

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a major risk factor for liver cancer; therefore, its prevention is an important clinical goal. Ablation of phosphatase and tensin homolog (PTEN) or the protein kinase Hippo signaling pathway induces liver cancer via activation of AKT or the transcriptional regulators YAP/TAZ, respectively; however, the potential for crosstalk between the PTEN/AKT and Hippo/YAP/TAZ pathways in liver tumorigenesis has thus far remained unclear. Here, we have shown that deletion of both PTEN and SAV1 in the liver accelerates the development of NAFLD and liver cancer in mice. At the molecular level, activation of YAP/TAZ in the liver of Pten–/– Sav1–/– mice amplified AKT signaling through the upregulation of insulin receptor substrate 2 (IRS2) expression. Both ablation of YAP/TAZ and activation of the Hippo pathway could rescue these phenotypes. A high level of YAP/ TAZ expression was associated with a high level of IRS2 expression in human hepatocellular carcinoma (HCC). Moreover, treatment with the AKT inhibitor MK-2206 or knockout of IRS2 by AAV-Cas9 successfully repressed liver tumorigenesis in Pten–/– Sav1–/– mice. Thus, our findings suggest that Hippo signaling interacts with AKT signaling by regulating IRS2 expression to prevent NAFLD and liver cancer progression and provide evidence that impaired crosstalk between these 2 pathways accelerates NAFLD and liver cancer.
Keywords: Hepatology, Metabolism
Keywords: Mouse models
Introduction
NAFLD is characterized by an excessive accumulation of fat in the liver. The most severe form of NAFLD, NASH, often progresses to liver cancer (1–3). Tracking with the increasing prevalence of general obesity, 30% of the US population is now estimated to have NAFLD, and 25% of these individuals will develop NASH (1, 3). Currently, there are no effective therapies to prevent the incidence and progression of NAFLD or NASH (4). This makes clarification of the detailed mechanisms of disease progression using appropriate animal models that much more urgent.
Insulin signaling begins with the binding of insulin to the insulin receptor (IR). This then phosphorylates insulin receptor substrates (IRSs) and subsequently triggers the recruitment of PI3K and the activation of AKT (5–9). Deletion of IRS1 and IRS2 in the mouse liver reduces AKT activity and gives rise to insulin resistance (10). Excessive AKT activation leads to the development of NAFLD by promoting the maturation of the transcription factor SREBP1c (11, 12). In its mature form, SREBP1c contributes to the induction of fatty acid synthase (FAS) and acetyl-CoA carboxylase (ACC), which are key enzymes in de novo lipogenesis (13). Phosphatase and tensin homolog (PTEN) is a negative regulator of AKT signaling, and several human cancers are associated with mutations or downregulation of the PTEN gene (14–16). Liver-specific PTEN-knockout mice progressively develop NAFLD, NASH, and HCC (17, 18) as a result of increased AKT signaling (19).
The Hippo signaling pathway has been implicated in the suppression of tissue regeneration, the proliferation of stem cells, and the development of cancer by inhibiting the oncogenic activity of the transcriptional coactivators YAP and TAZ (20, 21). In mice, liver-specific knockout of the Hippo pathway components MST1/2, SAV1, or NF2 induces the expansion of hepatic progenitor cells via YAP/TAZ activation and leads eventually to the development of liver cancer (HCC, cholangiocarcinoma [CC], or both) (22–25). Despite its importance in tumorigenesis, the role of Hippo signaling in the metabolic dysregulation that precedes the development of liver cancer remains unclear.
Previous studies have suggested that YAP regulates components of the AKT pathway (i.e., PI3K, PTEN, and AKT) and that the Drosophila Hippo ortholog MST1 binds and inhibits AKT directly (26–29). Increased YAP expression in human liver tumors is associated with high levels of p-AKT (30, 31). This suggests that crosstalk between the Hippo and AKT pathways may be important in the maintenance of functional liver homeostasis. The molecular coordination of these 2 pathways in liver tumorigenesis, however, has not been revealed. Using several mouse models, we now show that such crosstalk does indeed occur in the liver, that it is mediated at the level of YAP/TAZ and IRS2, and that it plays a key role in the development of NAFLD and its progression to liver cancer.
Results
Deletion of both PTEN and SAV1 in the liver promotes NAFLD, NASH, and tumorigenesis.
To investigate any potential crosstalk between the Hippo and AKT pathways in vivo, we first generated liver-specific PTEN and SAV1 double-knockout mice (Pten–/– Sav1–/–, referred to herein as DKO mice) (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI95802DS1). Consistent with previous observations (23), SAV1-deficient (Sav1fl/fl; albumin-Cre or Sav1–/–) mice had a slight enlargement of the liver, whereas PTEN-deficient (Ptenfl/fl; albumin-Cre or Pten–/–) mice developed fatty livers (Figure 1A). In contrast, the DKO mice had highly advanced liver tumors at 5 months of age (Figure 1A), with significantly increased liver weights at 2 and 4 months of age (Figure 1E). While Sav1–/– and Pten–/– mice developed liver tumors at 50 to 60 weeks, all DKO mice developed such tumors by 15 weeks (Figure 1B). Thus, DKO mice showed a marked acceleration of tumorigenesis with a corresponding reduction in survival (Figure 1, B and C). In addition, while Pten–/– mice developed only HCC (17, 18), Sav1–/– mice (23) and DKO mice developed both HCC and CC (Supplemental Figure 1B).
Figure 1. Liver-specific deletion of Pten and Sav1 accelerates the development of fatty liver and tumorigenesis.

(A) Livers from 5-month-old mice of the indicated genotypes. DKO (Pten–/– Sav1–/–) livers show advanced tumor development. (B and C) Tumor-free rate (B) and survival rate (C) for mice of the indicated genotypes. (D) Oil red O and PAS staining of livers from 1-month-old mice as well as H&E, Picrosirius red, TUNEL, and F4/80 staining of livers from 3-month-old mice. Scale bars: 100 μm (Oil red O, H&E, and F4/80), 50 μm (PAS), and 200 μm (Picrosirius red and TUNEL). (E) Liver-to-body weight ratio for mice at 2 and 4 months of age. (F) Liver enzymes (aspartate aminotransferase [AST] and alanine aminotransferase [ALT]) in the serum of 3-month-old mice of the indicated genotypes. (G) Quantification of apoptotic cells and macrophages following (D) TUNEL and F4/80 staining. Nuclei were stained with DAPI (blue fluorescence). Data represent the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 versus WT; †P < 0.05, ††P < 0.01, and †††P < 0.001 for the indicated comparisons (1-way ANOVA). (A–G) n = 5 WT mice; (A) n = 5 mice, (B–D) n = 8 mice, and (E–G) n = 3 mice of the other 3 genotypes.
Given that Pten–/– liver cancer proceeds through NAFLD and NASH (17, 18), we examined young DKO mice for phenotypic changes occurring prior to tumor development. One-month-old DKO livers showed excessive fatty acid accumulation (Oil red O), similar to what was observed in NAFLD. This phenotype, however, did not appear in Pten–/– mouse livers until 3 months of age (H&E) (Figure 1D). Acute deletion of Pten and Sav1 in the adult stage using a CRE-encoding adenovirus also consistently led to the development of NAFLD (Supplemental Figure 1, D and E). DKO mouse livers also had an excessive accumulation of glycogen (PAS) at 1 month of age (Figure 1D) that grew progressively worse with time (Supplemental Figure 1C). Moreover, 3-month-old DKO livers showed increased apoptosis (TUNEL) and macrophage accumulation (F4/80) (Figure 1D and Figure 1G). Because these are associated with advanced NAFLD (H&E) and fibrosis (Picrosirius red) (Figure 1D), they suggested progression to NASH. DKO mice showed significantly increased serum AST and ALT levels at 3 months of age (Figure 1F) as well as increased pan-cytokeratin–positive (pan-CK–positive) hepatic progenitor cells in nonductal regions at 5 months of age (Supplemental Figure 1C). Collectively, these results indicate that deletion of Sav1 accelerates the progression of Pten–/– livers through the steps of NAFLD, NASH, cirrhosis, and cancer. We therefore decided to focus on the development of NAFLD as a precursor of liver tumorigenesis.
