

Abstract
Recent findings have highlighted the role of microglia in orchestrating normal development and refining neural network connectivity in the healthy CNS. Microglia are not only vital cells in maintaining CNS homeostasis, but also respond to injury, infection, and disease by undergoing proliferation and changes in transcription and morphology. A better understanding of the specific role of microglia in responding to viral infection is complicated by the presence of nonmicroglial myeloid cells with potentially overlapping function in the healthy brain and by the rapid infiltration of hematopoietic myeloid cells into the brain in diseased states. Here, we used an inhibitor of colony-stimulating factor 1 receptor (CSF1R) that depletes microglia to examine the specific roles of microglia in response to infection with the mouse hepatitis virus (MHV), a neurotropic coronavirus. Our results show that microglia were required during the early days after infection to limit MHV replication and subsequent morbidity and lethality. Additionally, microglia depletion resulted in ineffective T cell responses. These results reveal nonredundant, critical roles for microglia in the early innate and virus-specific T cell responses and for subsequent host protection from viral encephalitis.
Keywords: Immunology, Virology
Keywords: Adaptive immunity, Innate immunity, Macrophages
Introduction
Microglia are CNS-resident mononuclear phagocytic cells typified by a unique ramified shape and distinctive gene expression (1, 2). Unlike blood-derived macrophages, however, most microglia are derived from a yolk sac progenitor and seed the brain early in development (3, 4). Recent work suggests that microglia serve their purpose by maintaining homeostasis in the brain. Indeed, microglia are important for neurogenesis, synapse maintenance, and neuronal function, indicating that these cells maintain a healthy brain by a multitude of functions (5–9).
As resident cells, microglia are assumed to help orchestrate the immune response to pathogen infection of the brain. While often touted as immune sentinels, little is known about how or if microglia engage in this function (10, 11). Microglia function is compromised in human genetic diseases such as Nasu-Hakola (TREM2-DAP12 mutations) and other adult-onset dementias (12, 13) and may be functionally impaired under certain physiological conditions (14). However, genetic disorders resulting in microglia dysfunction do not result in increased susceptibility to CNS infections and instead result in neurodegeneration (12, 13). While nonparenchymal macrophages of the brain may be the logical cells to respond to a pathogen introduced into the CNS through the bloodstream, these cells would not protect the brain from viruses that enter the CNS by transport through neurons. As the only myeloid cells present within the brain parenchyma, microglia are positioned to respond to a pathogen at this location. As such, the importance of microglia in the host response to virus infections may be partially dependent on the pathogen and route of entry.
Mice infected with the neuroattenuated rJ2.2 strain of the murine coronavirus, mouse hepatitis virus (MHV), develop mild acute encephalitis and acute and chronic demyelinating diseases (15). Approximately 90% of mice survive the acute infection, with demyelination occurring 14–21 days after infection in survivors (15, 16). Infection of the CNS with MHV results in the secretion of inflammatory cytokines and chemokines including type I IFNs (IFN-I), CCL2, TNF, and IL-6 (17–20). IFN-I are protective to the host, as treatment with exogenous IFN-α or IFN-β limits viral replication, and infection of mice genetically defective in IFN-I signaling converts a nonlethal coronavirus infection to one that is lethal (21–23). As a consequence of secretion of cytokines and chemokines, MHV infection of the CNS results in the recruitment of innate and adaptive immune cells to the brain. While large numbers of monocytes/macrophages infiltrate the CNS after infection, clodronate liposome–mediated depletion of these cells resulted in increased mortality but did not alter the viral load within the CNS, indicating that hematogenously derived monocytes/macrophages were not required for efficient viral clearance (24). Virus-specific CD8+ T cells, detectable within the brain by day 6 or 7 post infection (p.i.), are critical for viral clearance (25, 26). CD8+ T cells effect clearance by both cytolytic and noncytolytic mechanisms (27–29). Virus-specific CD4+ T cells are also important in viral clearance and enhance immune activity in the brain by secreting IFN-γ (28, 29) and promoting upregulation of MHC II on microglia (26). In addition to affording protection, MHV-specific CD4+ T cells are also pathogenic (30). How microglia affect this multifaceted T cell immune response to MHV is unknown.
The functions that microglia may use in responding to viruses are not well described. Microglia could function by initiating the IFN-I response, neutralizing virus by phagocytizing infected cells and/or virus, providing necessary signals to initiate the innate immune response via cytokine or chemokine secretion, and presenting antigen to, or stimulating T cells within, the brain. As an immunologically relevant cell type resident in the brain, microglia may be important for limiting replication of an invading pathogen. Complicating the study of microglia during infection is that, in cases of neuroinflammation, extensive monocyte/macrophage infiltration occurs in the brain. Studies separating the actions of microglia from those of infiltrating myeloid cells are difficult, because activated microglia undergo phenotypic changes that render them morphologically similar to infiltrating mononuclear phagocytic cells.
To understand the roles that microglia play in a neurotropic viral infection, how these roles affect the immune response to viruses, and how these roles may differ from those of infiltrating monocytes/macrophages, we used colony-stimulating factor 1 receptor (CSF1R) inhibitor–mediated depletion of microglia. Microglia are dependent on CSF1R signaling for survival, as mice deficient in the CSF1R or its ligand IL-34 lack microglia (3, 31). Treatment of mice with PLX5622, a specific and powerful inhibitor of CSF1R tyrosine kinase activity, results in near-complete depletion of microglia (32–34), facilitating studies of the role of these cells in the host response to a neurotropic virus infection.
Our results show that microglia are most crucial in the response to CNS pathogens at early time points after infection. In the absence of microglia, viral replication occurs more rapidly, allowing the virus to evade the immune response. Microglia are required between days 0 and 6 p.i., with depletion at later points having no effects on survival, suggesting that they play a role predominantly in the early stages of infection. While the absence of microglia does not affect overall inflammatory infiltration into the brain, microglia have an especially prominent role in orchestrating the virus-specific CD4+ T cell response.
Results
Microglia respond to CNS viral infection.
Injury, infection, and neurodegeneration can lead to morphological and molecular changes typical of microglia activation (35, 36). However, this activation profile differs depending on the stimuli received and can result in either harmful or beneficial effects (10, 11, 37). While microglia are often touted as the first responders to a CNS viral infection and are known to respond rapidly to injury (6), there is little direct evidence to show that microglia are needed for an optimal host response to pathogens. Microglia, which express CX3CR1 and can be readily identified in CX3CR1-GFP mice, responded to MHV infection, as evidenced by morphological changes and upregulation of IBA1, an activation marker, in brain locations where viral antigen was detected (Figure 1A). By day 4 p.i., microglia in the olfactory bulb, a region of the brain prominently infected by MHV (38–40), showed increased expression of IBA1, and by day 7 p.i., these cells showed a dramatic increase in IBA1 expression levels and exhibited a less ramified, more macrophage-like morphology (Figure 1A). While these microscopic analyses showed that microglia did indeed respond to viral infection, the molecular mechanisms associated with this response were unknown. To address this question, we used microarray analysis to compare gene expression in microglia harvested from naive mouse brains with those from day-4 p.i. brains. These analyses showed differential expression levels of 595 genes (Figure 1B). As shown by Ingenuity Pathway Analysis, the top 4 pathways changed after infection were IFN signaling, activation of IFN-regulatory factors by cytosolic pattern recognition receptors, granulocyte adhesion and diapedesis, and communication between the innate and adaptive immune system (Figure 1C). Individual genes in these pathways that were among the most upregulated in the array are shown in Figure 1D. These included IFN-induced genes such as Mx2, Ifit1, and Oas2. Genes encoding pattern recognition receptors and chemokines and genes associated with antigen presentation were also upregulated (Figure 1D). Aif1 (encoding IBA1) expression was upregulated in our microarray data set; however, it was upregulated by 1.85-fold, below our fold-change cutoff of 2. Together, these results show that microglia were activated and responded to viral infection of the CNS by changes in morphology and inflammation-associated gene expression.
