

Abstract
Dravet syndrome (DS) is a severe childhood-onset epilepsy commonly due to mutations of the sodium channel gene SCN1A. Patients with DS have a high risk of sudden unexplained death in epilepsy (SUDEP), widely believed to be due to cardiac mechanisms. Here we show that patients with DS commonly have peri-ictal respiratory dysfunction. One patient had severe and prolonged postictal hypoventilation during video EEG monitoring and died later of SUDEP. Mice with an Scn1aR1407X/+ loss-of-function mutation were monitored and died after spontaneous and heat-induced seizures due to central apnea followed by progressive bradycardia. Death could be prevented with mechanical ventilation after seizures were induced by hyperthermia or maximal electroshock. Muscarinic receptor antagonists did not prevent bradycardia or death when given at doses selective for peripheral parasympathetic blockade, whereas apnea, bradycardia, and death were prevented by the same drugs given at doses high enough to cross the blood-brain barrier. When given via intracerebroventricular infusion at a very low dose, a muscarinic receptor antagonist prevented apnea, bradycardia, and death. We conclude that SUDEP in patients with DS can result from primary central apnea, which can cause bradycardia, presumably via a direct effect of hypoxemia on cardiac muscle.
Keywords: Neuroscience
Keywords: Epilepsy, Respiration
Introduction
Patients with epilepsy have a high risk of sudden death (1). The underlying mechanisms of this sudden unexplained death in epilepsy (SUDEP) are unknown, but it is believed that most deaths occur immediately after a seizure (2, 3). To identify premorbid biomarkers of patients at highest risk for SUDEP, to develop targeted preventative treatments, and to recommend effective methods for rescuing patients from near-SUDEP, it is necessary to define the sequence of events that leads from a seizure to death.
A variety of mechanisms have been proposed for SUDEP (1, 2, 4–6), including cardiac arrhythmias (7–10), dysfunction of autonomic control (11–15), apnea/hypoventilation (3, 16–21), airway obstruction (22), pulmonary edema (23), brain stem spreading depolarization (BSD) (24), and postictal generalized EEG suppression (PGES) (25). Many investigators have focused on cardiac tachyarrhythmias as the cause of death, in part because of an association between SUDEP and mutations of genes expressed in the heart, such as those that underlie long QT syndrome (10, 26–28). In some cases these mutations may cause both arrhythmias and seizures (10, 29, 30), whereas in other cases the mutations may predispose patients to arrhythmias if they have seizures due to other reasons. In patients who are monitored in epilepsy monitoring units (EMUs), nonfatal seizures can sometimes induce bradycardia, asystole, O2 desaturation, or apnea (4, 5, 8, 16, 17). A small number of patients (n = 11) who died of SUDEP while being monitored in EMUs was recently reported (2). In those patients, the typical sequence of events was a generalized tonic-clonic seizure followed by bradycardia and asystole (2), along with decreased respiratory rate leading to terminal apnea. For those patients, measurements of blood pressure, ventilation, CO2 levels, or O2 saturation were not made, so there are still important unanswered questions about mechanisms, such as whether there was hypotension or whether lung inflation was effective. It is also not clear what happened during the seizures, as respiratory movements and the ECG were obscured by convulsive movements. Most of the monitored patients who experienced SUDEP had temporal lobe epilepsy, so it is not clear whether those data are relevant to SUDEP in patients with other types of epilepsy. However, all of these SUDEP cases involved terminal apnea first, followed by terminal asystole.
Dravet syndrome (DS) is an intractable epileptic encephalopathy in which febrile seizures typically appear in the first 6 to 12 months of life, followed by other types of severe, refractory seizures and cognitive impairment (31). In 70%–95% of cases, DS is due to mutations of SCN1A (32–34), which encodes the sodium channel Nav1.1. SUDEP is particularly common in patients with DS, causing death in 5%–10% of cases, most commonly during the first few years of life (31, 35, 36).
Clinical data have pointed to cardiac mechanisms being important for SUDEP in DS. For example, mutations of SCN1A in patients with DS cause increased QT and P wave dispersion and a decrease in heart rate variability (HRV) (12, 37). However, there are no published reports of ECG changes after fatal or nonfatal generalized seizures in patients with DS, other than a single case report after status epilepticus (38). Peri-ictal breathing has not been measured in patients with DS.
Mouse models of DS have been generated by knockout of Scn1a, or with mutations such as R1407X, which is seen in some patients and leads to truncation of Nav1.1 and functional inactivation (39). The phenotype of the Scn1aR1407X/+ mutation and the Scn1a knockout are similar and recapitulate the phenotype of DS in humans (31), including seizures that occur spontaneously and in response to hyperthermia (7, 39, 40). Heat-induced seizures in DS mice are often followed by death (7, 40). In addition to the brain, Nav1.1 is expressed in the heart, including the sinoatrial node (41) and cardiac T tubules (42). Mice with an Scn1aR1407X/+ mutation have cardiac myocyte dysfunction and die spontaneously at a young age after severe bradycardia, which has been interpreted as indicating that these mice die from intrinsic cardiac dysfunction (7). However, selective knockout of Scn1a only in brain interneurons results in seizures and spontaneous death (43). In mice with global knockout of Scn1a, heat-induced seizures are associated with progressively worsening bradycardia followed by terminal asystole (40). Bradycardia and death can be prevented by pretreatment with atropine (40). These data led to a different conclusion — that death in this DS mouse model is due to increased vagal output to the heart (40). However, breathing measurements were not made during spontaneous or heat-induced seizures in either of these DS mouse models, so it is unclear whether abnormal breathing contributes to SUDEP. Here we studied patients with DS and mice with an Scn1aR1407X/+ mutation (39) to evaluate a potential role of postictal breathing dysfunction in SUDEP.
Results
Peri-ictal respiratory dysfunction was common in patients with DS.
Video recordings of generalized seizures from 7 patients with DS and 7 patients with localization-related epilepsy were reviewed by an investigator (G.B. Richerson) blind to the cause of epilepsy. Visualization of chest and abdomen movements, aided by Eulerian video magnification (44), was used to evaluate breathing frequency and pattern (see Methods; Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI94999DS1). Patients with DS were significantly more likely to have peri-ictal abnormal breathing consistent with disturbances of respiratory rhythm generation or patterned output (Table 1; P = 0.018, χ2 test), including paradoxical breathing, inspiratory efforts with 2 or 3 peaks, ataxic breathing, or apnea of 5 seconds or longer (Figure 1, Supplemental Figure 2, and Supplemental Videos 1 and 2). Many patients produced loud upper airway sounds consistent with obstruction, but this was not significantly more common in either group (Table 1). In all of these cases, breathing abnormalities were transient.
Table 1. Patients with DS had abnormal breathing after seizures compared with patients with localization-related epilepsy (P = 0.018, χ2 test).
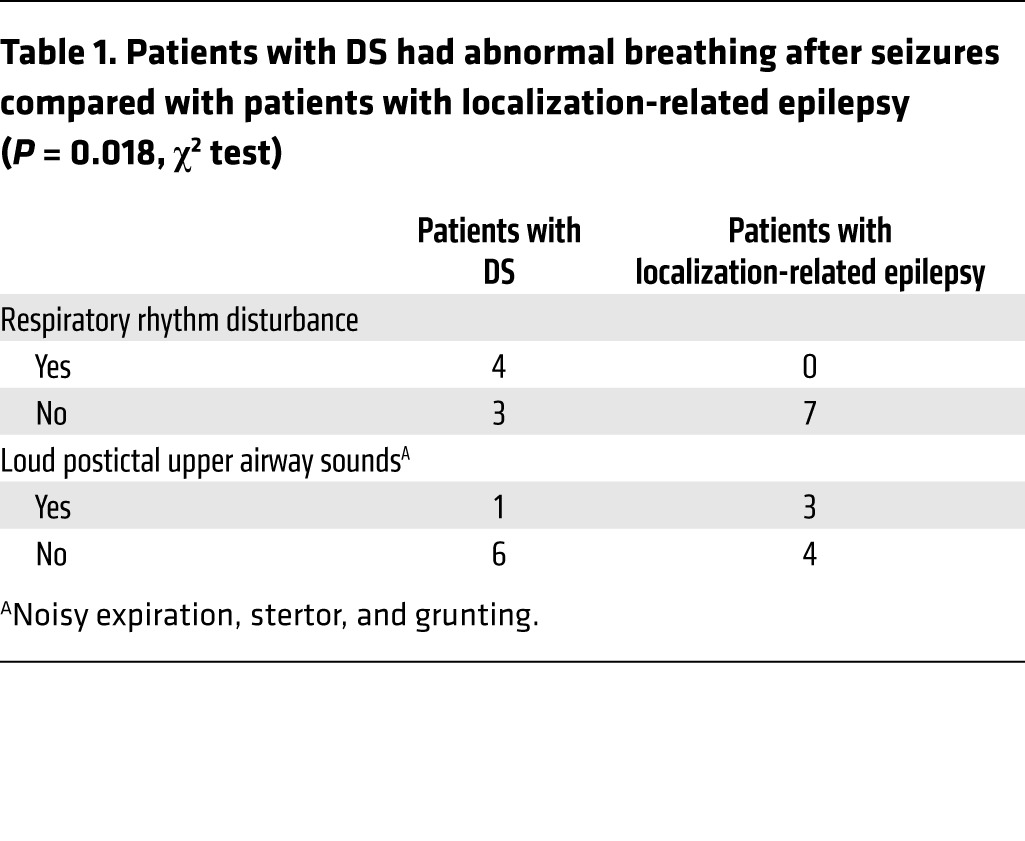
Figure 1. Seizures induce ataxic breathing in a patient with DS.

