estimated prevalence in industrialized nations of 1 in 2,500 live births (1–3). The cardinal feature of the disease is a prolonged QT interval on surface electrocardiograms that signals elevated risk for a life-threatening cardiac arrhythmia with clinical manifestations including palpitations, syncope, and an increased predisposition for sudden cardiac death (4). Specifically, mutations in KCNH2, which encodes the rapid component (IKr) of the delayed rectifier potassium channel (hERG), cause long QT syndrome type 2 (LQT2), which accounts for about 30% of all LQTS diagnoses (5).
As is commonly observed in many autosomal dominant cardiac channelopathies, the pattern of inheritance and clinical phenotypes of LQTS patients are complex and often display incomplete penetrance and/or variable expressivity; some individuals positive for disease-causing mutations are asymptomatic, and there are varying degrees of phenotypic severity (6). Specifically, at one end of the spectrum, mutation-positive individuals within the same family manifest QT prolongation, repeated syncopal attacks, and sudden cardiac death, while at the other end, mutation-positive individuals exhibit a relatively normal QT interval with no symptoms (7). The causes of this variable clinical expressivity are not well understood. We investigated this question by testing the hypothesis that modifier genes contribute to the variable clinical expressivity.
The original seminal description of generation of induced pluripotent stem cells (iPSCs) from somatic cells by Yamanaka and colleagues (8) ushered in a new era of in vitro disease modeling in myriad fields, including cardiac channelopathies. This discovery has led to the generation of patient-specific iPSC-derived cardiomyocytes (iPSC-CMs), which revolutionized cardiac arrhythmia disease modeling (9, 10). While heterologous expression systems and animal models paved the way for the fundamental understanding of LQTS pathogenesis, they could not address the variable expressivity seen in LQTS. However, iPSC-CM–based disease modeling platforms are poised to generate previously indeterminable mechanistic insights by providing an ultra-versatile in vitro platform that most closely mimics the native cardiomyocyte setting of the patients.
Furthermore, the impact of modeling cardiac disease in iPSC lines can be synergistically enhanced with the recent advent of next-generation sequencing technologies such as whole exome sequencing. While the protein coding regions of the genome constitute approximately 1% of the human genome (50 million bp), they can account for up to 85% of mutations reported in Mendelian disorders such as LQTS (11). Historically, Mendelian diseases such as LQTS are notoriously challenging to diagnose, risk-stratify, and treat because of their phenotypic heterogeneity and incomplete penetrance. However, the advancement of whole exome sequencing has exponentially accelerated the ability to screen for novel disease-modifying genes with base pair resolution (11–13).
For 20 years, an LQT2 family has been studied extensively by our multidisciplinary group (14, 15). Initial genotype testing of living family members following the sudden death of a 31-year-old female with a history of syncope along with the subsequent LQT2 diagnosis in a paternal aunt revealed a C→T nucleotide substitution at position 2,254 in exon 9 of KCNH2 resulting in the hERG R752W missense mutation. Further genotyping and ECG testing of 101 family members identified 26 additional hERG R752W mutation–positive individuals, of whom only 6 have a severely affected phenotype (Supplemental Tables 1 and 2; supplemental material available online with this article; https://doi.org/10.1172/JCI94996DS1). Previous studies showed that diagnosing congenital LQTS is difficult because of variable expressivity and genetic heterogeneity. However, it was posited that a combination of clinical and ECG techniques could identify gene-positive individuals within a single family with congenital LQTS. Yet, a study performed on this large Cleveland LQT2 family carrying the hERG R752W mutation demonstrated that even in this homogeneous LQTS population, the phenotype was so variable that detailed clinical and ECG analyses did not permit an accurate diagnosis of gene carrier status. In fact, there was substantial overlap (78%) of QTc among subjects with and without the mutation, suggesting the presence of 1 or more modifier genes (14). We deployed a novel strategy combining the use of patient-derived iPSC-CMs alongside exome sequencing and genome editing to unravel novel electrophysiological arrhythmogenic mechanisms and identify new disease-modifying gene variants that suggest a plausible explanation for the variable expressivity seen in this LQT2 family. Moreover, this work showcases the innovative and high-yield approach of using 2 nascent technologies in an additive manner to unravel the underlying mechanism of familial cardiac channelopathy.
Results
Patient-specific iPSC-CMs recapitulate genotype-phenotype discordance.
Two pairs of hERG R752W mutation–positive first-degree relatives (a father/son pair [III-3 and IV-15, respectively] and a sister/sister pair [IV-3 and IV-4]) with discordant phenotypes along with a healthy mutation-negative control subject (IV-17) were selected from this large LQT2 family (Figure 1). iPSC-CMs were generated from these 5 individuals. We recorded action potential duration (APD90) from the patient-specific iPSC-CMs to determine whether these cellular models could recapitulate the genotype-phenotype discordance observed in an LQT2 family. Cells derived from the severely-affected-phenotype father (III-3) exhibited significantly longer APD in comparison with a mutation-negative first-degree-relative control (IV-17). By contrast, cells derived from his hERG R752W mutation–positive son (IV-15) with a mildly affected phenotype did not exhibit a prolonged APD90 despite harboring the same pathogenic hERG mutation (Figure 2A and Supplemental Table 3). Similarly, APD90 recorded from the severely affected sister (IV-3) was significantly longer than that from her mildly affected sister (IV-4) and the mutation-negative-relative control, IV-17 (Figure 2B and Supplemental Table 3). Notably, APD50 was also significantly prolonged for the 2 severely affected individuals (III-3, IV-3) compared with their respective mildly affected first-degree relatives (Figure 2, A and B, and Supplemental Table 3).
Figure 1. Clinical details of carrier pairs in Cleveland LQT2 family.

Zoomed-in snapshot of the family pedigree that focuses on 5 individuals of the family we used to generate the carrier pairs along with their relevant patient history (see Methods for in-depth explanation of patient selection and phenotype binning criteria and Supplemental Table 2 for clinical details on all 26 R752W mutation–positive individuals). Individuals are referenced first by the generation number and then by the family member number, read from left to right (e.g., IV-3 is generation 4, family member 3). This reference system is used throughout the paper. The black square is the hERG mutation negative healthy male control. Blue circles and squares are females and males, respectively, who are mildly-affected-phenotype hERG R752W mutation–positive relatives. Red circles and squares are severely-affected-phenotype hERG R752W mutation–positive relatives. Hatched circles and squares are females and males, respectively, who are hERG R752W-carrying individuals who are not fully characterized in this study.
Figure 2. LQT2 genotype-phenotype discordance reproduced in patient-specific iPSC-CMs.

(A) Representative action potential traces from the control (IV-7), a severely-affected-phenotype LQT2 male (III-3), and his son, a hERG R752W mutant–positive, mildly-affected-phenotype male (IV-15). Summary APD90 (90% of repolarization) and APD50 (50% of repolarization) data are also shown (below traces). (B) Representative action potential traces from the control (IV-17) and the second pair (sister pair), a severely affected LQT2 female (IV-3) and a hERG R752W mutant–positive, mildly affected female (IV-4). Summary APD90 and APD50 data are shown (below traces). (C) Representative IKr traces and respective summary IKr tail current density from each patient-derived iPSC-CM depicted in A. (D) Representative IKr traces and respective summary IKr tail current density for each patient-derived iPSC-CM depicted in B. (E) A 1-Hz paced action potential train from IV-17 and IV-3 iPSC-CMs. Stars denote early afterdepolarizations. Dashed line in APD traces denotes 0 mV. Between 30 and 203 cells from 9 different iPSC clones (3 each from IV-17, III-3, IV-15 paired trio) were analyzed in A and C. Between 77 and 134 cells from 9 different iPSC clones (3 each from IV-17, IV-3, IV-4 paired trio) were analyzed in B and D. Exact numbers of replicate measures (n) for each are listed in Supplemental Table 3. Results are shown as mean ± SEM. *Statistical significance (P < 0.05) as determined by ANOVA in the summary data for A–D.
We recorded IKr and quantified the tail current density. As expected, iPSC-CMs from mutation-positive severely affected individuals (III-3 and IV-3) had significantly smaller IKr density compared with those from mutation-negative IV-17 (Figure 2, C and D). Interestingly, mutation-positive but mildly affected individuals (IV-15 and IV-4) also had significantly smaller IKr tail current density compared with IV-17 despite not having prolonged APD90. Finally, the recording of paced (1 Hz) action potential trains from III-3 and IV-3 elicited early afterdepolarizations that were never observed in cells from mildly affected (IV-15, IV-4) or mutation-negative (IV-17) family members (Figure 2E). This arrhythmogenic substrate is consistent with an LQT2 phenotype caused by a diminished repolarization capacity due to reduced hERG current density (16, 17). Collectively, these data demonstrated the capacity of patient-specific iPSC-CMs to recapitulate the clinical genotype-phenotype discordance in vitro.