Figure 1. Microglia respond to CNS infection.

(A) Olfactory bulb sections from CX3CR1GFP/+ mice were immunostained for IBA1 and with DAPI nuclear stain at the indicated time points relative to infection. Images are representative of 3 mice per group. Scale bars: 100 μm. (B) Microglia from naive and day-4 p.i. brains were isolated by flow cytometry and subjected to microarray analysis. Heatmap shows the 595 differentially expressed genes and log2 expression values from blue to red. (C) Pathways most altered in microglia after infection as assessed by Ingenuity Pathway Analysis. Orange bars indicate pathway upregulation; blue bars indicate downregulation of the pathway. Gray bars indicate that no direction of pathway dysregulation was detected. (D) Fold change of selected differentially expressed genes in microglia after infection. Data in B–D were derived from 4 independent samples per group and were analyzed as described in Methods. dpi, days post infection.
A CSF1R inhibitor, PLX5622, depletes microglia.
Next, to separate the roles of microglia and infiltrating macrophages, PLX5622, a CSF1R inhibitor previously reported to deplete microglia (34), was formulated into feed that was provided to mice. Microglia were nearly completely depleted by 3 days after the start of PLX5622 treatment (Figure 2A, gating strategy is shown in Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI97229DS1). We also assessed whether PLX5622 depleted other brain-resident macrophages or myeloid cells in the peripheral lymphoid organs, because these cells express CSF1R. Most CD45+ cells isolated from the naive mouse brain are microglia; consequently, total CD45+ cell numbers isolated from the brain after PLX5622 treatment were greatly decreased (Figure 2B). Neither brain-resident macrophages (Figure 2C), CD11b– immune cells, including T cells and B cells, in the brain (Figure 2D), nor CD11b+ cells in the peripheral lymphoid organs (Figure 2E) were diminished in number by PLX5622 treatment. Additionally, no changes in the number of T cells, B cells, or neutrophils were observed in the peripheral lymphoid organs (Supplemental Figure 2).
Figure 2. PLX5622 treatment specifically depletes microglia.

After PLX5622 treatment, flow cytometry was used to analyze cells from brain (A–D) and spleen (E). (A) Representative flow plot gated on CD45+ cells shows gating for microglia (CD45intCD11b+), macrophages (CD45hiCD11b+), and lymphocytes (CD45+CD11b–). Graph shows a summary of the number of microglia isolated per brain at the indicated time points after initiation of PLX5622 treatment. Number of total CD45+ cells (B), macrophages (C), and lymphocytes (D) found in the brain after 7 days of PLX5622 treatment. Number of CD11b+ cells (E) within the spleen after 7 days of PLX5622 treatment. Data in B–E represent combined results from 3 independent experiments, with a combined total of 8 mice per group. Statistical significance was determined using Mann-Whitney U tests. **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Microglia protect against MHV infection.
To investigate when microglia were required for an optimal outcome, we administered PLX5622 before and during MHV infection. Mice treated with PLX5622 beginning 7 days before infection and continuing to the end of the experiment uniformly died, compared with a 10%–20% mortality rate in infected mice fed normal chow (Figure 3, A and B). To assess the period of microglia action after infection, we varied the timing of drug administration relative to infection (Figure 3, A and B). PLX5622 administration starting on day 0 p.i. and continuing throughout infection resulted in a 90% mortality rate. When PLX5622 was started 3 days p.i. and remained in the feed for the duration of the experiment, approximately 40% of the mice survived. PLX5622 administration, commencing on day 6 p.i., had no effect on survival. Published studies have shown that microglia depletion after treatment and repletion after PLX5622 removal occurred rapidly (34). We confirmed that this is the case by showing that microglia could be replenished within 3 days after removal of PLX5622 (Supplemental Figure 3). When PLX5622 was given 7 days before infection and removed 3 days p.i., all mice succumbed to infection, as was observed with treatment for the duration of the experiment (Figure 3, C and D). Survival rates increased to approximately 30% if the drug was removed on the day of infection, while removal of the drug 2 days before infection resulted in 60% survival. Collectively, these results indicate that microglia provide critical antiviral roles between days 0 and 6 p.i.
Figure 3. Microglia protect mice from CNS infection.

Schematic describing setup of survival experiments (A and C) assessing PLX5622 treatment in relation to intracranial infection with 700 PFU MHV (day 0). Survival curve in B corresponds to data in A; survival curve in D corresponds to data in C. Data are representative of 2 to 5 independent experiments, with 5 to 15 mice per group. P < 0.05, by log-rank test when compared with control survival curves, except when PLX5622 treatment began on day 6. (E) Mice were treated with PLX5622 or control for 7 days and then inoculated intranasally with 3 × 104 PFU rJ or N1347A. Data are combined from 2 independent experiments and represent a total of 10 mice per group. Survival curves were assessed for statistical significance by log-rank test. PLX5622-treated, rJ-infected mice had significantly shorter survival than did mice in all the other groups, while mock-treated, N1347A-infected mice survived significantly longer (P < 0.01) than did mice in the other 3 groups.
To assess the effects of microglia depletion on infection with a highly neurovirulent virus, we infected PLX5622-treated mice with the JHM strain of MHV (recombinant version, referred to herein as rJ). Mice treated with PLX5622 had a decreased time-to-death after rJ infection (Figure 3E). To further assess the role of microglia in limiting viral replication, we infected PLX5622-treated mice with another neuroattenuated virus, N1347A, an rJ mutant lacking macrodomain enzymatic activity (41). Mice infected with N1347A had minimal mortality and weight loss as well as diminished viral titers compared with those infected with WT virus (41). When treated with PLX5622, the lethality of N1347A was increased to WT rJ levels (Figure 3E). Together, these results indicate that microglia are critical for limiting the lethality of neurotropic viruses.
A lack of microglia leads to increased viral replication in the brain.
To assess the potential role for microglia in limiting virus replication, we determined viral loads within the brain. A lack of microglia resulted in increased viral titers by day 3 p.i. compared with those in control mice (Figure 4A). Staining of sections from the olfactory bulb and brainstem with nucleocapsid (N) protein mAb was consistent with the increased titers, demonstrating a larger number of infected cells in the PLX5622-treated mice (Figure 5, A and B). There was a substantial amount of inter-mouse variability, such that the differences were not statistically significant. Peak viral replication occurred on day 5 p.i. in MHV-infected mice and then declined. However, in microglia-depleted mice, viral titers peaked on day 3 p.i. and remained elevated on day 5 p.i. Additionally, the absence of microglia diminished the kinetics of virus clearance; viral titers decreased only marginally by day 7 p.i. in PLX5622-treated mice compared with titers in untreated control mice (Figure 4A).