(A and B) Irregular breathing during and after a seizure. Video frame (A) from Supplemental Video 1 showing region of interest (vertical line pointed to by arrow) used to visualize respiratory movements. Using the methods illustrated in Supplemental Figure 1A, respiratory movements were plotted during and after a generalized seizure (B; traces are contiguous). During the seizure (first 13 seconds), most inspirations had more than one peak. Immediately after the seizure, there was a respiratory pause followed by continued abnormal inspirations lasting until 32 seconds postictal. A normal breathing pattern then resumed with regular, monophasic inspiratory efforts.
Video recordings can reveal valuable information about peri-ictal respiratory patterns, but as was true for previously reported EMU recordings (2), this approach leaves many unanswered questions (see Introduction). To more accurately characterize the abnormalities of peri-ictal breathing, a 9-year-old girl with DS and an associated de novo missense mutation of SCN1A was admitted for recording of video EEG, which was supplemented with recordings of respiratory impedance plethysmography and transcutaneous CO2 (tcCO2) (Figure 2). The patient had 3 consecutive generalized tonic-clonic seizures. The seizures on EEG lasted 48 seconds, 89 seconds, and 90 seconds, separated by 1–3 minutes (Figure 2A). Rectal diazepam (0.5 mg/kg) was given during the second seizure. Between seizures the patient’s eyes were open, but she had a limited response to verbal and tactile stimuli, and the EEG showed continuous slowing with frequent interictal epileptiform discharges. Epileptiform discharges and EEG slowing decreased after the third seizure.
Figure 2. Prolonged postictal hypoventilation in a patient with DS.

(A) Schematic of events while recording cardiorespiratory activity during and after a seizure in a 9-year-old girl with DS. The numbers above the upper line in A are measured values of tcCO2. The upper line shows the approximate time course of the changes in tcCO2. Arrows labeled “b-e” denote the time of recordings shown in parts B–E, respectively. Arrows also denote when supplemental O2 was administered or discontinued, and when the patient was stimulated to cause arousal. Red shading denotes convulsive seizures and orange shading denotes periods of decreased responsiveness and EEG slowing. (B–E) Respiratory impedance plethysmography (Pleth) and ECG during normal breathing when tcCO2 was 43 mmHg (B), between convulsive seizures (C), after the seizures when tcCO2 had risen to 53 mmHg (D), and 44 minutes after the seizures when tcCO2 had risen to 68 mmHg (E). At 2 hours, tcCO2 decreased when the patient was stimulated to arouse, but did not return to baseline until 4 hours.
Before the first seizure, plethysmography revealed normal, regular breathing (Figure 2B), and tcCO2 was 43–44 mmHg. During and after the seizures, the patient developed labored breathing with periods of apnea lasting up to 13 seconds (Figure 2, C–E). After the first seizure, oxygen desaturation occurred and the patient was treated with O2 delivered by a nonrebreather mask at an initial rate of 15 LPM and subsequently adjusted to keep O2 saturation greater than 92%. O2 was removed after the third seizure. tcCO2 increased during the seizures to 53–55 mmHg. Approximately 45 minutes after the third seizure, the patient developed labored breathing. Oxygen desaturation occurred and O2 was given again. At the time, tcCO2 had increased to 68 mmHg and remained at 70 mmHg for the next hour. At 2 hours, a painful stimulus was given that caused the patient to arouse, and tcCO2 declined to just above 50 mmHg where it remained for an additional 2 hours before returning to baseline. During most of the time after the seizures, the patient continued breathing at a rate of 18–22 breaths per minute without apnea (Figure 2E). However, the elevated tcCO2 indicated there was marked hypoventilation, apparently due to decreased tidal volume. It is not clear whether diazepam contributed to the hypoventilation. This case was remarkable for impairment of respiratory CO2 chemoreception that continued long after the seizure. It also demonstrated that supplementary oxygen is not adequate treatment when the primary problem is hypoventilation. Three years after the patient was recorded, she was found face down in bed, blue and pulseless, and could not be resuscitated. An autopsy was performed and the cause of death was given as SUDEP.
Taken together, these data indicate that many patients with DS have peri-ictal respiratory dysfunction that can extend long into the postictal period, including defects in respiratory rhythm generation, impaired central CO2 chemoreception, and loss of airway patency. In the nonfatal episodes documented here, these changes were transient, but the severe defects in breathing in the patient who later died of SUDEP suggest that respiratory changes may be biomarkers of patients at high risk.
Spontaneous seizures caused death in Scn1aR1407X/+ mice due to respiratory arrest.
Heterozygous Scn1aR1407X/+ knockin mice that express a mutation described in 3 unrelated patients with DS were used to define the cause of death after seizures (39). Video recordings were made continuously of 78 Scn1aR1407X/+ mice between the ages of P16 and P60. During that time, 38% of the mice spontaneously died (n = 30 of 78) (Figure 3). There was no difference in mortality between male and female mice (Supplemental Figure 3). Review of the video recordings revealed that all spontaneous deaths occurred after a generalized seizure characterized by tonic hind limb extension. All spontaneous seizures with tonic hind limb extension were fatal.
Figure 3. Survival curve of Scn1aR1407X/+ mice.

Video monitoring revealed that 38% (n = 30 of 78) of Scn1aR1407X/+ mice spontaneously died between P20 and P60. All of these spontaneous deaths occurred after Racine scale 5 generalized seizures with tonic hind limb extension.
To better understand the cause of postictal death in Scn1aR1407X/+ mice than is possible using video recording alone, a custom-designed mouse EMU was used to continuously monitor EEG, ECG, electromyography (EMG), video, and plethysmography. In 4,680 hours of recording from 19 Scn1aR1407X/+ mice, 2 mice died from spontaneous seizures. One mouse that was monitored for 408 hours from P23 to P40 (Figure 4) died after a seizure with tonic hind limb extension. Breathing and heart rate were both normal prior to the seizure. During the seizure, the mouse suddenly developed complete apnea that continued into the postictal period, and it never recovered. There was also a rapid decrease in heart rate shortly after the onset of apnea, but the heartbeat remained above 25%–51% of control for 60 seconds after the onset of apnea and continued for almost 4 minutes after the end of the seizure, with slowly worsening bradycardia eventually leading to terminal asystole. A similar sequence of events was seen in a second mouse monitored for 33 hours starting at P23 (Supplemental Figure 4). These observations suggest that respiratory arrest played a key role in the death of these animals. For both mice, recordings revealed central apnea, as there was no evidence for airway obstruction in the video recording, EMG, or plethysmography trace. Notably, the EEG rapidly became flat after the seizure, consistent with PGES (25). This same observation was reported previously after maximal electroshock (MES) induced seizures in Lmx1bf/f/p mice, which lack over 99% of 5-hydroxytryptamine (5-HT) neurons in the CNS (19).
Figure 4. Death of an Scn1aR1407X/+ mouse (P40) after a spontaneous seizure was due to respiratory arrest followed by secondary bradycardia.

(A) Events leading to death. Traces from top down are EEG, ECG, EMG, and whole-animal plethysmography (Pleth), in each case with paired power spectrum heatmaps. A spontaneous seizure occurred at the time indicated, and this was followed by PGES in the EEG. Shortly after the onset of the seizure, breathing became disrupted. Respiratory activity ceased halfway through the seizure. In contrast, the ECG did not change until near the end of the seizure, after which the frequency and amplitude slowly decreased over the next 4 minutes. Note there was some movement artifact on the plethysmography trace half way through the seizure. (B) Expanded traces of EEG, ECG, and plethysmography at the times labeled 1–5. (C) Respiratory rate and heart rate for the data shown in A and B. Data are discontinuous during the baseline period, because accurate measurements could be made only when the mouse was resting quietly. Data points are continuous from the onset of the seizure, except for one data point during which breathing was obscured by movement artifact.
Auerbach et al. (7) reported episodes of bradycardia and increased HRV in Scn1aR1407X/+ mice up to 48 hours prior to death. They did not measure breathing or EEG during those episodes, so it is not known whether the bradycardia occurred during seizures or was accompanied by apnea. During our experiments, we measured the EEG and breathing, along with the ECG, so we were able to evaluate whether spontaneous episodes of bradycardia were due to seizures or apnea. During a total of 441 hours monitoring EEG, ECG, and plethysmography from the 2 mice that died spontaneously, one had 3 spontaneous nonfatal seizures and the other had 4. All 7 of these seizures were generalized with clonic limb contractions and were classified as Racine scale 5 seizures. Three of these nonfatal seizures included transient episodes of apnea and bradycardia that were less severe than during fatal seizures (Supplemental Figure 5). Bradycardia never occurred without apnea or severely decreased breath amplitude.
Heat-induced seizures also caused respiratory arrest in Scn1aR1407X/+ mice.
As in patients with DS (31), an increase in body temperature can induce seizures in Scn1aR1407X/+ mice (45) and Scn1a-knockout mice (40). It has been reported that heat-induced seizures in DS mice lead to bradycardia followed by death (40). Here we verified that a rise in body temperature induced seizures in Scn1aR1407X/+ mice, with the first seizure occurring at an average of 39.67°C ± 0.97°C (n = 21) and leading to death in 18 of 21 mice after 3.53 ± 0.68 seizures. As reported previously with fatal heat-induced seizures in Scn1a-knockout mice (40), electrographic seizures accompanied by tonic hind limb extension or clonic limb contractions were followed by a decrease in heart rate that eventually resulted in terminal asystole (Supplemental Figure 6). However, we found that fatal seizures also always involved terminal apnea. This apnea occurred precipitously during the seizure or within seconds of the end of the seizure. In contrast, mild to moderate bradycardia often occurred shortly after the onset of apnea, but a heartbeat continued, slowly decreasing over time with terminal asystole not occurring until 3.89 ± 0.92 minutes after the end of seizures. Death could be prevented if mechanical ventilation was initiated within 5–10 seconds of the onset of apnea using a rodent ventilator (MiniVent type 845; Harvard Apparatus) connected to plastic tubing placed over the mouse’s nostrils (n = 5 mice). Death in this group of mice always occurred due to apnea, and bradycardia only occurred if there was apnea.