Because the IKr density is similar among all hERG mutation carriers (Figure 2, C and D), and the APDs between the control and the mildly affected individual are also similar, this suggests the presence of compensatory repolarizing currents in the mildly affected individuals. To assess this, we measured action potential variability in the presence of a hERG channel blocker, E-4031. Application of E-4031 to all 5 patient iPSC-CM cell lines at 10 and 100 nM prolonged APD90 as expected (Figure 3). Notably, however, E-4031 at 100 nM significantly exacerbated the existing IKr deficiency in the severely affected iPSC-CM lines (III-3 APD90 at 100 nM [mean ± SEM], 587 ± 79 ms, vs. no treatment, 368 ± 15 ms; and IV-3 APD90 at 100 nM, 617 ± 50 ms, vs. no treatment, 413 ± 32 ms). Comparatively, the mildly affected patient iPSC-CMs did not show the same drastic prolongation at 100 nM (IV-15 at 100 nM, 291 ± 18 ms, vs. no treatment, 208 ± 14 ms; and IV-4 at 100 nM, 275 ± 20 ms, vs. no treatment, 186 ± 13 ms), even though the IKr density was similar between the severely and mildly affected patient iPSC-CMs (Figure 2, C and D). This suggests the presence of a compensatory repolarizing current in the mildly affected individuals.
Figure 3. Effects of IKr blockade on severely and mildly affected patient iPSC-CMs.

(A and B) The effects of E-4031 (hERG channel blocker) at 2 different concentrations on the IV-17, III-3, and IV-15 trio as well as the IV-17, IV-3, and IV-4 trio. Between 8 and 32 cells were analyzed per E-4031 treatment condition for APD in A and B. NT, no treatment. *Statistical significance (P < 0.05) as determined by ANOVA in A and B.
Patient-specific iPSC-CMs reveal divergent calcium channel activity.
Initial electrophysiological analysis of action potential characteristics also revealed a significant prolongation in the APD50 in iPSC-CMs derived from severely affected LQT2 patients compared with their respective mildly affected first-degree relatives and the mutation-negative-relative control IV-17 (Figure 2, A and B, and Supplemental Table 3). A prolongation in APD50 is usually a reflection of enhanced calcium current. While there were no known clinical indications of calcium dysfunction in the LQT2 patients, we further investigated this observation by recording L-type Ca2+ current (ICaL) from the patient-specific iPSC-CMs. We observed in cells from the severely affected LQT2 patient iPSC-CMs (III-3 and IV-3) a significantly greater ICaL in comparison with IV-17 and the mildly affected relatives (IV-4 and IV-15, respectively; Figure 4, A–C).
Figure 4. L-type calcium current density increase revealed by patient-specific iPSC-CMs.

(A) Representative macroscopic whole-cell L-type Ca2+ (ICaL) traces from the IV-17, III-3, and IV-15 paired trio. (B) ICaL from the IV-17, IV-3, and IV-4 paired trio. (C) Summary data from A and B. Between 24 and 95 cells from 9 different iPSC clones (3 each from IV-17, III-3, IV-15 paired trio) were analyzed in A. Between 20 and 47 cells from 9 different iPSC clones (3 each from IV-17, IV-3, IV-4 paired trio) were analyzed in B. *Statistical significance (P < 0.05) as determined by ANOVA in C.
Pharmacological blockade supports Ca2+-mediated LQT2 phenotype.
We deployed a pharmacological strategy to further elucidate the contribution of enhanced ICaL activity in the severely affected patient iPSC-CM lines. The administration of nisoldipine (a Cav1.2 blocker) at the same concentration across all cell lines revealed a differential response to Cav1.2 blockade (Figure 5 and Supplemental Figure 1). In the severely affected patients, 0.05 μM nisoldipine shortened APD90 close to that of the family control from the same differentiation (Supplemental Table 4). A similar trend was observed in parallel with the effects of 0.05 μM nisoldipine on L-type calcium current in the severely affected patients where a larger reduction in current was observed, bringing the ICaL current density back to the control (Figure 5 and Supplemental Table 4). These results suggest that enhanced ICaL activity contributes to the action potential prolongation observed in the severely affected patients.
Figure 5. Nisoldipine reveals increased ICaL sensitivity in severely affected iPSC-CMs.

(A–E) The effect of 0.05 μM nisoldipine on both trios. Data are depicted as macroscopic action potential record, APD summary data, and ICaL comparison between no drug and nisoldipine. Between 8 and 11 cells were analyzed for the effects of nisoldipine on APD and 5–8 cells for the effects of nisoldipine on ICaL in A–E. See Supplemental Table 4 for exact numbers of replicate measures (n) and statistical analysis (paired Student’s t test). Dose-response relationship for nisoldipine (Supplemental Figure 1) was determined before selection of the concentration used in these experiments (a concentration lower than EC50 was chosen because the objective was to shorten the APD and ICaL to levels more like those of control iPSC-CMs). *P < 0.05; as determined by paired 2-tailed Student’s t test.
Identification of 2 novel disease-modifying gene variants by whole exome sequencing and functional electrophysiological interrogation.
Since the original report of variable expressivity (14, 15) in this LQT2 family (Figure 1 and Supplemental Tables 1 and 2), we hypothesized that the presence of disease-modifying gene variants might explain the genotype-phenotype discordance. Specifically, we postulated that mildly-affected-phenotype mutation-positive family members (IV-15 and IV-4) were protected from the disease phenotype by a compensatory gene. Conversely, we hypothesized the existence of a gene variant that exacerbated the primary hERG mutation in severely-affected-phenotype individuals (III-3 and IV-3). This conjecture was strongly corroborated by the incidental discovery of increased ICaL identified in severely affected LQT2 patient iPSC-CM lines (Figure 4). To investigate these possibilities, we performed exome sequencing on the same closely related severely affected/mildly affected phenotype pairs from which we generated iPSC lines.
We used a sequential prioritization strategy in analyzing exome data to identify potential modifier alleles. This strategy involved dividing the sequenced pairs into severely affected and mildly affected cohorts, filtering for mutually exclusive nonsynonymous and small insertion/deletion coding variants, then selecting variants in cardiac-expressed genes. Special attention was paid to variants in cardiac-expressed ion channel genes. Next, we filtered for genes identified either by genome-wide association studies (GWAS) seeking modulators of QT interval duration (18–21) or by the computational network analysis approach used by Berger and colleagues (22) that grouped genes and nodes into a repository known as the LQTS neighborhood (Supplemental Figure 2). Implementing this approach, we identified 12 SNPs and 4 insertion/deletions that could potentially contribute to genotype-phenotype discordance (Supplemental Table 5) but specifically pursued 2 gene variants: one found in the mildly affected mutation-positive cohort and one found in the severely affected mutation-positive subjects.
One variant, KCNK17-p.Ser21Gly, found in mildly affected mutation-positive individuals (IV-15 and IV-4; Supplemental Table 5) was in a gene (KCNK17) encoding a 2-pore-domain potassium channel (K2p17.1). KCNK17-p.Ser21Gly is a known common variant with a minor allele frequency of 0.41 in the Exome Aggregation Consortium (ExAC) database. This was the only variant identified in an ion channel. Single-cell reverse transcription PCR (RT-PCR) and immunohistochemistry demonstrated KCNK17 expression in patient-derived iPSC-CMs (Figure 6, A and B). Coexpression of KCNK17-Gly21 and KCNK17-Ser21 (mimicking the heterozygous state observed in family members) in Chinese hamster ovary cells revealed significantly greater potassium current compared with expression of the common allele only (Figure 6, C and D). These findings indicate that KCNK17-p.Ser21Gly is a gain-of-function variant.
Figure 6. Two-pore potassium channel KCNK17 variant produces significant increase in K2p current.

