

Abstract
Botryosphaeriaceae are an important fungal family that cause woody plant diseases worldwide. Recent studies have established a correlation between environmental factors and disease expression; however, less is known about factors that trigger these diseases. The current study reports on the 43.3 Mb de novo genome of Lasiodiplodia theobromae and five other genomes of Botryosphaeriaceae pathogens. Botryosphaeriaceous genomes showed an expansion of gene families associated with cell wall degradation, nutrient uptake, secondary metabolism and membrane transport, which contribute to adaptations for wood degradation. Transcriptome analysis revealed that genes involved in carbohydrate catabolism, pectin, starch and sucrose metabolism, and pentose and glucuronate interconversion pathways were induced during infection. Furthermore, genes in carbohydrate-binding modules, lysine motif domain and the glycosyl hydrolase gene families were induced by high temperature. Among these genes, overexpression of two selected putative lignocellulase genes led to increased virulence in the transformants. These results demonstrate the importance of high temperatures in opportunistic infections. This study also presents a set of Botryosphaeriaceae-specific effectors responsible for the identification of virulence-related pathogen-associated molecular patterns and demonstrates their active participation in suppressing hypersensitive responses. Together, these findings significantly expand our understanding of the determinants of pathogenicity or virulence in Botryosphaeriaceae and provide new insights for developing management strategies against them.
Keywords: de novo assembly, Lasiodiplodia theobromae, low-depth sequencing, RNASeq, virulence factors
1. Introduction
The fungal family Botryosphaeriaceae (Ascomycetes) includes saprobes, endophytes and phytopathogens.1–4 Members of this family are among the most widely geographically distributed opportunistic plant pathogens4 with an extensive host range, infecting many economically important plants worldwide.1–5 These pathogens can establish successful infections via wounds or natural openings, such as lenticels and stomata.2,3 Once inside the host tissues, they may remain latent, live as endophytes and then shift into pathogens when the host is stressed. Hence, taxa in this family have been described as latent pathogens or plant opportunistic fungal pathogens.4,5 These pathogens can cause serious diseases, such as dieback, cankers, shoot blights, leaf spots, fruit and seed rots, and witches’ brooms, in a broad range of woody plants.1,2,5 This shift from an endophyte to a pathogen is related to host biochemical and epigenetic changes in response to environmental stresses.4,5
Among the currently accepted 23 genera of Botryosphaeriaceae, Botryosphaeria Ces. & De Not., Diplodia Fr., Lasiodiplodia Ellis & Everh. and Neofusicoccum Crous, Slippers & A.J.L. Phillips are considered the most virulent.1,2 These botryosphaeriaceous pathogens attack the perennial organs of important woody plants, including apples, eucalyptus, grapes, peaches, pears and poplars, compromising the translocation of water and nutrients, and ultimately leading to death.3,5 To establish a successful infection, these woody pathogens overcome both preformed and inducible host defences by producing enzymes and/or toxins.5–10 During the last decade, several studies have confirmed the functions and effects of virulence factors, such as cell wall–degrading enzymes, effectors, secreted proteins and phytotoxins of these pathogens on their hosts.3–11 For example, most of these botryosphaeriaceous pathogens produce a wide array of secreted proteins that target the components in the primary and secondary cell walls of plants, such as endo-β-1,4-cellulases [glycosyl hydrolase family 5 (GH5)], β-glucosidases (GH3), xyloglucan transglucosylase/hydrolases (GH16) and β-xylosidases (GH43).3,5,7 These glycosyl hydrolase family proteins assist in the hydrolysis of glycosidic bonds in complex sugars and are involved in diverse functions, including the degradation of biomass, antibacterial defence strategies, pathogenesis mechanisms and a few typical cellular functions.12,13 Even though this knowledge can be used to identify proteins related to pathogenesis and virulence, a better understanding of their potential virulence functions is required to develop effective disease management strategies.
Although many genomic and transcriptomic studies have been conducted for different phytopathogens, only a few have focused on botryosphaeriaceous pathogens. These genomic studies, combined with transcriptome analyses, have significantly advanced our knowledge on the pathogenesis of these organisms. Recent studies on common botryosphaeriaceous woody plant pathogens have presented draft genomes for Diplodia sapinea,14Diplodia seriata3 and Neofusicoccum parvum.11 Functional annotation of sequenced genomes has provided a wide range of potential virulence factors, many of which are associated with lignocellulose degradation, host colonization, cellular transporters, nutrient uptake, secondary metabolism and toxin production. Some of the virulence genes predicted by Morales-Cruz et al.3 and Paolinelli-Alfonso et al.5 have been identified as virulence factors in other fungal pathogens, such as Eutypa lata, Phaeoacremonium aleophilum, Phytophthora sp. and Togninia minima.11,15,16 Although single-genome analysis facilitates better insights into the biology of a pathogen, comparative analysis of multiple genomes can often reveal a significantly greater amount of information on the physiology and evolution of a pathogen.17 Indeed, comparative genomic analyses of related taxa have facilitated the identification of both unique and shared genes and pathways related to virulence in fungal pathogens.
In this study, we sequenced and analysed the genomes of three botryosphaeriaceous pathogens, Botryosphaeria dothidea, Lasiodiplodia theobromae and N.parvum. Of these, L. theobromae has been recorded as the most virulent on grapes in China. Compared with L. theobromae, N. parvum and B. dothidea are considered to be mildly virulent and weakly virulent, respectively, on grapes in China.2 The main objectives of this study were to (i) identify the genomic characteristics of these fungi, (ii) understand the genetic variation among the sequenced species and genera, (iii) identify genes or gene families involved in virulence, with a special focus on genes that are unique to each of these genera, as well as those shared among these genera, and (iv) understand the opportunistic infections identified in these genera. Whole-genome, transcriptome and comparative genome analyses revealed that their genomes encode a diverse range of genes related to virulence, including carbohydrate-active enzymes (CAZymes), peroxidases, cytochrome P450s, effector proteins, transcription factor genes and genes involved in secondary metabolism. Our analyses identified obvious expansion of gene families related to transporters and the synthesis of secondary metabolites in Botryosphaeriaceae. We also identified different infection strategies utilized by L. theobromae to adapt to different lifestyles. Functional characterization of several genes confirmed their roles in pathogenicity as candidate virulence factors. Our study provides a solid foundation from which to explore the molecular processes responsible for virulence in L. theobromae as well as in other botryosphaeriaceous taxa.
2. Materials and methods
2.1. Strains, culture conditions and DNA isolation
Fungal strains were collected from Vitis vinifera in different provinces and used for sequencing (see Supplementary Table S1). The strains were deposited at the Beijing Academy of Agricultural and Forestry Sciences, Beijing, China. Cultures were grown in potato dextrose agar (PDA: 200 g potato, 20 g dextrose and 20 g agar) medium at 28 °C and stored at −80 °C. Total DNA was isolated from the collected mycelium using the Cetyltrimethylammonium bromide method.18
2.2. Genome sequencing and assembly
The genomic DNA of L.theobromae CSS-01s was sequenced with the Illumina platforms at the Beijing Genomics Institute (BGI) in Shenzhen, China, and a 500-bp paired-end DNA library and a 6-kb mate-pair DNA library were constructed. Data from the Illumina libraries were first filtered by removing low-quality sequences, base-calling duplicates and adaptor contamination. A total of 3.1 Gb of paired-end reads, with an average read length of 90 bp and a sequencing coverage of approximately 90×, was achieved for L. theobromae strain CSS-01s. The genome sequence was assembled using SOAPdenovo software. First, short reads from the paired-end library (500 bp) were assembled into contigs using sequence overlap information. Then, information from the mate-pair library was used to join the contigs into scaffolds. Finally, SOAP Gapcloser version 1.12 (http://soap.genomics.org.cn/soapdenovo.html (10 May 2016, date last accessed)) was used to close the gaps inside the scaffolds.
Two other botryosphaeriaceous taxa (B. dothidea and N. parvum) with two isolates for each species and another L. theobromae isolate (Supplementary Table S1) were subjected to low-depth sequencing. Genomic DNA extracted from all of the above isolates was sequenced using Solexa sequencing technology (Beijing Novo Gene Bioinformatics Technology Co., Ltd, Beijing, China). These libraries, with 500-bp inserts, were assembled using SOAPdenovo software (Supplementary Table S2).