MES also induced generalized seizures in Scn1aR1407X/+ mice, but these seizures were less likely to cause apnea (4 episodes of apnea after seizures in 34 mice). When apnea did occur, death was prevented by mechanical ventilation in 3 of 4 mice.
High-dose atropine prevented peri-ictal apnea in Scn1aR1407X/+ mice.
It has previously been shown that atropine prevents postictal bradycardia and death in Scn1a-knockout mice after hyperthermia-induced seizures (40). This led to the conclusion that seizures cause fatal bradycardia due to an increase in vagal parasympathetic output and activation of cardiac muscarinic acetylcholine receptors (40). To examine the mechanism by which atropine prevents postictal death, we induced seizures in Scn1aR1407X/+ mice using hyperthermia 30 minutes after pretreatment with 1 mg/kg (i.p.) atropine and compared the seizures to a matched cohort of Scn1aR1407X/+ mice. Atropine (1 mg/kg) did not reduce the likelihood of Racine scale 5 seizures, or the duration or severity of seizures as determined by EEG analysis (see Methods), but did reduce the likelihood of tonic hind limb extension. Death occurred in only 2 of 9 mice treated with atropine (1 mg/kg) compared with 6 of 6 mice treated with vehicle (Figure 5A). Unexpectedly, atropine (1 mg/kg) also prevented apnea in all mice that survived (n = 7 of 9) (Figure 5B). For mice treated with atropine that died (n = 2 of 9), the sequence of events was identical to that of mice treated with vehicle, with immediate and complete apnea beginning near the end of a generalized seizure, followed by slowly progressive bradycardia.
Figure 5. Postictal bradycardia is due to apnea, not increased parasympathetic drive.

(A) Effect of muscarinic antagonists on postictal heart rate after heat-induced seizures (HISs). Untreated mice all died in response to HIS (n = 6). Atropine prevented bradycardia in 7 of 9 mice when given at a dose of 1 mg/kg (i.p.). Methylatropine (1 mg/kg, i.p.) also prevented bradycardia in 6 of 6 mice. Treatment with N-methylscopolamine at very low dose (0.3 μg/kg, i.c.v.) prevented bradycardia in all mice (n = 5). In contrast, atropine (1 mg/kg, i.p.) did not prevent bradycardia or death in 2 of 9 mice, or when given at 0.03 mg/kg (n = 6 of 6; i.p.), a dose selective for blockade of peripheral muscarinic receptors. Bradycardia also occurred after spontaneous seizures (n = 2) and after pretreatment with N-methylscopolamine (1 mg/kg, i.p.; n = 6 of 6). All mice with postictal bradycardia also had apnea, whereas bradycardia did not occur in any mouse without apnea. T = 0 represents the time that apnea began (always during or at the end of seizures) or the end of seizures (if apnea did not occur). Error bars are not shown for the 2 groups with n = 2. (B) Heat-induced seizure in an Scn1aR1407X/+ mouse, 30 minutes after pretreatment with atropine (1 mg/kg, i.p.). During this seizure, there was no apnea or bradycardia, and the mouse did not die.
It is common to use atropine at a dose of 1 mg/kg to block peripheral muscarinic receptors in rodents. However, that dose is 20–50 times higher than is recommended for specific blockade of peripheral muscarinic receptors in mice (0.02–0.05 mg/kg, i.p.) (46–48). Atropine can readily cross the blood-brain barrier (49, 50). After 30 minutes, levels of atropine in the CNS reach more than 35% of serum levels (49). After a dose of 1 mg/kg, atropine levels would be more than enough to block central muscarinic receptors. Muscarinic acetylcholine receptors in the medulla and pons play an important role in respiratory control (51, 52) and central CO2 chemoreception (53, 54), and those receptors in the forebrain alter seizure activity (55).
Quaternary ammonium derivatives of muscarinic receptor antagonists, such as methylatropine and N-methylscopolamine, are used as selective antagonists of peripheral muscarinic receptors, as they are less likely to penetrate the blood-brain barrier (56, 57). N-methylscopolamine (1 mg/kg, i.p.) has previously been shown to prevent bradycardia and death after heat-induced seizures in Scn1a-knockout mice, supporting the hypothesis that postictal bradycardia is due to an increase in vagal tone (40). We replicated this experiment in Scn1aR1407X/+ mice (n = 6), but found that pretreatment with N-methylscopolamine (1 mg/kg, i.p.) 30 minutes prior to inducing seizures with hyperthermia did not prevent apnea, bradycardia, or death (Figure 5A). In contrast, methylatropine (1 mg/kg, i.p.) (n = 6) did prevent bradycardia and death (Figure 5A), but also prevented apnea. Selective blockade of peripheral muscarinic receptors would not be expected to prevent apnea, because the neural circuitry involved in generation of respiratory output is contained entirely within the CNS, as are descending projections from the forebrain that cause apnea during and/or after seizures (18, 20). Instead, the effect of methylatropine is likely to be due to penetration into the CNS. After equivalent doses of radioactive atropine or methylatropine, both drugs accumulate in the CNS (58). Systemic methylatropine has effects on cortical activity that are dependent on the dose and treatment duration, with 1 mg/kg (i.v.) inhibiting EEG activity by 70% within 20–30 minutes (59). Our data also indicate that high doses of quaternary ammonium anticholinergic agents can have central effects, and these high doses are protective against apnea.
Selective blockade of peripheral muscarinic receptors does not prevent postictal bradycardia.
An atropine dose of 0.02–0.04 mg/kg (i.p.) is recommended to block peripheral parasympathetic activity in mice (46–48). Measurement of HRV can be used to determine whether muscarinic receptor antagonists are effective at blocking vagal parasympathetic output to the heart. The most common type of HRV, respiratory sinus arrhythmia, is due to changes in parasympathetic output caused by breathing (60–64). This results in part from an increase in intrathoracic pressure during expiration, which leads to an increase in systemic blood pressure and reflex bradycardia (and opposite changes during inspiration). Blocking parasympathetic output eliminates this form of HRV (60–64). To verify that muscarinic receptor antagonists were effective in blocking peripheral muscarinic receptors, Poincaré plots (65) were used to analyze variability of the R-R interval on the ECG (Figure 6). Compared with WT mice, Scn1aR1407X/+ mice had significantly greater long-term (SD2) HRV (Figure 6, A and B, and Supplemental Figure 7, C and D). Atropine reduced SD2 in Scn1aR1407X/+ mice and was equally effective at 1 mg/kg and 0.03 mg/kg (Figure 6, C and D, and Supplemental Figure 7, C and D). Despite the ability of the lower dose of atropine to block peripheral muscarinic receptors, all Scn1aR1407X/+ mice in which seizures with tonic hind limb extension were induced by hyperthermia after pretreatment with 0.03 mg/kg atropine still developed bradycardia and died (Figure 5A; n = 6 of 6), but they also developed apnea (data not shown). N-methylscopolamine (1 mg/kg; n = 6) and methylatropine (1 mg/kg; n = 6) both caused a decrease in short-term (SD1) and long-term HRV (SD2) (Supplemental Figure 7). Despite the ability of all of these treatments to block vagal parasympathetic output, only some prevented postictal bradycardia and death. Those treatments that were protective prevented peri-ictal apnea, and those that were not protective did not prevent apnea.
Figure 6. Low-dose and high-dose atropine were equally effective at blocking HRV.

(A and B) Poincaré plots demonstrated greater short-term and long-term HRV (SD1 and SD2, respectively) in an Scn1aR1407X/+ mouse compared with a WT mouse. (C) High-dose atropine (1 mg/kg) reduced HRV in an Scn1aR1407X/+ mouse, consistent with blockade of parasympathetic output. (D) Low-dose atropine (0.03 mg/kg) was equally effective in reducing HRV in an Scn1aR1407X/+ mouse. The plots in the 4 parts of this figure are from 4 individual animals. Summary data for replicate animals for these experiments are shown in Supplemental Figure 7. Number of replicates for each group was 7 (A), 10 (B), 6 (C), and 6 (D).
Selective blockade of central muscarinic receptors prevented peri-ictal apnea and bradycardia.
Scn1aR1407X/+ mice (n = 5) were treated with i.c.v. N-methylscopolamine at a dose of only 0.3 μg/kg. This agent was the least permeable across the blood-brain barrier (see above), so it should have remained restricted to the CNS. However, even if the drug freely crossed the blood-brain barrier and was distributed throughout the body, this dose would still be only 1% of that required to block peripheral muscarinic receptors (0.03 mg/kg) and thus would not reach levels high enough to block parasympathetic drive to the heart. Thirty minutes after i.c.v. injection of N-methylscopolamine (0.3 μg/kg), hyperthermia was induced to at least 42.5°C, resulting in an average of 3.33 ± 0.89 seizures per mouse. Generalized seizures were induced in 5 of 5 mice at a body temperature of 40.7°C ± 0.53°C. All of the Scn1aR1407X/+ mice had generalized seizures with tonic hind limb extension or clonic limb contractions (n = 5), but none developed apnea or bradycardia, or died (Figure 7, A and B). Poincaré plots were used to evaluate ECG changes. There was no change in mean heart rate or HRV after i.c.v. N-methylscopolamine (0.3 μg/kg) (Figure 7C). Thus, i.c.v. N-methylscopolamine had no effect on peripheral muscarinic receptors, but prevented bradycardia by blocking apnea via an effect in the CNS.