(A) Single-cell RT-PCR confirms KCNK17 expression and ventricular lineage in electrophysiology-verified ventricular patient iPSC-CMs. MLC-2v, myosin light chain 2 ventricle (ventricular lineage marker); HCN4, hyperpolarization-activated cyclic nucleotide-gated potassium channel 4 (pacemaker lineage marker). (B) Immunohistochemistry confirms KCNK17 expression in iCell ventricular cardiomyocyte. Blue, nucleus; green, KCNK17. Scale bar: 50 μm. (C) Representative macroscopic current-voltage traces obtained from homozygous WT (KCNK17 S21 only, mimics severely affected patient alleles) and heterozygous state (KCNK17 S21 + G21, mimics mildly affected patient alleles). (D) Mean current density analyzed at 0 mV between WT and heterozygote conditions from 3 independent experiments. Relative current KCNK17 S21: 2.27 ± 0.30 (n = 29); and KCNK17 S21 + G21: 4.28 ± 0.70 (n = 32). *P = 0.01 as determined by unpaired Student’s t test. Results normalized to capacitance and shown as mean ± SEM. (E) Representative macroscopic action potential traces following KCNK17 siRNA silencing (gray) compared with scrambled control (black) in III-3 (P = 0.42, n = 15 in each group) and IV-4 (P = 0.01, n = 21 in control, n = 27 in siRNA) as determined by unpaired Student’s t test. (F) Summary APD90 and APD50 data from III-3 and IV-4 in control versus KCNK17 siRNA groups. *P < 0.02 as determined by unpaired Student’s t test. No significant changes were observed in mean diastolic potential (MDP) or action potential amplitude (APA) between siRNA and control recordings from III-3. MDP was unchanged in IV-4, but APA was higher in the siRNA group (100.26 ± 2.73 mV) compared with control (87.24 ± 2.18 mV) (Supplemental Table 6).
Furthermore, to assess the macroscopic contribution of KCNK17 variants to APD, we transfected either a KCNK17 siRNA or a scrambled control in 1 severely affected LQT2 cell line (III-3) and 1 mildly affected line (IV-4). Supplemental Figure 3 illustrates the silencing efficiency of our siRNA on KCNK17 transcript. Action potential recordings from III-3 (common KCNK17 allele) revealed no difference in APD in siRNA-treated cells compared with scrambled control (Figure 6, E and F, and Supplemental Table 6). However, KCNK17 suppression in IV-4 (heterozygous for KCNK17-p.Ser21Gly) resulted in significant prolongation of APD compared with controls (Figure 6, E and F, and Supplemental Table 6). This suggests that a KCNK17 gain-of-function variant can be LQTS protective by promoting APD shortening.
With regard to the severely affected mutation-positive patients, our fundamental approach was to investigate variants that could potentially explain the unexpected difference in ICaL (Figure 4) observed in iPSC-CMs from the severely affected family members. Specifically, we looked for adaptor proteins or known channel-modulating entities of ICaL. A common variant in REM2 (p.Gly96Ala; Supplemental Table 5) with a minor allele frequency of 0.1 (ExAC) was identified only in our severely affected mutation-positive subjects. REM2 encodes a member of the Ras superfamily, which are well-known modulators of voltage-gated calcium channels. The framework for the identification of this variant was originally suggested by a computational network analysis of novel genes or “nodes” related to known congenital LQTS genes (22). In this previous analysis, a member of the Ras superfamily known as REM was identified as a candidate gene in what was termed the LQTS neighborhood. Implementing the LQTS neighborhood as a filter in our own exome sequencing approach and mining our final data set, we identified REM2, a functionally homologous ICaL modifier to Rem and also a member of the Ras superfamily. Thus this gene became a biologically plausible candidate for explaining enhanced ICaL observed in the severely affected LQT2 patient iPSC-CMs. Functionally vetting this novel gene variant, we first demonstrated expression of REM2 in patient-derived iPSC-CMs by single-cell RT-PCR and immunohistochemistry (Figure 7, A and B). Overexpressing REM2-p.Gly96Ala in iCell cardiomyocytes resulted in a significantly greater level of ICaL activity in comparison with cells expressing the common REM2 allele (Figure 7, C and D). This variant could therefore explain the enhanced ICaL seen in cells derived from severely affected individuals (Figure 4).
Figure 7. REM2 variant found in severely affected individuals drives significant increase in ICaL, but is reverted back to WT levels with CRISPR/Cas9–mediated gene correction.

(A) Single-cell RT-PCR confirmation of REM2 expression and ventricular lineage in electrophysiology-verified ventricular patient iPSC-CMs (IV-3–derived cell shown here). MLC-2v, myosin light chain 2 ventricle (ventricular lineage marker); HCN4, hyperpolarization-activated cyclic nucleotide-gated potassium channel 4 (pacemaker lineage marker). (B) Immunohistochemical confirmation of REM2 expression in an iCell ventricular cardiomyocyte. Blue, nucleus; red, REM2. Scale bar: 50 μm. (C) Macroscopic L-type Ca2+ currents obtained from iCell ventricular cardiomyocytes transfected with either REM2 wild type or REM2 G96A (mimicking the SNP found in severely affected hERG R752W mutation–positive individuals, III-3 and IV-3). (D) Summary data of REM2 WT overexpression (2 μg cDNA) in iCell ventricular cardiomyocytes compared with REM2 G96A. REM2 WT and REM2 G96A are composed of 3 independent experiments (REM2 WT n = 12, REM2 G96A n = 11). Relative current for REM2 WT at 0 mV, –2.21 ± –0.34, compared with REM2 G96A, –3.60 ± –0.39. *P = 0.002 as determined by unpaired Student’s t test. (E) The effects of CRISPR/Cas9–mediated correction of the REM2 G96A mutant allele back to the wild type (from A96 to G96) on APD90 (macroscopic action potential record shown). (F) Summary APD90 and APD50 data from IV-3 compared with CRISPR-corrected IV-3. (G) Macroscopic raw traces of L-type calcium current comparing IV-3 against CRISPR-corrected IV-3. (H) Summary L-type calcium current data comparing IV-3 against CRISPR-corrected IV-3. See Supplemental Table 8 for exact numbers of replicate measures (n) and statistical analysis. Three separate CRISPR/Cas9–corrected differentiations were characterized for data shown in E–H. *Statistical significance (P < 0.05) as determined by unpaired Student’s t test in F and H.
To further assess the impact of this REM2 variant on APD and ICaL, we reduced the expression of REM2 using a REM2 siRNA as we did above for KCNK17. Supplemental Figure 3 illustrates the silencing efficiency of our siRNA on REM2 transcript. Notably, silencing REM2 had the most significant impact on APD and ICaL for both severely affected iPSC-CMs that carried the REM2 variant allele (III-3 and IV-3) (Supplemental Figure 4 and Supplemental Table 7).
CRISPR/Cas9 genome editing confirms role of REM2 G96A as driver of APD and ICaL phenotype.
Finally, we used CRISPR/Cas9–mediated genome editing to revert the variant REM2 allele back to wild type (A96 to G96) in the IV-3 (severely affected) iPSC line. Cardiomyocytes differentiated from the genome-edited IV-3 cell line exhibited a shorter APD90 (edited, 171 ± 13 ms, vs. original, 304 ± 20 ms, P < 0.0001) and lower levels of ICaL (edited, 4.54 ± 0.67 pA/pF, vs. original, –8.17 ± 0.72 pA/pF, P < 0.0001) (Figure 7, E–H, and Supplemental Table 8), which more closely resemble values observed in cardiomyocytes from the IV-17 cell line (APD90, 184 ± 13 ms; ICaL, –3.7 ± 0.38 pA/pF). These results strongly support REM2 as a genetic modifier of the cellular phenotype associated with symptomatic LQT2.