Figure 4. Delayed virus clearance in microglia-depleted animals.

Mice were treated with PLX5622 for 7 days prior to intracranial MHV infection. (A) Brain virus titers were determined on the indicated days p.i. by plaque assay. (B) Protein levels of IFN-α1, IFN-β, and IL-6 in the brain were determined 5 days p.i. (C) Expression levels of viral genomic RNA, IFN-α4, IFN-β, and IL-6 in the brains of infected mice as assessed by qPCR on the indicated days p.i. IFN-I and IL-6 levels generally correlated with those of viral genomic RNA (Table 1). Data represent the mean ± SEM. Data in A were combined from 3 independent experiments, with a combined total of 8 to 11 mice per group. Data shown in B were combined from 2 independent experiments. Data in C are representative of 3 independent experiments, with 10 mice per group. *P < 0.05 and **P < 0.01, by Mann-Whitney U test.
Figure 5. Increased viral protein in neurons of microglia-depleted brains.

Mice were treated with PLX5622 for 7 days prior to intracranial infection with MHV. (A) Viral N protein and IBA1 immunostaining of the brainstem and (B) olfactory bulb in control and PLX5622-treated mice on day 5 p.i. (C) NeuroTrace and N protein staining of the brainstem and olfactory bulb on day 5 p.i. and quantification of N protein–positive neurons. (D) NeuroTrace and N protein staining of brainstem on day 5 p.i. Yellow arrow indicates an N protein–positive, NeuroTrace-negative cell; white arrows indicate double-positive cells. Original magnification, ×20 (A–C) and ×40 (D). Images are representative of 4 to 5 mice.
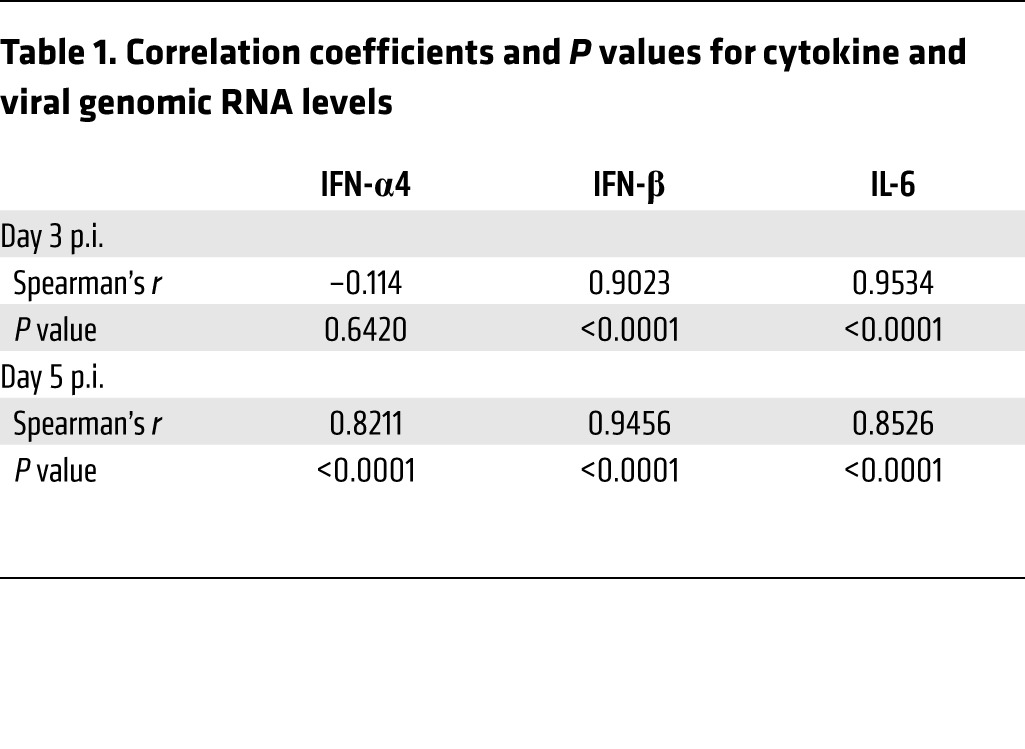
Given that microglia respond to infection by upregulating pathways associated with immune activation (microarray analyses in Figure 1), we next determined protein and mRNA levels of IFN-I and IL-6. IFN-I is critical for initiating the antiviral immune response, and IL-6 is a pleiotropic, proinflammatory immune mediator. After infection, IFN-I protein levels were modestly higher in PLX5622-treated mice, but these changes were not statistically significant. Protein levels of IL-6 were unchanged in the absence of microglia (Figure 4B). Assessment of these cytokines by quantitative PCR (qPCR) showed an increase in expression of IFN-β, IFN-α4, and IL-6 in PLX5622-treated animals on days 3 and 5 p.i. (Figure 4C). Together, these results indicate that the absence of microglia did not prevent the host from initiating an innate immune response and that other cell types produced these cytokines when microglia were depleted. The increase in viral load seen in Figure 4A was additionally confirmed by qPCR of genomic viral RNA (Figure 4C). Notably, the increased cytokine expression seen in PLX5622-treated mice was probably a consequence of increased viral load in the brain; statistical analysis using Spearman’s correlation showed that mRNA levels of these cytokines were positively correlated with those of viral genomic RNA on days 3 and 5 p.i., except for IFN-α4 on day 3 p.i. (Table 1).
Table 1. Correlation coefficients and P values for cytokine and viral genomic RNA levels.
To further investigate the consequences of microglia depletion, we next examined whether there were differences in the extent of neuronal infection, which could contribute to enhanced morbidity. Using NeuroTrace, a fluorescent Nissl stain, to identify neurons, we identified increased numbers of infected neurons in the brainstem but not the olfactory lobe of PLX5622-treated mice (Figure 5, C and D). These differences did not reach statistical significance, reflecting inter-mouse variability. Collectively, these results indicate that functional microglia are required for controlling virus replication and for minimizing disease development in the infected brain.
Macrophages are affected by CSF1R inhibitor treatment.
Next, to further address how microglia function to protect mice from MHV infection, we assessed the effect of their depletion on inflammatory cell infiltration into the brain. On day 3 p.i., total numbers of CD45+ cells were decreased, reflecting microglia depletion. However, by day 7 p.i., no differences were observed, indicating that blood-derived cells became predominant and that recruitment was not microglia dependent (Figure 6, A and D).
Figure 6. Immune cells infiltrate the brain after microglia depletion.

Mice were treated with PLX5622-containing or control feed for 7 days prior to intracranial infection with 700 PFU MHV. Cells from brains and DCLNs were assessed on the indicated days p.i. (A and D) Representative flow plots show CD45+ cell populations in the brain on the indicated days p.i. Graphs show a summary of flow cytometric data on cells isolated from mouse brains (A, B, D, and E) and DCLNs (C and F) on day 3 (A–C) or day 7 (D–F) p.i. Data represent the mean ± SEM and are from 3 independent experiments, with a combined total of 8 to 9 mice per group. Statistical significance was determined using Mann-Whitney U tests. *P < 0.05.