2.3. Transcriptome sequencing and analyses
Lasiodiplodia theobromae conidial suspension (1 ml, 1 × 105 conidial·ml−1) was inoculated onto the 27 wounded stems of V.vinifera var. Summer Black and maintained at two temperatures (25 °C and 35 °C) for 48 h in the green house at Beijing Academy of Agricultural and Forestry Sciences, Beijing, China. Wounded stems inoculated with sterile water were maintained as controls at the same conditions. Simultaneously, L. theobromae cultures were maintained on PDA at same conditions. Then, infected host and fungal samples were collected from hosts at 8- and 12-h post-inoculation (hpi) at 25 °C. Simultaneously, fungal mycelia were collected from PDA at 8- and 12-h post-culturing at both temperatures. All the collected samples were immediately frozen in liquid nitrogen and then stored at −70 °C for RNA isolation. Total RNA was isolated from in vitro cultures and from diseased samples using Trizol according to the manufacturer’s instructions (Invitrogen, USA). Total RNA from nine stems with three biological replicates for each sample at 8 and 12 hpi were used for library construction. All procedures including mRNA purification, cDNA preparation, end repair of cDNA, adapter ligation and cDNA amplification were carried out by following Novo gene company methodologies for preparing Illumina RNASeq libraries. The paired-end reads with 150 bp were generated via Illumina HiSeq™ by Novogene company. All the sequence data were filtered to remove adaptor sequences and low-quality sequences. The quality trimmed reads were mapped to L. theobromae genome using Botwie2, allowing up to two base mismatches per read. The reads per kb per million reads was calculated to reflect the expression level. The differentially expressed analysis was done by DESeq and DEGseq software. In addition, RNASeq data assembled by Trinity were used to confirm the completeness of the draft genome of L. theobromae strain CSS-01s. RNASeq reads were assembled into 24,537 contigs longer than 300 bp, and all the contigs were then searched using blast against the L. theobromae genome.
2.4. Annotation of repetitive elements
The Tandem Repeats Finder19 was used to search for tandem repeats in the L. theobromae genome. Transposable elements were predicted by combining the results from RepeatMasker (version 4.0), RepeatProteinMasker (version 4.0) and RepeatModeler version (1.0.4).20 Homology-based discovery of transposable elements was conducted by RepeatMasker and RepeatProteinMasker against the sequences in the RepBase database version 19.10 (RM database version 20140131). RepeatModeler integrated two de novo repeat finding programs, RECON (version 1.08)21 and RepeatScout (version 1.0.5),20 to identify repeat element boundaries and family relationships from the sequence data.
2.5. Annotation of protein-coding genes
Protein-coding genes in L. theobromae strain CSS-01s were identified by integrating ab initio prediction with homology-based gene prediction and transcriptome sequencing approaches. Repeat sequences were masked throughout the genome by RepeatModeler and RepeatMasker. Then, gene models were predicted from these repeat-masked genomes using three ab initio predictors, Augustus (version 3.0.3),22 SNAP23 and GeneMark-ES (version 2.3f)24. The accuracy of the ab initio annotation was improved using the proteomes of ten other fungi (Alternaria brassicicola, Aspergillus fumigatus, Blumeria graminis, Colletotrichum graminicola, C. higginsianum, Fusarium graminearum, Neurospora crassa, Pyricularia oryzae, Saccharomyces cerevisiae and Schizosaccharomyces pombe) and the RNASeq data. These 10 downloaded fungal proteomes were aligned to the L. theobromae genome by genBlastA (version 1.0.4). Proteins with more than 70% sequence identity were used as templates to predict the gene structure of L. theobromae with Genewise (version 2.4.1).25 Then, predicted genes with incomplete open reading frames were removed. The RNASeq data were aligned to the L. theobromae genome by TopHat (version 2.0.11),26 and potential transcripts in the L. theobromae genome were predicted using Cufflinks (version 2.2.1).27 TransDecoder (version 2.0.1) was used to identify candidate coding regions within the predicted transcript sequences. Finally, all the outputs of the above methods were converted to Gene Finding Format (GFF) files with local PERL scripts. The resultant GFF files from each of the prediction programs were used as inputs into the EVM (version 2012-06-25) program to integrate GFF files obtained from the three gene prediction methods. Homology gene prediction was used to predict genes in five other sequenced botryosphaeriaceous taxa as listed in Supplementary Table S1, using the predicted genes of L. theobromae CSS-01s as templates.
2.6. Functional annotation of predicted genes
Basic functional annotations of all predicted proteins were searched against the NCBI non-redundant sequence database and UniProt/Swiss-Prot databases under the threshold parameter of E-value cut-off ≤1e−5. Secreted proteins were predicted using a combination of tools. Potential secreted proteins should contain a signal peptide without any transmembrane domain. The availability of a secretion signal peptide evaluated by both SignalP (version 4.1)28 and TargetP (version 1.1),29 and the unavailability of a transmembrane domain predicted by TMHMM (version 2.0)30 were used to define potential secreted proteins. SignalP predicts the presence of signal peptide cleavage sites and their locations in the amino acid sequence. TargetP predicts the subcellular location of eukaryotic proteins based on the predicted presence of any N-terminal pre-sequences such as chloroplast transit peptides, mitochondrial targeting peptides or secretory pathway signal peptides. Among the predicted secretory proteins, secreted proteins ≤400 amino-acids in size and with ≥4 cysteine residues were defined as putative effectors.
Searches against the protein family database PFAM and Gene Ontology (GO) annotation of each genome were performed using InterProScan (http://www.ebi.ac.uk/Tools/pfa/iprscan5/ (1 June 2016, date last accessed)) analysis. Kyoto Encyclopaedia of Genes and Genomes (KEGG) annotation was performed to assign molecular interaction networks and metabolic pathways using the online KEGG Automatic Annotation Server (KAAS) (http://www.genome.jp/kegg/kaas/ (1 June 2016, date last accessed)). The transcription factors in the genome were annotated by mapping the IPRSCAN terms to the Fungal Transcription Factor database (http://ftfd.snu.ac.kr/tf.php?a=interpro (10 June 2016, date last accessed)), and transporters were annotated by searching against the TCDB database.
The family-specific HMM profiles of CAZymes were downloaded from the dbCAN database, and the hmmscan program in the HMMER 3.0 package31 was used to search the fungal proteomes against the downloaded HMM profiles. Then, the primary results were parsed by the hmmscan-parser scripts supplied by the dbCAN database.
2.7. Comparative genomics
The protein sequences of the L. theobromae CSS-01s genome were compared with the protein sequences of 10 other fungi genomes, including five pathogenic taxa (Blumeria graminis, Colletotrichum graminicola, C. higginsianum, F.graminearum and P.oryzae), two opportunistic pathogens (A.brassicicola and A.fumigatus) and three non-pathogenic fungi (N.crassa, S.cerevisiae and S.pombe), as well as five other isolates of the Botryosphaeriaceae family.32,33 OrthoMCL (version 2.0)34 was used to find the orthologues, co-orthologues and in-paralogue pairs (Supplementary Table S3), as well as reciprocal best hits for each proteome. The tools were executed with an E-value <10−5 with the All-versus-All BLASTP search and the inflation parameter 2.0 in the MCL algorithm.
2.8. Ka/Ks calculation
For Ka/Ks calculation, 1:1 orthologous genes were identified among each isolate pair of each Botryosphaeriaceae species. Subsequently, the proteins between the two isolates were aligned in MUSCLE (version 3.8) using the default parameters and then converted to the corresponding nucleotide sequence alignment. The FASTA files containing the aligned sequences were converted into AXT files by a local PERL script, and the ratios of Ka and Ks were calculated between orthologous coding regions with Ka/Ks_Calculator v2.0 using a maximum-likelihood YN model.
2.9. Divergence time estimate
OrthoMCL was used to identify 1,029 single-copy gene families. The proteins of these single-copy gene families were aligned using MAFFT (version 7.187) with the default parameters and then converted to the corresponding nucleotide sequence alignment. A phylogenomic tree was constructed using MEGA7 based on the concatenated alignments of these 1,029 single-copy orthologous families. The phylogenomic tree consists of 15 other fungal taxa including eight pathogenic taxa (Blumeria graminis, Botrytis cinera, Colletotrichum graminicola, C. higginsianum, D.seriata, E.lata, Fusarium graminearum and Pyricularia oryzae), three opportunistic pathogens (Alternaria alternata, Alternaria brassicicola and Aspergillus fumigatus) and three non-pathogenic fungi (Neurospora crassa, Saccharomyces cerevisiae and S.pombe), as well as five other isolates of the Botryosphaeriaceae family. Bootstrap support for internal branches was evaluated from 1,000 full heuristic searches. Divergence times between species were calculated using the MCMC tree program in the PAML.
2.10. Functional studies on putative effector proteins
To understand the functions of these putative effector genes identified from the current study, 27 randomly selected effectors, of which 18 are specific, were translocated into plant cells to identify effectors that suppress hypersensitive responses (HRs) in the host. These putative effector genes (without signal peptide regions) of Botryosphaeriaceae were amplified using the primer sets listed in Supplementary Table S4. The PCR products were digested with the corresponding restriction enzymes and subcloned into effector detector vector (pEDV).35Burkholderia glumae competent cells were prepared as described, with minor modifications.36 Each pEDV construct was transformed into B. glumae using electroporation (Bio-Rad MicroPulserTM). Transformed strains were spread on LB agar medium containing 25 mg·l−1 gentamycin. At least four glycerol stocks were prepared for each transformant and stored at −80 °C for further use. Leaves of 4-week-old Nicotiana benthamiana plants were infiltrated with bacterial inocula of OD600=0.1 using a 1 ml needleless syringe. A half-section of each leaf was injected with B. glumae containing the pEDV empty vector, and the other half-section was injected with B. glumae containing the effector gene construct. Plants were maintained at 25 °C. The timing of the hyposensitive responses was recorded at 2 days post-inoculation.