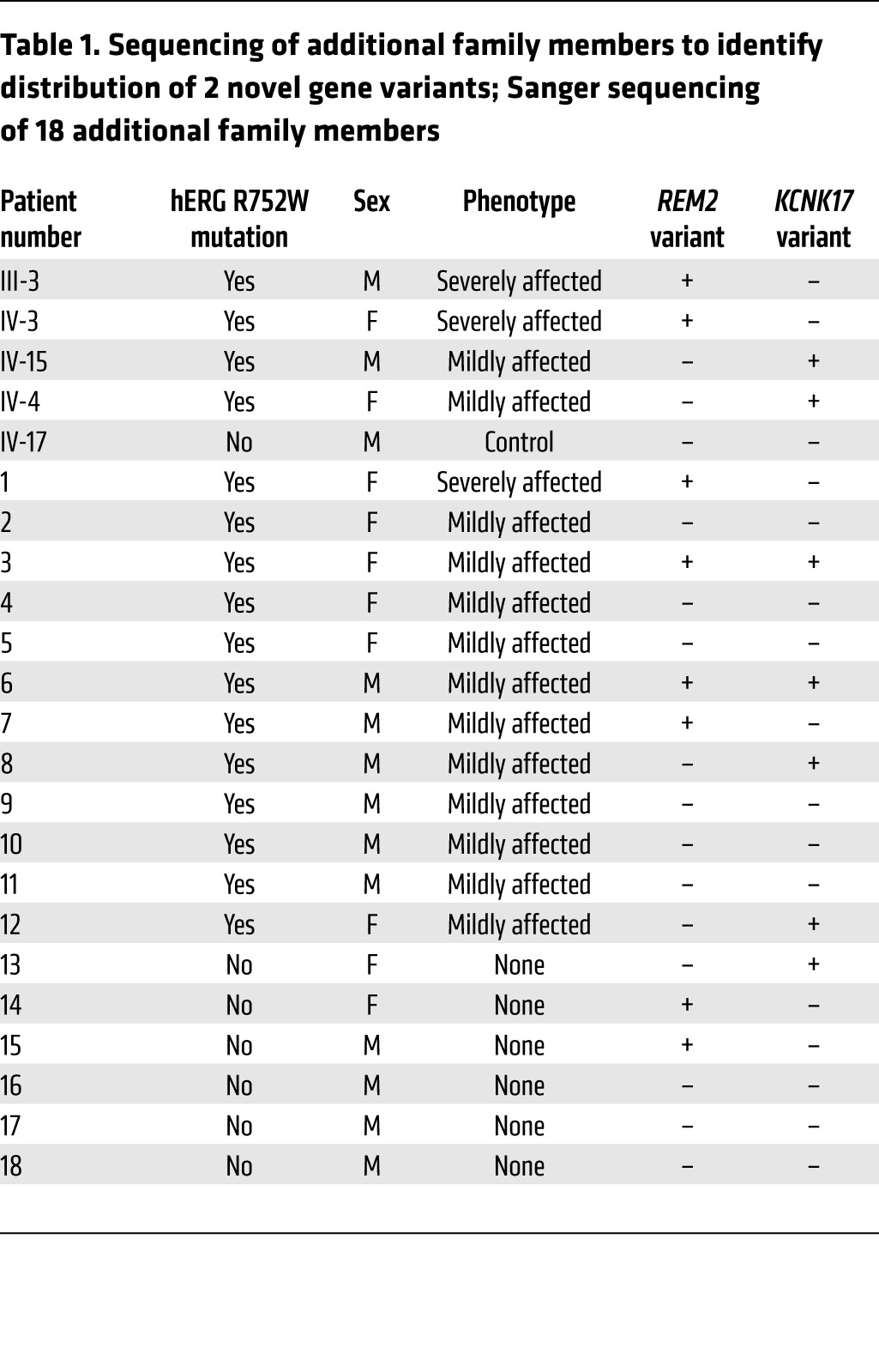
Genotyping analysis of variant distribution in hERG R752W mutation–positive family members.
From a translational perspective, we also genotyped an additional 18 patients from this family. Twelve were hERG R752W mutation carriers diagnosed with varying severities of LQT2, and 6 were healthy nonmutation carriers. Specifically, we assessed whether the REM2 variant identified through our exome sequencing strategy segregated with severely-affected-phenotype patients and conversely whether the KCNK17 variant distributed in a manner consistent with a protective role (Table 1). The REM2 G96A variant was found in all (3/3, 100%) severely-affected-phenotype individuals but only 3 of 13 (23%) mildly-affected-phenotype patients. Notably, 2 patients with a mildly affected/minimal phenotype (patients 3 and 6 in Table 1) carry not only the REM2 variant but also the KCNK17 variant. In these instances, we believe the KCNK17 variant is able to lessen the severity of the phenotype caused by the hERG mutation and REM2 variant (as supported by these patients’ mildly affected phenotypes). This strongly corroborates our initial wave of exome sequencing and electrophysiological data (Figure 7) indicating REM2 as a modifier gene that exacerbates a preexisting repolarization deficiency in these patients, particularly in the patients with the most extreme (severely affected) phenotypes. Conversely, KCNK17 was present in 6 of 13 (46%) mildly-affected-phenotype patients. Importantly, none of the severely-affected-phenotype patients inherited the KCNK17 variant. This distribution only in the mildly-affected-phenotype bin also strengthens our rationale and in vitro iPSC-CM electrophysiological data (Figure 6) indicating that KCNK17 participates as a protective modifier gene by mitigating the severity of the primary hERG mutation as a repolarization reserve. Collectively, the exome sequencing, electrophysiological, and genotyping data are holistically consistent with REM2 acting as a disease-promoting modifier gene (especially in the most severely affected phenotypes) and KCNK17 as a protective gene (as evidenced by the significant distribution rate in mildly-affected-phenotype individuals).
Table 1. Sequencing of additional family members to identify distribution of 2 novel gene variants; Sanger sequencing of 18 additional family members.
Discussion
Here we report the discovery of 2 novel gene variants that provide a physiologically plausible explanation for variable expressivity in a large subset of patients in a multigenerational LQTS type 2 family. This discovery was made through the synergistic pairing of 3 emergent technologies: induced pluripotent stem cell–derived cardiomyocytes (iPSC-CMs), next-generation whole exome sequencing, and CRISPR/Cas9–mediated genome editing. To our knowledge this is the first time all 3 platforms have been used in tandem and corroborated by electrophysiological analyses to elucidate a molecular mechanism of variable expressivity for a monogenic disorder.
We have confirmed early seminal reports (9, 10) of the capacity of patient-specific iPSC-CMs to faithfully reproduce disease phenotypes in vitro. While all individuals harboring the hERG R752W mutation display the same level of IKr deficiency, only the severely-affected-phenotype individuals had a significant prolongation in APD, consistent with their clinical phenotype. The similar current density in IKr observed for severely affected and mildly affected hERG R752W mutation–positive individuals indicated that the modifier gene(s) was not rescuing hERG R752W channel expression and was not responsible for the discordance observed in APD90. However, a serendipitous discovery occurred when we observed a pathophysiological increase in the amount of L-type Ca2+ current in the symptomatic patients’ iPSC-CMs even though we had no clinical indication that these patients had a concomitant calcium channel dysfunction. Importantly, our discovery of abnormal L-type Ca2+ current would not have been possible in a heterologous expression system or transgenic animal model. Our use of iPSC-CMs not only recapitulated the known LQT2 phenotype, but exclusively enabled the aberrant calcium current revelation although we had no prior clinical indications of calcium current abnormality, further validating the unique power of the patient-specific iPSC-CM platform.
Furthermore, classic efforts to link genes to Mendelian disease are gradually being displaced by next-generation sequencing platforms (23–25), and exome sequencing has proved itself as an indispensable technology to identify disease-causing coding variants in congenital arrhythmia syndromes (26, 27). Thus, we used a strategy seeking to identify gene variants affecting either ion channels, associated channel proteins, or loci associated with QT interval variability that could explain variable expressivity in an LQT2 family. Importantly, since our patient iPSC-CMs revealed a potential calcium dysfunction, we also hypothesized that this strategy might reveal a mechanistic modifier gene driving the increased L-type Ca2+ current. This approach led us to the identification and functional vetting of 2 novel disease-modifying genes.
The first modifier gene we interrogated was KCNK17. Two-pore-domain potassium channel (K2p) genes such as KCNK17 (encoding K2p17.1, also known as TASK-4 and TALK-2) are known to be robustly expressed in myriad tissues, including cardiac and central nervous system, where they traditionally have been thought to regulate background cell excitability as leak channels (28, 29). However, a wealth of transgenic animal studies support the notion that TASK family K2p channels also strongly contribute to cardiac repolarization (30–32). Recently, Friedrich et al. (27) used exome sequencing to identify a gain-of-function mutation in KCNK17 that in tandem with an SCN5A mutation underpinned the molecular mechanism of a severely affected arrhythmia phenotype. Furthermore, work by Schmidt et al. (33) showed that pharmacological inhibition of a K2p (TASK-1) is a novel potential pharmacological strategy for treatment of chronic atrial fibrillation. Additionally, Decher et al. identified a mechanism in a patient whereby increased sodium permeability through a K2p (TREK-1) predisposed the individual to right ventricular outflow tract ventricular tachycardia (34). Finally, recent work from our laboratory corroborates the notion that K2p channels are more than simply background leak channels, acting as active participants in ventricular repolarization (35). Collectively, this evidence supports the idea that K2p channels may modulate cardiomyocyte excitability and contribute to disease phenotypes as well as providing a therapeutic target. Although our results suggest that KCNK17 is not a universally protective gene (Table 1), we believe that the p.Ser21Gly variant identified in select LQT2 family members may promote enhanced repolarization reserve that conceivably protects mildly affected/minimal hERG mutation–positive individuals from a severely affected phenotype, as supported by 2 individuals (patients 3 and 6 in Table 1) who coinherited the REM2 variant whose deleterious effects are “canceled out” by KCNK17. Furthermore, in addition to the 2 original patients in whom we identified the KCNK17 variant via exome sequencing, 2 additional patients (patients 5 and 8 in Table 1) were categorized as having mildly affected phenotype plausibly attributable to the protective effect of increased KCNK17 current density. Again, this was strongly supported by the significant prolongation in APD observed when KCNK17 was silenced in the individuals carrying the variant with larger 2-pore-domain potassium current but not in the individuals carrying the variant with smaller 2-pore-domain potassium current (Figure 6, E and F). So while not all mildly-affected-phenotype individuals carried the KCNK17 S21G variant, our evidence strongly supports the role of KCNK17 as a protective modifier gene in 6 of 13 LQT2 patients (46%).
