

Abstract
The melon (Cucumis melo) genome and genetic maps with hundreds to thousands of single nucleotide polymorphism markers were recently released. However, a high-resolution genetic map was lacking. Gummy stem blight (Gsb) is a destructive disease responsible for considerable economic losses during melon production. We herein describe the development of an ultra-dense genetic map consisting of 12,932 recombination bin markers covering 1,818 cM, with an average distance of 0.17 cM between adjacent tags. A comparison of the genetic maps for melon, watermelon, and cucumber revealed chromosome-level syntenic relationships and recombination events among the three Cucurbitaceae species. Our genetic map was useful for re-anchoring the genome scaffolds of melon. More than 92% assembly was anchored to 12 pseudo-chromosomes and 90% of them were oriented. Furthermore, 1,135 recombination hotspots revealed an unbalanced recombination rate across the melon genome. Genetic analyses of the Gsb-resistant and -susceptible lines indicated the resistance phenotype is mediated by a single dominant gene. We identified Gsb-resistance gene candidates in a 108-kb region on pseudo-chromosome 4. Our findings verify the utility of an ultra-dense genetic map for mapping a gene of interest, and for identifying new disease resistant genes.
Keywords: ultra-dense genetic map, re-sequencing, gummy stem blight, Cucumis melo
1. Introduction
Gummy stem blight (Gsb) is a destructive disease caused by the ascomycetous fungus Didymella bryoniae (Auersw.) Rehm, which can infect many cucurbit species.1 Because of the agricultural importance of this disease, cucurbit sources of genetic resistance were selected several decades ago.2 Gsb of melon has increased in importance because it is responsible for considerable economic losses, especially in humid tropical and sub-tropical regions. Many Gsb-resistant melon varieties have been identified, and genetic analyses revealed several independent Gsb-resistance loci, including five monogenic dominant resistance loci (Gsb-1, Gsb-2, Gsb-3, Gsb-4, and Gsb-6) and one monogenic recessive resistance locus (gsb-5) from six resistant PIs (PI140471, PI157082, PI511890, PI482398, PI420145, and PI482399), respectively.3,4 Different molecular markers linked to Gsb resistance have been used for marker-assisted selection of Gsb-resistant germplasms.5 However, a functional Gsb-resistance gene has not been identified.
Several genetic linkage maps from different populations have been constructed based on relatively few molecular markers, which were used to map quantitative trait loci (QTLs) for agronomic and disease-resistance traits in melon.6–9 After the whole melon (Cucumis melo) genome sequence was published,10 those available simple sequence repeat and single nucleotide polymorphism (SNP) markers were integrated to build preferably genetic maps useful for anchoring contigs/scaffolds or locating QTLs.11 High-throughput re-sequencing has been proposed as a viable option for detecting markers, genotyping and constructing genetic maps.12 It has been used to construct ultra-dense genetic maps based on the availability of advanced recombinant inbred lines or an F2 population.13–16 Many important QTLs or genes have been identified using mapping-by-sequencing methods, which have accelerated forward-genetics research even in non-model species.17,18
During the co-evolution of plants and microbes, host-adapted pathogens evolved effectors to suppress pathogen-associated molecular patterns-triggered immunity (PTI), which is the first basal defence against infections by non-adapted pathogens. Effector-triggered immunity (ETI) conferred by plant resistance (R) genes contributes to a highly effective defence system, which is deployed by plants as a second, largely intracellular, layer of immunity.19–22 A coordinated relay of information between the cytoplasm and nucleus by intracellular signalling systems is required for both PTI and ETI to ensure defence-related transcripts are transferred from nucleus to the cytoplasmic protein synthesis machinery.
In this study, we constructed an ultra-dense genetic map with 12,932 recombination bin markers comprising 1,188,159 SNPs. This map may be useful for updating the melon genome and for mapping C. melo genes. For example, we mapped a 108-kb region associated with Gsb resistance on pseudo-chromosome 4. This ultra-dense genetic map may be useful for future molecular analyses of melon.
2. Materials and methods
2.1. Plant materials and Didymella bryoniae inoculation
An inbred line of Cucumis melo spp. conomon (var. conomon) (HS) and an inbred line of Cucumis melo spp. melo (var. inodorus) (XH) were crossed to generate F1, F2, and BC1 populations. The HS and XH lines as well as their F1, F2, and BC1 populations were used for genetic analyses. The F2 population was used to construct a genetic map and map the candidate Gsb-resistance gene.
Plants were inoculated with D. bryoniae using a modified version of a published method.23 The D. bryoniae spores collected from infected melon plants were cultured on potato dextrose agar medium in Petri dishes using a mycelial plug inoculation. The Petri dishes were incubated at 25 °C in the darkness for 7 days and then under ultraviolet irradiation conditions (40 w, 12 h light/12 h dark) for 4 days. Inocula were prepared by flooding the Petri dishes with 5–10 ml acidified distilled water. The resulting solution was acidified to pH 4.0 using lactic acid, and Tween-20 was added as a surfactant (20 drops/l). The spore suspension was adjusted to 5 × 105 spores/ml using a haemocytometer, and then applied to 3- to 4-week-old plants in a foliar spray. Inoculated plants were covered by a plastic tent to maintain a relative humidity of 92%. The plastic wrap was removed 3 days later. Plants were re-inoculated to ensure there were no escapees or false-positives.