2.11. Functional studies on putative lignocellulase encoding genes
Maximum parsimony analysis was conducted for all the annotated glycosyl hydrolase family genes in PAUP (phylogenetic analysis using parsimony) v.4.0b1037 using heuristic search option with tree bisection–reconnection branch swapping and 1,000 random sequence additions. The full-length coding sequences of candidate genes were isolated by PCR using the specific primers listed in Supplementary Table S4. The PCR products were digested with HindIII and EcoRI and inserted into the same sites behind the PtrpC promoter in a modified pBluescript II KS vector. The wild-type isolate, CSS-01s, was cultured at 28 °C on PDA plates. Mycelia collected from the culture were used to isolate fungal genomic DNA and protoplasts. Protoplasts were isolated and transformed using the PEG/CaCl2 method.38 Selection medium was supplemented with 1,100 μg/ml neomycin (Amresco, USA) to screen for neomycin-resistant transformants. Putative overexpression mutants were identified by PCR and further confirmed by qRT-PCR. Pathogenicity tests were conducted on detached ‘Summer Black’ grape shoots as described previously.2 The inoculated shoots were maintained on an alternating dark/light cycle under moist conditions in the greenhouse for 8–10 days at 25 °C. All treatments were performed with at least five replicates, and all experiments were repeated three times. Data were analysed by Student’s t-test using SAS software.
3. Results
3.1. Genome sequencing, assembly and annotation
The genome of the highly virulent L.theobromae strain CSS-01s (as shown in Fig. 1A) was assembled using sequencing data generated by Illumina technology (Supplementary Table S2). Approximately 342 million Illumina reads (∼90× coverage of the genome) were used for assembly in SOAPdenovo software, resulting in a genome sequence of 43.3 Mb. The assembled genome accounts for 95.7% of the total genome size (∼45.2 Mb), as estimated by JELLYFISH (Supplementary Fig. S1).39 The current genome assembly of L. theobromae strain CSS-01s consists of 29 scaffolds (≥ 1 kb) and 60 contigs with N50 lengths of 4.7 and 1.7 Mb, respectively. The overall GC content of the L. theobromae genome is approximately 54.77%. Furthermore, all RNASeq reads were assembled into 24,210 contigs longer than 300 bp, and 95.8% of these aligned with the predicted gene models, confirming the completeness and accuracy of the genome assembly. Compared with the virulence of L. theobromae, N.parvum and B.dothidea were mildly virulent and weakly virulent, respectively, in China (Fig. 1A). The genome assembly results for all botryosphaeriaceous isolates are shown in Table 1.
Figure 1.

Differences in the virulence and the phylogenomic analyses of the fungi used in the current study. (A) Differences in the virulence of isolates used for whole genome analyses on grapevine. Results indicate Lasiodiplodia theobromae as the most virulent among the three species. CK indicate the control for which normal PDA plug was used without any fungal inoculum. Figures showed the results of the pathogenicity test conducted for all the isolates used in the study. (B) Phylogenomic analyses for evolutionary relationships among different fungi. Time scale of the neighbour-joining phylogenomic tree was shown by million years ago at each node. Estimates of divergence times (millions of years) calculated from the rate of sequence similarity are indicated at each node. The red dots are the fossil calibrations from the Time Tree database.
Table 1.
Major features of L. theobromae, B. dothidea and N. parvum genome assemblies from Illumina data
|
L. theobromae |
B. dothidea |
N. parvum |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CSS-01S |
HBQJZ-02S |
Sxg01s |
LNXCEYGS-1 |
CDZ1M1s3 |
Gxg-01s |
|||||||
| Scaffold | Contig | Scaffold | Contig | Scaffold | Contig | Scaffold | Contig | Scaffold | Contig | Scaffold | Contig | |
| Total number | 29 | 60 | 338 | 1,594 | 960 | 2,203 | 1,010 | 2,190 | 1,520 | 2,757 | 1,149 | 2,534 |
| Total length (bp) | 43,283,415 | 43,280,627 | 43,092,220 | 42,975,261 | 43,081,153 | 43,048,860 | 43,005,517 | 42,846,751 | 41,659,988 | 41,636,448 | 42,600,755 | 42,560,320 |
| N50 (bp) | 4,775,223 | 1,738,941 | 272,785 | 48,918 | 134,448 | 67,019 | 123,568 | 71,603 | 52,961 | 39,937 | 102,721 | 57,603 |
| N90 (bp) | 1,285,992 | 409,362 | 68,335 | 14,359 | 30,311 | 15,690 | 25,957 | 16,444 | 13,679 | 10,166 | 22,655 | 14,005 |
| Maximum length (bp) | 6,723,813 | 3,030,172 | 1,438,296 | 232,370 | 476,208 | 428,425 | 620,772 | 400,854 | 282,031 | 179,773 | 703,251 | 276,000 |
| Minimum length (bp) | 1,230 | 410 | 1,005 | 500 | 1,000 | 31 | 1,000 | 39 | 1,001 | 25 | 1,001 | 31 |
| Sequence GC (%) | 54.77 | 54.77 | 54.93 | 54.94 | 54.77 | 54.77 | 54.76 | 54.76 | 57.05 | 57.05 | 56.91 | 56.91 |
Integrated data from the ab initio prediction, homology-based search and RNASeq facilitated the prediction of 12,902 high-confidence protein-coding genes in L. theobromae (CSS-01s), with an average sequence length of 1,629 bp. The number of predicted genes in the L. theobromae genome is similar to those in other fungal genomes (Supplementary Table S5). On average, each predicted gene contains 2.8 exons, with 9,902 of the predicted genes containing introns. The coding regions of these predicted genes constitute 43.6% of the genome. The genomic features for L. theobromae CSS-01s is shown in Table 2. The genome features of the other five Botryosphaeriaceae isolates are shown in Supplementary Table S6.
Table 2.
Genome features of L. theobromae CSS-01S
| Features | L. theobromae CSS-01s |
|---|---|
| Size (Mb) | 43.3 |
| Coverage | 90 x |
| % G+C content | 54.77 |
| % Repeat | 3.02 |
| Protein-coding genes | 12,902 |
| Average gene length (bp) | 1,629 |
| Gene density (# gene per Mb) | 297 |
| Exons per gene | 2.8 |
| Average exon length (bp) | 506 |
| Introns per gene | 1.8 |
| Average intron length (bp) | 88 |
| tRNA | 177 |
The predicted genes were further annotated based on comparisons with several databases, as indicated in Supplementary Table S7. An interproScan analysis identified 7,733 proteins belonging to 3,570 protein families in L. theobromae. Functional annotation showed that among the 12,902 predicted genes, 7,149 (55.4%) could be assigned with GO terms (Supplementary Fig. 2), and 5,520 genes could be mapped to the KEGG pathway database. Our analyses predicted 937 secreted proteins, accounting for 6.9% of the proteome, with 359 proteins identified as potential effectors. Similar numbers of secreted proteins were found in D.seriata3 (910) and N.parvum11 (1097). However, the corresponding number of secreted proteins found in the Pyricularia oryzae32 (739), Valsa mali40 (782) and V. pyri40 (774) genomes were lower than that of L. theobromae. Of all the secreted proteins identified, 269 (including 101 potential effectors) were specific to L. theobromae. Among the protein-coding genes revealed by the different annotation systems, the major functional groups in L. theobromae include degrading CAZymes (820 genes, of which 198 are specific to L. theobromae), cellular transporters (2,419 genes, of which 307 are specific), transcription factors (528 genes, of which 118 are specific) and secondary metabolites (54 genes, none were specific) (see Fig. 2C and D). Genome synteny analysis revealed an overall correlation between the homologous chromosomes of the six isolates (Supplementary Fig. S3). Repetitive elements identified in this study are shown in Supplementary Table S8.
Figure 2.

Overview of the gene family size comparison between the L. theobromae (CSS-01s) and other fungi. (A and B) Venn diagram of predicted gene families in (A) L. theobromae, B. dothidea and N. parvum and (B) Botryosphaeriaceae family with other fungi. The values explain the union counts of orthologous gene families shared among different fungi groups. Botryosphaeriaceae taxa share the highest number of unique gene families with opportunistic pathogens. (C) Top 20 Pfam domains in L. theobromae CSS-01s genome, compared with those in other fungi used in the comparative analysis. (D) Pair-wise comparison of Pfam gene family sizes between L. theobromae CSS-01s and other fungi used in the comparative analysis.