Increased AKT signaling accelerates the development of fatty liver in Pten–/– Sav1–/– mice.
We next performed gene expression profiling on the DKO mice to explore the mechanism underlying their early development of NAFLD. We found that DKO livers had increased expression of genes related to insulin signaling (i.e., Pdk4, Igfbp1, and Irs2) and reduced expression of genes related to carbohydrate or glucose metabolism (i.e., Pygb, Slc2a2, Pklr, and Aacs) (Figure 2A). In a gene set enrichment analysis (GSEA) of DKO livers relative to Pten–/– livers, we found enrichment of gene signatures related to lipogenesis and insulin signaling, including IRS targets and gene sets related to steroid biosynthesis, lipid biosynthesis, and fatty acid metabolism (Figure 2B and Table 1). Given these observations, we asked whether AKT acts as an effector molecule in the dysregulation of liver metabolism we observed in DKO mice.
Figure 2. Deletion of SAV1 potentiates AKT signaling in PTEN-deficient livers.

(A) Heatmap of differentially expressed genes in the livers of 3-month-old mice as revealed by microarray analysis. (B) GSEA for Pten–/– Sav1–/– (DKO) livers compared with Pten–/– livers. Upregulated gene signatures in DKO mouse livers are depicted using MSigDB (Broad Institute). NES, normalized enrichment score; NOM p-val, nominal P value; FDR q-val, FDR q value. (C) Immunoblot analysis of AKT signaling components and lipogenesis-related proteins in the livers of 3-month-old mice. α-Tubulin was used as a loading control. mSREBP1c, mature form of SREBP1c; pSREBP1c, precursor form of SREBP1c. (D) Immunohistochemical staining of p-AKT (Ser473) and immunofluorescence staining of PIP3 (green fluorescence) in the livers of 3-month-old mice. Scale bars: 50 μm (left) and 100 μm (right). (E–G) qPCR analysis of relative mRNA levels for lipogenesis- or inflammation-related genes in the livers of 3-month-old mice. Data represent the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the corresponding value for WT mice; †P < 0.05, ††P < 0.01, and †††P < 0.001 for the indicated comparisons (1-way ANOVA). (A and B) n = 2 mice; (C–G) n = 3 or 5 mice.
Table 1. List of upregulated GSEA gene signatures for Pten–/– Sav1–/– livers compared with Pten–/– livers.
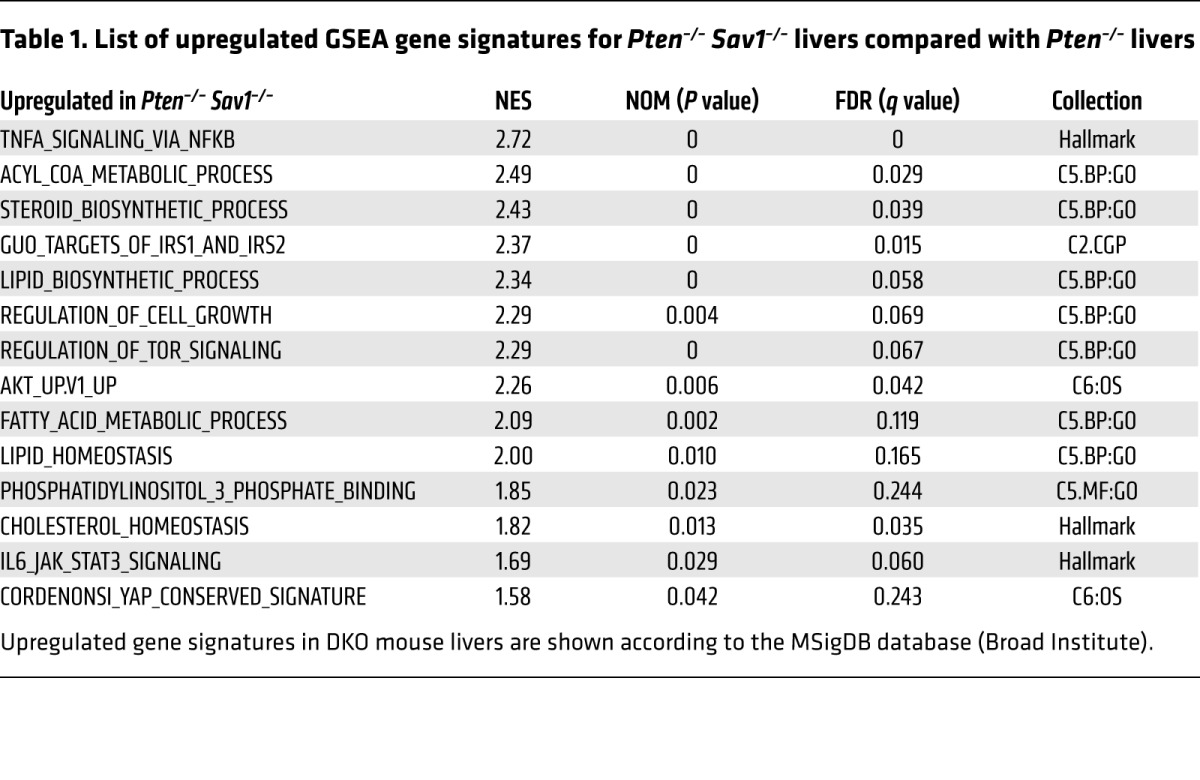
As expected, Pten deletion led to activation of AKT, as evidenced by increased levels of p-AKT (Figure 2C). Surprisingly, DKO livers showed even greater AKT activation than did Pten–/– livers, while neither Sav1–/– nor WT livers showed any such activation, presumably because of the presence of PTEN (Figure 2C). Consistent with this result, DKO livers also showed greater increases downstream of AKT signaling than did Pten–/– livers, including increased phosphorylation of glycogen synthase kinase 3β (p-GSK3β), increased processing of SREBP1c, and upregulation of FAS and ACC (Figure 2C). This increase in p-GSK3β in DKO livers is also consistent with their excessive accumulation of glycogen (18) (Figure 1D, PAS staining). Using IHC, we also observed increases in p-AKT and PIP3 in DKO hepatocytes but not biliary cells or infiltrated immune cells (Figure 2D), suggesting that the dysregulation of liver metabolism in DKO mice depends on cell-autonomous signaling. DKO livers showed increased expression of lipogenesis-related genes (i.e., Fasn, Acc1, and Scd1) and SREBP family genes (i.e., Srebp1a, Srebp1c, and Srebp2) (Figure 2, E and F). They also showed increased expression of inflammation-related genes such as IL-6 (Il6) and TNF-α (Tnfa), indicating the progression of chronic NAFLD to NASH (Figure 2G). In summary, DKO livers showed increased AKT activation, lipogenesis, glycogenesis, and inflammation.
YAP/TAZ enhance AKT activation and the development of fatty liver in the absence of PTEN.
To identify the mechanism underlying the enhanced AKT activation we observed in DKO livers, we examined the status of the various Hippo pathway components. Consistent with the role of SAV1 as an upstream regulator of LATS (23), SAV1-deficient livers (from both Sav1–/– and DKO mice) showed reduced LATS activation (p-LATS) that was also associated with increased levels of YAP but low levels of p-YAP (Figure 3A). Both Pten–/– and Sav1–/– mice showed increased levels of TAZ compared with expression levels in WT mice, but the increase in TAZ in DKO mice was even greater (Figure 3A). We confirmed the increased levels of YAP and TAZ in DKO livers via IHC (Figure 3B). Interestingly, we found high levels of YAP expression only in the nuclei of DKO mouse liver cells compared with liver cells from the other groups, but we found abundant TAZ in both the cytoplasm and nuclei (Figure 3, C and D). The increased expression of the YAP/TAZ targets CTGF and CYR61 provided further confirmation of the upregulation of YAP/TAZ activity in DKO livers (Figure 3A).
Figure 3. Increased TAZ or YAP activity promotes NAFLD via AKT activation.