2.2. Sequencing library construction and high-throughput sequencing
Genomic DNAs was extracted from the leaves of the parental lines (HS and XH) and 150 individual lines from the F2 population according to the CTAB method. The genomic DNAs were sheared into ∼500-bp fragments using the S2/E210 Ultrasonicator (Covaris, USA) for the subsequent paired-end (PE125) sequencing using the HiSeq2500 system (Illunima, USA).
2.3. SNP identification and genotyping
Raw reads were filtered to generate clean reads, which were mapped to the genome to estimate the insert sizes in the sequencing library, calculate the reads depth, and determine the distribution of reads in the melon genome (http://www.ncbi.nlm.nih.gov/assembly/GCF_000313045.1/ (23 August 2017, date last accessed))10 using the Burrows–Wheeler Aligner programme.24 Duplicated reads were removed using SAMtools.25 Potential SNPs between all lines and the genome were detected using the GATK toolkit.26 The SNPs identified between the parents were considered as polymorphic for a subsequent bin calling. The F2 genotypes were identified based on the SNP positions. The ‘pileup’ function of the SAMtools programme was used to merge the SNP datasets, in which only biallelic SNPs were kept. Short reads that were matched to multiple locations in either genome, or that did not match perfectly with at least one of the parental genomes were discarded. The VCF format document was used for genotyping according to the parental SNPs, and only the aa × bb genotype was kept. The SNPs with less than 4× coverage in either parent or with fewer than 15 SNPs in a single scaffold were eliminated. Additionally, the SNPs with an extreme segregation distortion (P < 0.01) were initially excluded by the χ2 test for a subsequent bin calling.
2.4. Bin map and linkage map construction
A modified slide window method was adopted for bin calling.13 The genotype of each window was called with a window size of 15 SNPs and a step size of 1 SNP. Windows containing more than 13 ‘aa’ or ‘bb’ types were genotyped as ‘aa’ or ‘bb’, respectively. Fifteen adjacent SNP intervals with the same genotype across the entire F2 population were combined into a recombination bin. A final set of bin markers was used to construct linkage groups (LGs) with the HighMap programme.27 Marker loci were partitioned primarily into LGs based on their locations in the C. melo genome. Next, the modified logarithm of odds (MLOD) scores between markers were calculated to verify the robustness of the markers in each LG. Markers with MLOD scores <5 were filtered prior to ordering. The error correction strategy of SMOOTH was then used according to the parental contributions to genotypes, and a k-nearest neighbour algorithm was applied to impute missing genotypes. Skewed markers were then added into the map by applying a multipoint maximum likelihood mapping. Map distances were estimated using the Kosambi mapping function. We then used the ALLMAPS programme to construct melon pseudo-chromosomes based on a comparison between the constructed genetic map and the map of the melon genome.28 Finally, we constructed pseudo-chromosomes referred to the published genome of melon.
2.5. Analysis of co-linearity and recombination hotspots
The sequences of the bin markers included in the linkage map were aligned to the physical sequences of C. lanatus (ftp://www.icugi.org/pub/genome/watermelon/97103/v1/ (23 August 2017, date last accessed)) and C. sativus (http://www.ncbi.nlm.nih.gov/genome/1639?genome_assembly_id=228904 (23 August 2017, date last accessed)) using the BLAT programme to confirm their physical positions in the genome. Spearman’s rank-order correlation coefficient was calculated to assess the co-linearity between C. melo and C. lanatus, and between C. melo and C. sativus. Recombination hotspots (RHs) were predicted based on the recombination rate of markers and the anchored C. melo genome. If the value that the genetic distance between adjacent markers was divided by a value >20 cM/Mb, the region between the two adjacent markers was considered to be an RH.29
2.6. Re-anchoring scaffolds
Filtered markers were mapped to genome assembly scaffolds, and the markers on scaffolds that were mapped to pseudo-chromosomes were counted. If the value of the following equation was >0.5, we kept the scaffold mapping results, otherwise, they were eliminated: [number of markers in the largest pseudo-chromosome − number of markers in the second largest pseudo-chromosome]/number of markers in the largest pseudo-chromosome. We then determined the position of each marker relative to each other. If the relative position in the scaffold was identical to the relative position in the genetic map, the scaffold was considered to be oriented in the forward direction. Additionally, if more markers were in the forward position than in the reverse position, the scaffold was considered to be facing the forward direction. Finally, we selected one clearly oriented scaffold as the initial scaffold, and iteratively anchored other scaffolds with higher scores.
2.7. Mapping of candidate Gsb-resistance genes
The rQTL package was used to map the candidate Gsb-resistance genes. The IM method of the rQTL package was applied for qualitative trait mapping.30 The significance thresholds were determined using 1,000 permutations, in which the threshold value was set as 2.5. The hk algorithm was selected as the me parameter, while 1 and 3 were used as the win and cofactor parameters, respectively.
2.8. RNA isolation, cDNA preparation, and quantitative real-time PCR
Leaves from HS and XH plants after inoculation (0, 0.5, 1, 2, 3, 5, and 7 days) were sampled and frozen in liquid nitrogen. Total RNA was isolated from leaves using mirVanaTM miRNA Isolation Kit (Ambion) according to the manufacturer’s instructions. RNA concentration and quality were assessed using a Thermo 2000 Bioanalyzer with an RNA NanoDrop (Thermo Scientific, USA; http://www.thermo.com (23 August 2017, date last accessed)). Samples showing A260/A230 ratio of 2.0–2.2 and A260/A280 ratio of 1.8–2.0 were used for further analysis. For quantitative real-time PCR, 1 µg of total RNA was reverse transcribed to first-strand cDNA in a final reaction volume of 20 µl using miScript II RT Kit (Qiagen). The transcriptional patterns of the candidate Gsb-resistance genes were analysed using a quantitative real-time PCR (RT-qPCR). Details regarding the primers used in this study are provided in Supplementary Table S10.