Figure 7. Peri-ictal apnea, bradycardia, and death were prevented by N-methylscopolamine administered into the lateral cerebral ventricle at very low dose (0.0003 mg/kg).

(A) Heart rate and breathing did not change in response to a heat-induced Racine scale 5 seizure in an Scn1aR1407X/+ mouse. A seizure occurred at the time indicated. Insets to the right were obtained at the times indicated with the vertical lines on the left. Respiratory activity and the ECG did not change during the seizure. Note that there was movement artifact seen intermittently on the respiratory trace. Sudden changes in amplitude of the ECG were also an artifact due to movements of the animal. (B) Summary of results from Scn1aR1407X/+ mice with heat-induced Racine scale 5 seizures after N-methylscopolamine (0.3 μg/kg, i.c.v.; n = 5). Breathing and heart rate were not affected in any of the mice. (C) Poincaré plots demonstrated that N-methylscopolamine (0.3 μg/kg, i.c.v.) did not block peripheral muscarinic receptors as seen by the lack of an effect on R-R interval. The Poincaré plots shown here are from a single animal. Summary data for replicate animals for this experiment are shown in Supplemental Figure 7. Number of replicates was 4 animals.
Hypoxia caused bradycardia due to a direct effect on the heart.
Based on the above results, we hypothesized that seizures caused apnea, and the resulting hypoxia had a direct negative chronotropic effect on cardiac tissue. Consistent with this possibility, during vascular perfusion of the isolated rat heart ex vivo, it has been shown that hypoxia causes a decrease in heart rate and cardiac contractility due to a direct effect on the heart (66). WT (n = 5) and Scn1aR1407X/+ (n = 6) mice were anesthetized with isoflurane (1%) and placed in a whole-body plethysmograph. After recording baseline ECG and breathing, ambient gas was switched from normoxia (21% O2 / 79% N2) to anoxia (0% O2 / 100% N2). This led to bradycardia with a time course similar to that seen after seizures in Scn1aR1407X/+ mice (Figure 8). It also caused a decrease in amplitude of the QRS complex, as also occurred after fatal seizures, presumably a surrogate of decreased cardiac contractility (66). Pretreatment of WT mice with atropine at 1 mg/kg (n = 5) did not prevent the bradycardia induced by anoxia (Figure 8), indicating that the decrease in heart rate was not mediated by an increase in vagal output.
Figure 8. Anoxia caused bradycardia due to a direct effect on the heart.

WT mice exposed to anoxia developed bradycardia (n = 5). Pretreatment with atropine (1 mg/kg, i.p.) caused an increase in baseline heart rate, but did not prevent this bradycardia (n = 5), indicating that bradycardia was not due to increased vagal output. Scn1aR1407X/+ mice also developed bradycardia in response to anoxia (n = 6).
Discussion
Here we show that patients with DS have peri-ictal breathing dysfunction, including ataxic respiratory output, paradoxical breathing, airway obstruction, and prolonged depression of CO2 chemoreception that lasted as long as 4 hours after the end of seizures. The respiratory abnormalities we recorded were transient. However, a child with particularly severe and prolonged postictal hypoventilation later died of SUDEP. These results suggest that postictal ventilatory abnormalities may play a major role in SUDEP in patients with DS and may be a biomarker for those at highest risk.
We also studied seizures in a DS mouse model and found that postictal death was due to immediate fatal apnea. In contrast, Kalume et al. (40) reported that postictal death after heat-induced seizures in Scn1a-knockout mice is due to severe bradycardia and this was proposed to be due to increased vagal parasympathetic tone. This conclusion was supported by data showing that death was prevented by treatment with atropine (40). Auerbach et al. (7) reported spontaneous death preceded by bradycardia in 3 Scn1aR1407X/+ mice, and in that study, death was attributed to abnormal intrinsic electrophysiological properties of cardiac myocytes (7). However, breathing was not measured in either the study by Auerbach et al. (7) or the study by Kalume et al. (40). Here we report postictal bradycardia in Scn1aR1407X/+ mice with a similar time course to that previously reported (7, 40), but found that central apnea occurred prior to bradycardia. Apnea rapidly causes hypoxemia in mice and rats within seconds (67, 68). Therefore, the long duration of apnea we saw in response to seizures would be expected to cause a severe drop in blood oxygen. Hypoxemia has a negative chronotropic effect on the heart (66), explaining why atropine did not prevent bradycardia in response to exposure to hypoxic gas (Figure 8).
We found that atropine was protective against peri-ictal bradycardia, but this was not due to blockade of parasympathetic output. Instead, at high doses, atropine prevented peri-ictal apnea due to a central effect. Peri-ictal apnea and bradycardia were not prevented by selective blockade of peripheral muscarinic receptors using atropine at a low dose (0.03 mg/kg, i.p.) or the relatively membrane-impermeable N-methylscopolamine at a high dose (1 mg/kg, i.p.). In both cases, blockade of peripheral muscarinic receptors was verified by suppression of HRV. In contrast, peri-ictal apnea and bradycardia were both prevented by selective blockade of central muscarinic receptors using a very low dose of N-methylscopolamine (0.3 μg/kg, i.c.v.). Mice were protected from death even though peripheral muscarinic receptors were not blocked, as verified by a lack of effect on HRV. These results indicate that muscarinic receptor antagonists prevent postictal death by blocking central apnea rather than blocking vagal output.
Our results with systemic N-methylscopolamine were different from those of Kalume et al. (40), who found protection from death despite using the same concentration and route of delivery. The reason for this difference is unclear, but it is possible that the drug was able to penetrate into the CNS more easily in their study, perhaps because the blood-brain barrier was leakier due to a larger number of seizures. There may also be strain differences in sensitivity to the drug. Although relatively membrane impermeable, N-methylscopolamine can still cross the blood-brain barrier. However, given its equipotency with atropine, systemic N-methylscopolamine should be able to block peripheral muscarinic receptors at a dose of only 0.02–0.04 mg/kg. If its target was peripheral receptors, a systemic dose of 1 mg/kg should not be necessary. When delivered directly into the lateral ventricle, a 3,000-fold lower dose of only 0.3 μg/kg was sufficient to protect from seizure-induced death, supporting the hypothesis that the pharmacological target is in the brain.
These results indicate that atropine analogues have effects in the CNS that prevent seizure-induced apnea. High-dose systemic atropine did not reduce the duration or severity of heat-induced seizures as determined by EEG analysis or decrease the likelihood of Racine scale 5 seizures, but it did decrease the likelihood of tonic hind limb extension, suggesting there were effects on forebrain seizure activity. It is not known whether an effect on seizures in the forebrain protected against apnea or whether protection was due to inhibition of forebrain projections to the brain stem or to an effect on the respiratory network. Muscarinic receptors in the medulla play an important role in respiratory control (51, 52) and central respiratory CO2 chemoreception (53, 54). Depending on the site of action, muscarinic receptor activation can either stimulate or inhibit breathing (51, 52, 54, 69, 70). Acetylcholinesterase inhibitors block central respiratory rhythm generation via an effect in the brain stem (69, 71), and this central apnea can be prevented by atropine (72). It is possible that similar muscarinic mechanisms contribute to peri-ictal apnea in Scn1aR1407X/+ mice. It is also possible that muscarinic receptors in the forebrain alter seizures (55) so they are less likely to cause apnea. Defining the specific mechanisms of apnea that are blocked by atropine analogues may help to identify methods for prevention of SUDEP.
There are many open questions about the mechanisms of death in DS mice. For example, it is not clear whether Scn1a mutations increase the likelihood of invasion of the brain stem by seizures or make the respiratory network more likely to fail in response to descending input. It is not known which neurons in the brain stem express Scn1a and whether those neurons are involved in breathing. Seizures and death can be prevented in heterozygous Scn1a-knockout mice with GS967, a drug that inhibits persistent sodium current (73), but it is not clear whether persistent sodium current plays a role in peri-ictal apnea.
The relationship among BSD (24), PGES (25), and peri-ictal apnea remains unclear. It is not known whether BSD occurs in our model. PGES always occurred in Scn1aR1407X/+ mice that died, as was previously reported in Lmx1bf/f/p mice after MES seizures (19). BSD, PGES, and apnea could all be reflective of some underlying causative mechanism. One may cause the other 2, or they could be independent processes. It does not appear that PGES caused apnea, since apnea often occurred before PGES (Figure 4) and because generation of the automatic respiratory rhythm in the brain stem does not require descending input from the forebrain. Further work is required to define the sequence of these and other ictal and postictal events. What is clear is that when seizures are fatal, the apnea is immediate and severe. There is even complete suppression of gasping, or “autoresuscitation,” which usually occurs as a terminal event and is induced by severe hypoxia, indicating that there is such profound inhibition of the brain stem respiratory network that gasping does not even occur.
We observed that Scn1aR1407X/+ mice had greater HRV than WT mice, whereas Kalume et al. (40) observed that Scn1a-knockout mice had less HRV than WT mice. This difference is likely to be methodological, as our measurements of HRV were designed for a different purpose than those of Kalume et al. (40). In our study we examined the R-R interval over a short time period (30–40 seconds) when mice were lying in their cage motionless but awake, whereas Kalume et al. (40) examined the R-R interval over a long period (8 hours) during a variety of normal behaviors and activity levels. There are differences in HRV under different levels of animal activity (63). We chose to use the former method, as our goal was to verify block of peripheral muscarinic receptors. Kalume et al. (40) measured HRV over a long period to test the hypothesis that decreased HRV is a risk factor for SUDEP. The existing evidence does not yet support the latter hypothesis (74), although a decrease in HRV has been linked to a high risk of sudden cardiac death in humans (75). There is no evidence that the increase in HRV for short periods reported here in DS mice confers a higher risk of SUDEP, nor was that the purpose of these measurements.