3.2. Selection pressures on orthologues of L. theobromae and other botryosphaeriaceous isolates
Adaptive evolution is often associated with changes in protein-coding genes in the genome41, and the Ka/Ks ratio is commonly used to reveal the direction and magnitude of selection upon these genes. Among the orthologous pairs between the L. theobromae strains, there were significant divergences for 809 orthologues. The Ka and Ks rates as well as their ratios were all calculated (Supplementary Fig. S4). Among these, four pairs of orthologues have Ka/Ks ratios > 1, and 13 have Ka/Ks rates ranging from 0.5 to 1. The Ka/Ks ratios between L. theobromae CSS-01s and the four other isolates of botryosphaeriaceous taxa are listed in Supplementary Table S9. Orthologous pairs with a Ka/Ks value > 1 indicate positive selection, whereas Ka/Ks value between 0.5 and 1 indicate weak purifying selection. Interestingly, all four genes (evm.model.scaffold_15.78, evm.model.scaffold_4.947, evm.model.scaffold_9.354 and evm.model.scaffold_9.168) showing positive selection were newly discovered genes with unknown functions. However, two of the genes have similar sequences in N. parvum11 and Macrophoma phaseolina42. The similar gene found in N. parvum contains a W40 domain with a wide variety of functions involved in regulatory modules in signal transduction, pre-mRNA processing and cytoskeleton assembly.11
3.3. Comparative genomics and evolution
Comparative genomics revealed a broad range of gene families available in the genome with potential functions related to virulence. In addition to the six botryosphaeriaceous isolates sequenced in this study, 15 other fungal genomes were also included for comparative analyses.32,33 These taxa includes pathogens, opportunistic pathogens and non-pathogenic fungi. According to the analysis, these species shared 1,029 single-copy orthologous groups. Phylogenetic analysis (Fig. 1B) revealed that the species belonging to Botryosphaeriaceae are more closely related to A. brassicicola and A. alternata, than to the other taxa. This is acceptable, because they all belong to the same class, Dothideomycetes. When compared with all other genomes, 2,813 gene families containing 1,129 species-specific groups (1,156 genes) were found to be specific to Botryosphaeriaceae, most of which were single-copy genes (Fig. 2A and B). These genes are enriched in cytochrome P450s and GO functional categories, such as hydrolase activity, lyase activity, metabolic processes, oxidation-reduction processes, oxidoreductase activity, iron ion binding, heme binding and transporter activity (FDR <0.05, Supplementary Table S10).
Based on their lifestyles, botryosphaeriaceous taxa share 221 unique orthologous gene families with pathogenic fungi, 1,049 with opportunistic fungal pathogens and 36 with non-pathogenic fungi (Fig. 2A and B). Orthologous families shared between pathogenic fungi and Botryosphaeriaceae are enriched in phosphorylation, signal transduction and enzyme inhibitor activity functions (FDR <0.05, Supplementary Table S11), whereas no significant functional enrichment was found in the gene families shared between the non-pathogenic taxa and botryosphaeriaceous taxa. Among all other fungal groups, Botryosphaeriaceae taxa shared the most orthologous gene families (1,049) with opportunistic fungal pathogens (Fig. 2A and B), with enriched hydrolase, catalytic, cellulose binding, oxidoreductase, transmembrane transporter, and transcription factor activities, among many others (FDR <0.05, see Supplementary Table S12). These genes are mainly involved in the metabolism of starch, sucrose, fructose and mannose, as well as secondary metabolite synthesis.
3.4. Gene family expansions and contractions
The expansion of gene families in fungal genomes is mainly associated with host adaptations5,43 and functions linked to virulence, such as host cell wall degradation, transport functions and secondary metabolite synthesis.5,44 This study used the Computational Analysis of gene Family Evolution (CAFÉ)45 program to identify gene families that have potentially undergone significant expansions or contractions in botryosphaeriaceous taxa. When compared with all other fungal genomes analysed in this study, five gene families (51 genes) in L. theobromae have significantly expanded (P ≤0.05, Supplementary Table S13), whereas one family has contracted. According to Clusters of Orthologous Group (COG) annotation, these expanded gene families in botryosphaeriaceous taxa are functionally related to secondary metabolism, plant cell wall degradation and cellular transporters such as non-ribosomal peptide synthetases (NRPS), cytochrome P450, polyketide synthase (PKS), ATP-binding cassette (ABC)–type multidrug transport system, ATPase component, and sulphite reductase and alpha subunit (flavoprotein).
3.5. Secondary metabolism genes
Secondary metabolites are believed to be involved in the development of certain disease symptoms and, in some cases, cause the host to die.5,6,46 Genes encoding a biosynthetic pathway are co-regulated at the transcriptional level and are often physically clustered on fungal chromosomes.46 A biosynthetic gene is often surrounded by genes encoding transporters and others involved in post-synthesis modification of the metabolite.46,47 In this study, two computational programs, SMURF (http://jcvi.org/smurf/index.php (20 June 2016, date last accessed))48 and antiSAMSH (http://antismash.secondarymetabolites.org/ (20 June 2016, date last accessed)),49 were used to identify genes involved in the biosynthesis of secondary metabolites. The L. theobromae genome contains a total of 58 secondary metabolite gene clusters, categorized into eight main groups. The majority of the clusters belong to type 1 polyketide synthases (t1 PKS, 11 clusters), followed by 12 non-ribosomal peptide synthetases (NRPS), 10 terpene synthases, four non-ribosomal peptide synthetase–t1 PKS hybrids, three putative fatty acid clusters, a type 1 PKS-non-ribosomal peptide synthetase hybrid, a non-ribosomal peptide synthetase–putative fatty acid hybrid and another putative saccharide cluster. In addition to these eight known clusters, there were nine other clusters encoding secondary metabolite related proteins.
3.6. Host-pathogen interactions revealed during infection
A comprehensive understanding of the L. theobromae transcriptome should significantly facilitate the discovery of new genes and their expression profiles. To understand pathogenicity-related genes, the transcript profiles of L. theobromae in V.vinifera were analysed at two early infection stages (Fig. 3A). When compared with axenic cultures, 75 and 283 genes were up-regulated, whereas 89 and 232 genes were down-regulated, at 8 and 12 hpi, respectively (Fig. 3C). During the early infection stages of L. theobromae, a total of 285 up-regulated genes and 243 down-regulated genes were identified. Among these genes, genes that exhibited significant expression during infection are listed in Supplementary Tables S14 and S15. According to GO enrichment analysis, the up-regulated genes at 8 hpi were enriched in molecular function and biological process terms, concentrated in ferric iron binding, the hydrolysis of O-glycosyl compounds and polygalacturonase activity. However, more genes were up-regulated at 12 hpi than at 8 hpi. At 12 hpi, most of the up-regulated genes were involved in pectin degradation, polygalacturonase activities and carbohydrate catabolic processes. This finding indicates that during infection, these up-regulated genes facilitate the degradation of cell walls by L. theobromae. KEGG pathway analysis confirmed that the genes up-regulated at 8 and 12 hpi were significantly enriched in starch and sucrose metabolism pathways, as well as in the pentose and glucuronate interconversion pathways.
Figure 3.

Difference in the virulence of Lasiodiplodia theobromae and their gene expression at different stages of growth and at different temperatures. (A) Difference in the virulence of L. theobromae at different infection stages on grape shoots. Figure shows the progress of infection of L. theobromae isolate CSS-01s at (i) 8, (ii) 12 and (iii) 24 hpi. (B) Differences in the virulence of L. theobromae isolate CSS-01s at different temperatures on grape shoots and their growth differences on the axenic cultures at respective temperatures: (i) control test with PDA without inoculum, (ii) virulence at 25 °C (iii) virulence at 35 °C, (iv) axenic culture growth at 25 °C and (v) culture growth at 35 °C. (C) Venn diagram showing in planta differentially expressed genes of L. theobromae at different time points after inoculation. Numbers indicate differentially expressed genes that are specific to each time point, or shared by two different time points. Differentially expressed genes at 8 and 12 hpi are represented with circles. (D) Up-regulation of LysM domain genes of L. theobromae during transcriptome analysis. L indicates samples collected from axenic cultures, M indicates samples of fungus and host tissues. Two different temperatures of 25 °C and 35 °C indicated by numbers 25 and 35. Two sample collection time points were indicated by 8 and 12 hpi.
From a total of 75 genes up-regulated at 8 hpi, 40 were secreted proteins, of which 20 were predicted to be putative effectors. Among the 283 up-regulated genes at 12 hpi, 119 were secreted proteins, of which 51 were predicted to be putative effectors. These putative effectors contain conserved functional domains such as glycoside hydrolase, pectate lyase or fungal-specific extracellular EGF-like domains (CFEM) to enhance their invasion and pathogenesis. Unlike in other pathogenic taxa,42 secondary metabolism genes were not up-regulated at both 8 and 12 hpi.
3.7. Expansion and expression of membrane transporters
Among the wide array of membrane transporters, two main families are abundant in fungal genomes, the ABC family and the major facilitator super (MFS) family.50 The L. theobromae genome encodes 917 membrane transporters. Searches against the transporter classification database confirm that L. theobromae has the largest number of membrane transporters compared with the other taxa (Supplementary Table S16). The number of membrane transporters in the L. theobromae genome is similar to that in A. fumigatus. Several membrane transporters in the L. theobromae genome are significantly expanded, including amino acid/choline transporters, anion:cation symporters, sugar porters, drug:H+ antiporter-2, multidrug resistance exporters (a.k.a. ABCB) and the pleiotropic drug resistance (PDR) transporters (Fig. 4A). Upon host contact, 17 membrane transporters were significantly up-regulated, including amino acid transporters, PDRs, sugar porters and triose-phosphate transporters. The multidrug resistance proteins in the PDR family confer resistance to cycloheximide and antifungal agents such as azoles and terbinafine51, and both were up-regulated. Two genes in the sugar porter family were significantly up-regulated; one of the two genes is a glucose/xylose: H+ symporter and the other is an MFS permease. These sugar transporters may allow the fungus to grow under different pH conditions.52
Figure 4.