2.9. Genotyping analysis of MELO03C012987 gene
Full-length sequence of MELO03C012987 gene (except for stop codon) was amplified by PCR from leaves of six resistant inbred lines (HP9818, HB6, HB11, HB13, CNZ, and QDH) and five susceptible inbred lines (RM, MW, DZX, M14086, and M15019) to analyse the SNP further. The primers are listed in Supplementary Table S10. The resulting PCR products were cloned into a pEASY-Blunt Zero Cloning Vector according to the manufacturer’s instructions (TRANs, Beijing, China; www.transgen.com.cn (23 August 2017, date last accessed)) and amplified in E coil overnight. Subsequently, eight clones of each line were sequenced by ABI3730xl sequencer. Few clones with sequence that did not match perfectly with reference full length were discarded.
3. Results
3.1. Gsb resistance in C. melo is controlled by a single dominant gene
The HS and XH inbred lines were used to construct a genetic map and evaluate Gsb resistance (Fig. 1A). Plants growth and the development of disease symptoms in seedlings inoculated with D. bryoniae were used to assess Gsb resistance or susceptibility. Accordingly, the HS and XH inbred lines were resistant and susceptible to Gsb, respectively (Fig. 1B and Table 1). A phenotypic analysis revealed that the F1 population from the XH × HS cross was resistant to Gsb, which is suggestive of a dominant resistance trait (Fig. 1A and Table 1). A phenotypic analysis of the F2 population derived from the XH × HS reciprocal cross revealed a 1:3 segregation ratio for the F2 population and a 1:1 segregation ratio for the BC1 population, which is consistent with a monogenic resistance trait (Table 1).
Figure 1.

Phenotypic analysis of the two inbred lines used to construct a genetic map and for mapping the Gsb-resistance gene. (A) Phenotypes of C. melo spp. conomon (HS) and C. melo spp. melo (XH) under normal conditions. (B) Phenotypes of the two inbred lines and their F1 generation after being inoculated with Didymella bryoniae.
Table 1.
Inheritance of Gsb resistance in C. melo
| Crosses | Generation | Resistant | Susceptible | Expected ratio (R:S) | Chi-square | Pa |
|---|---|---|---|---|---|---|
| HS | P1 | 20 | 0 | |||
| XH | P2 | 0 | 20 | |||
| XH×HS | F1 | 23 | 0 | |||
| XH×HS | F2 | 165 | 54 | 3:1 | 0.013 | 0.91 |
| HS×XH | F2 | 81 | 32 | 3:1 | 0.663 | 0.42 |
| (XH×HS)×XH | BC1 | 47 | 73 | 1:1 | 5.663 | 0.017 |
Phenotypic observations for the whole plant 21 days post-inoculation were used to determine resistance/susceptible to Gsb.
Observed segregation ratios are statistically consistent with the expected ratios (χ2 test, P < 0.05).
3.2. Construction of a sequence-based ultra-dense C. melo genetic map
In total, 12.94 and 13.14 Gb clean reads were generated for the XH and HS inbred lines (26× genome coverage), respectively. Meanwhile, 217.7 Gb clean reads were generated for the 150 F2 population individuals (2× genome coverage), with more than 85% of the bases higher than Q30 (Supplementary Table S1). The well-distributed clean reads coverage on assembly scaffolds as well as the distribution of SNP mutation and SNP quality were indicative of the stochastic nature of resequencing (Supplementary Table S1 and Figs S1 and S2). The co-segregating SNPs were clustered in recombination bins (marked as Block), which were used to construct genetic linkage map. We constructed an ultra-dense genetic map consisting of 12 pseudo-chromosomes based on 12,932 recombination bin markers comprising 1,188,159 C. melo SNPs (Supplementary File S1), which covered 1,818 cM, with an average distance of 0.17 cM between adjacent bin markers (Table 2, Supplementary Table S3).
Table 2.
Construction of an ultra-dense C. melo genetic map
| Pseudo- chromosome | Total maker | Total distance (cM) | Average distance (cM) | Max gap (cM) |
|---|---|---|---|---|
| Chr1 | 969 | 167.29 | 0.17 | 2.402 |
| Chr2 | 1,041 | 148.75 | 0.14 | 3.74 |
| Chr3 | 1,597 | 179.22 | 0.11 | 8.07 |
| Chr4 | 1,073 | 226.08 | 0.21 | 12.461 |
| Chr5 | 632 | 79.69 | 0.13 | 7.933 |
| Chr6 | 1,031 | 164.23 | 0.16 | 4.209 |
| Chr7 | 1,408 | 170.29 | 0.12 | 2.736 |
| Chr8 | 1,469 | 122.15 | 0.08 | 3.138 |
| Chr9 | 950 | 153.95 | 0.16 | 2.402 |
| Chr10 | 170 | 79.69 | 0.47 | 20.059 |
| Chr11 | 1,562 | 169.69 | 0.11 | 3.807 |
| Chr12 | 1,030 | 157.15 | 0.15 | 3.071 |
| Total/average | 12,932 | 1818.18 | 0.17 | 20.059 |
Haplotype maps and linkage relationships heat maps were used to evaluate the quality of the genetic map. Haplotype maps, which indicate genotyping errors, were generated for each of the 150 lines in the F2 population as well as the parental lines. These maps presented the recombination events (pseudo-chromosome 10 haplotype map in Supplementary Fig. S3 and all haplotype maps for the 12 pseudo-chromosomes in Supplementary File S2). Almost all the recombination blocks were completely defined, with only pseudo-chromosome 7 missing 0.01% of its data. The linkage relationships heat maps, reflected the relationship between recombination markers in one pseudo-chromosome, and were used for ordering errors. These maps were created based on pair-wise recombination values for the 12,932 bin markers (pseudo-chromosome 10 linkage relationship heat map in Supplementary Fig. S4 and all linkage relationship heat maps for the 12 pseudo-chromosomes in Supplementary File S3). The constructed pseudo-chromosomes generally performed well according to the generated the heat maps.