(A) Immunoblot analysis of Hippo pathway components in livers from 3-month-old mice of the indicated genotypes. (B–D) IHC analysis of YAP/TAZ (B), quantification of YAP/TAZ localization from B and C, and nuclear/cytoplasmic fractionation (D) in livers from 1-month-old mice of the indicated genotypes. N, nucleus; C, cytoplasm. Scale bars: 50 μm. (E and F) Macroscopic appearance and H&E and Oil red O staining of livers from 6-week-old WT (E) and Pten–/– (F) mice 4 days after injection with adenoviruses (Ad) encoding GFP, TAZ4SA, or YAP5SA at 6 weeks of age. Scale bars: 50 μm. Arrows indicate hepatocytes with excessive lipid droplets. (G and H) Immunoblot analysis of livers as in E and F. (A–D) n = 5; (E–H) n = 3. s.e., short exposure; l.e., long exposure.
To determine whether YAP and TAZ directly promote fatty liver development via AKT activation, we used an adenovirus to induce overexpression in the liver of active TAZ (TAZ4SA) or a version of YAP (YAP5SA) that cannot be inhibited by LATS (32). In WT mice, we found that liver size and morphology remained unaffected 4 days after injection of the viruses inducing the expression of TAZ4SA or YAP5SA (Figure 3E, and Supplemental Figure 2, A and B). On the other hand, in Pten–/– mice under the same experimental conditions, the expression of TAZ4SA or YAP5SA induced hepatomegaly, promoted the development of NAFLD without fibrosis or inflammation (Figure 3F, and Supplemental Figure 2, A and C), and elicited marked increases in p-AKT and FAS (Figure 3, G and H). The fact that TAZ4SA and YAP5SA did not affect p-AKT levels in WT livers (Figure 3, G and H) suggests that PTEN was able to rapidly convert PIP3 to PIP2 and thereby prevent AKT activation. This observation suggests that PTEN deficiency is a suitable genetic background in which to study the role of YAP/TAZ in AKT signaling and metabolic dysfunction. These results also support the finding that the upregulation of YAP/TAZ promotes fatty liver development in the Pten–/– background by increasing AKT activity.
Transcriptional regulation of Irs2 by YAP/TAZ.
The next question we addressed was how YAP and TAZ potentiate AKT activity in Pten–/– livers. In DKO livers, we observed a marked increase in PIP3, a direct upstream activator of AKT (Figure 2E). We also observed increased levels of p-ERK (Figure 4A), a downstream target of PIP3. We therefore hypothesized that YAP and TAZ may serve as upstream regulators of AKT. DKO mice showed dramatic increases in IRS2, but not IR, IRS1, or PI3K (Figure 4A). In addition, the increase in Irs2 mRNA we observed in DKO mice was more significant than the increase we observed in Sav1–/– or Pten–/– mice (Figure 4B). This is reminiscent of the pattern we observed for YAP/TAZ expression in these same animals (Figure 3, A and B) and is consistent with our microarray analysis showing that DKO mice expressed higher levels of Irs2 mRNA and IRS target genes than did Pten–/– mice (Figure 2, A–C). Together, these results suggest that YAP/TAZ activation in DKO livers increases insulin signaling by upregulating IRS2.
Figure 4. YAP/TAZ directly activate Irs2 transcription through TEAD binding.

(A) Immunoblot analyses of insulin signaling molecules in the livers of 3-month-old mice. (B) qPCR analysis of Irs2 mRNA levels in the same livers as in A. (C–G) Representative immunoblot analyses of AML12 cells infected with lentiviruses encoding shPten and/or shSav1 (C)and their insulin-induced AKT activity (D). AML12 cells were infected with a retrovirus encoding IRS2 or a control (CTL) (E), and shPten- and shSav1-expressing AML12 cells in C were transfected with siIrs2 or control (siCtl) (F). AML12 cells were infected with retrovirus encoding TAZ4SA or TAZ4SA/S51A (G). In D and F, the cells were deprived of serum for 16 hours and then treated with insulin (100 nM) for the indicated durations. (H and I) Luciferase reporter assay (I) for 293T cells expressing TAZ4SA or TAZ4SA/S51A as well as luciferase (Luc) reporter constructs that include regions of the Irs2 distal promoter and first intron (H) containing WT or deleted (TBSsΔ). Ex, exon. (J) ChIP-qPCR analysis of the binding of TAZ to the Irs2 promoter analyzed in I. AML12 cells infected with control or TAZ4SA retroviruses and subjected to immunoprecipitation with antibodies recognizing TAZ or IgG. Quantitative data in B, I, and J represent the mean ± SEM. **P < 0.01 and ***P < 0.001 versus the corresponding CTL; †P < 0.05, ††P < 0.01, and †††P < 0.001 for the indicated comparisons. One-way ANOVA (B) and Student’s t test (I and J). (A and B) n = 3 for each group. (C–J) n = 3 independent experiments; (I) n = 5 independent experiments.
Next, we used the normal mouse hepatocyte cell line AML12 to further clarify the molecular relationship between YAP/TAZ and IRS2 in vitro. We found slight increases in IRS2 expression associated with the depletion of either PTEN or SAV1 in this cell line, but an even greater increase in IRS2 expression associated with the simultaneous knockdown of both PTEN and SAV1 (Figure 4C). While SAV1 depletion did not affect insulin-induced AKT activation, depletion of both SAV1 and PTEN enhanced AKT activation by dramatically increasing IRS2 expression (Figure 4D). We also found that overexpression of IRS2 induced AKT activation (Figure 4E) and knockdown of IRS2 in PTEN and SAV1-depleted cells attenuated their insulin-induced activation of AKT (Figure 4F).
Since the transcription factor TEAD binds YAP/TAZ for target gene transcription (33), we asked whether YAP/TAZ can directly regulate Irs2 expression through TEAD. We found that TAZ4SA, but not the TEAD-binding deficient mutant (TAZ4SA/S51A), could enhance IRS2 expression and AKT phosphorylation (Figure 4G). There are 6 potential TEAD-binding sites (TBSs) within the distal promoter and first intron of Irs2 (Figure 4H). TAZ4SA, but not TAZ4SA/S51A, activated reporter constructs containing TBS2, TBS4, or TBS5, but it could not activate reporters lacking these binding sites (TBSsΔ) (Figure 4I). By performing ChIP–quantitative PCR (qPCR) analysis, we found that TAZ binds TBS2 and TBS5 of Irs2 (Figure 4J). These results indicate that the YAP/TAZ-TEAD complex directly induces the transcription of Irs2, thereby promoting AKT signaling.
Deletion of YAP/TAZ or activation of Hippo signaling attenuates fatty liver development by downregulation of IRS2.
To confirm the role of YAP/TAZ in the DKO mouse phenotype, we generated DKO mice that also carried conditional Yap and/or Taz alleles (Supplemental Figure 3A). We observed mild improvement in the fatty liver phenotypes of 4-week-old Pten–/– Sav1–/– Taz–/– (PST triple-knockout [TKO]) and Pten–/– Sav1–/– Yap–/– (PSY TKO) mice compared with that seen in DKO mice (Figure 5A). In contrast, deletion of both Yap and Taz in DKO mice (PSYT quadruple-knockout [QKO]) rescued the DKO fatty liver phenotype (Figure 5A). QKO mice also had a reduced abundance of IRS2, p-AKT, p-GSK3β, and FAS (Figure 5B), and less upregulation of Irs2 mRNA than did DKO mice (Figure 5C). DKO and either PST or PSY TKO livers did not differ in their p-AKT levels (Figure 5B), presumably because of some sort of compensatory regulation of YAP or TAZ in TKO livers (34). We detected a reduced p-YAP/YAP ratio in PST TKO mice compared with DKO mice, suggesting that PST TKO mice have more active YAP. In PSY TKO mice, it was TAZ that appeared in greater abundance than was observed in DKO mice (Figure 5B). Unexpectedly, although we did not see any reduction in the liver-to-body weight ratio of PSYT QKO mice compared with that of DKO mice, QKO mice developed severe liver fibrosis, and their serum was much yellower than that of WT mice (Supplemental Figure 3, B–D). As previously reported (25, 35), Yap–/– Taz–/– mice do not form biliary ducts (CK19-negative) and have chronic liver damage and fibrosis (Supplemental Figure 4, A–F). The fibrotic changes we observed in PSYT QKO livers were likely due to the toxicity that arose because of the YAP/TAZ ablation–induced malformation of the biliary duct cells (Supplemental Figure 3C). Despite this complication, our results indicate that loss of Yap/Taz can rescue the NAFLD phenotype in hepatocytes of DKO mice.