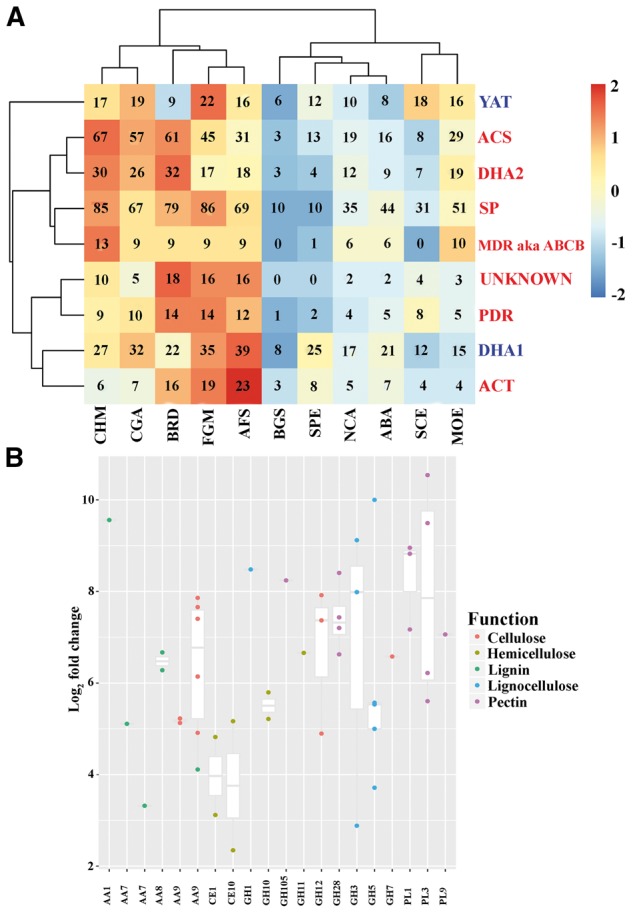
Gene family expansions and contractions of L. theobromae, and up-regulation of gene expressions during infection. (A) Comparison of membrane transporter families in L. theobromae (CSS-01s). The families with red colour mean expansion in the L. theobromae genome, whereas the blue means contraction. (B) Plots of up-regulated CWDE gene expressions in each family during the infections of L. theobromae. Transcriptomic analysis revealed several CWDE belonging to different families up-regulated during infection. These gene families were involved in pathogenicity processes of the fungi. PL indicates pectin lyases.
3.8. Lasiodiplodia theobromae secretome
Proteins secreted by pathogenic fungi are essential for host–pathogen interactions, including the successful establishment of infections.53 Therefore, identification and functional annotation of the secretome is important for identifying potential virulence factors. The L. theobromae genome encodes a total of 937 secreted proteins, accounting for 7.2% of its proteome, as predicted by SignalP-4.1,28 TargetP-1.1,54 and TMHMM-2.0.55 Among the secreted proteins identified, 417 were assigned with GO terms. Among the 283 genes up-regulated during infection, 105 are secreted proteins. Functional enrichment analysis showed that proteins involved in carbohydrate metabolism are significantly overrepresented in the up-regulated L. theobromae secretome (Supplementary Table S17).
3.9. Prediction of putative effectors and their roles in suppressing HRs
Plant pathogenic fungi secrete effectors that suppress the first layer of inducible defences, known as pathogen-associated molecular pattern (PAMP) triggered immunity, which detects conserved pathogen molecules, such as flagellin, Elongation Factor Thermo unstable, peptidoglycan and chitin by host receptors.56 However, very little is known about the effector proteins that contribute to Botryosphaeriaceae virulence. Among the 937 secreted proteins predicted from the L. theobromae genome, we identified 359 putative effectors. Searches for conserved motifs among these effectors showed that 157 contain Y/F/WxC motifs, 45 contain L/IxAR motifs, 1 contains YxSL[R/K] motifs, and 13 contain a single RxLR motif each. Among these, 77 putative effectors were specific to L. theobromae. Five effectors (BR_1.280, BR_2.1361, BR_2.1420, BR_4.711 and BR_10.166) exhibited a strong suppressive effect, whereas BR_1.397, BR_1.899 and BR_5.292 had a weak effect in N. benthamiana (Fig. 5). Two of the effectors, BR_1.280 and BR_2.1361, encode hypothetical proteins (Fig. 5). These results significantly expand our understanding of the determinants of pathogenicity or virulence in Botryosphaeriaceae.
Figure 5.

Putative effectors and their hypersensitive responses in N. benthamiana. Putative Botryosphaeriaceae effectors suppressed B. glumae-induced hyposensitive response in N. benthamiana. Representative cell death symptoms were photographed at 2 days after B. glumae inoculation. The right half leaf sections were injected with B. glumae with the pEDV empty vector and the left half sections were injected with B. glumae with effector gene constructs. Black circle represents pEDV empty vector, red circle represents effectors specific to Botryosphaeriaceae, orange circle represents commons effectors in Botryosphaeriaceae and the purple circle represents leaves injected with 0.9%NaCl.
3.10. Potential adaptations of the L. theobromae genome and transcriptome for plant cell wall degradation
Phytopathogenic fungi encode many CAZymes. During colonization and infection, CAZymes play an important role in the disassembly of plant cell walls, including the breakdown, biosynthesis or modification of glycoconjugates and oligo- and polysaccharides.3,4 CAZymes are grouped into five classes (glycoside hydrolases, GH; glycosyl transferases, GT; polysaccharide lyases; carbohydrate esterases, CE and auxiliary activities, AA) and an associated module (carbohydrate-binding modules, CBMs).
To understand the potential genes involved in the adaptation of L. theobromae to different habitats and substrates, we identified the repertoire of CAZymes known to be involved in organic matter degradation. A total of 820 CAZymes (mapped to 763 predicted L. theobromae genes) (Supplementary Table S18) were identified (see Fig. 2C). From the identified CAZymes, 10 (AA3, AA7, CBM18, CE1, CE10, GH109, GH3 GH43, GH78 and GT1) were expanded in the L. theobromae genome. Among the 283 genes up-regulated during infection, 66 are annotated as cell wall-degrading enzymes (CWDEs), involved in lignin degeneration (6 genes), cellulose degeneration (11 genes), hemicellulose degeneration (8 genes) and pectin degeneration (13 genes) (Fig. 4B). GHs represented the largest super family involved in carbohydrate degradation processes.3,5 For a successful infection, pathogens must encode pectin degrading enzymes to break down plant pectin. The GH28 (polygalacturonases) family plays a critical role in pectin degradation in fungal pathogens such as C. higginsianum, V. mali and V. pyri5,40,57 and has expanded in the genomes of the two Valsa species.3 When compared with the other fungal genomes, the genome of L. theobromae has the most GH28 members (14 genes, of which 4 were up-regulated during infection), suggesting that L. theobromae has a high capacity for pectin degradation.
Lignocellulose, which is composed of cellulose, hemicellulose and lignin, is one of the barriers that pathogens must surpass in order to colonize host tissues. Since L. theobromae is mainly a trunk pathogen, lignocellulose degrading enzymes likely play a major role during infection.4 Indeed, GH subfamilies involved in the degradation of cellulose and hemicellulose were the most abundant among all the fungal genomes. The degradation of cellulose requires the collaboration of endoglucanases (GH6, GH9, GH12, GH44, GH45, GH51, GH74, GH124, CBM65 and CBM72), cellobiohydrolases (GH6, GH7, GH19, GH48 and GH74) and β-1,4-glucosidase (GH3). The L. theobromae genome encodes many of these CWDEs. Furthermore, among all the fungal pathogens studied, L. theobromae possesses the highest number of GH3s in its genome. Xylanases in the GH3 sub-family degrade xylan, and the L. theobromae genome contains a complete suite of enzymes to degrade xylan (Supplementary Fig. S5). During infection, three GH12, one GH7 and three GH3 genes encoding lignocellulose-degrading enzymes were up-regulated in L. theobromae, indicating their ability to effectively degrade cellulose, hemicellulose and lignin. Similar up-regulation of these genes were observed in L. theobromae.5 Compared with L. theobromae, the GH12 family is expanded in C. higginsianum, and similar CAZyme modules are available in L. theobromae, A. fumigatus and F. graminearum.3,5,6 Among the fungi analysed so far, the L. theobromae genome encodes the highest number of xyloglucan transglucosylases (GH16, 24 genes), followed by F. graminearum, C. higginsianum and C. graminicola. Endo-β-1,4-cellulases (GH5) represent another gene family with a high number of encoded genes in L. theobromae, and these genes are commonly found in highly pathogenic species, such as E.lata and N. parvum.3,5,6 Our analyses identified the highest number of GH5s in C. higginsianum, followed by L. theobromae and C. graminicola.
CBMs are an important gene family with carbohydrate binding activity.58 According to our comparative analysis, the highest number of CBMs was found in the highly pathogenic P. oryzae (99 genes), followed by C. graminicola (70 genes), C. higginsianum (80 genes), F. graminearum (68 genes) and L. theobromae (54 genes). In contrast, all the non-pathogenic species, including S. pombe (8 genes) and S. cerevisiae (8 genes), have fewer reported CBMs in their genomes.