We then compared our genetic map (HS × XH) with a published melon genetic map (PS × SC).31 Of the 580 SNPs in the PS × SC F2 genetic map, 558 were mapped to the HS × XH F2 genetic map. Most of the pseudo-chromosomes were consistent with co-linearity (Fig. 2 and Supplementary Fig. S5). We likewise compared the HS × XH F2 genetic map with the melon genetic map compiled by the International Cucurbit Genomics Initiative.6 This comparison uncovered similar co-linear relationships among pseudo-chromosomes.
Figure 2.

Constructed pseudo-chromosome (Chr9) and the co-linearity between the genetic map constructed from the HS × XH population in this study and a published genetic map for the PS × SC-9 population.
3.3. Genome re-anchoring
The HS × XH F2 genetic map was used to re-anchor the genome scaffolds to the 12 pseudo-chromosomes. We obtained all assembled scaffolds from the melon reference genome.10 Based on the genetic map, we anchored 92% of the scaffolds assembly (344.9 Mb of 374.8 Mb; 153 scaffolds) onto the 12 pseudo-chromosomes (Fig. 3 and Supplementary Table S4). We determined the orientation of 97.75% (336 Mb) of the anchored scaffolds, and detected chimeric scaffolds, with each anchored to different pseudo-chromosomes in the genome. For example, in scaffold NW_007546289.1, 264 Blocks were mapped onto pseudo-chromosome 5, and 72 Blocks were anchored onto pseudo-chromosome 8. In addition, in scaffold NW_007546312.1, 79 Blocks were mapped to pseudo-chromosome 8, and 41 Blocks were anchored to pseudo-chromosome 11. These results indicate that this ultra-dense genetic map may be used to improve the published melon genome assembly.
Figure 3.

Re-anchored scaffolds based on the ultra-dense genetic map constructed in this study. Yellow columns represent the 12 pseudo-chromosomes of melon. Bin markers are located according to genetic distance (cM). Scaffolds were displayed in blue and grey lines represent corresponding genetic markers between each pseudo-chromosome and scaffolds.
3.4. Analysis of RHs
We identified 1,135 RHs, which were unequally distributed to all 12 pseudo-chromosomes. pseudo-chromosome 9 had the most RHs (153), while pseudo-chromosome 10 had the fewest (30) (Fig. 4 and Supplementary Table S5). Most RHs were located at the telomeres of pseudo-chromosome, which suggested the pericentromeric regions lacked recombination events (Fig. 4). The fact that almost no RHs were detected on one arm of pseudo-chromosome 10, may have been because very few markers were distributed in this region (Fig. 4), or might be related to the acrocentric morphology of this chromosome.31
Figure 4.

Genetic positions of the recombination hotspots in the 12 melon pseudo-chromosome.
3.5. Comparison of the Cucurbitaceae pseudo-chromosomes
A comparison between our genetic map of melon and the genetic maps of other Cucurbitaceae species (i.e., watermelon and cucumber) revealed a relatively weak syntenic relationship between the genomes (Fig. 5, Supplementary Fig. S6 and Table S6). In a comparison with cucumber chromosomes, Spearman’s rank-order correlation coefficients for 12 melon pseudo-chromosomes varied from 0.63 to 0.07. Additionally, in a comparison with watermelon chromosomes, Spearman’s rank-order correlation coefficients for 12 melon pseudo-chromosomes ranged from 0.67 to 0.01 (Supplementary Table S6). Of the 12 melon pseudo-chromosomes, pseudo-chromosome 10 exhibited a closer syntenic relationship with watermelon Chr 5 and cucumber Chr 5 than the other pseudo-chromosomes (Fig. 5 and Supplementary Table S6). These results imply that the genomic structures of pseudo-chromosome 10 from melon, Chr 5 from watermelon and cucumber are relatively conserved.
Figure 5.