It is not known how commonly death in other mouse models of SUDEP is due to the mechanisms described here. Similarly, it is not known whether the mechanisms of SUDEP in patients with DS are shared with those in other types of epilepsy. However, the results presented here may also be relevant to SUDEP in other types of epilepsy, since peri-ictal apnea has previously been reported as the cause of death in Lmx1bf/f/p, DBA, and 5-HT2c knockout mice (19, 21, 76). For a seizure to induce apnea, forebrain activity must propagate to the lower brain stem where breathing is controlled. The likelihood for this may vary depending on the type of seizure, whether the relevant descending projections are engaged, and how sensitive the respiratory network is to descending input.
Cardiac abnormalities clearly play an important role in many cases of SUDEP, likely including some cases of DS, but it can be misleading to focus attention on cardiac changes without simultaneously evaluating breathing and blood gases, since changes in O2, CO2, and pH can induce a variety of cardiovascular abnormalities, including bradycardia and tachyarrhythmias. This is of practical clinical importance, since a cardiac pacemaker would not be appropriate for preventing SUDEP in a patient with bradycardia secondary to central apnea. Instead, interventions that restore ventilation would be more logical for rescuing patients with epilepsy who have peri-ictal respiratory arrest. Patients with epilepsy at high risk of apnea would be prudent to avoid alcohol and medications that depress breathing. Stimulating arousal to reinitiate breathing or use of ventilatory assistance with a bag valve mask or mouth-to-mouth resuscitation may be more effective than cardiac interventions, as they would reverse a primary defect of hypoventilation.
Methods
Monitoring of patients with DS with video EEG telemetry.
Patients were admitted to the pediatric EMU at Lurie Children’s Hospital (Chicago, Illinois, USA) for routine video-EEG monitoring of refractory seizures. Seven patients with DS and convulsive seizures were selected for video analysis along with 7 patients with convulsive seizures from localization-related epilepsy. Respiratory movements were difficult to visualize in some video recordings. Therefore, visualization of movements was enhanced using phase-based Eulerian video magnification motion processing (44) with open-source software (http://people.csail.mit.edu/mrub/vidmag/) written for MATLAB (Mathworks Co.). Video motion magnification processing was performed on video recordings with amplification of ×4 or ×10, applying a bandpass filter of 0.05–3.0 Hz.
To better visualize breathing in video images, a vertical line was drawn through a part of the body that showed large movements (the umbilicus in Figure 1A and Supplemental Figure 1A). Using a custom-written MATLAB script, the color value was determined at each point along the vertical line for each video frame. These values were used to create a graph with time (T; consecutive video frames) represented on the x axis, vertical location on the y axis (Y), and color in relative units on the z axis (Supplemental Figure 1B). The breathing trace in this spatiotemporal YT slice was rendered more apparent by converting the image to grayscale and enhancing the contrast. The most visible single breathing trace was selected by cropping the spatiotemporal YT image (Supplemental Figure 1, C and D) and drawing a line along the center of the trace (Supplemental Figure 1E) using image editing software (Corel PhotoPaint).
In a single patient (Figure 2), measurements of breathing were made using respiratory impedance plethysmography (Piezo Crystal Respiration Effort Sensors, SleepSense), tcCO2, SaO2 (SenTec Digital Monitoring System, SenTec AG), ECG, and video EEG.
Mouse husbandry and genotyping.
A pair of Scn1aR1407X/+ heterozygous male mice on a C3HFeB/HeJ background were provided by Miriam Meisler (University of Michigan, Ann Arbor, Michigan, USA), and were bred with C3HFeB/HeJ female mice (Jackson Laboratory) to establish a breeding colony. Breeding and genotyping of these mice have been previously described (7). Briefly, DS mice were genotyped by PCR amplification with the primers DS-F (5′ CAATGATTCCTAGGGGGATGTC 3′) and DS-R (5′ GTTCTGTGCACTTATCTGGATTCAC 3′). Digestion of the PCR product with HpaII generated 2 fragments (295 bp and 223 bp) from the WT allele and an uncut fragment (518 bp) from the mutant allele. Genomic DNA was PCR amplified, digested with HpaII, and separated on 2% agarose gels containing 0.15 μg/ml ethidium bromide.
EEG/EMG/ECG headmount and activity/temperature telemetry implants.
EEG/EMG/ECG custom headmounts (8400-K1; Pinnacle Technology Inc.) were implanted in P16–P22 DS mice as previously described (77). Briefly, mice received preoperative analgesia (meloxicam, 0.5 mg/kg, i.p.; or buprenorphine, 0.1 mg/kg, i.p.). Anesthesia was induced and maintained with 0.5%–3% inhaled isoflurane. A midline skin incision was made over the skull, 4 holes were drilled into the skull, and a headmount was attached with two 0.1-inch (anterior) and two 0.125-inch (posterior) stainless steel machine screws (0.8 × 4.2; stainless steel nose pad screw; QTE North America Inc.). EMG leads extending from the posterior portion of the headmount were sutured into nuchal muscles bilaterally 3 mm from midline. ECG leads were tunneled under the skin and sutured to the anterior thoracic wall in an approximate lead II configuration. The base of the headmount, screw heads, and EMG and ECG leads were anchored with dental acrylic (Jet Acrylic; Lang Dental) and the skin was sutured closed leaving only the headmount socket exposed. Temperature/activity telemetry probes (G2 E-Mitter; Mini-Mitter Inc.) were inserted into the peritoneal cavity to measure body temperature as previously described (77). A sterile surgical prep was made on the abdomen, and temperature probes were inserted into the peritoneum through a midline incision. The incision was closed with suture or Vetbond (3M Animal Care Product). Animals received postoperative analgesia with meloxicam (0.5 mg/kg, i.p.) or buprenorphine (0.1 mg/kg, i.p.) for 3 days and were allowed to recover for at least 5 days before recordings were made.
Long-term, continuous mouse EMU/plethysmography data acquisition.
A preamplifier (8406-SE31M-C; Pinnacle Technology Inc.) was inserted into the implanted headmount connector for recording EEG, ECG, and EMG (gain ×10; high pass filter for ECG 0.5 Hz; EEG 1 Hz; EMG 10 Hz). The animal was placed into a mouse EMU/plethysmography chamber custom-designed and built to comply with requirements for continuous housing described in the Guide for the Care and Use of Laboratory Animals (78). The floor was circular and 5 inches in diameter. The walls were semiconical with a roof that was 5.5 inches high and 3 inches in diameter. The chamber volume was 815 ml. There were ports for air in and air out, and for pressure and temperature/humidity monitoring. The chamber was supplied with a continuous flow of room air at approximately 400 ml/min via supply and exhaust air pumps (MK-1504 Aquarium Air Pump; AQUA Culture) balanced to maintain chamber pressure near atmospheric. Mice had access to a continuous supply of water and food.
The EMU/plethysmography chamber pressure was measured using a digital differential pressure transducer (SDP610-25Pa; Sensirion AG). Chamber temperature and relative humidity were also continuously monitored (HIH-6130 sensor; Honeywell International Inc.). Signal outputs from the pressure transducer and temperature/humidity sensor were connected to a microcontroller (Teensy 3.1; PJRC.com) using a custom-designed printed circuit board. The digital transducers and microcontroller communicated through an inter-integrated circuit (I2C) interface via the Arduino platform (ARDUINO 1.6.9; Arduino.cc, and Teensyduino 1.28; PJRC.com) to upload configuration code of the digital sensors to the microcontroller board. Chamber temperature, relative humidity, and pressure were acquired continuously at 100 Hz through a USB port with data acquisition software custom written in MATLAB. Breathing caused changes in pressure in the chamber due to heating and humidification of inspired air based on well-established physical principles, and these were used to calculate changes in lung volume (79).
EEG, EMG, and ECG leads were tethered to a commutator (catalog 8408; Pinnacle Technology Inc.) and connected to an amplifier (Model 440 Instrumentation Amplifier; Brownlee Precision Co.) where signals were amplified ×10 and bandpass filtered (EEG, 0.3–400 Hz; EMG, 10–500 Hz; ECG, 1–500 Hz). These signals were digitized with an analog-to-digital converter (PCI-6225; National Instruments Corp.) in a desktop computer and acquired with the same custom-written MATLAB data acquisition software. Body temperature and activity were captured by a telemetry receiver (ER-4000, Energizer Receiver; Mini-Mitter, Inc.) placed beneath the recording chamber, transmitted to the computer via a serial port, and sampled every 10 seconds with the same custom-written MATLAB data acquisition software. All digitized data were saved on external hard drives.
Monitoring of spontaneous deaths in mice.
One cohort of Scn1aR1407X/+ mice (n = 78) was monitored for spontaneous deaths using video recording only (Figure 3). Uninstrumented mice were housed in their home cages and continuous video monitoring was performed from P16–P60. Video recordings were made at 30 frames per second using a night vision web camera (FL8910W; Foscam Digital Technologies). Up to 32 cameras were connected to a single computer, and video recordings were stored on an external hard drive using commercial video webcam software (Blue Iris 4; Foscam Digital Technologies). When a mouse was found dead in a cage, the video was reviewed to determine the time of death and whether death was preceded by a behavioral seizure.