AAs are families of redox enzymes that act in conjunction with CAZymes to degrade lignocellulosic material. They represent ligninolytic enzymes and lytic polysaccharide mono-oxygenases involved in lignin breakdown.5,59 Our analyses found that both AA3 (38 genes) and AA7 (50 genes) families were expanded in the L. theobromae genome. The AA3 family is related to cellobiose dehydrogenase activity, and AA7 oxidizes glycosyl residues.59 These AA3 genes were also abundant in the N. parvum and Stereum hirsutum genomes.3 However, only one or two AA family genes were present in the non-pathogenic fungal genomes.
3.11. Lasiodiplodia theobromae gene expression induced by infection and high temperature exhibits characteristics of opportunistic phytopathogens
Transcriptome analysis conducted at two temperatures and at two time points after host inoculation identified a large number of differentially expressed genes. Differences in virulence at the different temperatures on grape shoots and growth on axenic cultures are shown in Fig. 3B. According to the transcriptomic data of samples collected from L. theobromae cultures on PDA plates, 361 genes were up-regulated, whereas 248 were down-regulated at the higher temperature. These up-regulated genes include genes with LysM (see Fig. 3D), allergens, GH 61 family genes, cytochrome P450s and heat shock proteins. Of these, few were related to primary pathogenicity mechanisms.
Phylogenetic analysis was conducted for randomly selected genes belong to glycosyl hydrolase family (Fig. 6A). According to the analysis, all of the genes clade together with related GH family genes identified from previous studies. Figure 6B demonstrates the gene expression of these genes. To investigate the roles of these up-regulated genes in the virulence of Botryosphaeriaceae, a GH 5 family putative cellulase gene (GH 7.188) and a GH 61 family putative lignocellulase gene (GH 2.1698) were selected for further study. These two genes were overexpressed using a strong PtrpC promoter. The overexpressing transformants showed no effect on vegetative growth and conidiation on PDA medium. We confirmed the elevated expression levels of these over-expressing transformants of GH 2.1698 and GH 7.188. Intriguingly, pathogenicity assays showed that both overexpressing transformants had significantly enhanced virulence on grape shoots compared with the wild-type strain CSS-01 (Fig. 6C and D). These results suggest that lignocellulose degrading enzymes play an important role during the colonization of Botryosphaeriaceae canker development. Furthermore, our results suggest that virulence in this family is temperature dependent, a typical characteristic of opportunistic infection.
Figure 6.

Phylogentic analyses, gene expression and pathogenicity assays conducted for glycosyl hydrolase family genes. (A) One of the 1000 most parsimonious trees obtained from a heuristic search of glycosyl hydrolase family genes and 36 related sequences from closely related species. Parsimony bootstrap support values ≥ 50% are indicated at the nodes and branches and the sequences from this study are given in bold. (B) Gene expression analysis of selected glycosyl hydrolase family genes. This includes the gene expression at different conditions (on axenic cultures and on grape shoots after inoculation at 25 °C and at 35 °C) and at different infection stages (8 and 12 hpi). (C) Lesions on stems were photographed at 3 days post-inoculation of the above L. theobromae transformants. WT indicates the wild-type strain of L. theobromae CSS-01s and numbers 1–10 represent the replicates of each overexpressing transformants conducted for each gene. First 10 stems were inoculated with the GH 2.1698 overexpressing transformants of L. theobromae, and the second 10 stems were inoculated with the GH 7.188 overexpressing transformants of L. theobromae. (D) Bar chart showing the mean lesion lengths of the overexpressing transformants. WT indicates the wild-type strain of L. theobromae CSS-01s. H2-OV1–H2-OV10 represents the mean lesion lengths of the GH 2.1698 overexpressing transformants of L. theobromae, and H7-OV1–H7-OV10 represents the GH 7.188 overexpressing transformants of L. theobromae.
4. Discussion
In this study, we developed fundamental genomic and transcriptomic resources for functional characterization of three botryosphaeriaceous pathogens, B.dothidea, L.theobromae and N.parvum. These three species have similar genome sizes (42.1 Mb to 43.3 Mb), and the assemblies of B. dothidea and N. parvum were similar to those reported in previous studies. Indeed, all the sequenced genomes in Botryosphaeriaceae have similar sizes, ranging from 36.9 to 43.5 Mb, with similar genome coverage.3,11,14 In addition to these botryosphaeriaceous taxa, the genome sizes for other pathogenic fungi compared here were similar to other common plant pathogens with similar lifestyles (Table 3).32,40,60,61 Both the JELLYFISH analysis,39 which estimated genome size based on k-mer distributions,62 and the tRNA analyses indicated a high degree of completeness in the assembled genomes. Furthermore, transcriptomic data obtained through RNASeq confirmed the accuracy and completeness of the assembled genome and facilitated the refinement of the predicted gene models of L. theobromae.63
Table 3.
Comparison of genome sizes between L. theobromae and other pathogenic fungi
| Pathogenic taxa | Genome size (Mb) |
|---|---|
| Neofusicoccum parvum26 | 42.50 |
| Neofusicoccum parvum (Current study) | 42.60 |
| Botryosphaeria dothidea (Current study) | 43.10 |
| Botryosphaeria dothidea30 | 43.50 |
| Diplodia sapinea30 | 36.97 |
| Diplodia seriata5 | 37.10 |
| Lasiodiplodia theobromae (Current study) | 43.30 |
| Eutypa lata25 | 54.40 |
| Pyricularia oryzae39 | 40.30 |
| Diaporthe ampelina5 | 47.40 |
| Neurospora crassa40 | 39.90 |
| Ustilaginoidea virens41 | 39.40 |
| Valsa pyri21 | 35.74 |
| Valsa mali21 | 44.74 |
Compared with the protein-coding genes predicted for other pathogens,3,14,40,60,61 our combined approach allowed for the prediction of the highest number of protein-coding genes in any fungal pathogen. Identifying repetitive elements is important for understanding the genome architecture of a pathogen and, ultimately, its evolution.3,4 Repetitive elements present in the genomes of Botryosphaeriaceae taxa were much lower, ranging from 3.02% to 3.84%. This ratio is much lower than those reported for other pathogens such as P. oryzae (11.75%),64N. crassa (12.72%),60Ustilago virens (25%)61 and V. mali (14.05%).40 This finding confirms the high genetic similarity observed among Botryosphaeriaceae species in previous studies.65
Comparative genome analysis revealed data on genetic variation, differential gene expression, and evolutionary dynamics between species and their specific adaptations to different environmental conditions and lifestyles. This study revealed that Botryosphaeriaceae species are phylogenetically closer to opportunistic pathogens, indicating that they may share traits common to opportunistic fungal pathogens. This was clearly evidenced from the phylogenetic study as well as from Fig. 2B, which shows that Botryosphaeriaceae fungi share the highest number of unique orthologous gene families with opportunistic fungal pathogens (1,049) than to pathogenic (221) and non-pathogenic fungi (36). It was further confirmed by the inventory of genes identified by comparative genome analysis, which are involved in the infection and the colonization of the host by L. theobromae and other Botryosphaeriaceae pathogens. Similar results were discovered in a recent transcriptomic study conducted for L. theobromae.5 Functional annotation of these genomes provides additional evidence for the hypothesis that Botryosphaeriaceae fungi are more closely related to opportunistic fungi. According to the GO categories of the overrepresented genes, Botryosphaeriaceae species shared only three overrepresented genes with primary fungal pathogens (Supplementary Table S11), whereas they shared 27 with opportunistic phytopathogens (Supplementary Table S12). The categories of genes shared with opportunistic pathogens include those related to cell wall degradation, carbohydrate and protein metabolism, gene expression regulation and the biosynthesis of secondary metabolites. Many of these genes have been shown to be involved in the pathogenicity of many fungal pathogens.3,5,14,40,61 Based on the phylogenetic analysis, the highest number of unique orthologous gene families shared with other opportunistic fungal pathogens, and the highest number of overrepresented gene categories shared with other opportunistic pathogens, we confirmed that Botryosphaeriaceae fungi are more closely related to opportunistic pathogens than to other pathogens. Gene categories shared between Botryosphaeriaceae species and other pathogenic fungi were also enriched in core elements essential for virulence in many pathogens. Phosphorylation is an essential post-translational modification that regulates a broad spectrum of biological processes through the activity of numerous protein kinases.66 Signal transduction is another important gene category for a pathogen to respond to environmental changes during host infection and regulate a network of cellular pathways.66. All of these virulence-related genetic elements are commonly found in the genomes of fungal pathogens such as P. oryzae, Fusarium oxysporum,66Botrytis cinera67 and many other pathogenic fungi.68
During the comparative genome analysis, we observed significant expansions and contractions in many gene families. Such changes can provide adaptive advantages for the organisms at certain ecological niches during natural selection.69 These expanded gene families include those responsible for virulence and nutrient uptake. In addition to the cellular transporters and cytochrome P450s, several gene families related to secondary metabolism have also undergone significant expansion. Among the 58 secondary metabolite clusters observed, NPRS gene clusters are responsible for the synthesis of toxic polypeptides, whereas PKS clusters are responsible for the production of naphthalenone pentaketides in botryosphaeriaceous fungi, D. seriata, L. theobromae5 and N. parvum, as well as other pathogens such as A. fumigatus, A. nidulans, D. ampelina, E. lata, Phaeomoniella chlamydospora and T.minima.9 Even though secondary metabolite gene clusters are not subjected to regulation during infection, their significant expansion in the genome suggests that they may be involved in producing disease symptoms and host adaptations. Cytochrome P450s are a super family of mono-oxygenases involved in the post-synthesis modification of diverse metabolites.3,9,46,70 Furthermore, these proteins facilitate fungal adaptation to specific ecological niches by modifying potentially harmful environmental chemicals.70 Having a genome enriched in species-specific cytochrome P450s may help explain the broad geographic distribution of botryosphaeriaceous taxa. Expansion of this gene family observed in this study further confirms their high capacity for adapting to different hosts, which may explain the wide host range of Botryosphaeriaceae species. Because these genes are also related to several physiological traits, their expansion has likely facilitated the evolution of pathogenicity.71
Cellular transporters have significant roles in fungal pathogenesis as well as in protecting against plant defence compounds.72 Comparative genome analysis revealed that L. theobromae encode the highest number of cellular transporters among all the analysed genomes. Genes encoding (ABC)–type transport systems showed significant expansion in L. theobromae, and their expression is significantly up-regulated during infection. The importance of these multidrug facilitator transporters in drug resistance and pathogenesis has been confirmed in B. cinera, Gibberella pulicaris, P. oryzae, Nectria haematococca and other pathogenic fungi.15 They are also responsible for polysaccharide, lipid and amino acid transport. Similarly, the expanded and up-regulated transporter family genes identified in this study could also play an important role in the fungal pathogenesis of L. theobromae during infection by providing protection through the detoxification of antifungal phytotoxins. Sugar transporters also showed a high level of regulation during host infection in L. theobromae, allowing it to adapt to different host environments with varying pH conditions52 and various plant–pathogen interactions.52,60,73 These sugar symporters belong to the MFS family responsible for sporulation, cell-to-cell communication and pathogenesis in fungi.74 Because these families are up-regulated during infection, both ABC-type transporters and MFS transporters are considered to be virulence or pathogenicity factors in L. theobromae.