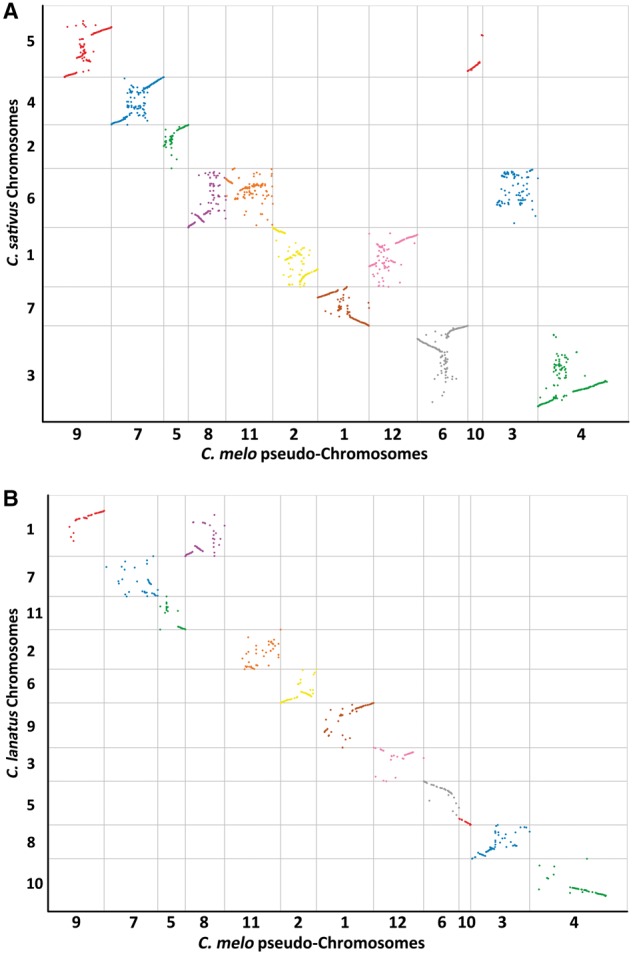
Comparison of the syntenic relationships among the pseudo-chromosomes of C. melo, and chromosomes of C. sativus and C. lanatus. (A) Syntenic relationships between C. melo and C. sativus. (B) Syntenic relationships between C. melo and C. lanatus.
3.6. Mapping of C. melo Gsb-resistance gene candidates
The 12 pseudo-chromosomes as well as the genotyping and phenotyping data related to Gsb resistance were used to map the candidate Gsb-resistance gene (GsbR). We identified a 0.667-cM region on pseudo-chromosome 4 with two bin markers (Block13188 and Block13179) that satisfied the LOD threshold of 2.5, implying the region is linked to Gsb resistance. The LOD values of Block13188 and Block13179 were 2.528 and 2.737, respectively, while the contribution rates were 9.36 and 10.72, respectively (Fig. 6A and Supplementary Table S7). The additive effects and dominant effects were 0.177 and 0.12, respectively (Supplementary Table S7). Furthermore, a scaffold (CM3.5_scaffold00018) with 71 well-distributed SNPs was identified during the genotyping of the HS (Gsb resistant) and XH (Gsb susceptible) parent lines (Supplementary Table S8). Eight candidate genes were annotated in this region based on the melon reference genome, GO, NR, Swiss-Prot, COG, and KEGG databases (Fig. 6A, Table 3, and Supplementary Table S9).
Figure 6.

Mapping of C. melo Gsb-resistance candidate genes. (A) Mapping of Gsb-resistance candidate genes. (B) Analysis of Gsb-resistance candidate gene expression levels in plants inoculated with Didymella bryoniae. (C) DUF761 domain in MEL03C012987 gene. (D) A non-synonymous SNP in the MELO03C012987 gene. (E) Genotyping of the non-synonymous substitution of MEL03C012987 in resistant and susceptible lines of C. melo.
Table 3.
Annotation of Gsb-resistance candidate genes
| Gene ID | Location | Direction | Annotation | Expression |
|---|---|---|---|---|
| MELO3C012986 | 3934220–3943507 | + | Similar to VHS domain-containing protein | Expressed |
| MELO3C012987 | 3957629–3958543 | − | Similar to uncharacterized Avr9/Cf-9 rapidly elicited protein 146 | Expressed |
| MELO3C012988 | 3960154–3961860 | − | Similar to putative ribonuclease H protein | ND |
| MELO3C012989 | 3962492–3963050 | − | LINE-1 retrotransposable element ORF2 protein | ND |
| MELO3C012990 | 3963801–3965186 | − | uncharacterized protein | ND |
| MELO3C012991 | 3967157–3970154 | + | Similar to Protein SEC13 homolog | Expressed |
| MELO3C012992 | 3979703–3980781 | + | Similar to GATA transcription factor 9 | Expressed |
| MELO3C012993 | 4015998–4027273 | + | Similar to Biotin–protein ligase | Expressed |
We analysed the expression patterns of these candidate genes in HS and XH plants inoculated with D. bryoniae. We were unable to detect the expression of MELO3C012988, MELO3C012989, and MELO3C012990 in control and inoculated plants, implying these genes may not directly influence Gsb-resistance. In contrast, MELO03C012987 expression was considerably upregulated in HS and XH plants inoculated with D. bryoniae (Fig. 6B). The expression levels of the remaining candidate genes (i.e., MELO3C012986, MELO3C012991, MELO3C012992, and MELO3C012993) were not induced by D. bryoniae inoculation (Fig. 6B). These results indicated that MELO03C012987 is most likely the gene responsible for Gsb-resistance in melon. A Pfam BLAST analysis indicated that MELO03C012987 includes a DUF761 domain (Fig. 6C). We then compared the mapped region between the HS and XH lines. A non-synonymous SNP was identified in the MELO03C012987 gene, which resulted in a Lysine being replaced by a glutamic acid in the encoded protein (Fig. 6D). Genotyping MELO03C012987 from six resistant and five susceptible inbred lines of C. melo revealed that all five susceptible lines (RM, MW, DZX, M14086, and M15019) exhibited Adenine at the non-synonymous SNP, which is same as that found in XH plant. Of the six resistant lines genotyped, five (HB6, HB11, HB13, CNZ, and QDH) had the Guanine at this non-synonymous substitution as that in HS plant. Whereas, HP9818 exhibited either Guanine or Adenine, representing a heterozygous genotype (Fig. 6E and Supplementary Table S11).