A second cohort of Scn1aR1407X/+ mice (n = 19) was monitored for spontaneous deaths over 195 days using video recording as well as EEG, EMG, ECG, and measurements of breathing, temperature, and activity. Each recording session lasted up to 24 hours, after which the EMU was opened and cleaned. Mice were then returned to the EMU for continued monitoring or to their home cage. While in the EMU, mice had free access to food and water. Long-term mouse EMU data were continuously stored on an external hard drive (DRDR5A21-20TB; DroboWorks.com). In 4,223 total hours of recording 19 mice between P23 and P40, 2 mice died spontaneously. When these mice died, monitoring was terminated and data were reviewed to assess cause of death.
Heat-induced seizures in mice.
Mice were instrumented with headmounts and telemetry probes to allow recordings of all the signals described for monitoring spontaneous deaths. After at least 5 days, mice were lightly anesthetized with 1% isoflurane for 1–2 minutes to connect a preamplifier to the headmount. Any experimental drugs were injected at that time. Mice were then placed in the plethysmography chamber and allowed to recover from anesthesia and acclimate for 30 minutes. Seizures were then induced by hyperthermia using a protocol previously reported for a DS mouse model (40). Using a heat lamp, body temperature in Scn1aR1407X/+ mice and WT littermates was increased from 36°C to 42.5°C at a rate of 0.5°C per minute or until a generalized seizure with tonic hind limb extension was induced. To maximize the amplitude of the breathing signal, the plethysmography chamber was maintained at 25°C–27°C by a thermodynamic cooling system made in-house that cooled the incoming air. In some cases mice were resuscitated after a seizure by placing Tygon tubing (United States Plastic Corp.) over the snout and inflating the lungs with air using a mechanical ventilator (MiniVent type 845; Harvard Apparatus).
Effect of muscarinic receptor antagonists on bradycardia.
To determine the role of parasympathetic output in postictal bradycardia, muscarinic receptor antagonists were administered 30 minutes prior to heat-induced seizures. All antagonists given via i.p. injection were dissolved in a volume of 50 μl in bacteriostatic 0.9% NaCl solution. In a separate set of experiments, N-methylscopolamine was given via injection into the lateral ventricle dissolved to a volume of 50 nl in artificial cerebrospinal fluid (aCSF) containing 125 mM NaCl, 5.0 mM KCl, 24 mM NaHCO3, 1.25 mM KH2PO4, 0.8 mM CaCl2, 1.25 mM MgSO4, and 30 mM glucose. Coordinates used for i.c.v. injection were 0 mm AP to bregma, 1.0 mm lateral, and 2.5 mm below the skull surface. Atropine, methylatropine nitrate, and N-methylscopolamine bromide were obtained from Sigma-Aldrich. At a dose of 1 mg/kg (i.p.), atropine had no effect on the likelihood of Racine scale 5 seizures, or the duration or severity of seizures based on EEG analysis. In the cohort of control mice shown in Figure 5A for fatal seizures (n = 7), seizure duration was 25.7 ± 9.2 seconds and the mean EEG power between 0.5 and 20 Hz (as a measure of seizure severity) was 0.083 ± 0.026 s–1. In contrast, for the mice treated with atropine shown in Figure 5A for nonfatal seizures (n = 14 seizures), seizure duration was 21.9 ± 8.3 seconds and the mean EEG power was 0.121 ± 0.055 s–1. These differences were not statistically significant (P > 0.05; Mann-Whitney U test). After treatment with atropine (1 mg/kg, i.p.) or N-methylscopolamine (0.3 μg/kg, i.c.v.), some seizures were characterized by clonic limb contractions instead of tonic hind limb extension, although in both cases they met criteria for Racine scale 5 seizures.
HRV was assessed using Poincaré plots (65) before and 30 minutes after administering muscarinic receptor antagonists. This method graphs the time between one pair of QRS complexes on an ECG (R-R′ interval) versus the time between the next pair of QRS complexes (R′-R″). The SD of the points along the unity line is termed long-term adaptation (SD2), and the standard deviation of the points perpendicular to the unity line is referred to as short-term adaptation (SD1). HRV is primarily due to vagus nerve–mediated parasympathetic inhibition of the sinoatrial node (60–65). For all Poincaré plots, 500 pairs of R-R intervals were plotted and analyzed at baseline and after drug injection. Data were not analyzed during any animal movements to eliminate artifacts.
Exposure to hypoxia.
Scn1aR1407X/+ mice and WT littermates were instrumented with ECG leads and temperature probes as above. After at least 5 days of recovery, mice were placed in a plethysmography chamber where the ECG and body temperature were continuously monitored. The chamber was flushed with gas flowing at 700 ml/min, first with medical grade 21% O2/79% N2/1% isoflurane. After establishing a baseline, the gas was switched to 100% N2/1% isoflurane. Isoflurane levels were maintained at 1% using a precision vaporizer (Summit Anesthesia Solutions). The time course for the change of PO2 was measured using an O2 analyzer (Model S-3A; AEI Technologies) in a separate set of experiments without isoflurane to avoid damage to the O2 analyzer. Mice were studied either with or without injection of atropine (1 mg/kg, i.p.) 30 minutes before exposure to anoxia. Control mice were injected with the same volume of saline solution. The plethysmography chamber was maintained at 30°C using a heat lamp and feedback controller (TCAT-2AC; Physitemp Instruments).
Data analysis and statistics.
Time-frequency power spectrum analysis was performed with MATLAB based on a short-time fast Fourier transform. The time-dependent power spectrum density of the signal was computed and displayed with discrete-time epochs calculated for 0.5-second intervals. Sections overlapped by 20% to smooth the signal. The ECG sampling rate was 1 kHz, whereas EEG, EMG, and plethysmography were sampled at 100 Hz.
Analyses of respiratory rate and heart rate were performed using software custom written in MATLAB and/or Clampfit Software (Molecular Devices). Inspiratory peaks were detected automatically from plethysmography data by applying a bandpass filter of 1–10 Hz and using peak detection verified manually. R-wave detection of ECG data was performed by applying a continuous wavelet transform and peak detection in MATLAB. The time interval between breath peaks (interbreath interval) and the R-R interval from detected R waves were computed as the mean for 10-second epochs.
Abnormal breathing in patients with DS was compared with breathing in patients with localization-related epilepsy (Table 1) using a χ2 test. Severity of seizures was measured by computing the mean EEG intensity from power spectra generated using MATLAB. Statistical comparisons of seizure severity and duration, and Poincarè plots (SD1 and SD2), were made using a Mann-Whitney U test with an overall significance level set to P < 0.05 (GraphPad Prism V6.01). When data are presented as X ± Y, X is the group mean and Y is the standard deviation. All error bars on graphs represent the SEM.
Clinical phenotype of a patient with DS with prolonged postictal hypoventilation.
The patient shown in Figure 2 had her first seizure at 5.5 months of age. Seizure types prior to the EMU admission had included generalized convulsions, hemiconvulsions, partial seizures with variable semiology, atypical absence seizures, and myoclonic jerks. At the time of EMU admission, seizure types included (a) feeling “spacey” for 5–10 seconds with blank staring, slackening of facial muscles, and slurred speech, and (b) repetitive asynchronous generalized convulsions in series lasting 1–2 minutes each, interrupted by a fixed gaze for up to 5 minutes. The patient’s seizures had worsened in frequency and severity during the previous 6 months. There was concern for a decrease in alertness and cognitive ability. The EMU study was performed to assess EEG background activity and to clarify the types of seizures. The patient was treated at the time of admission with levetiracetam, clobazam, stiripentol, and a ketogenic diet. The first event for the episode shown in Figure 2 began off camera in the bathroom after the mother reported that the patient had an increase in staring episodes. During the seizures, the EEG showed generalized onset of high amplitude, diffuse, spike/polyspike wave discharges that evolved in amplitude and frequency. These ictal abnormalities terminated abruptly with generalized slowing. Interictal discharges would quickly reappear and coalesce just prior to the next convulsive seizure.
Study approval.
All experimental protocols used in human studies were approved by the Northwestern University Institutional Review Board and performed under the direct supervision of a faculty neurologist. Written informed consent was obtained from participants or their guardians.
All procedures and experiments involving mice were carried out with approval of the University of Iowa Institutional Animal Care and Use Committee, and in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals (78). The minimum possible number of animals was used, and care was taken to reduce any discomfort.
Author contributions
YJK designed and built the experimental apparatus used for mouse experiments, carried out the spontaneous death and mouse survival experiments, wrote data acquisition and analysis software, analyzed data, instantiated the Eulerian video magnification, and wrote the manuscript. EB designed and carried out the mouse experiments on hyperthermia-induced seizures, peripheral parasympathetic blockade, i.c.v. injection of N-methylscopolamine and hypoxia, analyzed data, and edited the manuscript. CKT and LASM carried out the mouse survival experiments and edited the manuscript. XZ carried out all genotyping and contributed to animal husbandry. BKG contributed to the design, analysis, and interpretation of cardiorespiratory data from humans and mice, and edited the manuscript. SHK, DRN, and LCL collected data from the patients with DS, analyzed data, and edited the manuscript. GBR oversaw the work, helped design all experiments, analyzed data, and edited the manuscript.
Supplementary Material
Acknowledgments
NIH U01 NS 090414 (to YJK, EB, CKT, LASM, BKG, and GBR), and the Beth L. Tross Epilepsy Research Fund (GBR).
Version 1. 01/11/2018
In-Press Preview
Version 2. 02/12/2018
Electronic publication
Version 3. 03/01/2018
Print issue publication
Footnotes
DRN’s present address is: Children’s Hospital Los Angeles, Keck School of Medicine, University of Southern California, Los Angeles, California, USA.
SHK’s present address is: Division of Pediatric Neurology, Severance Children’s Hospital, and Department of Pediatrics, Yonsei University College of Medicine, Seoul, South Korea.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(3):1141–1153.https://doi.org/10.1172/JCI94999.