To assess the potential role of microglia in initiating the immune response in the deep cervical lymph nodes (DCLNs), the draining LNs of the brain, we measured total cell numbers. We detected no significant differences in total cell numbers in the DCLNs after PLX5622 treatment (Figure 6, C and F), suggesting that the lack of microglia does not grossly affect the initiation of the immune response. We also measured PLX5622 effects on macrophage numbers in the brain, since, while macrophages were not depleted by PLX5622 (Figure 2), they express CSF1R and might be affected by the drug (42). We detected no differences in the number of monocytes/macrophages on day 3 p.i. compared with numbers in control-treated mice (Figure 6B). However, by day 7 p.i., monocyte/macrophage numbers in the infected brain were increased (Figure 6E), suggesting that microglia depletion resulted in a compensatory increase in hematogenously derived macrophages. Monocytes/macrophages within the infected, microglia-depleted CNS, compared with those in untreated mice, had lower expression of MHC II and higher expression of Ly6C (Figure 7, A and B), indicating that they were less mature (43). To further assess changes in macrophages caused by PLX5622 treatment, we performed microarray analysis of day-4 p.i. brain-derived macrophages. Microarray analysis of monocytes/macrophages in control and PLX5622-treated groups showed differential expression of 235 genes (Figure 7C). Among the upregulated genes were S100a8/a9, upregulated in multiple inflammatory conditions (44), and the chemokines CCL4 (Ccl4) and CCL5 (Ccl5) (Figure 7D) (45). The changes in gene expression could be due to indirect or direct effects of PLX5622 on macrophages, or may reflect changes in macrophages that compensated for the lack of microglia.
Figure 7. Macrophages are functionally altered by PLX5622 treatment.

Mice were treated with PLX5622 for 7 days prior to intracranial MHV infection. (A and B) Decreased MHC II (A) and increased Ly6C (B) levels on microglia and macrophages from PLX5622-treated mice on day 7 p.i. Data represent the mean ± SEM. (C) Macrophages were isolated from 3 mouse brains per group on day 4 p.i. and subjected to microarray analysis. A heatmap was generated from all genes with significant differences in expression, with 235 genes differentially expressed (log2 expression values are shown from blue to red). (D) Fold change of selected differentially expressed genes in macrophages after PLX5622 treatment. Data in A and B are representative of 3 independent experiments, with 3 to 5 mice per group. *P < 0.05 and ****P < 0.0001, by Mann-Whitney U test.
Microglia depletion alters the CD4+ T cell response to viral infection, especially in the brain.
PLX5622 treatment decreased survival, even when treatment began as late as day 3 p.i., indicating that microglia contributed to the antiviral immune response after the innate response was initiated and suggesting an effect on the adaptive immune response. Priming of T cells occurs predominantly in the DCLNs, leading to the initial proliferation of virus-specific T cells and subsequent recruitment of virus-specific T cells to the brain (46). As such, we next examined the adaptive immune response in the DCLNs. In PLX5622-treated mice, CD8+ T cell numbers were unaltered in the DCLNs (Supplemental Figure 4A), as were the numbers of virus-specific CD8+ (Supplemental Figure 4C) and CD4+ T cells (Supplemental Figure 4D). However, total numbers of CD4+ T cells and FOXP3+ Tregs in the DCLNs were decreased (Supplemental Figure 4, B and E) in microglia-depleted mice, although the difference in Tregs did not reach statistical significance. These results suggested that, while priming of the virus-specific T cell response was not substantially altered after microglia depletion, PLX5622 treatment diminished aspects of the CD4+ T cell response.
These differences between control and PLX5622-treated mice were accentuated in the brain, the site of infection. Flow cytometric analysis showed increases in the frequency and total number of CD8+ T cells in the brains of PLX5622-treated animals (Figure 8A), but these differences were not statistically significant. Strikingly, the percentage and total numbers of CD4+ T cells in the brain were decreased (Figure 8B), as were Treg frequencies and numbers (Figure 8C). We next assessed whether the virus-specific T cell response was similarly affected. There were no differences in the frequencies of brain-derived CD8+ T cells responding to the MHV-specific immunodominant S510 epitope. The total number of virus-specific CD8+ T cells showed a trending increase (Figure 8D) after microglia depletion, reflecting the increased total number of CD8+ T cells in the brain. The virus-specific CD4+ T cell response, evaluated by measuring IFN-γ expression after stimulation with M133 peptide, was greatly diminished in frequency and number (Figure 8E). While the total number of virus-specific CD4+ T cells was decreased, the remaining cells remained polyfunctional, as PLX5622 treatment did not affect the frequency or number of IFN-γ–expressing CD4+ or CD8+ T cells that dually expressed TNF or IL-10 (Figure 8, F–I).
Figure 8. Altered T cell response in the brains of microglia-depleted mice.

Mice were treated with PLX5622 for 7 days prior to viral inoculation. In A–I and L, mice were inoculated via intracranial injection with MHV. In A–I, immune cells in the brain were assessed by flow cytometry on day 7 p.i. Frequency and number of CD8+ T cells (A), CD4+ T cells (B), and Tregs (C) in the brain. (D) Virus-specific CD8+ T cell frequency and number were determined by IFN-γ ICS after stimulation with S510 peptide. (E) Virus-specific CD4+ T cell frequency and number were determined by IFN-γ ICS after stimulation with M133 peptide. IL-10 (F and I) and TNF (G and H) expression in virus-specific (IFN-γ+) CD4+ and CD8+ T cells, respectively. (J and K) Virus-specific CD4+ and CD8 + T cells in the spleen on day 7 after i.p. injection of rJ. (L) Survival of Rag1–/– mice. Data indicate the mean ± SEM and represent combined data from 3 to 5 independent experiments, with a combined total of 9 to 17 mice per group. Statistical significance determined using Mann-Whitney U tests. *P < 0.05, **P < 0.01, and ***P < 0.001. The significance in differences in survival curves was assessed by log-rank test.
This dramatic decrease in the number of virus-specific CD4+ T cells could result from the effects of PLX5622 on microglia, or alternatively, and less likely, on macrophages. To address the latter possibility, PLX5622-treated mice were intraperitoneally injected with rJ, resulting in a subclinical infection but also in the generation of a virus-specific T cell response in the spleen (47). PLX5622 treatment did not change the number of virus-specific CD4+ or CD8+ T cells after intraperitoneal infection with rJ (Figure 8, J and K), indicating that the drug did not significantly impact antigen presentation after peripheral infection and suggesting that the lack of microglia was essential in the modified and possibly dysfunctional CD4+ T cell response after MHV infection.
Together, these results support the notion that microglia are required for an optimally protective T cell response. To assess this directly, mice deficient in recombination-activating gene 1 (Rag1–/–), which lack B and T cells, were given control feed or feed containing PLX5622 prior to MHV infection. Rag1–/– mice treated with PLX5622 died slightly more rapidly after infection than did mice given control feed. While these results were not statistically significant (Figure 8L), they were consistent with a role for microglia in the innate immune response. However, the results also highlight the key role that the virus-specific T cells have in protection and, together with the other results described above, demonstrate that microglia are required for the development of an optimal antiviral T cell response.