Figure 5. Deletion of Yap/Taz rescues the NAFLD phenotype via downregulation of IRS2/AKT signaling.

(A) Representative H&E and Oil red O staining of livers from 1-month-old mice (n = 5) of the indicated groups. Scale bars: 100 μm (top) and 50 μm (bottom). (B and C) Immunoblot analysis of IRS2/AKT and YAP/TAZ signaling (B) and qPCR analysis of relative Irs2 mRNA levels (C) in the livers of mice (n = 3) as in A. Quantitative data represent the mean ± SEM. *P < 0.05 versus DKO; ††P < 0.01 for the indicated comparisons (1-way ANOVA).
Next, to determine whether the components of the Hippo pathway upstream of YAP/TAZ inhibit AKT signaling, we generated Pten–/– mice expressing transgenes encoding either a WT (MSTWTTg) or kinase-dead mutant (MSTkdTg) form of human MST1, which is a binding partner of SAV1 and an activator of the Hippo pathway (Supplemental Figure 5A). Pten–/– MSTWTTg livers, but not Pten–/– MSTkdTg livers, were smaller and had less lipid droplet accumulation than did Pten–/– livers (Figure 6, A and B, and Supplemental Figure 5, B and C). In contrast to Pten–/– MSTkdTg livers, Pten–/– MSTWTTg livers also showed lower AKT activation and IRS2 expression than did Pten–/– livers (Figure 6C and Supplemental Figure 5D). In addition, overexpression of MST1WTTg, but not MSTkdTg, increased LATS activation and decreased YAP/TAZ abundance (Figure 6, B and C). Together, these data indicate that enhanced Hippo pathway activity inhibits AKT signaling, probably by inhibiting the YAP/TAZ-mediated regulation of IRS2 and, consequently, attenuates the development of NAFLD.
Figure 6. Activation of the Hippo pathway rescues the NAFLD phenotype through suppression of IRS2/AKT signaling.

(A–C) Macroscopic appearance (A), H&E, Oil red O, TAZ, and YAP immunohistochemical staining (B), as well as immunoblot analysis (C) of livers from 2-month-old Pten–/– and Pten–/– MSTWTTg mice (n = 4). Scale bars: 50 μm (top 2 rows) and 100 μm (bottom 2 rows). β-Actin was used as a loading control in C.
YAP/TAZ and IRS2/p-AKT are positively correlated in HCC patients’ specimens.
To extend our results in mice to humans, we examined the hepatic expression of YAP, TAZ, and IRS2 in HCC patients’ samples. The human HCC and cirrhosis databases showed positive correlations between IRS2 and YAP or TAZ mRNA levels (36–38) (Figure 7A). Furthermore, we also found significantly positive correlations between IRS2 levels and the downstream targets of YAP/TAZ (e.g., CTGF and CYR61) (Figure 7B). IRS2 protein levels were also positively correlated with TAZ/YAP levels in human HCC samples: 77% of specimens with high TAZ levels and 81% of specimens with high YAP levels also expressed high levels of IRS2 (Figure 7, C and D). More important, HCC specimens with associated NAFLD show significantly higher IHC intensities for TAZ, YAP, IRS2, and p-AKT(Ser473) than did HCC samples without NAFLD (Figure 7, E and F, and Supplemental Figure 6, A–C). These results suggest that YAP and TAZ are critical in the development of fatty liver and its progression to liver cancer in humans via the upregulation of IRS2 and subsequent AKT activation.
Figure 7. Correlation of TAZ/YAP and IRS2–p-AKT expression in patients with liver cancer.

(A and B) Scatter plots of log2 (mRNA abundance) values for IRS2 versus TAZ or YAP1 (A) and for IRS2 versus CTGF or CYR61 (B) in tissue specimens from patients with liver cancer or cirrhosis (n = 47) (36), HCC (n = 91) (38), or both HCC and cirrhosis (n = 96) (37). These data were obtained from the Oncomine database and were compared with one another by calculation of the Pearson’s r correlation coefficient. (C and D) Representative IHC staining of TAZ and IRS2 (C) or YAP and IRS2 (D) in HCC patients’ specimens and a comparison of their corresponding levels of expression using the χ2 test (P < 0.001 for C and D). Scale bars: 100 μm. (E and F) Representative images of HCC specimens with associated NAFLD that had high TAZ, YAP, IRS2, and p-AKT (Ser473) IHC intensities compared with HCC specimens not associated with NAFLD (Non-NAFLD) (E). Quantification of the percentage of specimens from E and F. ***P < 0.001, by χ2 test. Scale bars: 50 μm. ND, not detected.
Inhibition of AKT signaling or silencing of IRS2 attenuates the development of liver cancer.
Given the lack of treatments for NASH (4), we next asked whether pharmacological inhibition of AKT could ameliorate fatty liver and slow cancer progression in DKO mice. To this end, we administered the pan-AKT inhibitor MK-2206 (phase II clinical trials) intraperitoneally to 3-week-old DKO mice. We found that MK-2206–treated DKO mice had less fatty liver, decreased liver weight, and reduced liver fibrosis as well as improved liver function and lower expression of lipogenesis-related genes (i.e., Fasn, Acc1, and Srebp1c) than did vehicle-treated DKO (control) mice (Figure 8, A–D, and Supplemental Figure 7A). While 80% of 12-week-old DKO mice had liver tumors (Figure 1B), MK-2206–treated DKO mice showed reduced fatty liver and tumorigenesis, as revealed by their reduced liver size, normalized liver color, and reduction in tumor nodule size, despite similar tumor nodule numbers and serum chemistry (Figure 8, E–H). MK-2206–treated DKO livers had fewer hepatocytes with excessive lipid droplets (H&E), reduced fibrosis (Picrosirius red), fewer Ki-67–positive cells, and lower levels of hepatocyte TAZ expression without a corresponding change in YAP expression (Figure 8, I and J, and Supplemental Figure 7B). These effects were also accompanied by reduced expression of genes related to fibrosis (i.e., Acta2, desmin, and Tgfb), cell death or injury (i.e., Hmox1 and Gadd153), and inflammation (i.e., Tnfa) (Figure 8K). Together, these results indicate that, like Hippo pathway activation (Figure 6, A–C), AKT inhibitor treatment markedly attenuates the development of NAFLD and liver cancer in DKO mice.
Figure 8. Treatment with the AKT inhibitor MK-2206 attenuates the development of NAFLD and liver tumor progression.

(A–D) Experimental design used to treat 3-week-old DKO mice for 2 weeks with MK-2206 (n = 3 or 5) or vehicle (control, n = 3). Macroscopic appearance of the liver as well as H&E and Oil red O staining (A), liver-to-body weight ratio (B), analysis of liver enzymes in the serum (C), and qPCR analysis of lipogenesis-related gene expression in the liver (D). Scale bars: 50 μm (A). (E–K) Experimental design used to treat 12-week-old DKO mice for 2 weeks with MK-2206 (n = 3 or 4) or vehicle (n = 3). Macroscopic appearance of the liver (E), liver-to–body weight ratio (F), liver tumor number and size (G), serum analysis (H), H&E, Picrosirius red, Ki-67, and TAZ immunohistochemical staining (I), quantification of Ki-67+ hepatocytes (J), and qPCR analysis of the expression of genes related to fibrosis (Acta2, desmin, and Tgfb), cell death or injury (Hmox1 and Gadd153), or inflammation (Tnfa) (K). Arrowheads in E indicate tumor nodules. Scale bars: 300, 400, 100, and 100 μm (I, top to bottom, respectively). Chol, cholesterol; TG, triglycerides. Quantitative data in F–H, J, and K represent the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 versus control (Student’s t test).