4. Discussion
The melon genome sequence10 and several genetic maps7,8,11,31 have recently been published. However, functional genomics studies involving melon are still restricted by the fact only a few hundred or thousand markers have been explored. Based on a F2 population of double-haploid DHL92 used for melon genome sequence,10 a genetic map was developed composing of 580 SNPs, which anchored the scaffolds into a chromosome-scale pseudomolecules.31 This work not only provided a significantly updated melon genome, but also indicated that high-resolution genetic map could be an efficient approach for the improvement of genome draft sequence. Rapid advances in high-throughput sequencing have revolutionized SNP marker discovery and genotyping analyses. Additionally, high-throughput sequencing-based approaches are now routinely used to construct genetic maps.13,15,16 An important strength of genotyping-by-sequencing (GBS) is that the detection and genotyping of genome-wide SNP markers occur simultaneously. Moreover, a GBS approach does not require a reference genome for SNP calling and genotyping. However, an available reference genome is useful because it enables the proper alignment and ordering of sequenced tags and helps to impute low-coverage data.32 These two sub-species are commonly used in breeding programmes because they are resistant to multiple diseases and exhibit desirable traits regarding sugar content and flavour. After a genotyping analysis, we obtained 12,932 bin markers, which included more than one million SNPs. Using these bin markers, we constructed the first ultra-dense genetic map that can be used as a reference in future studies of melon. Recently, 354.98 Mb of melon scaffolds were anchored and 325.4 Mb of sequences were oriented by a new set of targeted SNP markers with better distribution in the parents of the DHL92 melon reference genome.31 In our study, several missed scaffolds were assembled into melon pseudo-chromosome. Furthermore, more sequence (336.05 Mb) were oriented in our new scaffold genome, suggesting that our ultra-dense genetic map could further improve the anchoring and orienting of melon scaffold genome assembly.
A GBS-based high-resolution genetic map can be efficiently used to identify genetic regions and candidate genes underlying agronomically important traits.17,33,34 We mapped a 0.667 cM region on pseudo-chromosome 4, and identified eight candidate genes for Gsb resistance in this region. Thus, an ultra-dense genetic map can be constructed and candidate genes underlying Gsb resistance can be mapped using a GBS approach. Genetic map-based high-quality reference genome sequences provide useful information not only for identifying genes and regulatory elements, but also for characterizing genomic diversity.35 Our ultra-dense genetic map improved the re-anchoring of scaffolds to the reference genome. It may also contribute fundamental information for the de novo assembly of a high-quality oriental melon genome. Moreover, the improved anchoring of scaffolds may help researchers analyse chromosomal structures to improve the accuracy of gene mapping. Comparisons of genetic maps can reveal the genetic basis for conserved and variable sequences within species or among related species. We observed a strong co-linearity, but also rearrangements between our HS × XH F2 genetic map and the genetic maps of whole-genome sequenced melons, especially in pseudo-chromosomes 3–5. A chromosome-level comparison between the C. melo genetic map and the C. sativus and C. lanatus genomes revealed syntenic relationships among these three Cucurbitaceae species. The complex syntenic patterns presented as segmented chromosome-to-chromosome orthologous relationships were consistent with the notion that the chromosomal evolution and rearrangement among these three Cucurbitaceae species is very complex.10,36,37 However, it remains possible that such differences are derived from different mapping populations being used for various genotypes associated with a chromosome-level reorganization.
Among the detected Gsb-resistance candidate genes, MELO3C012986 encodes a protein similar to a VHS domain-containing protein, which is involved in clathrin-related endomembrane trafficking in plants.38MELO3C012991 encodes a Sec13, which is part of a nuclear pore complex with subunits that are involved in the responses to pathogens, cold stress, auxin, and ethylene.39 We identified an RT-like gene (i.e., encoding a reverse transcriptase) upstream of Sec13. An RT gene is usually associated with a mobile element, such as a retrotransposon, retrovirus, group II intron, retron, or retroplasmid, as well as an occasional retro (pseudo) gene.40–42MELO3C012992 encodes a GATA zinc finger transcription factor family gene (GATA TF4-like), which has been implicated in light-responsive, circadian-regulated, and nitrate-dependent control of transcription.43,44
Plants are often attacked by pathogens and have evolved a multi-layered defence system. An essential defence mechanism involves the expression of resistance (R) genes, which enables plants to detect pathogens carrying the corresponding avirulence (Avr) genes.21 The R protein-mediated pathogen recognition can be direct or indirect via the action of effectors (Avr proteins) on host targets. The R gene-based resistance is usually considered to be related to gene-for-gene resistance, which often involves a hypersensitive response (HR). MELO3C012987 encodes a protein that is similar to the uncharacterized Avr9/Cf-9 rapidly elicited (ACRE) protein 146, which is related to the gene-for-gene dominant resistance to fungal pathogens.45,46 Many ACRE genes encode putative signalling components, suggesting they are essential for early defence responses.45 Four genes (i.e., ACRE74, ACRE189, ACRE264, and ACRE276) have been confirmed as essential for both Cf-9- and Cf-4-mediated HR. Previous studies determined that ACRE74, ACRE264, and ACRE276 are critical for the Cf-9-mediated resistance to C. fulvum.47–49 Our data suggested that MELO3C012987, which encodes a protein similar to ACRE146, might be the Gsb-resistance gene in melon.