Discussion
Our results show that microglia play nonredundant and unique roles in host survival during infection with a neurotropic virus infection. Because of their residence in the CNS, microglia potentially serve as sentinel cells able to detect invasive pathogens. They may also contribute to host defense by phagocytosing virus or virus-infected cells, facilitating an early host response to virus replication. Using a drug that primarily depletes microglia, we showed that these cells are most important in the earliest stages of infection, consistent with the idea that they play a key role in host protection. In the absence of microglia, virus replication was poorly controlled, and infection of neurons was increased (Figure 4A).
Microarray analysis demonstrated that IFN signaling was the most upregulated pathway in the data set (Figure 1C). However, in experiments in which microglia were depleted, we observed no decrease in the protein levels of IFN-I (Figure 4) within the brain, indicating that microglia were not required for IFN-I production after infection. However, this does not preclude a role for IFN-I signaling in establishing an antiviral environment, especially since microglia are among the first cells to detect and respond to viruses.
Microglia function was required for many, but not all, aspects of the host response. While the numbers of infiltrating, hematogenously derived phagocytic cells such as neutrophils and macrophages were not diminished by treatment with PLX5622, infiltrating macrophages were phenotypically more immature, as evidenced by decreased MHC class II antigen and increased Ly6C expression (Figure 7, A and B). Thus, these cells, which might be expected to provide partial compensation for the absence of microglia, were phenotypically less potent antigen-presenting cells, possibly contributing to the observed suboptimal CD4+ T cell response (Figure 8E).
While the results described above and elsewhere (32, 34, 48) show that PLX5622 has profound effects on microglia survival, CSF1R, the receptor affected by PLX5622, is also expressed on other myeloid cells, including brain perivascular macrophages and infiltrating hematopoietic monocytes/macrophages. At the same time that microglia were depleted, other brain-resident myeloid cells (Figure 2C) and circulating myeloid cells (Figure 2E) were not decreased in numbers after PLX5622 treatment. Rather, after PLX5622 treatment, the number of infiltrating macrophages was increased after MHV infection (Figure 6E), perhaps as a compensatory response to the lack of microglia. To further address the effect of CSF1R inhibition on infiltrating macrophage function, we compared the transcriptional profile of macrophages entering the brain after infection, with or without PLX5622 treatment. Expression of genes associated with macrophage activation was augmented in macrophages obtained from PLX5622-treated nice compared with those from control mice. These results, together with the decreased levels of MHC II and increased levels of Ly6C on macrophages in PLX5622-treated mice (Figure 7), indicate that macrophage maturation and probably function were compromised in the absence of microglia. These results were likely a consequence of microglia depletion, but we cannot rule out the possibility of a direct effect of PLX5622 on the function of macrophages.
Whereas altered macrophage function may contribute to poor outcomes in PLX5622-treated mice, this altered function is unlikely to be the primary cause of increased virus titers in the brain (Figure 4A). In a previous study, systemic treatment with mannosylated liposomes encapsulating dichloromethylene diphosphonate (Cl2MDP, clodronate) depleted peripheral monocytes/macrophages without affecting microglia numbers or virus titers in the brain (24, 49). Additionally, we measured the potential effects of PLX5622 on macrophages by analyzing clinical disease and T cell responses after intraperitoneal injection of neurovirulent rJ (Figure 8L). We found that intraperitoneal injection of rJ resulted in a subclinical infection and observed no differences in clinical disease or virus-specific T cell responses, again demonstrating minimal direct effects of PLX5622 treatment on cells other than microglia.
Microglia continued to be required for a protective immune response, even after the first few days of infection, because mice given PLX5622 beginning 3 days p.i. had decreased survival. This may reflect the absence of antigen presentation by microglia. Microglia upregulated Tap1 and Itgax, genes involved in antigen presentation (Figure 1D). Additionally, depletion of microglia not only resulted in loss of the major MHC II–expressing cell type in the brain, but also resulted in decreased expression of MHC II on incoming monocytes/macrophages. As MHC II is required for CD4 T cell restimulation and activation, this decrease in MHC II in the brain probably contributed to the diminished virus-specific CD4+ T cell response observed in the absence of microglia. Additionally, the virus-specific CD8+ T cell response in the brain was increased in PLX5622-treated mice compared with that observed in control mice (Figure 8D, P = 0.0559). This modest increase in CD8+ T cell numbers stands in contrast to results observed in suckling mice infected intracerebrally with dengue virus, in which microglia depletion using liposomes containing clodronate resulted in diminished CD8+ T cell recruitment to the brain (50). While both of these studies show effects of microglia depletion on T cell responses in the brain, the differences in effects on specific T cell subsets may reflect the virus under study, the method of microglia depletion (clodronate liposomes versus PLX5622), and/or the age of the mice at the time of infection.
Both virus-specific CD4+ and CD8+ T cells are required for efficient viral clearance in MHV-infected mice (51, 52), with CD8+ T cells effecting viral clearance (27, 28, 53) and CD4+ T cells facilitating CD8+ T cell entrance into the parenchyma (54). The frequency and number of FOXP3+ Tregs were also decreased. Tregs have multiple roles in modulating the inflammatory response including, in the MHV-infected mouse, inhibiting exuberant T cell responses in the brain (46, 55, 56). In the absence of microglia, a diminished Treg response might contribute to increased effector CD4+ and CD8+ T cell responses, which, while ineffective in clearing virus, contributed to increased host damage in our study. The MHV-specific CD4+ T cell response has both protective and pathogenic roles, and the latter is specifically modulated by virus-specific Tregs (46).
The importance of the T cell response in viral clearance was further demonstrated using MHV-infected Rag1–/– mice. Microglia depletion resulted in a small increase in time to death in these mice (Figure 8L), consistent with an effect on the innate immune response. However, these differences were minimal compared with the effects of drug treatment in immunocompetent mice (Figure 3), in which virus was cleared by the antivirus T cell response. Notably, PLX5622-treated B6 and Rag1–/– mice succumbed to the infection at similar rates, highlighting the key role that microglia play in the development of a normal T cell response (compare Figure 3, B and D, with Figure 8L). Specifically, the decreased virus-specific CD4+ T cell response in the absence of microglia indicates that these cells are important for initiating the CD4+ T cell response or in restimulating CD4+ T cells that enter the brain.
Microglia have multiple functions in the CNS, including surveillance of the brain for damaged neurons and neuronal synapses that require pruning (9). Microglia also play a role in autoimmune demyelination (experimental autoimmune encephalomyelitis [EAE]), with a specific role in clearing myelin debris (37). Notably, microglia differed phenotypically and in terms of gene expression from infiltrating macrophages in mice with EAE. Our results also suggest that infiltrating macrophages had functions that were nonredundant with those of microglia and were unable to compensate for their absence. The importance of microglia phagocytosis has been demonstrated in the context of prion disease, in which microglia depletion resulted in accelerated disease progression, probably due to diminished prion clearance (57).
In summary, our results demonstrate a critical role for microglia in the context of viral encephalitis, affecting the initiation of the innate and adaptive immune responses and the kinetics of virus replication and clearance. Viral encephalitis tends to be a disease with sporadic incidence that occurs in only a small percentage of infected patients. Our results raise the possibility that microglia dysfunction contributes to the development of clinical encephalitis in the minority of patients who develop this disease after infection.
Methods
Mice.