To clarify the role that IRS2 plays in the development of NAFLD and cancer in DKO mice in vivo, we generated an adeno-associated virus (AAV) encoding Staphylococcus aureas Cas9 (saCas9) and single-guide RNA (sgRNA) against Irs2 (sgIrs2) to abrogate IRS2 expression in the DKO mouse liver (39). Importantly, we found that deletion of Irs2 in DKO livers rescued the cancerous phenotypes of control virus–injected (sgCtl-injected) DKO mice. sgIrs2-injected DKO livers showed significant reductions in the number and size of cancer nodules by reducing their levels of p-AKT (Figure 9, A–D). We did not, however, detect any attenuation of NAFLD progression by injection of sgIrs2 into 5-week-old mice, which already had advanced NAFLD (Figure 9E). Because the rapid progression of tumor formation in DKO livers stems from their enhanced development of fatty liver, the suppression of IRS2 significantly prevents disease progression from NAFLD to cancer (Figure 9E). Collectively, these results suggest that increased IRS2 expression in DKO liver is the key factor promoting liver cancer progression.
Figure 9. Suppression of Irs2 attenuates the development of liver tumor progression.

(A–E) Injection of AAV virus encoding Cas9 and an sgRNA against Irs2 (sgIrs2) (n = 3) or an sgCtl (n = 3) into 5-week-old DKO mice. Macroscopic appearance of the liver (A), immunoblot analysis (B), liver-to-body weight ratio (C), liver tumor number and size (D), and H&E staining (E). Arrowheads in A indicate tumor nodules. Magnification ×40 (E). Quantitative data in C and D represent the mean ± SEM. *P < 0.05 versus control (Student’s t test).
Discussion
In this study, we show that the Hippo and AKT signaling pathways function cooperatively to maintain liver homeostasis. Importantly, we have shown that this hyperactivation of AKT in the liver arises from a positive feedback loop that links YAP/TAZ and IRS2/AKT signaling (Figure 10). This positive feedback loop functions as follows: (a) Loss of PTEN induces AKT activation and the consequent stabilization of TAZ via GSK3b/bTrCP signaling (40). The loss of SAV1 also activates YAP/TAZ. (b) Activated YAP/TAZ then directly induces IRS2 transcription. (c) The resulting upregulation of IRS2 promotes the activation of PI3K, further enhancing the AKT activity already increased by PTEN deletion. This increased activation of AKT further stabilizes TAZ, thereby completing the positive feedback loop that leads to hyperactivation of AKT and development of NAFLD.
Figure 10. Schematic for the positive feedback loop linking Hippo/YAP/TAZ and IRS2/AKT signaling in the liver of DKO (Pten–/– Sav1–/–) mice.

Hippo signaling interacts with AKT signaling by regulating IRS2 expression and prevents NAFLD and liver cancer progression.
Our results identify new approaches to the prevention and/or treatment of NAFLD and liver cancer. We found that treatment with the pan-AKT inhibitor MK-2206 attenuated the development of NAFLD and reduced tumor growth in DKO mice. Of note, MK-2206 has anticancer activity in humans (41–43) and has been evaluated for the treatment of liver cancer in clinical trials (https://ClinicalTrials.gov; NCT01425879 and NCT01239355). Our finding that the amplification of AKT activity stems from the function of a positive feedback loop linking YAP/TAZ and AKT suggests that AKT inhibitors may be particularly effective against liver tumors that show high levels of YAP/TAZ and AKT activity. In contrast to treatment with MK-2206, treatment with verteporfin did not block the progression of the NAFLD phenotype of DKO mouse livers (data not shown). It is possible that the inhibitory action of verteporfin was insufficient to counteract the hyperactivated YAP/TAZ in DKO mice. Given that DKO mice develop NAFLD and NASH that lead to liver cancer at an early age, a progression that resembles that of human liver disease, we expect that the DKO mouse model will prove valuable in the validation of drugs targeting AKT-dependent liver disease.
Our discovery of the link between the Hippo-YAP/TAZ and IRS2/AKT signaling pathways may explain the positive correlation between nuclear YAP1 abundance and AKT activity that was previously identified in human liver tissue specimens (30, 31) as well as the correlation between IRS2 and YAP1 or TAZ expression we observed in this study. These 2 pathways were previously linked by the observation that YAP-mediated induction of miR-29 inhibits PTEN (28), but we were unable to detect any difference in PTEN abundance between Yap–/– Taz–/– and WT livers (Supplemental Figure 4G). In addition, we found that DKO mice developed HCC/CC mixed-type tumors in the liver, despite the presence of highly active AKT and PIP3 in their hepatocytes (Figure 2D and Supplemental Figure 1B). Consistent with a previous report that YAP/TAZ activation can convert hepatocytes to progenitor/ductal cells (35), our results suggest that, in addition to inducing HCC, hyperactivated YAP/TAZ in DKO hepatocytes can also induce the development of ductal cell–derived CC by altering liver cell types.
The effects of TAZ on inflammation were recently implicated in the progression of NASH (44). Luo et al. suggested that the liver microenvironment is a key factor for NASH-derived cancer development in Pten and Shp2 DKO mice (45). Here, we found that the increased levels of TAZ in DKO livers were associated with enhanced immune cell infiltration and increased levels of inflammatory cytokines in the liver (Figure 1D, Figure 2G, and Table 1), as well as with the rapid development of NASH. The rescue models in our study (Pten–/– MSTWTTg and AKT inhibitor treatment) are also consistent with the hypothesis that suppression of AKT attenuates the development of advanced liver disease by reducing inflammation. It remains unclear, however, whether the increased inflammation observed in DKO livers depends primarily on AKT signaling, Hippo signaling, or another signaling pathway. Moreover, considering the reciprocal effects observed in the hepatic microenvironment, further studies of the relationship between accelerated disease progression and the regulation of DKO liver tissue microenvironments are necessary.
Although several studies have identified unidirectional interactions between the Hippo and AKT signaling pathways, our results revealed bilateral crosstalk between these pathways. We found that AKT signaling regulates the Hippo pathway by affecting TAZ protein stability in the liver. Pten–/– livers showed increased TAZ, the detailed mechanism of which was characterized in vitro (40). Most important, we observed that AKT inhibitor treatment significantly reduced TAZ abundance. On the other hand, the Hippo pathway also regulated AKT signaling through the transcriptional control of IRS2. In other words, both pathways must be fine-tuned to maintain liver homeostasis, because their dysregulation leads to liver disease. Furthermore, although it is well known that both Pten–/– and Sav1–/– livers develop cancer, our findings revealed that Pten–/– Sav1–/– mice developed NAFLD faster than did Pten–/– mice. In fact, Pten–/– mice developed NAFLD at a later age, while Sav1–/– mice did not develop NAFLD. We therefore suggest that the Hippo and PTEN/AKT pathways may not work in parallel. Instead, the Hippo and PTEN/AKT pathways seem to converge in the development of NAFLD, thus producing synergistic effects in the development of liver cancer.
We have demonstrated the importance of a positive feedback loop linking YAP/TAZ with IRS2/AKT signaling in the development of NAFLD, NASH, and liver cancer. We believe our identification of this link between these 2 pathways will facilitate the development of new therapeutic approaches for liver disease. Treatments combining AKT inhibitors and YAP/TAZ inhibitors may produce synergistic effects to prevent liver disease and/or inhibit liver tumorigenesis. Moreover, IRS2 is regulated by transcriptional regulators such as SREBP1c, STAT3, and HIF2α (46–48). We can now expand this list to include YAP/TAZ, which we have shown to be a bona fide transcriptional regulator of IRS2, and we anticipate that this expansion of the known regulators of IRS2 will also facilitate the development of new therapeutic approaches to treat liver disease (8, 49, 50).
Methods
Generation of liver-specific knockout and transgenic mice.