The second modifier gene identified in our study is REM2, which belongs to the RGK family (Rem/REM2/Gem/Kir) of GTPases. We pursued REM2 after the fortuitous finding that iPSC-CMs derived from severely-affected-phenotype patients had significantly enhanced L-type Ca2+ (ICaL) current compared with control and mildly-affected-phenotype mutation-positive relatives (Figure 4). Cav1.2 channels in the heart are under precise physiological modulation from a host of intracellular proteins (36). One of these regulators is the Ras superfamily of GTPases (37), which have been shown to inhibit voltage-gated calcium channels through putative interactions with auxiliary Cav β subunits (38, 39). Experimentally, pharmacological blockade of Cav1.2 using nisoldipine further revealed the role of ICaL in the patient-specific iPSC-CMs (which was especially pronounced in the severely affected cell lines; Figure 5). Based on these observations and our iPSC-CM electrophysiological data, we posited that REM2-p.Gly96Ala identified in the whole exome sequencing data set explains the robust increase in ICaL. In fact, expression of this REM2-p.Gly96Ala variant in control iPSC-CMs (iCells) increased ICaL to a greater degree than WT REM2 (Figure 7). Furthermore, silencing REM2 using siRNA drastically shortened APD and reduced ICaL in severely affected iPSC-CMs (Supplemental Figure 4). More conclusively, the use of CRISPR/Cas9–mediated genome editing to correct the REM2 G96A mutant allele to wild type in the severely affected IV-3 iPSC line returned the APD and ICaL to near control-like levels (Figure 7, E–H, and Supplemental Table 8). Finally, the additional genotyping of 18 family members strongly corroborates this variant as a plausible explanation of variable expressivity in a subset of family members. Similarly to KCNK17, while REM2 is not the universal driver of disease in this family, we show here that it segregates with 100% (3 of 3) of severely-affected-phenotype LQTS patients in this family (Table 1). Collectively, the experimental and patient observations confirm the physiological role of REM2, a member of the Ras superfamily of GTPases, which has previously been described to regulate voltage-gated calcium channels (39, 40). And while the seminal report of Splawski et al. (41) originally reported and linked CACNA1C mutations to Timothy syndrome (with long QT syndrome type 8 overlap), here we report a novel role for a known voltage-gated calcium channel regulatory protein to exacerbate a canonical LQT2 mutation. To our knowledge this is the first description of a voltage-gated calcium channel–associated protein behaving as a modifier gene for LQT2. But importantly, it is also the first report implicating REM2 as a regulator of cardiac electrophysiology, making it a potential new LQT gene.
The hypothesis of a “double hit” that exacerbates or results in a proarrhythmic phenotype as described in this report is neither unprecedented nor unfounded. While historically LQTS is considered a “one gene, one disease” etiology, the genetic landscape is quite complex, and evidence is mounting for more oligogenic or oligo-SNP–based drivers of disease. The compounding effects of 2 mutations in the same gene (e.g., compound heterozygosity) or 2 separate genes (digenic heterozygosity) are well documented in not only LQTS (42, 43), but other cardiac pathologies as well (44–46). Consistent with the findings in these reports, we propose here a new take on the double hit hypothesis: a primary mutation in a gene that dictates the bulk of ventricular repolarization (hERG) in tandem with an SNP in a gene that encodes a voltage-gated calcium channel–associated protein (REM2). This combination results in exacerbation of a primary deficiency in repolarization responsible for the final disease phenotype. We also show that a second SNP in KCNK17, which encodes a 2-pore-domain potassium channel, can protect patients by behaving as a compensatory current in the presence of a primary repolarizing deficiency (including 2 patients who have both a primary hERG mutation and the REM2 SNP, in which case the KCNK17 SNP serves as the “tie breaker”).
Study limitations.
While various other modifier gene candidates were also identified through our exome sequencing prioritization strategy, we elected to focus on 2 rapidly assayable and biologically plausible targets that could directly influence QTc and explain the increase in ICaL from severely-affected-phenotype mutation-positive patients. Other candidates were excluded because either their primary functions are described in a noncardiac setting (e.g., GRIN3A, ref. 47; and CAMKK2, ref. 48) unrelated to calcium channel function or these variants were deemed nonpathogenic by in silico predictors (PolyPhen, SIFT). Conceptually, our exome sequencing filtering approach was designed to identify modifier genes in an LQT2 family and not de novo mutations. KCNK17 was pursued because it is the only ion channel that turned up in either cohort, and REM2 is a known modulator of voltage-gated calcium channels that could explain the increase in ICaL observed in Figure 4. However, we do not exclude the likelihood of some permutation of these other variants identified in Supplemental Table 5 to also contribute to either the protective effect in the mildly affected individuals or the additive effect in the severely affected individuals. In fact, this is probably the cause for individual 7 in Table 1, who, even though he carries the hERG mutation and the REM2 variant, displays a mildly affected phenotype. However, pursuing the identification of additional modifiers in an efficient and clinically translational approach will be the focus of further studies.
Additionally, while filtering using GWAS runs the risk of missing rare variants, we used this filter to ensure that we cast a wide enough net so as to not exclude previously identified common modulators of QTc. Another consideration with these diseases is that while the mutations set baseline risk, the exact timing of the lethal events depends on triggering factors yet to be identified, suggesting that the variable expressivity could be ascribed to environmental or other acquired influences. However, it has been clinically observed with this specific family that in carriers with multiple ECG recordings documented over 20 years, variation in QTc with time was minimal (14). Hence, the variable expressivity of QTc in these family members carrying the same mutation with stable QTc values over time suggests an important influence of modifier genes with environmental factors playing a minor role. Here, we have not identified the universal mechanism of disease expressivity in this family, but rather isolated and vetted 2 disease-modifying genes that provide a plausible explanation for increased or reduced risk of severely affected LQT2 phenotype.
Conclusions.
In summary, we demonstrate the synergistic usage of iPSC technology, next-generation exome sequencing, and CRISPR/Cas9 genome editing to identify plausible genetic explanations for variable phenotypic expressivity in a large LQTS type 2 family. We postulate that REM2-driven increased ICa,L in combination with a primary KCNH2 (hERG, IKr) haploinsufficiency is the permutation that produces the full-blown disease phenotype and that KCNK17 can promote a compensatory repolarization reserve. Importantly, we have uncovered 2 new genes that could be linked to arrhythmias. Finally, we advance that this strategy can be deployed to screen for novel genetic modifiers in at-risk populations extending beyond the cardiac channelopathy landscape. All 3 technologies accelerate the efficiency and resolution of informing on candidates underlying genotype-phenotype discordance. We show that the combinatorial usage of these platforms can pinpoint novel and potentially actionable targets, thus laying the framework for a tractable approach to precision medicine.
Methods
Patient IRB approval and informed consent.
Study subjects were ascertained following written informed consent procedures approved by the Institutional Review Board of Case Western Reserve University and in accordance with the MetroHealth Medical Center Human Investigation HIPAA Authorization Policy. Comprehensive clinical analyses in the enrolled family members (white, n = 101) include history, physical examination, and ECG recording.
Selection of patients in the study and clinical diagnosis parameters.
Patients described in this study were initially identified and diagnosed by a clinical cardiac electrophysiologist who has been tracking and treating this family for 20 years and previously reported on the variable expressivity of LQTS in this family (14). Furthermore, molecular work has previously elucidated the disease-causing mechanism for the hERG R752W mutation (15). Phenotype binning (severely affected and mildly affected) was based on previously determined clinical diagnostic criteria (ECG morphology abnormalities and arrhythmic syncope).
Two pairs of hERG R752W mutation–positive severely-affected-phenotype versus mildly-affected-phenotype first-degree relatives (a father/son pair [III-3 and IV-15, respectively] and a sister/sister pair [IV-3 and IV-4]) along with a healthy mutation-negative control subject (IV-17) were selected from this large LQT2 family (Figure 1). We proceeded with these individuals to drive the crux of the study based on their close genetic relationship, but divergent clinical phenotypes.
Generation of patient-specific iPSCs.