Specific pathogen–free C57Bl/6 mice were purchased from Charles River Laboratories, and B6.129P(Cg)-Ptprca Cx3cr1tm1Litt/LittJ (CX3CR1-GFP), and Rag1tm1Mom mice were purchased from The Jackson Laboratory. Mice were maintained in specific pathogen–free facilities at The University of Iowa. Five- to six-week-old male mice were used in all experiments.
PLX5622 treatment.
PLX5622, formulated in an AIN-76A rodent diet at a dose of 1,200 mg/kg standard chow, was prepared by Research Diets and provided by Plexxikon Inc. Mice were provided PLX5622 in their chow beginning 7 days before viral infection, except where indicated otherwise.
Viral infection.
A neuroattenuated variant of the JHMV strain of MHV, rJ2.2 (a recombinant version of the J2.2-V-1 virus, referred to herein as MHV) was propagated in mouse 17Cl-1 cells and titered on HeLa-MHVR cells (58). Five- to six-week-old mice were inoculated intracerebrally with 700 PFU rJ2.2 in 30 μl DMEM. Where specified, 2 × 104 PFU recombinant parental JHMV (rJHMV, referred to herein as rJ) was inoculated into mice intraperitoneally. In other experiments, 3 × 104 PFU rJ or N1347A, an rJ macrodomain point mutant virus (41), was inoculated into mice intranasally. After viral inoculation, mice were examined and weighed daily.
Virus plaque assay.
To titer virus from infected animals, mice were sacrificed and perfused with PBS. Brain tissue was homogenized into PBS using a manual homogenizer and frozen. After thawing, cellular debris was removed by centrifugation, and virus titers in the supernatant were determined on HeLa-MHVR cells as previously described (59).
Isolation of immune cells from brain tissue.
Brains harvested after PBS perfusion were digested with 1 mg/ml collagenase D (Roche) and 0.1 mg/ml DNase I (Roche) at 37°C for 30 minutes after mechanical dissociation. Dissociated, collagenase-treated brain was passed through a 70-μm cell strainer, followed by 37% Percoll gradient centrifugation. Cells were collected from the pellet, washed, and resuspended in culture medium for further analysis.
Antibodies and flow cytometry.
The following antibodies were used in this study: CD4–eFluor 450 (clone RM4-5; eBioscience); CD8-FITC (clone 53-6.7; BD Biosciences); CD11b–eFluor 450 (clone M1/70; eBioscience); CD16/CD32-PerCP-Cy5.5 (clone 93; BioLegend); CD45-PE-Cy7 (clone 30-F11; BioLegend); FOXP3-FITC (clone FJK-16s; eBioscience); I-A/I-E-PerCP-Cy5.5 (clone M5/114.15.2; BioLegend); IFN-γ–APC (clone XMG1.2; BioLegend); IL-10–PE (clone JES5-16E3; BD Biosciences); Ly6C-PE (clone AL-21; BD Biosciences); and TNF-PE, -PE-Cy7 (clone MP6-XT22; BD Biosciences). Data were acquired with a BD FACSVerse and analyzed using FlowJo software (Tree Star). Cells were sorted using a BD FACSAria.
Intracellular cytokine and transcription factor staining.
To detect IFN-γ, TNF, and IL-10 production by virus-specific T cells, immune cells isolated from the LNs or brain were stimulated with cognate peptides in complete RPMI 1640 medium for 6 hours at 37°C. Golgiplug (1 μl/ml; BD) and 2 × 106 antigen-presenting cells (CHB3 cells, B cell line, H-2Db, I-Ab) were included in the culture. MHV-specific peptides encompassing residues 133–147 of the transmembrane (M133) protein and residues 510–518 of the surface (S510) glycoprotein (60, 61) (Bio-Synthesis Inc.) were used at final concentrations of 5 μM (M133) or 1 μM (S510). A FOXP3 Staining Buffer Set (eBioscience) was used for analysis of FOXP3 expression; otherwise, BD Cytofix/Cytoperm and Perm/Wash buffers (BD Biosciences) were used in intracellular cytokine staining (ICS) assays.
RNA extraction, PCR, and primers.
Brains were collected and manually homogenized into TRIzol (Thermo Fisher Scientific). RNA was extracted according to the manufacturer’s instructions. RNA was transcribed into cDNA using Moloney murine leukemia virus reverse transcriptase (MMLV RT; Thermo Fischer Scientific), and expression levels were measured on a QuantStudio qPCR 3 system (Thermo Fisher Scientific). The primer sets used have been previously described (62), and the primer sequences are listed in Supplemental Table 1. The expression levels were normalized to hypoxanthine-guanine phosphoribosyltransferase (HPRT) by the following threshold cycle (Ct) equation: ΔCt = Ct of the gene of interest − Ct of HPRT. All results are shown as a ratio to HPRT calculated as 2−ΔCt.
Measurement of cytokine protein levels.
Mouse brains were flash-frozen in liquid nitrogen before homogenization in T-PER Tissue Protein Extract Reagent (Thermo Fisher Scientific). Samples were centrifuged at 10,000 g for 10 minutes after homogenization. Supernatants were used in protein assays. Protein levels of IFN-α, IFN-β, and IL-6 were determined using Luminex Protein Assays (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Brain histology and immunohistochemistry.
For H&E staining, brains were removed, fixed in zinc formalin, and embedded in paraffin. Tissue sections were stained with H&E.
For immunohistochemical staining, frozen sections were obtained by fixing brains in 4% paraformaldehyde solution followed by immersion in 10%, 20%, and 30% sucrose solutions for cryoprotection prior to freezing by heat displacement. Sections were immersed in PBS before treatment with 0.1% Triton-X in PBS. Sections were then rinsed in PBS. Next, samples were incubated in CAS block (Invitrogen, Thermo Fisher Scientific) followed by incubation in primary antibody diluted in 1% goat serum in PBS overnight at 4°C in a humidity chamber. Primary antibodies against IBA1 (Wako) at 1:2,000 and viral N protein (provided by Michael Buchmeier, University of California Irvine) were used at 1:10,000. Sections were rinsed before incubation with a 1:200 dilution of an appropriate A568- or A488-conjugated goat anti-mouse or anti-rabbit antibody (Thermo Fisher Scientific). After rinsing with PBS, slides were mounted with Vectashield Antifade Reagent containing DAPI (Vectashield Laboratories). NeuroTrace (Thermo Fisher Scientific) staining was performed according to the manufacturer’s protocol. Images were obtained using an Olympus BX61 light microscope. Three images were taken per section, with each section representing different mice. Microscopy images were quantified using ImageJ 64 (NIH) software and manual counting.
Microarray analysis.
Microglia and macrophages were stained with anti-CD45 antibody after isolation from CX3CR1GFP/+ mice as described above. Microglia were sorted on the basis of intermediate expression of CD45 and high expression of GFP. Macrophages were sorted on the basis of high expression of CD45 and intermediate expression of GFP. Transcriptome profiles of FACS-sorted microglia and macrophages were analyzed using Affymetrix Mouse Clariom S arrays. Sample processing and Affymetrix array hybridization were completed by the Genomics Division of the Iowa Institute of Human Genetics using manufacturer-recommended protocols. Briefly, 1 ng total RNA was converted to amplified ds-cDNA using the 3′ IVT Pico Reagent Kit (catalog 902789; Affymetrix). Following fragmentation (fragment size, ~40–70 nt), the samples were labeled with biotin. Biotin-labeled ds-cDNA was then mixed with Affymetrix Eukaryotic Hybridization Buffer, placed onto the Affymetrix Mouse Clariom S Array (part no. 902918), and incubated at 45°C for 18 hours with 60 rpm rotation. The arrays were then washed, stained, and scanned using an Affymetrix Model 3000 scanner (with 7G upgrade). Data were collected using Affymetrix Command Console operating software (AGCC), version 4.1.2. Normalization and signal summarization were performed with Affymetrix Expression Console (EC) Software Build 1.4.4.46.