Transcriptome profiling identified a set of putative effectors that were up-regulated during L. theobromae infection. These include conserved functional domains important for fungal pathogenesis mechanisms and other effector domains such as CFEM. Effectors enable pathogens to evade or suppress plant defence systems.56,75 Genetic manipulation of fungal pathogens for candidate effector genes is difficult and cumbersome. This makes the whole process of effector functional analysis time consuming and laborious. Translocation of effector proteins to host using a type III secretion system (T3SS) of bacterial pathogens proven to be a fast alternative to the traditional method of generating fungal transformants. This efficient effector delivery (pEDV) system utilizes the N-terminal portion of AvrRps4, which is required for Type III secretion system (T3SS)-dependent delivery and in planta processing. In this study, we have experimentally shown that several randomly selected putative effectors can suppress B.glumae triggered hypersensitivity in N.benthamiana. These results are consistent with the hypothesis that these putative effectors play crucial roles in the pathogenesis of L. theobromae on its hosts by identifying PAMPs. This system has been successfully used in screening candidate non-bacterial effectors predicted in the genome Hyaloperonospora arabidopsidis,76P. oryzae,77 and Ustilaginoidea virens.64
In addition to the biotrophic stage, most phytopathogens also have hemi-biotrophic or necrotrophic stages, in which the fungi destroy the cell wall, enter the cell, and kill and destroy host cell structures.72 Proteins secreted by these pathogens play crucial roles in early colonization and pathogenesis by targeting all components of the primary and secondary plant cell walls.3 The current study reveals a broad range of gene families with potential virulence functions in L. theobromae, similar to the botryosphaeriaceous taxa D. seriata3, L. theobromae5 and N. parvum3. Previous studies have identified a broad range of key genes responsible for wood degradation and host colonization in phytopathogens.3,5,78,79 Similar to highly pathogenic B. cinera,80L.theobromae possesses the highest number of genes encoding pectolytic enzymes among the sequenced Botryosphaeriaceae genomes. The significant induction of these genes during infection is consistent with their critical roles in fungal pathogenesis. Because most of the dicotyledon cell walls consist of ∼35% pectin, these polygalacturonases facilitate the breakdown of cell wall.81 This localized degradation of cell wall is required for the fungus to access the plant cytoplasm and to spread across host tissues. 81,82 Their importance has been experimentally confirmed in many other species, including Colletotrichum gloeosporioides, C. higginsianum, C. lindemuthianum, V. mali and V. pyri.3,5,40,57,82 In addition, the L. theobromae genome encodes a wider array of enzymes (GH5, GH3, GH16, GH43) targeting cellulose and hemicellulose, similar to other pathogens such as D. seriata, E. lata, N. parvum and P. chlamydospora.3,5,78,80,82 Their expression levels were significantly induced during infection, and the current study confirmed the enhanced pathogenicity of several putative lignocellulase genes by overexpressing them in L. theobromae. Together, the up-regulation of genes encoding CAZymes targeting pectin, cellulose, hemicellulose and xylan during infection explains the rapid colonization and infection of L. theobromae on woody plants.
Carbohydrate binding modules have been linked to the evolution of fungal pathogenicity in many fungi. During the infection of L. theobromae, CBM 18 family genes were significantly up-regulated. This subclass domain of chitin recognition has been confirmed for fungal protection from host defences by preventing plant factors from degrading fungal chitin.83 Hence, during fungal infection, this family together with auxiliary enzymes participates in fungal pathogenesis. In this study, the AA3 and AA7 families showed significant expansion in the L. theobromae genome. AA7-domain families are responsible for oxidizing a variety of carbohydrates and contributing to lignin degradation,59 whereas AA3-domain families participate in the degradation of cellulose, hemicellulose and lignin.45 Similar expansions of AA7s and AA3s have been found in many other fungal pathogens, such as B. cinera, C. graminicola, C. higginsianum, D. seriata, E. lata, F. graminearum, F. solani, P. oryzae, N. parvum and S. hirsutum.3,59,84 In addition, this study showed the significantly induced expression of peroxidases and oxidoreductases in L. theobromae during infection. These two classes of enzymes are known to be involved in pathogenicity-related processes such as the degradation of lignin and the detoxification of reactive oxygen species produced by the host.3,5,78
Our pathogenicity analysis revealed that a higher temperature increased the virulence of L. theobromae on grapevine. Consistent with this result, transcriptome analysis also revealed that several genes were up-regulated at a higher temperature. Furthermore, overexpression of two genes belonging to GH 5 and GH 61 families in L. theobromae significantly enhanced its virulence on Vitis shoots. The GH 5 and GH 61 families encode cellulases and lignocellulases, respectively. These genes are involved in cell wall degradation during infection and colonization. Transcriptome analysis revealed several genes corresponding to fungal pathogenicity, such as chitin-binding CBM family genes, cytochrome P450s and heat shock proteins, which were induced at high temperature. This increased virulence at a higher temperature is a typical characteristic of an opportunistic infection.4 Hence, we can confirm that L. theobromae can cause opportunistic infections at high temperatures. Transcriptomic study conducted on L. theobromae also confirmed the up-regulation of virulence related genes under heat stress conditions.5 Recently few studies have been conducted to determine the influence of environmental conditions such as high temperature and high humidity, on the establishment of the pathogen and the development of numerous diseases on plants.5,85
In summary, this study revealed a high similarity between L. theobromae and other opportunistic fungal phytopathogens. We established genomic and transcriptomic resources for L. theobromae and incorporated the data into a comparative analysis to catalogue genes and gene families with putative virulence functions. L.theobromae employs multiple virulence strategies to adapt to its diverse lifestyles, including CWDEs, secreted proteases, secondary metabolites, putative effectors and membrane transporters. Specific gene families of pectinases, secondary metabolism enzymes and transporters related to multidrug resistance are uniquely expanded in the L. theobromae genome and are frequently up-regulated during infection. In addition, large gene inventories were involved in cell wall degradation, nutrient uptake and secondary metabolism in L. theobromae. These results suggest potential adaptations for colonization and infection of the host and reflect their hemi-biotrophic lifestyle. Functional studies, together with transcriptomic data, indicate the active role of putative effectors and some lignocellulases in virulence, pathogenicity and host adaptation. Transcriptomic analysis revealed that several pathogenicity-related genes were induced at high temperature. This discovery, together with the phylogenomic analysis, indicates that L.theobromae is an opportunistic phytopathogen. This study provides valuable genomic and transcriptomic resources for future studies of the evolution and pathogenesis of L. theobromae.
Supplementary data
Supplementary data are available at DNARES online.
Supplementary Material
Acknowledgements
The work is supported by CARS-30, NSFC (31501567), KJCX20140402 and Beijing Nova Program (Z141105001814047). We are grateful to Dr Ricardo Dias for comments on the manuscript and Dr Wenxian Sun (China Agricultural University, China) for providing the vectors for the study.
Accession numbers
MDYX00000000, SRP108461, SRP107819.
Data availability
The whole genome shotgun project for Lasiodiplodia theobromae CSS-01S has been deposited at DDBJ/ENA/GenBank under the accession code MDYX00000000. The version described in this paper is version MDYX01000000. Whole genome sequences of the other five isolates of Lasiodiplodia theobromae (HBQJZ-02S), Botryosphaeria dothidea (Sxg01s, LNXCEYGS-1), Neofusicoccum parvum (CDZ1M1s3, Gxg-01s) has been deposited at GenBank under the accession code SRP108461. All Total mRNA and Polysome-bound mRNA sequencing data generated in this study for Lasiodiplodia theobromae CSS-01S have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm. nih.gov/sra/) under accession number SRP107819.