Contributor Information
YuJaung Kim, Email: yujaung-kim@uiowa.edu.
Eduardo Bravo, Email: eduardo-bravo@uiowa.edu.
Caitlin K. Thirnbeck, Email: caitlinthirnbeck@gmail.com.
Lori A. Smith-Mellecker, Email: lori-a-smith@uiowa.edu.
Se Hee Kim, Email: she33435@gmail.com.
Brian K. Gehlbach, Email: brian-gehlbach@uiowa.edu.
Linda C. Laux, Email: LLaux@luriechildrens.org.
Xiuqiong Zhou, Email: xiuqiong-zhou@uiowa.edu.
Douglas R. Nordli, Jr., Email: dnordli@chla.usc.edu.
George B. Richerson, Email: george-richerson@uiowa.edu.
References
- 1.Devinsky O, Hesdorffer DC, Thurman DJ, Lhatoo S, Richerson G. Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol. 2016;15(10):1075–1088. doi: 10.1016/S1474-4422(16)30158-2. [DOI] [PubMed] [Google Scholar]
- 2.Ryvlin P, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. 2013;12(10):966–977. doi: 10.1016/S1474-4422(13)70214-X. [DOI] [PubMed] [Google Scholar]
- 3.Langan Y, Nashef L, Sander JW. Sudden unexpected death in epilepsy: a series of witnessed deaths. J Neurol Neurosurg Psychiatry. 2000;68(2):211–213. doi: 10.1136/jnnp.68.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Surges R, Thijs RD, Tan HL, Sander JW. Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nat Rev Neurol. 2009;5(9):492–504. doi: 10.1038/nrneurol.2009.118. [DOI] [PubMed] [Google Scholar]
- 5.Massey CA, Sowers LP, Dlouhy BJ, Richerson GB. Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol. 2014;10(5):271–282. doi: 10.1038/nrneurol.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moseley B, Bateman L, Millichap JJ, Wirrell E, Panayiotopoulos CP. Autonomic epileptic seizures, autonomic effects of seizures, and SUDEP. Epilepsy Behav. 2013;26(3):375–385. doi: 10.1016/j.yebeh.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 7.Auerbach DS, et al. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One. 2013;8(10):e77843. doi: 10.1371/journal.pone.0077843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sundström J, Lind L, Andrén B, Lithell H. Left ventricular geometry and function are related to electrocardiographic characteristics and diagnoses. Clin Physiol. 1998;18(5):463–470. doi: 10.1046/j.1365-2281.1998.00126.x. [DOI] [PubMed] [Google Scholar]
- 9.Surges R, et al. Pathologic cardiac repolarization in pharmacoresistant epilepsy and its potential role in sudden unexpected death in epilepsy: a case-control study. Epilepsia. 2010;51(2):233–242. doi: 10.1111/j.1528-1167.2009.02330.x. [DOI] [PubMed] [Google Scholar]
- 10.Johnson JN, Hofman N, Haglund CM, Cascino GD, Wilde AA, Ackerman MJ. Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology. 2009;72(3):224–231. doi: 10.1212/01.wnl.0000335760.02995.ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30(15):5167–5175. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delogu AB, et al. Electrical and autonomic cardiac function in patients with Dravet syndrome. Epilepsia. 2011;52 Suppl 2:55–58. doi: 10.1111/j.1528-1167.2011.03003.x. [DOI] [PubMed] [Google Scholar]
- 13.DeGiorgio CM, et al. RMSSD, a measure of vagus-mediated heart rate variability, is associated with risk factors for SUDEP: the SUDEP-7 Inventory. Epilepsy Behav. 2010;19(1):78–81. doi: 10.1016/j.yebeh.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moseley BD, Wirrell EC, Nickels K, Johnson JN, Ackerman MJ, Britton J. Electrocardiographic and oximetric changes during partial complex and generalized seizures. Epilepsy Res. 2011;95(3):237–245. doi: 10.1016/j.eplepsyres.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 15.Surges R, Henneberger C, Adjei P, Scott CA, Sander JW, Walker MC. Do alterations in inter-ictal heart rate variability predict sudden unexpected death in epilepsy? Epilepsy Res. 2009;87(2-3):277–280. doi: 10.1016/j.eplepsyres.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Nashef L, Walker F, Allen P, Sander JW, Shorvon SD, Fish DR. Apnoea and bradycardia during epileptic seizures: relation to sudden death in epilepsy. J Neurol Neurosurg Psychiatry. 1996;60(3):297–300. doi: 10.1136/jnnp.60.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bateman LM, Li CS, Seyal M. Ictal hypoxemia in localization-related epilepsy: analysis of incidence, severity and risk factors. Brain. 2008;131(Pt 12):3239–3245. doi: 10.1093/brain/awn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhan Q, et al. Impaired serotonergic brainstem function during and after seizures. J Neurosci. 2016;36(9):2711–2722. doi: 10.1523/JNEUROSCI.4331-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buchanan GF, Murray NM, Hajek MA, Richerson GB. Serotonin neurones have anti-convulsant effects and reduce seizure-induced mortality. J Physiol (Lond) 2014;592(19):4395–4410. doi: 10.1113/jphysiol.2014.277574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dlouhy BJ, et al. Breathing inhibited when seizures spread to the amygdala and upon amygdala stimulation. J Neurosci. 2015;35(28):10281–10289. doi: 10.1523/JNEUROSCI.0888-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng HJ, Faingold CL. Abnormalities of serotonergic neurotransmission in animal models of SUDEP. Epilepsy Behav. 2017;71(Pt B):174–180. doi: 10.1016/j.yebeh.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakase K, et al. Laryngospasm, central and obstructive apnea during seizures: Defining pathophysiology for sudden death in a rat model. Epilepsy Res. 2016;128:126–139. doi: 10.1016/j.eplepsyres.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Seyal M, Bateman LM, Albertson TE, Lin TC, Li CS. Respiratory changes with seizures in localization-related epilepsy: analysis of periictal hypercapnia and airflow patterns. Epilepsia. 2010;51(8):1359–1364. doi: 10.1111/j.1528-1167.2009.02518.x. [DOI] [PubMed] [Google Scholar]
- 24.Aiba I, Noebels JL. Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med. 2015;7(282):282ra46. doi: 10.1126/scitranslmed.aaa4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lhatoo SD, Faulkner HJ, Dembny K, Trippick K, Johnson C, Bird JM. An electroclinical case-control study of sudden unexpected death in epilepsy. Ann Neurol. 2010;68(6):787–796. doi: 10.1002/ana.22101. [DOI] [PubMed] [Google Scholar]
- 26.Goldman AM. Mechanisms of sudden unexplained death in epilepsy. Curr Opin Neurol. 2015;28(2):166–174. doi: 10.1097/WCO.0000000000000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aurlien D, Leren TP, Taubøll E, Gjerstad L. New SCN5A mutation in a SUDEP victim with idiopathic epilepsy. Seizure. 2009;18(2):158–160. doi: 10.1016/j.seizure.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 28.Hata Y, Yoshida K, Kinoshita K, Nishida N. Epilepsy-related sudden unexpected death: targeted molecular analysis of inherited heart disease genes using next-generation DNA sequencing. Brain Pathol. 2017;27(3):292–304. doi: 10.1111/bpa.12390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parisi P, et al. Coexistence of epilepsy and Brugada syndrome in a family with SCN5A mutation. Epilepsy Res. 2013;105(3):415–418. doi: 10.1016/j.eplepsyres.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 30.Tiron C, et al. Further evidence of the association between LQT syndrome and epilepsy in a family with KCNQ1 pathogenic variant. Seizure. 2015;25:65–67. doi: 10.1016/j.seizure.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52 Suppl 2:3–9. doi: 10.1111/j.1528-1167.2011.02994.x. [DOI] [PubMed] [Google Scholar]
- 32.Parihar R, Ganesh S. The SCN1A gene variants and epileptic encephalopathies. J Hum Genet. 2013;58(9):573–580. doi: 10.1038/jhg.2013.77. [DOI] [PubMed] [Google Scholar]
- 33.Hani AJ, Mikati HM, Mikati MA. Genetics of pediatric epilepsy. Pediatr Clin North Am. 2015;62(3):703–722. doi: 10.1016/j.pcl.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 34.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68(6):1327–1332. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakauchi M, et al. Retrospective multiinstitutional study of the prevalence of early death in Dravet syndrome. Epilepsia. 2011;52(6):1144–1149. doi: 10.1111/j.1528-1167.2011.03053.x. [DOI] [PubMed] [Google Scholar]
- 36.Cooper MS, et al. Mortality in Dravet syndrome. Epilepsy Res. 2016;128:43–47. doi: 10.1016/j.eplepsyres.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Ergul Y, Ekici B, Tatli B, Nisli K, Ozmen M. QT and P wave dispersion and heart rate variability in patients with Dravet syndrome. Acta Neurol Belg. 2013;113(2):161–166. doi: 10.1007/s13760-012-0140-z. [DOI] [PubMed] [Google Scholar]
- 38.Daverio M, Ciccone O, Boniver C, De Palma L, Corrado D, Vecchi M. Supraventricular tachycardia during status epilepticus in Dravet syndrome: a link between brain and heart? Pediatr Neurol. 2016;56:69–71. doi: 10.1016/j.pediatrneurol.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 39.Ogiwara I, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27(22):5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalume F, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. 2013;123(4):1798–1808. doi: 10.1172/JCI66220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maier SK, et al. An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc Natl Acad Sci USA. 2003;100(6):3507–3512. doi: 10.1073/pnas.2627986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maier SK, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci USA. 2002;99(6):4073–4078. doi: 10.1073/pnas.261705699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheah CS, et al. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA. 2012;109(36):14646–14651. doi: 10.1073/pnas.1211591109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu HY, Rubinstein M, Shih E, Guttag J, Durand F, Freeman WT. Eulerian video magnification for revealing subtle changes in the world. ACM Transactions on Graphics. 2012. https://people.csail.mit.edu/mrub/papers/vidmag.pdf.