Patient-specific induced pluripotent stem cell (iPSC) lines (IV-17, III-3, IV-15, IV-3, and IV-4) were generated in the Case Western Reserve University Pluripotent Stem Cell Facility. Patients III-3 and IV-3 had recurrent episodes of syncope and were symptomatic for LQT2, while IV-15 and IV-4 were asymptomatic (Figure 1). Briefly, fibroblasts were harvested and cultured to generate human iPSC lines consistent with iPSC methodology described previously (49). Fibroblasts were plated at 1 × 104 cells/cm2 and infected with VSV-g pseudotyped hSTEMCCA lentivirus containing a polycistronic coding sequence for the human genes OCT3/4, SOX2, KLF4, and MYC. Cells were transitioned to human pluripotent stem cell medium consisting of DMEM-F12 supplemented with 20% knockout serum replacement, 20 ng/ml FGF2, 2 mM GlutaMAX (ThermoFisher Scientific), 1× nonessential amino acids, and 0.1 mM 2-mercaptoethanol. Nascent iPSC colonies were individually picked and expanded around day 30. Established lines were passaged as small clusters every 4–5 days with enzymes (1 mg/ml collagenase type IV and 1 mg/ml dispase). Irradiated mouse embryonic fibroblasts (MEFs) served as a feeder layer for iPSC derivation and subsequent expansion. iPSC lines were then plated on Matrigel-coated (BD Biosciences) 6-well plates in feeder-free medium (Applied Stem Cells Inc.). For characterization of validated iPSCs, alkaline phosphatase staining was carried out using the stem cell characterization kit (Millipore). Immunostaining was performed to immunolabel nuclear, cytoplasmic, and surface markers of stem cells, including OCT3/4 (1:500; Santa Cruz Biotechnology), SOX2 (1:200; Abgent), SSEA4 (1:50), TRA1–60 (1:50), and TRA1–81 (1:50) (Millipore). The patient-derived iPSCs expressed antigens of embryonic stem cells including OCT3/4, SOX2, SSEA4, TRA1–60, and TRA1–81 and were positive in alkaline phosphatase staining. iPSCs were dissociated and stored in vials in liquid nitrogen until ready for cardiomyocyte differentiation.
To control for intraclone variability, multiple clonal lines were generated per patient described in Figure 1. These lines were stored and continuously propagated and used for cardiomyocyte differentiation and electrophysiological characterization.
Preparation of iPSC lines for cardiomyocyte differentiation.
iPSC vials were thawed from liquid nitrogen storage and cultured on 6-well dishes that were coated with Matrigel prepared according to company protocol (Corning Matrigel hESC-qualified Matrix). Cells were pelleted by centrifugation and resuspended using 6–12 ml of mTeSR medium (STEMCELL Technologies) plus 5 mM ROCK inhibitor (StemRD) and plated on 6-well Matrigel-coated dishes. mTeSR medium was changed daily until iPSCs were ready for passing (5–8 days after plating). iPSC colonies that achieved confluence were incubated with Accutase (Stem Cell Technologies) to generate smaller cell aggregates. Cell scrapers were used to gently detach the colonies, which were centrifuged and dissociated by pipetting, then plated onto Matrigel-coated dishes in dilutions of 1:3 to 1:6.
In vitro cardiomyocyte differentiation from iPSCs.
Human iPSC lines were differentiated to cardiomyocytes using a previously described Wnt-based high-yield protocol (50, 51) and lactate purification step (52). Briefly, iPSC colonies were washed with 1 ml of PBS, and 1 ml of TrypLE Express (Gibco) was added to each well. Colonies were incubated in trypsin and dissociated to single cell by slow pipetting. Cell suspensions were pelleted by centrifugation (3×), then counted manually (Neubauer chamber). Singularized cells were seeded into Matrigel-coated 12-well dishes in concentrations of 500,000, 750,000, and 1 × 106 cells per well. Cells were cultured for 4 days, and monolayers were incubated in 2 ml solution of RPMI medium (Invitrogen) enriched with B27-Insulin supplement (Invitrogen) plus 12 μM CHIR99021 (Selleck) in DMSO for 24 hours (considered day 0). Twenty-four hours after addition of CHIR99021 molecule, medium was replaced with 2 ml of RPMI + B27-Insulin supplement. On day 3, 1 ml of the old medium from each monolayer was mixed with 1 ml of fresh RPMI + B27-Insulin supplement plus 5 μM IWP4 (Stemgent). On day 5, monolayers were expanded and washed with PBS (1 ml/well) to rinse IWP4 and cellular debris. Accutase was added to the cells, monolayers were titrated to singularize cells, and cell suspension was pelleted by centrifugation. Each monolayer was split 1:3 into fibronectin-coated dishes. Usually, cells started spontaneously beating at day 8. Cells were fed every 2 days with RPMI + B27 until day 20, when lactate purification was used as a metabolic stressor. Dead cells were flushed, and around day 28, beating cells were ready for harvest/dissociation and plating for electrophysiological analysis. Immunocytostaining of patient-derived iPSC-CMs for canonical sarcomeric proteins confirmed cardiomyocyte lineage (Supplemental Figure 5).
Mitigation of iPSC-CM cellular phenotype variability.
iPSC-CMs can exhibit large variability in APD between differentiations (e.g., interbatch). To mitigate this known phenomenon, our analysis is performed in a manner in which we never compare differentiations from different time points or from different batches. In other words, a control cell line (IV-17) is always differentiated concomitantly with a severe and mild iPSC-CM cell line, and all data collected are analyzed as a trio. Cross-comparison across groups and differentiations is never performed because each differentiation has intrinsic and systemic differences that must be uniformly maintained within the trio. Thus, each time we have compared data within a trio, we have always observed the same results: significantly prolonged APD for the severely affected individual compared with the control or mildly affected pair (although from differentiation to differentiation the absolute APD values might have varied). Using this methodology, we have observed a conservation of the observed trends over multiple differentiations over a 5-year period.
CRISPR/Cas9 genome editing.
Genome editing of cell lines was contracted through Applied Stem Cell. The REM2 A96 variant in IV-3 iPSC cells was converted to G96 using the CRISPR/Cas9 system. Briefly, guide RNAs (gRNAs) were designed and tested as transfection candidates based on the following criteria: (a) an assessment of gRNA activity as measured by deep sequencing; and (b) proximity to the corresponding point mutation insertion site. Based on off-target analysis, 2 gRNAs were chosen: g7 [EA102.REM2.g7 TGAACTTGACTGGCCACCTC (AGG)-PAM] and g12 [EA102.REM2.g12 CGTCTGACTCCTTGGGCTCA (GGG)-PAM), where next-generation sequencing data show 45% and 72% of gRNA activity, respectively. Each of the single-stranded oligodeoxynucleotide donors (ssODNs) was designed to be used as a repair template at each of the gRNA cut sites during the homology-directed repair process (ssODN sequence: CAGACGAAGAGGCAGTATGCCTGTCCCCTACAAGCACCAGCTCCGGCGGGCCCAGGCTGTAGATGAACTTGACTGGCCACCTCAGGCCTCATCCTCTGGCTCGTCTGACTCCTTGGGATCCGGAGAGGCAGCCCCTGCTCAAAAGGATGGCATCTTCAAGGTCATGCTAGTGGGGGAGAGCGGCGTGGGCAAGAGCAC). After electroporation of both gRNAs into the iPSCs, and since the PGK/Puro construct provided transient puromycin resistance, the transfected iPSCs were subjected to puromycin selection for 2 days after transfection; then single-cell colonies were grown for another few weeks. Single-cell colonies were picked and transferred into a 24-well plate for further growth. After colonies reached 90% confluence, cells were divided into 2 parts, 1 part was sent for genotyping, and remaining cells were seeded into the newly prepared 24-well plate with Matrigel. Screening indicated that 2 clones contained the predicted digestion pattern of Gly96 homozygous mutation. Sequence analysis of these clones confirmed the change of GlyAla96Gly (GCC→GGC). Differentiation of corrected iPSCs to cardiomyocytes was performed as described above under In vitro cardiomyocyte differentiation from iPSCs.
Electrophysiological assessments.