Sav1fl/fl, Ptenfl/fl, Yapfl/fl, Tazfl/fl, MSTWTTg, and MSTkdTg mice were generated as previously described (17, 23, 51–53). Mice were crossed as indicated to obtain the desired genotypes. Mice were genotyped by PCR analysis using the primers listed in Supplemental Table 1. Only male mice were used for the present study, and they were deprived of food for 16 hours before sacrifice. Adenovirus for overexpression of CRE, YAP5SA, TAZ4SA, or GFP (control) and AAV-saCas9-sgIrs2 or sgCtl in the mouse liver were injected into the tail vain of 6- and 5-week-old mice, respectively. The mice were deprived of food for 16 hours at day 4 (YAP5SA, TAZ4SA, and GFP) or week 4 (CRE and GFP) after adenovirus injection. MK-2206 (Selleckchem; S1078) was prepared in 30% Captisol (CyDex Pharmaceuticals; RC-0C7) and administered to DKO mice by intraperitoneal injection at a dose of 66 mg/kg every other day for 2 weeks, beginning at 3 or 12 weeks of age.
Histology, immunostaining, and TUNEL staining.
For immunohistochemical staining, 4-μm liver sections on slides were serially rehydrated with xylene and ethanol before heat-induced antigen retrieval (10 mM sodium citrate, pH 6.0; Duchefa). The antigen retrieval step was skipped for slides stained with the anti-IRS2 antibody. Blocking was performed with 0.3% BSA in PBS for the p-AKT–specific antibody and 5% goat serum in 3% BSA including 0.3% Triton-X for all other antibodies. After quenching endogenous peroxidases with hydrogen peroxide (Merck), the samples were incubated with a primary antibody in blocking solution. After washing and incubation with an HRP-conjugated anti-rabbit secondary antibody (Jackson Immunoresearch; 1:500), DAB (Vector Laboratories) was added for antigen detection. Finally, the slides were counterstained with hematoxylin. The antibodies used for immunohistochemical and immunohistofluorescence staining included those specific to Ser473 phosphorylated AKT (p-AKT) (Cell Signaling Technology; 4060); TAZ (MilliporeSigma; HPA007415); YAP (Cell Signaling Technology; 4912); Ki-67 (Abcam; 16667); pan-CK (Dako; Z0622); CK19 (Abcam; 15464); F4/80 (Abcam; 105155); PIP3 (Echelon; Z-P345); and IRS2 (Abcam; 84906). For Oil red O staining, cryosections (10-μm thickness) of liver tissue were fixed with cold 10% formalin, dehydrated with 100% propylene glycol (MilliporeSigma; 398039), washed with 85% propylene glycol, and then stained with 0.5% Oil red O (MilliporeSigma; O0625). The sections were counterstained with Mayer’s hematoxylin (MilliporeSigma; MHS1). Staining with Picrosirius red was performed with a solution of 0.1% Direct Red (MilliporeSigma; 365548) and 0.1% Fast Green (MilliporeSigma; F7252) in picric acid and subsequently incubated in 0.5% acetic acid. For PAS staining, the sections were incubated with 0.5% periodic acid followed by the Schiff reagent (MilliporeSigma; 3952016). TUNEL staining was performed with the In Situ Cell Death Detection Kit (Roche; 11684795910).
Immunoblot analysis.
Liver or AML12 cell lysates were prepared with Proprep Lysis Buffer (Intron Biotechnology) and NETN buffer (20 mM Tris-HCl [pH 7.4], 100 mM NaCl, 1 mM EDTA, 0.5% nonidet P-40), respectively. For nuclear/cytoplasmic fractionation analysis, frozen liver tissue was added to lysis buffer (10 mM HEPES [pH 7.8], 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, and protease inhibitors) for cytoplasmic extraction. After grinding the tissue with a hand pestle, the tissues were mixed with 0.3% NP-40 by vortexing for 5 seconds, and the cytoplasmic fraction was obtained from the supernatant after centrifugation. After 2 washes in PBS, the pellet was boiled in sample buffer and used as the nuclear fraction. The primary antibodies for the immunoblot analyses included those specific to p-AKT (catalog 4051 or 4056), AKT2 (catalog 3063), p-GSK3β (catalog 9336), p-S6K (catalog 9205), FAS (catalog 3189), p-ACC (catalog 3661), PTEN (catalog 9559), MST1 (catalog 3682), p-YAP (catalog 4911), YAP (catalog 4912), TAZ (catalog 4883), LATS2 (catalog 5888), p-LATS (catalog 8654), and p-ERK (catalog 4376) (all from Cell Signaling Technology); to IR (catalog 07-724), the p85 subunit of PI3K (catalog 06-195), IRS1 (catalog 06-248), and IRS2 (catalog 06-506) (all from MilliporeSigma); to SREBP1 (catalog 28481), FAS (catalog 196854), ACC (catalog 45174), and GAPDH (catalog 125247) (all from Abcam); to lamin B (catalogs 6217), CTGF (catalog 14939), and CYR61 (catalog 13100) (all from Santa Cruz Biotechnology); to β-actin (catalog A5316; MilliporeSigma); and to α-tubulin (catalog LF-PA0146; Abfrontier). The SAV1-specific antibody was developed in our laboratory. See complete unedited blots in the supplemental material.
Adenovirus and AAV preparation.
The CRE, human TAZ4SA, and YAP5SA cDNAs were cloned separately into the pAdtrack-CMV-GFP vector. The resulting vectors were then recombined with the pAdEasy-1 vector in BJ5183-AD-1 electroporation–competent cells (Agilent Technologies; 240005 and 200157). The recombinant DNA was linearized with PacI and introduced into 293AD cells by transfection with polyethylenimine (Polyscience; 23966). After checking the cells for GFP expression, we pelleted them with centrifugation, resuspended them with 10% glycerol in PBS, and lysed them with 4 freeze-thaw cycles (LN2 and a 37°C water bath) to release their viruses. To amplify the adenoviruses, we repeated this step with increasing numbers of cells. The adenoviruses were finally purified by ultracentrifugation at 46,000 × g for 2 hours at 4°C on a discontinuous gradient from 2.2 to 4.0 M CsCl (Amresco) in 10 mM HEPES (MilliporeSigma). The adenovirus-containing layer was removed with a syringe needle, and the viruses were washed twice in a solution containing 10 mM Tris-HCl (pH 8.0) and 2 mM MgCl2 using an Amicon Ultra Centrifugal Filter (MilliporeSigma; UFC810024). Virus titration was performed by counting exposed 293AD or target cells positive for GFP with a fluorescence microscope. A total of 1 × 109 to 1 × 1010 PFU were used for tail-vein injections. For the generation of an AAV encoding saCas9-sgRNA against Irs2, we used the pX602-AAV-TBG::NLS-SaCas9-NLS-HA-OLLAS-bGHpA;U6::BsaI-sgRNA vector, which was purchased from Addgene (plasmid 61593) (39). The steps for AAV generation, concentration, and purification were performed as previously described (39). Genomic copies of AAV (2 × 1010 to 2 × 1011) were used for tail-vein injections into 5-week-old mice that were analyzed 7 weeks later. The oligonucleotide sequences for sgIrs2 are listed in Supplemental Table 1.
Generation of stable knockdown or overexpression of cell lines.
To generate knockdown constructs, the plko.1 vector was digested with EcoRI and AgeI and ligated with annealed oligonucleotides encoding SAV1 or PTEN shRNAs (5′-CCGGCGGCTACATCTCTAGGGAATTCTCGAGAATTCCCTAGAGATGTAGCCGTTTTT-3′ and 5′-CCGGCAACCGATACTTCTCTCCAAACTCGAGTTTGGAGAGAAGTATCGGTTGTTTTT-3′, respectively). The shRNA constructs were transfected into 293T cells, together with psPAX2 and pMD2G. After 2 days, viral particles were harvested from the culture media by filtration. The viruses were then used to infect AML12 cells in the presence of polybrene (6 μg/ml) (MilliporeSigma; H9268), and stable cell lines were obtained via antibiotic selection with 10 μg/ml puromycin (Gibco, Thermo Fisher Scientific; A11138-03) or 50 μg/ml hygromycin B (Thermo Fisher Scientific; 10687010). We cloned TAZ4SA, TAZ4SA/S51A, and YAP5SA into pMSCV-puro vector (catalog 631461; Clontech), or purchased IRS2 construct (catalog DU4859; MRC PPU Reagents) for generating AML12 stable cell line expressing those genes, respectively. Next, the resulting constructs were used to prepare recombinant retroviruses for infection and subsequent puromycin selection of infected cells.