Data were normalized and median polished using Robust Multichip Average background correction with log2-adjusted values. Significance testing was performed on log2-adjusted values comparing the 2 groups. In the microglia microarray, changes in gene expression with a fold change greater than or equal to 2 and a P value of less than 0.05 after correction for FDR were considered significant. In the macrophage microarray, changes in gene expression with a fold change greater than or equal to 2 and a P value of less than 0.05 were considered significant. Analysis of data and pathway analysis were performed using Partek GS software and Ingenuity Pathway Analysis (QIAGEN Bioinformatics). Microarray data were deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE103380 [microglia] and GSE103379 [macrophages]).
Statistics.
Data are presented as the mean ± SEM unless otherwise indicated. Mann-Whitney U tests were used to analyze differences in means. Log-rank tests were used to determine significant differences in the survival of mice. A P value of less than 0.05 was considered significant. All statistical analysis except microarray analysis was performed using GraphPad Prism, version 7 (GraphPad Software).
Study approval.
All animal protocols were approved by the IACUC of the University of Iowa.
Author contributions
DLW and SP conceived the study; DLW and SP acquired and validated the data; DLW and AS executed the experiments; DLW and DKM analyzed the data; and DLW and SP wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Anthony Fehr (University of Iowa, Iowa City, IA) for critical review of this manuscript. This work was supported in part by grants from the NIH (RO1 N36592) and the National Multiple Sclerosis Society (RG 5340-A-7). The authors would like to acknowledge use of the University of Iowa’s Central Microscopy Research Facility, the Flow Cytometry Facility, the core resources of the Genomics Division of the Iowa Institute of Human Genetics, which are supported by the Vice President for Research and Economic Development, the Holden Comprehensive Cancer Center and the Carver College of Medicine.
Version 1. 01/29/2018
Electronic publication
Version 2. 03/01/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(3):931–943.https://doi.org/10.1172/JCI97229.
Contributor Information
Alan Sariol, Email: alan-sariol@uiowa.edu.
David K. Meyerholz, Email: david-meyerholz@uiowa.edu.
Stanley Perlman, Email: stanley-perlman@uiowa.edu.
References
- 1.Butovsky O, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17(1):131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hickman SE, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16(12):1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomez Perdiguero E, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson LH, Lenz KM. Microglia depletion in early life programs persistent changes in social, mood-related, and locomotor behavior in male and female rats. Behav Brain Res. 2017;316:279–293. doi: 10.1016/j.bbr.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 7.Paolicelli RC, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 8.Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. 2011;14(10):1227–1235. doi: 10.1038/nn.2923. [DOI] [PubMed] [Google Scholar]
- 9.Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crotti A, Ransohoff RM. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity. 2016;44(3):505–515. doi: 10.1016/j.immuni.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 11.Rock RB, et al. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17(4):942–64, table of contents. doi: 10.1128/CMR.17.4.942-964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rademakers R, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2011;44(2):200–205. doi: 10.1038/ng.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paloneva J, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71(3):656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erny D, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18(7):965–977. doi: 10.1038/nn.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergmann CC, Lane TE, Stohlman SA. Coronavirus infection of the central nervous system: host-virus stand-off. Nat Rev Microbiol. 2006;4(2):121–132. doi: 10.1038/nrmicro1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss SR, Leibowitz JL. Coronavirus pathogenesis. Adv Virus Res. 2011;81:85–164. doi: 10.1016/B978-0-12-385885-6.00009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pearce BD, Hobbs MV, McGraw TS, Buchmeier MJ. Cytokine induction during T-cell-mediated clearance of mouse hepatitis virus from neurons in vivo. J Virol. 1994;68(9):5483–5495. doi: 10.1128/jvi.68.9.5483-5495.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parra B, Hinton DR, Lin MT, Cua DJ, Stohlman SA. Kinetics of cytokine mRNA expression in the central nervous system following lethal and nonlethal coronavirus-induced acute encephalomyelitis. Virology. 1997;233(2):260–270. doi: 10.1006/viro.1997.8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rempel JD, Murray SJ, Meisner J, Buchmeier MJ. Differential regulation of innate and adaptive immune responses in viral encephalitis. Virology. 2004;318(1):381–392. doi: 10.1016/j.virol.2003.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun N, Grzybicki D, Castro RF, Murphy S, Perlman S. Activation of astrocytes in the spinal cord of mice chronically infected with a neurotropic coronavirus. Virology. 1995;213(2):482–493. doi: 10.1006/viro.1995.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minagawa H, Takenaka A, Mohri S, Mori R. Protective effect of recombinant murine interferon beta against mouse hepatitis virus infection. Antiviral Res. 1987;8(2):85–95. doi: 10.1016/0166-3542(87)90079-9. [DOI] [PubMed] [Google Scholar]
- 22.Smith AL, Barthold SW, Beck DS. Intranasally administered alpha/beta interferon prevents extension of mouse hepatitis virus, strain JHM, into the brains of BALB/cByJ mice. Antiviral Res. 1987;8(5-6):239–245. doi: 10.1016/S0166-3542(87)80002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cervantes-Barragan L, et al. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood. 2007;109(3):1131–1137. doi: 10.1182/blood-2006-05-023770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xue S, Sun N, Van Rooijen N, Perlman S. Depletion of blood-borne macrophages does not reduce demyelination in mice infected with a neurotropic coronavirus. J Virol. 1999;73(8):6327–6334. doi: 10.1128/jvi.73.8.6327-6334.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marten NW, Stohlman SA, Bergmann CC. Role of viral persistence in retaining CD8(+) T cells within the central nervous system. J Virol. 2000;74(17):7903–7910. doi: 10.1128/JVI.74.17.7903-7910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergmann CC, Parra B, Hinton DR, Chandran R, Morrison M, Stohlman SA. Perforin-mediated effector function within the central nervous system requires IFN-gamma-mediated MHC up-regulation. J Immunol. 2003;170(6):3204–3213. doi: 10.4049/jimmunol.170.6.3204. [DOI] [PubMed] [Google Scholar]
- 27.Lin MT, Stohlman SA, Hinton DR. Mouse hepatitis virus is cleared from the central nervous systems of mice lacking perforin-mediated cytolysis. J Virol. 1997;71(1):383–391. doi: 10.1128/jvi.71.1.383-391.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parra B, et al. IFN-gamma is required for viral clearance from central nervous system oligodendroglia. J Immunol. 1999;162(3):1641–1647. [PubMed] [Google Scholar]