Electrophysiological recordings in the voltage-clamp and current-clamp modes of the whole-cell patch clamp (53) were obtained using an Axopatch 200A amplifier and acquired with a Digidata 1440A digitizer. To generate voltage-clamp command pulses, pCLAMP version 10 was used (Molecular Devices). To minimize voltage-clamp errors, series resistance compensation of Axopatch 200A was performed to values greater than 85%. Patch electrodes were prepared from 8161 Corning borosilicate capillary glass (Dow Corning) and lightly polished to 1.5–2.5 MΩ for iCells, iPSC-CMs, and Chinese hamster ovary cells. The amphotericin-perforated patch technique was used to obtain whole-cell recordings of action potentials under current clamp conditions. Whole-cell voltage-clamp recordings of IKr, IK2p17.1, and ICaL were obtained using standard patch clamp technique. Action potentials and IKr were recorded at 37°C, whereas all other currents were recorded at 21°C. All ionic currents were normalized to cell capacitance. The cells were bathed in a chamber continuously perfused with Tyrode’s solution composed of (in mmol/l adjusted to pH 7.35 with NaOH) 137 NaCl, 5.4 KCl, 2 CaCl2, 1.0 MgSO4, 10 glucose, 10 HEPES. The intracellular pipette solution was composed of (in mmol/l adjusted to pH 7.3 with KOH) 120 aspartic acid, 20 KCl, 10 NaCl, 2 MgCl2, 5 HEPES, and 240 μg/ml of amphotericin B (Sigma-Aldrich). A gigaseal was rapidly formed. Typically, 10 minutes later, amphotericin B pores lowered the resistance sufficiently to current-clamp the cells. Myocytes were paced in the current-clamp mode using a 1.5- to 2-diastolic-threshold 5-ms current pulse at 1 Hz. Action potential trains were also recorded at 1 Hz and further analyzed using Clampfit software. The extracellular bath solution and intracellular pipette solution used for IKr were the same Tyrode’s as used for action potentials. The extracellular bath solution for ICaL contained (in mmol/l at pH 7.4 adjusted with HCl): 50 NaCl, 5.4 CsCl, 1.8 MgCl, 1.8 CaCl2, 10 HEPES, 10 glucose, 80 N-methyl-D-glucamine. The intracellular pipette solution contained (in mmol/l at pH 7.2 adjusted with CsOH): 130 cesium methanesulfonate, 20 tetraethylammonium chloride, 1 MgCl2, 10 EGTA, 10 HEPES, 4 Mg-ATP. This solution was used to record ICaL from iCell cardiomyocytes as well as iPSC-CMs. The extracellular bath solution for IK2p17.1 contained (in mmol/l at pH 7.4 adjusted with NaOH): 140 NaCl, 5 KCl, 1 MgCl2, 1.8 CaCl2, 10 HEPES, 10 glucose. The intracellular pipette solution used for IK2p17.1 (KCNK17, 2-pore-domain K+ current) contained (in mmol/l at pH 7.2 adjusted with KOH): 100 K-aspartate, 20 KCl, 2 MgCl2, 1 CaCl2, 10 EGTA, 2 ATP, 10 HEPES. IKr was elicited by 3-second depolarizing steps from a holding potential of –40 mV to potentials ranging from –20 to +20 in 20-mV increments. This was followed by a 2-second repolarization phase to –40 mV to elicit tail current. ICaL was elicited by 300-ms depolarizing steps from a holding potential of –40 mV to potentials ranging from –30 mV to +60 mV in 10-mV increments. IK2p was elicited by 500-ms depolarizing steps from a holding potential of –80 mV to potentials ranging from –120 mV to +80 mV.
Enrichment, library preparation, and exome sequencing.
Whole exome sequencing was performed on genomic DNA from the 2 first-degree relative pairs (pedigree generation and family member numbers: III-3, IV-15; and IV-3, IV-4) of mutation-positive subjects by the Vanderbilt Technologies for Advanced Genomics core facility. Exome enrichment was performed using the Agilent Sure Select Human All Exon 50 Mb capture reagent according to the supplier’s instructions. Paired-end (2 × 100 bp) sequencing was performed on an Illumina HiSeq2000 sequencer.
Exome sequence analysis.
After removal of low-quality reads, alignments to a reference human genome (UCSC Genome Browser assembly hg19) were performed with the Burrows-Wheeler Aligner (BWA) (11), and then sequences were processed with the Genome Analysis Toolkit (Broad Institute) to remove duplicate reads and to call variants. Default settings were used in the BWA alignments, including a maximum of 2 mismatches in the seed portion of reads (first 32 bp) and no more than 3 mismatches for the entire read. The threshold for detecting variants was set at a genotype quality score of 40.
Generation of KCNK17 and REM2 mutations.
The REM2 plasmid was provided by Henry Colecraft (Columbia University, New York, New York, USA), and the KCNK17 plasmid was provided by Dierk Thomas (Heidelberg University, Heidelberg, Germany). The REM2 G96A and KCNK17 S21G variants were created using Stratagene QuikChange XL Site-Directed Mutagenesis Kit in the REM2 background (PubMed accession no. NM 173527) expressed in the pCDNA4 vector (BD Biosciences Clontech) and the KCNK17 background (PubMed accession no. NM 031460) expressed in the pCDNA3.
Expression of KCNK17 in Chinese hamster ovary cells and REM2 in iCell cardiomyocytes.
KCNK17 and REM2 were expressed using transient transfections in Chinese hamster ovary (CHO) cells or iCell cardiomyocytes (Cellular Dynamics International), respectively. The total DNA transfected was equal to 2.0 μg for both conditions (1.8 μg of KCNK17 or REM2 DNA + 0.2 μg of yellow fluorescent protein). In the case of the KCNK17 heterozygote condition in CHO cells, the total DNA transfected was still 2.0 μg but divided evenly at a 1:1 ratio (0.9 μg per condition + 0.2 μg of yellow fluorescent protein). Transfection of both CHO cells and iCells was accomplished using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol, using 2.0 μg DNA and 35-mm dishes (CHO cells) or scaled for a 24-well plate (iCells).
KCNK17 and REM2 siRNA knockdown experiments.
iPSC-CMs from patients were transfected with either a TALK-2 (KCNK17) siRNA (Santa Cruz Biotechnology, sc-61641), a REM2 siRNA (Santa Cruz Biotechnology, sc-92154), or Ambion siRNA scrambled negative control (Ambion/Life Technologies) using Lipofectamine RNAiMax (Life Technologies) at 20 nM of final concentration in a 24-well plate. Cells were also cotransfected with 10 nM of Ambion FAM-labeled siRNA negative control (Ambion/Life Technologies) to identify transfected cells. Action potentials from transfected cells were recorded 48 hours after transfection.
REM2 and KCNK17 genotyping.
Genomic DNA was extracted from whole blood and REM2 and KCNK17 were sequenced in 18 family members from the LQT2 family. Exon 2 of REM2 and exon 1 of KCNK17 were sequenced in these individuals using specific primers (REM2 forward primer ACAGCCTGGATTGAGCCTTC, reverse primer CAGGTCACGAGCACTTCCTT; KCNK17 forward primer CCATTCCCCAACACTCCTCC, reverse primer CGAGGGTCTTTTCCTCCGAG). PCR reactions were performed using 50 ng of genomic DNA with primers corresponding to exon 2 of REM2 and exon 1 of KCNK17. PCR products were sent out for sequencing (Eurofins MWG Operon).
Immunohistochemistry.
Patient-specific iPSC-CMs were fixed with 4% paraformaldehyde for 15–30 minutes and immunostained with the following primary antibodies: sarcomeric α-actinin (mouse monoclonal clone EA-53, Sigma-Aldrich; 1:800), cardiac troponin T (rabbit polyclonal PA5, Pierce; 1:500), REM2 (mouse polyclonal, Abcam; 1:100), and KCNK17 (rabbit polyclonal, Abcam; 1:100). Alexa Fluor 488– and 594–conjugated secondary antibodies specific to the corresponding species were used (Invitrogen; 1:1,000 for mouse and 1:2,000 for rabbit). Nuclei were immunostained using Vector Laboratories Vectashield Antifade Mounting Medium with DAPI. Microscopy was performed using a Leica SP2 confocal. Images were processed using ImageJ software (NIH).
Single-cell RT-PCR.
Single-cell reverse transcription PCR (RT-PCR) was performed by patch-clamping of the cell to obtain the electrophysiological verification of myocyte type by action potential recording followed by harvesting of the cell via extended suction using the same pipette. The contents of the pipette were deposited into a tube containing a scaled reverse transcription mixture (High Capacity Reverse Transcription kit, Applied Biosystems). Following reverse transcription, PCR was carried out using 2 μl of the RT reaction.
Statistics.
All statistical analysis was performed using the standard statistics suite available in Origin 8.5.1 (OriginLab). Normality and homogeneity of all data sets were determined using the Shapiro-Wilk and Levene tests, respectively, and by visual inspection of distribution in our data sets. Multiple comparisons between severely-affected-phenotype LQT2 mutation–positive individuals and mildly-affected-phenotype mutation-positive individuals or non-hERG-mutation-carrying controls were performed using ANOVA. E-4031 and nisoldipine experiments were analyzed using the paired Student’s 2-tailed t test. Two-sided P values less than 0.05 were considered statistically significant. All results are reported as mean ± SEM unless otherwise noted.
Author contributions
ESK performed clinical assessment and referred the family. SC, EF, ALG, ESK, and ID contributed to study conceptualization and experimental design. SC and XW performed in vitro data collection. ARN and PJT performed iPSC generation and cardiomyocyte differentiation. ALG performed the exome sequencing. SC, ALG, ESK, and ID wrote and edited the manuscript.