In conclusion, our ultra-dense genetic map may be useful for future studies aimed at identifying the genes regulating agronomically important melon traits. Additionally, functionally characterizing MELO3C012987 may contribute to the development of new strategies to control Gsb in Cucurbitaceae species. However, gene-based disease resistance sometimes fails because of developmental and/or environmental changes. Incorporating Gsb resistance into field-grown crops will require the development of sustainable broad-spectrum disease resistance.
Availability
The re-sequence data can be available at NCBI under accessions SRP114921.
Supplementary Material
Acknowledgements
We would like to thank Prof Fengming Song from Institute of Biotechnology, Zhejiang University, to offer the Gsb isolate.
Supplementary data
Supplementary data are available at DNARES online.
Funding
This study was supported by grants from the earmarked fund for Modern Agro-Industry Technology Research System of China (CARS-26-17), the Science and Technology Program of Zhejiang Province (2017C32041), and the Breeding Alliance of Rice and Major Economic Crops of Zhejiang University.
Conflict of interest
None declared.
References
- 1. Keinath A.P. 2011, From native plants in Central Europe to cultivated crops worldwide: the emergence of Didymella bryoniae as a Cucurbit Pathogen. Hortscience, 46, 532–5. [Google Scholar]
- 2. Sitterly W.R. 1972, Breeding for disease resistance in Cucurbits. Annu. Rev. Phytopathol., 10, 471–90. [Google Scholar]
- 3. Frantz J.D., Jahn M.M.. 2004, Five independent loci each control monogenic resistance to gummy stem blight in melon (Cucumis melo L.). Theor. Appl. Genet., 108, 1033–8. [DOI] [PubMed] [Google Scholar]
- 4. Wolukau J.N., Zhou X.H., Chen J.F.. 2009, Identification of amplified fragment length polymorphism markers linked to gummy stem blight (Didymella bryoniae) resistance in Melon (Cucumis melo L.) PI 420145. Hortscience, 44, 32–4. [Google Scholar]
- 5. Bi Y., Xu B., Qi C., et al. 2015, Pryamiding disease resistance genes and variety improvment by molecular marker-assisted selection in melon (Cucumis melon L.). Sci. Agric. Sin., 48, 523–33. [Google Scholar]
- 6. Diaz A., Fergany M., Formisano G., et al. 2011, A consensus linkage map for molecular markers and quantitative trait loci associated with economically important traits in melon (Cucumis melo L.). BMC Plant Biol., 11, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harel-Beja R., Tzuri G., Portnoy V., et al. 2010, A genetic map of melon highly enriched with fruit quality QTLs and EST markers, including sugar and carotenoid metabolism genes. Theor. Appl. Genet., 121, 511–33. [DOI] [PubMed] [Google Scholar]
- 8. Yuste-Lisbona F.J., Capel C., Sarria E., et al. 2011, Genetic linkage map of melon (Cucumis melo L.) and localization of a major QTL for powdery mildew resistance. Mol. Breeding, 27, 181–92. [Google Scholar]
- 9. Chang C.W., Wang Y.H., Tung C.W.. 2017, Genome-wide single nucleotide polymorphism discovery and the construction of a high-density genetic map for melon (Cucumis melo L.) using genotyping-by-sequencing. Front. Plant Sci., 8, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garcia-Mas J., Benjak A., Sanseverino W., et al. 2012, The genome of melon (Cucumis melo L.). Proc. Natl. Acad. Sci. U.S.A., 109, 11872–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diaz A., Forment J., Argyris J.M., et al. 2015, Anchoring the consensus ICuGI genetic map to the melon (Cucumis melo L.) genome. Mol. Breeding, 35, 188. [Google Scholar]
- 12. Davey J.W., Hohenlohe P.A., Etter P.D., Boone J.Q., Catchen J.M., Blaxter M.L.. 2011, Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet., 12, 499–510. [DOI] [PubMed] [Google Scholar]
- 13. Huang X.H., Feng Q., Qian Q., et al. 2009, High-throughput genotyping by whole-genome resequencing. Genome Res., 19, 1068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xua X.Y., Zeng L., Tao Y., et al. 2013, Pinpointing genes underlying the quantitative trait loci for root-knot nematode resistance in palaeopolyploid soybean by whole genome resequencing. Proc. Natl. Acad. Sci. U.S.A., 110, 13469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang S., Chen J., Zhang W., et al. 2015, Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes. Genome Biol., 16, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mun J.H., Chung H., Chung W.H., et al. 2015, Construction of a reference genetic map of Raphanus sativus based on genotyping by whole-genome resequencing. Theor. Appl. Genet., 128, 259–72. [DOI] [PubMed] [Google Scholar]
- 17. Candela H., Casanova-Saez R., Micol J.L.. 2015, Getting started in mapping-by-sequencing. J. Integr. Plant Biol., 57, 606–12. [DOI] [PubMed] [Google Scholar]
- 18. Schneeberger K. 2014, Using next-generation sequencing to isolate mutant genes from forward genetic screens. Nat. Rev. Genet., 15, 662–76. [DOI] [PubMed] [Google Scholar]
- 19. Chisholm S.T., Coaker G., Day B., Staskawicz B.J.. 2006, Host-microbe interactions: shaping the evolution of the plant immune response. Cell, 124, 803–14. [DOI] [PubMed] [Google Scholar]
- 20. Dodds P.N., Rathjen J.P.. 2010, Plant immunity: towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet., 11, 539–48. [DOI] [PubMed] [Google Scholar]
- 21. Jones J.D.G., Dangl J.L.. 2006, The plant immune system. Nature, 444, 323–9. [DOI] [PubMed] [Google Scholar]