Knockdown of IRS2 with siRNA in AML12 cells.
An AML12 cell line stably expressing shPten and shSav1 was transfected with 20 nM siRNA (ST Pharm Oligo Center) using RNAiMAX (Invitrogen, Thermo Fisher Scientific; 13778-150) according to the manufacturer’s instructions. Two days later, the cells were deprived of serum for sixteen hours and then treated with insulin (100 nM). The oligonucleotide sequence information was provided by Calvin J. Kuo (Stanford University School of Medicine, CA, USA) (47). An AML12 cell line was purchased from ATCC and maintained in DMEM (Gibco, Thermo Fisher Scientific; 12100046) containing 10% FBS (Gibco, Thermo Fisher Scientific; 12483020), 1% penicillin-streptomycin (Gibco, Thermo Fisher Scientific; 15140122), 0.005 mg/ml insulin (Gibco, Thermo Fisher Scientific; 12585014), 0.005 mg/ml transferrin (MilliporeSigma; T8158), 5 ng/ml selenium (MilliporeSigma; S5261), and 40 ng/ml dexamethasone (MilliporeSigma; D4902) in a humidity-controlled environment (37°C, 5% CO2). The cell line was confirmed to be mycoplasma free with a Mycoplasma PCR Detection Kit (Intron; 25233).
Luciferase assay.
The indicated portions of the Irs2 genomic locus, including 6 potential TBSs, were cloned into the pGL3-Basic vector (Promega). Each mutant construct was generated by deletion of specific TBSs. 293T cells were cotransfected with a Renilla plasmid, a TEAD-encoding plasmid, and the constructs of interest. Twenty-four hours later, the cells were harvested, lysed, and assayed with the Dual Luciferase Reporter Assay System (Promega; E1960).
ChIP-qPCR analysis.
Two days after retrovirus infection, AML12 cells were fixed with 1% formaldehyde for 10 minutes and then neutralized with 125 mM glycine for 5 minutes at room temperature. The cells were washed with PBS and then lysed with ChIP dilution buffer (50 mM HEPES [pH 7.5], 155 mM NaCl, 1% Triton X-100, 0.1% sodium deoxycholate, 1 mM EDTA) containing 1% SDS. The DNA in the cell lysates was fragmented by sonication using a Bioruptor sonicator. The cell lysates were centrifuged at 20,000 × g for 15 minutes at 4°C, and the resulting supernatants were further diluted with ChIP dilution buffer. The supernatants were then incubated overnight at 4°C with either the TAZ antibody (MilliporeSigma; HPA007415) or IgG (Santa Cruz Biotechnology). The next day, protein A/G beads (Gendepot) were added, and the samples were incubated for an additional 3 hours at 4°C. The beads were then isolated with centrifugation, washed with ChIP wash buffer (10 mM Tris-HCl [pH 8.0], 250 mM LiCl, 0.5% nonidet P-40, 0.5% sodium deoxycholate, 1 mM EDTA), and suspended in SDS lysis buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 1% SDS) for overnight incubation at 65°C. The beads were then removed, and the remaining material was incubated for 2 hours at 55°C with proteinase K (20 mg/ml) and glycogen (20 mg/ml). After a final 1-hour incubation with RNaseA at 37°C, the DNA was purified using standard procedures and analyzed by qPCR using the primers listed in Supplemental Table 1.
Gene expression profiling and GSEA.
Total RNA was extracted from livers with Ribo-EX (GeneAll) and subjected to microarray analysis with MouseRef-8, version 2.0 BeadChip (Illumina). To identify differentially expressed genes, the raw values from all groups of mice were normalized to the mean values for each gene set and log2 transformed. For the heatmap, the values of 25,697 genes were rank-ordered by their average value for the Pten–/– Sav1–/– samples, and 500 high-rank and 500 low-rank values were selected. The Multi-Experiment Viewer program was used to generate the heatmap, and representative genes from the GO_Insulin receptor signaling pathway, KEGG_Insulin signaling pathway, GO_Glucose homeostasis, Reactom_Metabolism of carbohydrates, or Cordenonsi_YAP conserved signature gene clusters were selected from the Broad Institute’s Molecular Signatures Database (MSigDB; http://www.broadinstitute.org/gsea/msigdb/index.jsp). GSEA was performed using the GenePattern tool from the Broad Institute, with version 5.2 of the Molecular Signature Database libraries (http://www.broadinstitute.org/gsea/msigdb/index.jsp). Pten–/– Sav1–/– mice were compared with Pten–/– mice, and representative upregulated genes with a nominal P value of less than 0.05 and a FDR q of less than 0.25 are presented.
Statistics.
Because of individual variation between mice, we used at least 3 to 5 mice per group for all experiments (e.g., immunoblot analyses, qPCR, H&E staining, Oil red O staining, Picrosirius red staining, IHC, etc.). We used only 2 mice per group for the microarray analyses. Group sizes were determined in accordance with previous studies, and no mice were excluded. For the comparison of liver-specific mutant mice with WT mice, we used WT littermates for all experiments. We did not randomize the mouse experiments. The H&E staining experiments and microarray analyses were conducted by researchers who were blinded to the mouse genotypes. All data met the assumptions of each statistical test, and the variations between groups were similar. All in vitro experiments were replicated at least 3 times, except for the luciferase assay, which was replicated 5 times. Data are presented as the mean ± SEM and analyzed using 1-way ANOVA followed by Tukey’s multiple comparisons test, a 2-tailed Student’s t test, or a χ2 test, as appropriate. Statistical analyses were performed with GraphPad Prism 7 (GraphPad Software). P values of less than 0.05 were considered statistically significant. All graphs were generated with GraphPad Prism 7.
Microarray data availability.
Raw expression data for the microarray analyses have been deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE94797).
Study approval.
Human liver cancer tissue samples were obtained from 56 consecutive patients who underwent surgical resection of their liver tumors at the Chungnam National University Hospital between 2000 and 2003. Nine cases of NAFLD-associated HCCs were retrieved from the surgical pathology files of Seoul National University Bundang Hospital between 2007 and 2013. HCC samples from these patients were isolated and histologically confirmed. All patients provided written informed consent to undergo therapy and participate in the study. The IRB of Chungnam National University Hospital (IRB nos. 2017-02-026 and B-1704-391-301) approved the experiments. Paraffin blocks for the 56 human liver cancer samples were identified on the corresponding H&E-stained sections. The areas of interest representing HCC were identified and marked on the donor block. Then, 2-mm cores from the areas of interest were transferred to recipient master blocks using the Tissue Microarrayer (Meditech Industries). Animal care and experiments were performed in accordance with guidelines approved by Korea Advanced Institute of Science and Technology.
Author contributions
SHJ and DSL conceived the project and wrote the manuscript. SHJ performed most of the experiments, with contributions from HBK, MCK, JML, JHL, JBK, and HSC. JHK, WYP, and SYK performed and analyzed the microarray experiments. JWK provided the Ptenfl/fl mice and helped with the analysis. JMK and HK collected the human liver cancer tissue specimens and contributed to their analysis using H&E and immunohistochemical staining.
Supplementary Material
Acknowledgments
We thank Tae-Chang Yang (KAIST) for preparing the H&E staining slides; Eric N. Olson (UT Southwestern) for providing the YAPfl/fl TAZfl/fl mice; Kwang-Hee Bae and Kyoung-Jin Oh (KRIBB) for help in generating the adenoviral system; Gou Young Koh, David Eisenbarth, and Daehee Hwang (KAIST) for editing the manuscript; and Won-Il Jeong (KAIST) of the Korea Mouse Phenotyping Project (NRF-2014 M3A9D5A01073556) for technical support with the phenotypic analysis of mouse livers. This study was supported by grants from the National Creative Research Initiatives (NRF-2010-0018277, to DSL; NRF-2011-0018305, to HSC); the Korea Mouse Phenotyping Project (NRF-2014 M3A9D5A01075128, to SHJ); and the TJ Park Science Fellowship of the POSCO TJ Park Foundation (to SHJ).