Supplementary Material
Acknowledgments
We thank Henry Colecraft for providing the REM2 plasmid and Dierk Thomas for providing the KCNK17 plasmid. This study was supported by NIH/NHLBI grant 1R01HL124245 (to ID), American Heart Association Established Investigator Award 12EIA9300060 (to ID), an American Heart Association Pre-Doctoral Fellowship from the Great Rivers Affiliate 15PRE25700037 (to SC), and T32 Training Grant HL105338-01 (to SC). This publication was made possible by the Clinical and Translational Science Collaborative of Cleveland, grant UL1TR000439 from the National Center for Advancing Translational Sciences component of the NIH, and the NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Version 1. 02/12/2018
Electronic publication
Version 2. 03/01/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(3):1043–1056.https://doi.org/10.1172/JCI94996.
Contributor Information
Sam Chai, Email: sam.sl.chai@gmail.com.
Xiaoping Wan, Email: xwan@metrohealth.org.
Angelina Ramirez-Navarro, Email: arnavarro@metrohealth.org.
Elizabeth S. Kaufman, Email: ekaufman@metrohealth.org.
Eckhard Ficker, Email: eficker@metrohealth.org.
References
- 1.Priori SG, et al. Genetic and molecular basis of cardiac arrhythmias: impact on clinical management parts I and II. Circulation. 1999;99(4):518–528. doi: 10.1161/01.CIR.99.4.518. [DOI] [PubMed] [Google Scholar]
- 2.Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet. 2008;372(9640):750–763. doi: 10.1016/S0140-6736(08)61307-0. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz PJ, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120(18):1761–1767. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.George AL., Jr Molecular and genetic basis of sudden cardiac death. J Clin Invest. 2013;123(1):75–83. doi: 10.1172/JCI62928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80(5):795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 6.Bianchi L, et al. Mechanisms of I(Ks) suppression in LQT1 mutants. Am J Physiol Heart Circ Physiol. 2000;279(6):H3003–H3011. doi: 10.1152/ajpheart.2000.279.6.H3003. [DOI] [PubMed] [Google Scholar]
- 7.Benhorin J, et al. Variable expression of long QT syndrome among gene carriers from families with five different HERG mutations. Ann Noninvasive Electrocardiol. 2002;7(1):40–46. doi: 10.1111/j.1542-474X.2001.tb00137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 9.Moretti A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363(15):1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 10.Itzhaki I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471(7337):225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 11.Ng SB, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42(1):30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emond MJ, et al. Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet. 2012;44(8):886–889. doi: 10.1038/ng.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iossifov I, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaufman ES, et al. Electrocardiographic prediction of abnormal genotype in congenital long QT syndrome: experience in 101 related family members. J Cardiovasc Electrophysiol. 2001;12(4):455–461. doi: 10.1046/j.1540-8167.2001.00455.x. [DOI] [PubMed] [Google Scholar]
- 15.Ficker E, et al. Novel characteristics of a misprocessed mutant HERG channel linked to hereditary long QT syndrome. Am J Physiol Heart Circ Physiol. 2000;279(4):H1748–H1756. doi: 10.1152/ajpheart.2000.279.4.H1748. [DOI] [PubMed] [Google Scholar]
- 16.Volders PG, et al. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. Cardiovasc Res. 2000;46(3):376–392. doi: 10.1016/S0008-6363(00)00022-5. [DOI] [PubMed] [Google Scholar]
- 17.Milberg P, et al. Inhibition of the Na+/Ca2+ exchanger suppresses torsades de pointes in an intact heart model of long QT syndrome-2 and long QT syndrome-3. Heart Rhythm. 2008;5(10):1444–1452. doi: 10.1016/j.hrthm.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 18.Pfeufer A, et al. Common variants in myocardial ion channel genes modify the QT interval in the general population: results from the KORA study. Circ Res. 2005;96(6):693–701. doi: 10.1161/01.RES.0000161077.53751.e6. [DOI] [PubMed] [Google Scholar]
- 19.Pfeufer A, et al. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet. 2009;41(4):407–414. doi: 10.1038/ng.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeufer A, et al. Genome-wide association study of PR interval. Nat Genet. 2010;42(2):153–159. doi: 10.1038/ng.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newton-Cheh C, et al. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet. 2009;41(4):399–406. doi: 10.1038/ng.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berger SI, Ma’ayan A, Iyengar R. Systems pharmacology of arrhythmias. Sci Signal. 2010;3(118):ra30. doi: 10.1126/scisignal.2000723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singleton AB. Exome sequencing: a transformative technology. Lancet Neurol. 2011;10(10):942–946. doi: 10.1016/S1474-4422(11)70196-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bamshad MJ, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 25.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26(10):1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 26.Crotti L, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127(9):1009–1017. doi: 10.1161/CIRCULATIONAHA.112.001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friedrich C, et al. Gain-of-function mutation in TASK-4 channels and severe cardiac conduction disorder. EMBO Mol Med. 2014;6(7):937–951. doi: 10.15252/emmm.201303783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldstein SA, Bockenhauer D, O’Kelly I, Zilberberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci. 2001;2(3):175–184. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- 29.Gierten J, et al. Regulation of two-pore-domain (K2P) potassium leak channels by the tyrosine kinase inhibitor genistein. Br J Pharmacol. 2008;154(8):1680–1690. doi: 10.1038/bjp.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Putzke C, et al. The acid-sensitive potassium channel TASK-1 in rat cardiac muscle. Cardiovasc Res. 2007;75(1):59–68. doi: 10.1016/j.cardiores.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 31.Decher N, et al. Knock-out of the potassium channel TASK-1 leads to a prolonged QT interval and a disturbed QRS complex. Cell Physiol Biochem. 2011;28(1):77–86. doi: 10.1159/000331715. [DOI] [PubMed] [Google Scholar]
- 32.Donner BC, et al. Functional role of TASK-1 in the heart: studies in TASK-1-deficient mice show prolonged cardiac repolarization and reduced heart rate variability. Basic Res Cardiol. 2011;106(1):75–87. doi: 10.1007/s00395-010-0128-x. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt C, et al. Upregulation of K(2P)3.1 K+ current causes action potential shortening in patients with chronic atrial fibrillation. Circulation. 2015;132(2):82–92. doi: 10.1161/CIRCULATIONAHA.114.012657. [DOI] [PubMed] [Google Scholar]
- 34.Decher N, et al. Sodium permeable and “hypersensitive” TREK-1 channels cause ventricular tachycardia. EMBO Mol Med. 2017;9(4):403–414. doi: 10.15252/emmm.201606690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chai S, et al. Contribution of two-pore K+ channels to cardiac ventricular action potential revealed using human iPSC-derived cardiomyocytes. Am J Physiol Heart Circ Physiol. 2017;312(6):H1144–H1153. doi: 10.1152/ajpheart.00107.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 37.Colicelli J. Human RAS superfamily proteins and related GTPases. Sci STKE. 2004;2004(250):RE13. doi: 10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Béguin P, et al. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature. 2001;411(6838):701–706. doi: 10.1038/35079621. [DOI] [PubMed] [Google Scholar]
- 39.Finlin BS, Crump SM, Satin J, Andres DA. Regulation of voltage-gated calcium channel activity by the Rem and Rad GTPases. Proc Natl Acad Sci U S A. 2003;100(24):14469–14474. doi: 10.1073/pnas.2437756100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang T, Colecraft HM. Regulation of voltage-dependent calcium channels by RGK proteins. Biochim Biophys Acta. 2013;1828(7):1644–1654. doi: 10.1016/j.bbamem.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Splawski I, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 42.Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 2004;109(15):1834–1841. doi: 10.1161/01.CIR.0000125524.34234.13. [DOI] [PubMed] [Google Scholar]
- 43.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2(5):507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 44.Van Driest SL, et al. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44(3):602–610. doi: 10.1016/j.jacc.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 45.Bhuiyan ZA, et al. Desmoglein-2 and desmocollin-2 mutations in dutch arrhythmogenic right ventricular dysplasia/cardiomypathy patients: results from a multicenter study. Circ Cardiovasc Genet. 2009;2(5):418–427. doi: 10.1161/CIRCGENETICS.108.839829. [DOI] [PubMed] [Google Scholar]
- 46.Xu T, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55(6):587–597. doi: 10.1016/j.jacc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kehoe LA, Bernardinelli Y, Muller D. GluN3A: an NMDA receptor subunit with exquisite properties and functions. Neural Plast. 2013;2013:145387. doi: 10.1155/2013/145387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Racioppi L, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem. 2012;287(38):31658–31665. doi: 10.1074/jbc.R112.356485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sheng Y, et al. Using iPSC-derived human DA neurons from opioid-dependent subjects to study dopamine dynamics. Brain Behav. 2016;6(8):e00491. doi: 10.1002/brb3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lian X, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A. 2012;109(27):E1848–E1857. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lian X, et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat Protoc. 2013;8(1):162–175. doi: 10.1038/nprot.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tohyama S, et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013;12(1):127–137. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 53.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391(2):85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.