- 22. Tsuda K., Katagiri F.. 2010, Comparing signaling mechanisms engaged in pattern-triggered and effector-triggered immunity. Curr. Opin. Plant Biol., 13, 459–65. [DOI] [PubMed] [Google Scholar]
- 23. Kwon M.K., Hong J.R., Sun H.J., Sung K.Y., Cho B.H., Kim K.C.. 1997, Standardization of a mass production technique for pycnidiospores of Didymella bryoniae, gummy stem blight fungus of Cucurbits. Korean J. Plant Pathol., 13, 105–12. [Google Scholar]
- 24. Li H., Durbin R.. 2009, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li H., Handsaker B., Wysoker A., et al. 2009, The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DePristo M. A., Banks E., Poplin R., et al. 2011, A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu D.Y., Ma C.X., Hong W.G., et al. 2014, Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS One, 9, e98855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tang H.B., Zhang X.T., Miao C.Y., et al. 2015, ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biol., 16, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang J., Zhang Q.X., Cheng T.R., et al. 2015, High-density genetic map construction and identification of a locus controlling weeping trait in an ornamental woody plant (Prunus mume Sieb. et Zucc). DNA Res., 22, 183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Broman K.W., Sen S.. 2009, A guide to QTL mapping with Q/qtl. Springer, New York. [Google Scholar]
- 31. Argyris J.M., Ruiz-Herrera A., Madriz-Masis P., et al. 2015, Use of targeted SNP selection for an improved anchoring of the melon (Cucumis melo L.) scaffold genome assembly. BMC Genomics, 16, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Poland J.A., Rife T.W.. 2012, Genotyping-by-sequencing for plant breeding and genetics. Plant Genome-Us, 5, 92–102. [Google Scholar]
- 33. Verma S., Gupta S., Bandhiwal N., Kumar T., Bharadwaj C., Bhatia S.. 2015, High-density linkage map construction and mapping of seed trait QTLs in chickpea (Cicer arietinum L.) using genotyping-by-sequencing (GBS). Sci. Rep.-Uk, 5, 17512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee J., Izzah N.K., Choi B.S., et al. 2016, Genotyping-by-sequencing map permits identification of clubroot resistance QTLs and revision of the reference genome assembly in cabbage (Brassica oleracea L.). DNA Res., 23, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feuillet C., Leach J.E., Rogers J., Schnable P.S., Eversole K.. 2011, Crop genome sequencing: lessons and rationales. Trends Plant Sci., 16, 77–88. [DOI] [PubMed] [Google Scholar]
- 36. Guo S.G., Zhang J.G., Sun H.H., et al. 2013, The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet., 45, 51–82. [DOI] [PubMed] [Google Scholar]
- 37. Huang S.W., Li R.Q., Zhang Z.H., et al. 2009, The genome of the cucumber, Cucumis sativus L. Nat. Genet., 41, 1275–U1229. [DOI] [PubMed] [Google Scholar]
- 38. Zouhar J., Sauer M.. 2014, Helping hands for budding prospects: ENTH/ANTH/VHS accessory proteins in endocytosis, vacuolar transport, and secretion. Plant Cell, 26, 4232–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Parry G. 2013, Assessing the function of the plant nuclear pore complex and the search for specificity. J. Exp. Bot., 64, 833–45. [DOI] [PubMed] [Google Scholar]
- 40. Schulman A.H. 2013, Retrotransposon replication in plants. Curr. Opin. Virol., 3, 604–14. [DOI] [PubMed] [Google Scholar]
- 41. Gladyshev E.A., Arkhipova I.R.. 2011, A widespread class of reverse transcriptase-related cellular genes. Proc. Natl. Acad. Sci. U.S.A., 108, 20311–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Toro N., Nisa-Martinez R.. 2014, Comprehensive phylogenetic analysis of bacterial reverse transcriptases. Plos One, 9, e114083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reyes J.C., Muro-Pastor M.I., Florencio F.J.. 2004, The GATA family of transcription factors in Arabidopsis and rice. Plant Physiol., 134, 1718–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Manfield I.W., Devlin P.F., Jen C.H., Westhead D.R., Gilmartin P.M.. 2007, Conservation, convergence, and divergence of light-responsive, circadian-regulated, and tissue-specific expression patterns during evolution of the Arabidopsis GATA gene family. Plant Physiol., 143, 941–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stergiopoulos I., de Wit P.J.G.M.. 2009, Fungal effector proteins. Annu. Rev. Phytopathol., 47, 233–63. [DOI] [PubMed] [Google Scholar]
- 46. Grennan A.K. 2006, Plant response to bacterial pathogens. Overlap between innate and gene-for-gene defense response. Plant Physiol., 142, 809–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rowland O., Ludwig A.A., Merrick C.J., et al. 2005, Functional analysis of Avr9/Cf-9 rapidly elicited genes identifies a protein kinase, ACIK1, that is essential for full Cf-9-dependent disease resistance in tomato. Plant Cell, 17, 295–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nekrasov V., Ludwig A. A., Jones J. D. G.. 2006, CITRX thioredoxin is a putative adaptor protein connecting Cf-9 and the ACIK1 protein kinase during the Cf-9/Avr9-induced defence response. FEBS Lett., 580, 4236–41. [DOI] [PubMed] [Google Scholar]
- 49. van den Burg H.A., Tsitsigiannis D.I., Rowland O., et al. 2008, The F-box protein ACRE189/ACIF1 regulates cell death and defense responses activated during pathogen recognition in tobacco and tomato. Plant Cell, 20, 697–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.