- 29.González JM, et al. Inhibition of interferon-gamma signaling in oligodendroglia delays coronavirus clearance without altering demyelination. Am J Pathol. 2006;168(3):796–804. doi: 10.2353/ajpath.2006.050496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anghelina D, Pewe L, Perlman S. Pathogenic role for virus-specific CD4 T cells in mice with coronavirus-induced acute encephalitis. Am J Pathol. 2006;169(1):209–222. doi: 10.2353/ajpath.2006.051308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. 2012;13(8):753–760. doi: 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elmore MR, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spangenberg EE, et al. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain. 2016;139(Pt 4):1265–1281. doi: 10.1093/brain/aww016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dagher NN, et al. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation. 2015;12:139. doi: 10.1186/s12974-015-0366-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J. Myeloid Cells in the Central Nervous System. Immunity. 2017;46(6):943–956. doi: 10.1016/j.immuni.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salter MW, Beggs S. Sublime microglia: expanding roles for the guardians of the CNS. Cell. 2014;158(1):15–24. doi: 10.1016/j.cell.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 37.Yamasaki R, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. 2014;211(8):1533–1549. doi: 10.1084/jem.20132477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnett EM, Perlman S. The olfactory nerve and not the trigeminal nerve is the major site of CNS entry for mouse hepatitis virus, strain JHM. Virology. 1993;194(1):185–191. doi: 10.1006/viro.1993.1248. [DOI] [PubMed] [Google Scholar]
- 39.Schwob JE, Saha S, Youngentob SL, Jubelt B. Intranasal inoculation with the olfactory bulb line variant of mouse hepatitis virus causes extensive destruction of the olfactory bulb and accelerated turnover of neurons in the olfactory epithelium of mice. Chem Senses. 2001;26(8):937–952. doi: 10.1093/chemse/26.8.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Youngentob SL, Schwob JE, Saha S, Manglapus G, Jubelt B. Functional consequences following infection of the olfactory system by intranasal infusion of the olfactory bulb line variant (OBLV) of mouse hepatitis strain JHM. Chem Senses. 2001;26(8):953–963. doi: 10.1093/chemse/26.8.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fehr AR, Athmer J, Channappanavar R, Phillips JM, Meyerholz DK, Perlman S. The nsp3 macrodomain promotes virulence in mice with coronavirus-induced encephalitis. J Virol. 2015;89(3):1523–1536. doi: 10.1128/JVI.02596-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sasmono RT, et al. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003;101(3):1155–1163. doi: 10.1182/blood-2002-02-0569. [DOI] [PubMed] [Google Scholar]
- 43.Templeton SP, Kim TS, O’Malley K, Perlman S. Maturation and localization of macrophages and microglia during infection with a neurotropic murine coronavirus. Brain Pathol. 2008;18(1):40–51. doi: 10.1111/j.1750-3639.2007.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vogl T, Gharibyan AL, Morozova-Roche LA. Pro-inflammatory S100A8 and S100A9 proteins: self-assembly into multifunctional native and amyloid complexes. Int J Mol Sci. 2012;13(3):2893–2917. doi: 10.3390/ijms13032893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chernov AV, et al. The calcium-binding proteins S100A8 and S100A9 initiate the early inflammatory program in injured peripheral nerves. J Biol Chem. 2015;290(18):11771–11784. doi: 10.1074/jbc.M114.622316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao J, Zhao J, Perlman S. Virus-specific regulatory T cells ameliorate encephalitis by repressing effector T cell functions from priming to effector stages. PLoS Pathog. 2014;10(8):e1004279. doi: 10.1371/journal.ppat.1004279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu GF, Dandekar AA, Pewe L, Perlman S. CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J Immunol. 2000;165(4):2278–2286. doi: 10.4049/jimmunol.165.4.2278. [DOI] [PubMed] [Google Scholar]
- 48.Elmore MR, Lee RJ, West BL, Green KN. Characterizing newly repopulated microglia in the adult mouse: impacts on animal behavior, cell morphology, and neuroinflammation. PLoS ONE. 2015;10(4):e0122912. doi: 10.1371/journal.pone.0122912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma Y, et al. Macrophage depletion reduced brain injury following middle cerebral artery occlusion in mice. J Neuroinflammation. 2016;13:38. doi: 10.1186/s12974-016-0504-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsai TT, et al. Microglia retard dengue virus-induced acute viral encephalitis. Sci Rep. 2016;6:27670. doi: 10.1038/srep27670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Houtman JJ, Fleming JO. Dissociation of demyelination and viral clearance in congenitally immunodeficient mice infected with murine coronavirus JHM. J Neurovirol. 1996;2(2):101–110. doi: 10.3109/13550289609146543. [DOI] [PubMed] [Google Scholar]
- 52.Williamson JS, Stohlman SA. Effective clearance of mouse hepatitis virus from the central nervous system requires both CD4+ and CD8+ T cells. J Virol. 1990;64(9):4589–4592. doi: 10.1128/jvi.64.9.4589-4592.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parra B, Lin MT, Stohlman SA, Bergmann CC, Atkinson R, Hinton DR. Contributions of Fas-Fas ligand interactions to the pathogenesis of mouse hepatitis virus in the central nervous system. J Virol. 2000;74(5):2447–2450. doi: 10.1128/JVI.74.5.2447-2450.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phares TW, Stohlman SA, Hwang M, Min B, Hinton DR, Bergmann CC. CD4 T cells promote CD8 T cell immunity at the priming and effector site during viral encephalitis. J Virol. 2012;86(5):2416–2427. doi: 10.1128/JVI.06797-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao J, Zhao J, Fett C, Trandem K, Fleming E, Perlman S. IFN-γ- and IL-10-expressing virus epitope-specific Foxp3(+) T reg cells in the central nervous system during encephalomyelitis. J Exp Med. 2011;208(8):1571–1577. doi: 10.1084/jem.20110236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Savarin C, Bergmann CC, Hinton DR, Stohlman SA. Differential Regulation of Self-reactive CD4+ T Cells in Cervical Lymph Nodes and Central Nervous System during Viral Encephalomyelitis. Front Immunol. 2016;7:370. doi: 10.3389/fimmu.2016.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu C, et al. A neuroprotective role for microglia in prion diseases. J Exp Med. 2016;213(6):1047–1059. doi: 10.1084/jem.20151000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pewe L, et al. A severe acute respiratory syndrome-associated coronavirus-specific protein enhances virulence of an attenuated murine coronavirus. J Virol. 2005;79(17):11335–11342. doi: 10.1128/JVI.79.17.11335-11342.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eckerle LD, et al. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010;6(5):e1000896. doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castro RF, Perlman S. CD8+ T-cell epitopes within the surface glycoprotein of a neurotropic coronavirus and correlation with pathogenicity. J Virol. 1995;69(12):8127–8131. doi: 10.1128/jvi.69.12.8127-8131.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haring JS, Pewe LL, Perlman S. High-magnitude, virus-specific CD4 T-cell response in the central nervous system of coronavirus-infected mice. J Virol. 2001;75(6):3043–3047. doi: 10.1128/JVI.75.6.3043-3047.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Athmer J, Fehr AR, Grunewald M, Smith EC, Denison MR, Perlman S. In situ tagged nsp15 reveals interactions with coronavirus replication/transcription complex-associated proteins. MBio. 2017;8(1):e02320-16. doi: 10.1128/mBio.02320-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.