- 45.Cao D, et al. Efficacy of stiripentol in hyperthermia-induced seizures in a mouse model of Dravet syndrome. Epilepsia. 2012;53(7):1140–1145. doi: 10.1111/j.1528-1167.2012.03497.x. [DOI] [PubMed] [Google Scholar]
- 46.Gargiulo S, et al. Mice anesthesia, analgesia, and care, Part I: anesthetic considerations in preclinical research. ILAR J. 2012;53(1):E55–E69. doi: 10.1093/ilar.53.1.55. [DOI] [PubMed] [Google Scholar]
- 47.Flecknell PA. Anaesthesia of animals for biomedical research. Br J Anaesth. 1993;71(6):885–894. doi: 10.1093/bja/71.6.885. [DOI] [PubMed] [Google Scholar]
- 48. Hawk CT, Leary SL,Morris TH. Formulary for Laboratory Animals. Third Edition. Hoboken, NJ: Wiley-Blackwell; 2005. [Google Scholar]
- 49.Proakis AG, Harris GB. Comparative penetration of glycopyrrolate and atropine across the blood--brain and placental barriers in anesthetized dogs. Anesthesiology. 1978;48(5):339–344. doi: 10.1097/00000542-197805000-00007. [DOI] [PubMed] [Google Scholar]
- 50.Houze P, Pronzola L, Kayouka M, Villa A, Debray M, Baud FJ. Ventilatory effects of low-dose paraoxon result from central muscarinic effects. Toxicol Appl Pharmacol. 2008;233(2):186–192. doi: 10.1016/j.taap.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 51.Muere C, et al. Atropine microdialysis within or near the pre-Botzinger Complex increases breathing frequency more during wakefulness than during NREM sleep. J Appl Physiol. 2013;114(5):694–704. doi: 10.1152/japplphysiol.00634.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mallios VJ, Lydic R, Baghdoyan HA. Muscarinic receptor subtypes are differentially distributed across brain stem respiratory nuclei. Am J Physiol. 1995;268(6 Pt 1):L941–L949. doi: 10.1152/ajplung.1995.268.6.L941. [DOI] [PubMed] [Google Scholar]
- 53.Dev NB, Loeschcke HH. A cholinergic mechanism involved in the respiratory chemosensitivity of the medulla oblongata in the cat. Pflugers Arch. 1979;379(1):29–36. doi: 10.1007/BF00622901. [DOI] [PubMed] [Google Scholar]
- 54.Nattie EE, Wood J, Mega A, Goritski W. Rostral ventrolateral medulla muscarinic receptor involvement in central ventilatory chemosensitivity. J Appl Physiol. 1989;66(3):1462–1470. doi: 10.1152/jappl.1989.66.3.1462. [DOI] [PubMed] [Google Scholar]
- 55.Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA. Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse. 1989;3(2):154–171. doi: 10.1002/syn.890030207. [DOI] [PubMed] [Google Scholar]
- 56.Brezenoff HE, Xiao YF, Vargas H. A comparison of the central and peripheral antimuscarinic effects of atropine and methylatropine injected systemically and into the cerebral ventricles. Life Sci. 1988;42(8):905–911. doi: 10.1016/0024-3205(88)90389-X. [DOI] [PubMed] [Google Scholar]
- 57.Herz A, Teschemacher H, Hofstetter A, Kurz H. The importance of lipid-solubility for the central action of cholinolytic drugs. Int J Neuropharmacol. 1965;4:207–218. doi: 10.1016/0028-3908(65)90037-7. [DOI] [PubMed] [Google Scholar]
- 58.Witter A, Slangen JL, Terpstra GK. Distribution of 3H-methylatropine in rat brain. Neuropharmacology. 1973;12(9):835–841. doi: 10.1016/0028-3908(73)90036-1. [DOI] [PubMed] [Google Scholar]
- 59.Paul-David J, Riehl JL, Unna KR. Quantification of effects of depressant drugs on EEG activation response. J Pharmacol Exp Ther. 1960;129:69–74. [PubMed] [Google Scholar]
- 60.Taylor JA, Carr DL, Myers CW, Eckberg DL. Mechanisms underlying very-low-frequency RR-interval oscillations in humans. Circulation. 1998;98(6):547–555. doi: 10.1161/01.CIR.98.6.547. [DOI] [PubMed] [Google Scholar]
- 61.Fouad FM, Tarazi RC, Ferrario CM, Fighaly S, Alicandri C. Assessment of parasympathetic control of heart rate by a noninvasive method. Am J Physiol. 1984;246(6 Pt 2):H838–H842. doi: 10.1152/ajpheart.1984.246.6.H838. [DOI] [PubMed] [Google Scholar]
- 62.Thireau J, Zhang BL, Poisson D, Babuty D. Heart rate variability in mice: a theoretical and practical guide. Exp Physiol. 2008;93(1):83–94. doi: 10.1113/expphysiol.2007.040733. [DOI] [PubMed] [Google Scholar]
- 63.Tsai ML, Chen CC, Yeh CJ, Chou LM, Cheng CH. Frequency ranges of heart rate variability related to autonomic nerve activity in the mouse. Clin Exp Hypertens. 2012;34(3):182–190. doi: 10.3109/10641963.2011.577492. [DOI] [PubMed] [Google Scholar]
- 64.Gehrmann J, Hammer PE, Maguire CT, Wakimoto H, Triedman JK, Berul CI. Phenotypic screening for heart rate variability in the mouse. Am J Physiol Heart Circ Physiol. 2000;279(2):H733–H740. doi: 10.1152/ajpheart.2000.279.2.H733. [DOI] [PubMed] [Google Scholar]
- 65.Kamen PW, Krum H, Tonkin AM. Poincaré plot of heart rate variability allows quantitative display of parasympathetic nervous activity in humans. Clin Sci. 1996;91(2):201–208. doi: 10.1042/cs0910201. [DOI] [PubMed] [Google Scholar]
- 66.Chen KC, McGrath JJ. Response of the isolated heart to carbon monoxide and nitrogen anoxia. Toxicol Appl Pharmacol. 1985;81(3 Pt 1):363–370. doi: 10.1016/0041-008x(85)90408-9. [DOI] [PubMed] [Google Scholar]
- 67.Carreras A, Wang Y, Gozal D, Montserrat JM, Navajas D, Farré R. Non-invasive system for applying airway obstructions to model obstructive sleep apnea in mice. Respir Physiol Neurobiol. 2011;175(1):164–168. doi: 10.1016/j.resp.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 68.Kannurpatti SS, Biswal BB, Hudetz AG. Differential fMRI-BOLD signal response to apnea in humans and anesthetized rats. Magn Reson Med. 2002;47(5):864–870. doi: 10.1002/mrm.10131. [DOI] [PubMed] [Google Scholar]
- 69.Gaspari RJ, Paydarfar D. Dichlorvos-induced central apnea: effects of selective brainstem exposure in the rat. Neurotoxicology. 2011;32(2):206–214. doi: 10.1016/j.neuro.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lydic R, Baghdoyan HA. Pedunculopontine stimulation alters respiration and increases ACh release in the pontine reticular formation. Am J Physiol. 1993;264(3 Pt 2):R544–R554. doi: 10.1152/ajpregu.1993.264.3.R544. [DOI] [PubMed] [Google Scholar]
- 71.Gillis RA, Walton DP, Quest JA, Namath IJ, Hamosh P, Dretchen KL. Cardiorespiratory effects produced by activation of cholinergic muscarinic receptors on the ventral surface of the medulla. J Pharmacol Exp Ther. 1988;247(2):765–773. [PubMed] [Google Scholar]
- 72.Fleming NW, Henderson TR, Dretchen KL. Mechanisms of respiratory failure produced by neostigmine and diisopropyl fluorophosphate. Eur J Pharmacol. 1991;195(1):85–91. doi: 10.1016/0014-2999(91)90384-3. [DOI] [PubMed] [Google Scholar]
- 73.Anderson LL, Hawkins NA, Thompson CH, Kearney JA, George AL. Unexpected efficacy of a novel sodium channel modulator in Dravet syndrome. Sci Rep. 2017;7(1):1682. doi: 10.1038/s41598-017-01851-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harden C, et al. Practice guideline summary: sudden unexpected death in epilepsy incidence rates and risk factors: report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2017;88(17):1674–1680. doi: 10.1212/WNL.0000000000003685. [DOI] [PubMed] [Google Scholar]
- 75.Stein PK, Domitrovich PP, Huikuri HV, Kleiger RE, Cast Investigators Traditional and nonlinear heart rate variability are each independently associated with mortality after myocardial infarction. J Cardiovasc Electrophysiol. 2005;16(1):13–20. doi: 10.1046/j.1540-8167.2005.04358.x. [DOI] [PubMed] [Google Scholar]
- 76.Tecott LH, et al. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995;374(6522):542–546. doi: 10.1038/374542a0. [DOI] [PubMed] [Google Scholar]
- 77.Buchanan GF, Richerson GB. Central serotonin neurons are required for arousal to CO2. Proc Natl Acad Sci USA. 2010;107(37):16354–16359. doi: 10.1073/pnas.1004587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. American College of Physicans. Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academies Press; 2011. [PubMed] [Google Scholar]
- 79.Drorbaugh JE, Fenn WO. A barometric method for measuring ventilation in newborn infants. Pediatrics. 1955;16(1):81–87. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.