Version 1. 02/05/2018
Electronic publication
Version 2. 03/01/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(3):1010–1025.https://doi.org/10.1172/JCI95802.
Contributor Information
Sun-Hye Jeong, Email: sunhye8@kaist.ac.kr.
Han-Byul Kim, Email: hh310@kaist.ac.kr.
Min-Chul Kim, Email: merchikim@gmail.com.
Ji-min Lee, Email: jmajw@berkeley.edu.
Jae Ho Lee, Email: hisa3028@snu.ac.kr.
Jeong-Hwan Kim, Email: alternaria@naver.com.
Seon-Young Kim, Email: kimsy@kribb.re.kr.
Jae Bum Kim, Email: jaebkim@snu.ac.kr.
Haeryoung Kim, Email: haeryoung.kim@snu.ac.kr.
Jin-Man Kim, Email: jinmank@cnu.ac.kr.
Hueng-Sik Choi, Email: hsc@chonnam.ac.kr.
Dae-Sik Lim, Email: dlim@kaist.ac.kr.
References
- 1.Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10(11):656–665. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- 2.Marengo A, Rosso C, Bugianesi E. Liver cancer: connections with obesity, fatty liver, and cirrhosis. Annu Rev Med. 2016;67:103–117. doi: 10.1146/annurev-med-090514-013832. [DOI] [PubMed] [Google Scholar]
- 3.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313(22):2263–2273. doi: 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- 4.Chi KR. The NASH drug dash. Nat Rev Drug Discov. 2015;14(7):447–448. doi: 10.1038/nrd4667. [DOI] [PubMed] [Google Scholar]
- 5.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8(12):915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 6.Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12(3):159–169. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- 7.Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. 2012;55(10):2565–2582. doi: 10.1007/s00125-012-2644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 9.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 10.Kubota N, et al. Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab. 2008;8(1):49–64. doi: 10.1016/j.cmet.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Porstmann T, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8(3):224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol Metab. 2010;21(5):268–276. doi: 10.1016/j.tem.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. 1997;99(5):846–854. doi: 10.1172/JCI119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13(5):283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 15.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carracedo A, Alimonti A, Pandolfi PP. PTEN level in tumor suppression: how much is too little? Cancer Res. 2011;71(3):629–633. doi: 10.1158/0008-5472.CAN-10-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horie Y, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113(12):1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stiles B, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected] Proc Natl Acad Sci U S A. 2004;101(7):2082–2087. doi: 10.1073/pnas.0308617100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He L, et al. The critical role of AKT2 in hepatic steatosis induced by PTEN loss. Am J Pathol. 2010;176(5):2302–2308. doi: 10.2353/ajpath.2010.090931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014;13(1):63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163(4):811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou D, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009;16(5):425–438. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee KP, et al. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107(18):8248–8253. doi: 10.1073/pnas.0912203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu L, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A. 2010;107(4):1437–1442. doi: 10.1073/pnas.0911427107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang N, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19(1):27–38. doi: 10.1016/j.devcel.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cinar B, et al. The pro-apoptotic kinase Mst1 and its caspase cleavage products are direct inhibitors of Akt1. EMBO J. 2007;26(21):4523–4534. doi: 10.1038/sj.emboj.7601872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin Z, et al. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ Res. 2015;116(1):35–45. doi: 10.1161/CIRCRESAHA.115.304457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tumaneng K, et al. YAP mediates crosstalk between the Hippo and PI(3)K–TOR pathways by suppressing PTEN via miR-29. Nat Cell Biol. 2012;14(12):1322–1329. doi: 10.1038/ncb2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye X, Deng Y, Lai ZC. Akt is negatively regulated by Hippo signaling for growth inhibition in Drosophila. Dev Biol. 2012;369(1):115–123. doi: 10.1016/j.ydbio.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 30.Sohn BH, et al. Inactivation of Hippo pathway is significantly associated with poor prognosis in hepatocellular carcinoma. Clin Cancer Res. 2016;22(5):1256–1264. doi: 10.1158/1078-0432.CCR-15-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Straßburger K, Tiebe M, Pinna F, Breuhahn K, Teleman AA. Insulin/IGF signaling drives cell proliferation in part via Yorkie/YAP. Dev Biol. 2012;367(2):187–196. doi: 10.1016/j.ydbio.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 32.Zhao B, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29(6):783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moroishi T, et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015;29(12):1271–1284. doi: 10.1101/gad.262816.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yimlamai D, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157(6):1324–1338. doi: 10.1016/j.cell.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Archer KJ, Mas VR, David K, Maluf DG, Bornstein K, Fisher RA. Identifying genes for establishing a multigenic test for hepatocellular carcinoma surveillance in hepatitis C virus-positive cirrhotic patients. Cancer Epidemiol Biomarkers Prev. 2009;18(11):2929–2932. doi: 10.1158/1055-9965.EPI-09-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mas VR, et al. Genes involved in viral carcinogenesis and tumor initiation in hepatitis C virus-induced hepatocellular carcinoma. Mol Med. 2009;15(3–4):85–94. doi: 10.2119/molmed.2008.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiang DY, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68(16):6779–6788. doi: 10.1158/0008-5472.CAN-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ran FA, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang W, et al. The N-terminal phosphodegron targets TAZ/WWTR1 protein for SCFβ-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J Biol Chem. 2012;287(31):26245–26253. doi: 10.1074/jbc.M112.382036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yap TA, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29(35):4688–4695. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 42.Yap TA, et al. Interrogating two schedules of the AKT inhibitor MK-2206 in patients with advanced solid tumors incorporating novel pharmacodynamic and functional imaging biomarkers. Clin Cancer Res. 2014;20(22):5672–5685. doi: 10.1158/1078-0432.CCR-14-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molife LR, et al. Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J Hematol Oncol. 2014;7:1. doi: 10.1186/1756-8722-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2016;24(6):848–862. doi: 10.1016/j.cmet.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo X, et al. Dual Shp2 and Pten deficiencies promote non-alcoholic steatohepatitis and genesis of liver tumor-initiating cells. Cell Rep. 2016;17(11):2979–2993. doi: 10.1016/j.celrep.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Awazawa M, et al. Adiponectin enhances insulin sensitivity by increasing hepatic IRS-2 expression via a macrophage-derived IL-6-dependent pathway. Cell Metab. 2011;13(4):401–412. doi: 10.1016/j.cmet.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 47.Wei K, et al. A liver Hif-2α-Irs2 pathway sensitizes hepatic insulin signaling and is modulated by Vegf inhibition. Nat Med. 2013;19(10):1331–1337. doi: 10.1038/nm.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6(1):77–86. doi: 10.1016/S1097-2765(05)00010-9. [DOI] [PubMed] [Google Scholar]
- 49.Fukushima T, et al. Nedd4-induced monoubiquitination of IRS-2 enhances IGF signalling and mitogenic activity. Nat Commun. 2015;6:6780. doi: 10.1038/ncomms7780. [DOI] [PubMed] [Google Scholar]
- 50.Peterson SB, Hart GW. New insights: A role for O-GlcNAcylation in diabetic complications. Crit Rev Biochem Mol Biol. 2016;51(3):150–161. doi: 10.3109/10409238.2015.1135102. [DOI] [PubMed] [Google Scholar]
- 51.Xin M, et al. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal. 2011;4(196):ra70. doi: 10.1126/scisignal.2002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xin M, et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci U S A. 2013;110(34):13839–13844. doi: 10.1073/pnas.1313192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi J, et al. Mst1-FoxO signaling protects naïve T lymphocytes from cellular oxidative stress in mice. PLoS ONE. 2009;4(11):e8011. doi: 10.1371/journal.pone.0008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.