Conflict of interest
None declared.
Author contributions
J.Y.Y., W.S.Z., Z.C. and Q.K.X. contributed equally to this work as first authors. J.Y.Y., W.S.Z., Y.L.P. and X.H.L. managed the project and designed the experiment; J.Y.Y., Q.K.X., W.Z., and K.W.T.C. conducted the experiments; J.Y.Y., W.S.Z., Z.C., M.F.X., K.D.H., X.H.L. and J.P.X. analysed the data; Z.C. and K.W.T.C. wrote the paper. J.Y.Y., Z.C., K.W.T.C., K.D.H., J.P.X. and A.J.L.P. revised the paper. All of the other authors contributed by assisting in molecular experiments and pathogenicity tests.
References
- 1. Phillips A. J. L., Alves A., Abdollahzadeh J., et al. 2013, The Botryosphaeriaceae: genera and species known from culture, Stud. Mycol., 76, 51–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yan J., Xie Y., Zhang W., et al. 2013, Species of Botryosphaeriaceae involved in grapevine dieback in China, Fungal Divers., 61, 221–36. [Google Scholar]
- 3. Morales-Cruz A., Amrine K. C. H., Blanco-Ulate B., et al. 2015, Distinctive expansion of gene families associated with plant cell wall degradation, secondary metabolism, and nutrient uptake in the genomes of grapevine trunk pathogens, BMC Genomics, 16, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chethana K. W. T., Li X. H., Zhang W., Hyde K. D., Yan J. Y.. 2016. Trail decryption of molecular research on Botryosphaeriaceae in woody plants, Phytopathol. Mediterr., 55, 147–71. [Google Scholar]
- 5. Paolinelli-Alfonso M., Villalobos-Escobedo J. M., Rolshausen P., et al. 2016, Global transcriptional analysis suggests Lasiodiplodia theobromae pathogenicity factors involved in modulation of grapevine defensive response, BMC Genomics, 17, 615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramírez-Suero M., Chong J., Farine S., et al. 2012, Effect of Neofusicoccum parvum and Diplodia seriata extra-cellular compounds on defence gene expression in Vitis vinifera cv. Chardonnay and cv. Gewurztraminer, Phytopathol. Mediterr., 51, 410–52. [Google Scholar]
- 7. Fernandes I., Alves A., Correia A., Devreese B., Esteves A. C.. 2014, Secretome analysis identifies potential virulence factors of Diplodia corticola, a fungal pathogen involved in cork oak (Quercus suber) decline, Fungal Biol., 118, 516–23. [DOI] [PubMed] [Google Scholar]
- 8. Abou-Mansour E., Débieux J. L., Ramírez-Suero M., et al. 2015, Phytotoxic metabolites from Neofusicoccum parvum, a pathogen of Botryosphaeria dieback of grapevine, Phytochemistry, 115, 207–15. [DOI] [PubMed] [Google Scholar]
- 9. Andolfi A., Mugnai L., Luque J., Surico G., Cimmino A., Evidente A.. 2011, Phytotoxins produced by fungi associated with grapevine trunk diseases, Toxins, 3, 1569–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bertsch C., Ramírez‐Suero M., Magnin‐Robert M., et al. 2013, Grapevine trunk diseases: complex and still poorly understood, Plant Pathol., 62, 243–65. [Google Scholar]
- 11. Blanco-Ulate B., Rolshausen P., Cantu D.. 2013, Draft genome sequence of Neofusicoccum parvum isolate UCR-NP2, a fungal vascular pathogen associated with grapevine cankers, Genome Announc., 1, e00339–132013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parrent J. L., James T. Y., Vasaitis R., Taylor A. F.. 2009, Friend or foe? Evolutionary history of glycoside hydrolase family 32 genes encoding for sucrolytic activity in fungi and its implications for plant-fungal symbioses, BMC Evol. Biol., 9, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ma Z., Song T., Zhu L., et al. 2015, A Phytophthora sojae glycoside hydrolase 12 protein is a major virulence factor during soybean infection and is recognized as a PAMP, Plant Cell, 27, 2057–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van der Nest M. A., Bihon W., De Vos L., et al. 2014, Draft genome sequences of Diplodia sapinea, Ceratocystis manginecans, and Ceratocystis moniliformis, IMA Fungus, 5, 135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Han Y., Liu X., Benny U., Kistler H.C., VanEtten H. D.. 2014, Genes determining pathogenicity to pea are clustered on a supernumerary chromosome in the fungal plant pathogen Nectria haematococca, Plant J., 25, 305–14. [DOI] [PubMed] [Google Scholar]
- 16. Quinlan R. J., Sweeney M. D., Lo Leggio L., et al. 2011, Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components, Proc. Natl. Acad. Sci. USA, 108, 15079–084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guttman D., McHardy A. C., Schulze-Lefert P.. 2014, Microbial genome-enabled insights into plant-microorganism interactions, Nat. Rev. Genet., 5, 797–813. [DOI] [PubMed] [Google Scholar]
- 18. Harris P. J., Stone B. A.. 2008, Chemistry and molecular organization of plant cell walls, Water Res., 45, 1587–96. [Google Scholar]
- 19. Molina L., Kahmann R.. 2007, An Ustilago maydis gene involved in H2O2 detoxification is required for virulence, Plant Cell, 19, 2293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Talbot N. J., Ebbole D. J., Hamer J. E.. 1993, Karyotype variation within clonal lineages of the rice blast fungus Magnaporthe grisea, Appl. Environ. Microbiol., 59, 585–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Price A. L., Jones N. C., Pevzner P. A.. 2005, De novo identification of repeat families in large genomes, Bioinformatics, 21, 351–8. [DOI] [PubMed] [Google Scholar]
- 22. Bao W., Kojima K. K., Kohany O.. 2015, Repbase Update, a database of repetitive elements in eukaryotic genomes, Mob. DNA, 6 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bao Z., Eddy S. R.. 2002, Automated de novo identification of repeat sequence families in sequenced genomes, Genome Res., 12, 1269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stanke M., Morgenstern B.. 2005, AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints, Nucleic Acids Res., 33, W465–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lomsadze A., Ter-Hovhannisyan V., Chernoff Y. O., Borodovsky M.. 2005, Gene identification in novel eukaryotic genomes by self-training algorithm, Nucleic Acids Res., 33, 6494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. She R., Chu J. S., Wang K., Pei J., Chen N.. 2009, GenBlastA: enabling BLAST to identify homologous gene sequences, Genome Res., 19, 143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Birney E., Clamp M., Durbin R.. 2004, GeneWise and genomewise, Genome Res., 14, 988–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petersen T. N., Brunak S., von Heijne G., Nielsen H.. 2011, SignalP 4.0: discriminating signal peptides from transmembrane regions, Nat. Methods, 8, 785–6. [DOI] [PubMed] [Google Scholar]
- 29. Mortazavi A., Williams B. A., McCue K., Schaeffer L., Wold B.. 2008, Mapping and quantifying mammalian transcriptomes by RNA-Seq, Nat. Methods, 5, 621–8. [DOI] [PubMed] [Google Scholar]
- 30. Haas B. J., Salzberg S. L., Zhu W., et al. 2008, Automated eukaryotic gene structure annotation using evidencemodeler and the program to assemble spliced alignments, Genome Biol., 9, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moriya Y., Itoh M., Okuda S., Yoshizawa A. C., Kanehisa M.. 2007, KAAS: an automatic genome annotation and pathway reconstruction server, Nucleic Acids Res., 35, W182–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dean R. A., Talbot N. J., Ebbole D. J., et al. 2005, The genome sequence of the rice blast fungus Magnaporthe grisea, Nature, 434, 980–6. [DOI] [PubMed] [Google Scholar]
- 33. Islam Md. S., Haque Md. S., Islam M. M., et al. 2012, Tools to kill: genome of one of the most destructive plant pathogenic fungi Macrophomina phaseolina, BMC Genomics, 13, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saier M. H. Jr., Reddy V. S., Tamang D. G., Vastermark A.. 2014, The transporter classification database, Nucleic Acids Res., 42, 251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Katoh K., Misawa K., Kuma K., Miyata T.. 2002, MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform, Nucleic Acids Res., 30, 3059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kumar S., Stecher G., Tamura K.. 2016, MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets, Mol. Biol. Evol., 33, 1870–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Swofford D. L. 2002, PAUP* 4.0: phylogenetic analysis using parsimony (* and other methods). Sinauer Associates, Sunderland. [Google Scholar]
- 38. Yang Z. 2007, PAML 4: phylogenetic analysis by maximum likelihood, Mol. Biol. Evol., 24, 1586–91. [DOI] [PubMed] [Google Scholar]
- 39. Marcais G., Kingsford C.. 2011, A fast, lock-free approach for efficient parallel counting of occurrences of k-mers, Bioinformatics, 27, 764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Galagan J. E., Calvo S. E., Borkovich K. A., et al. 2003, The genome sequence of the filamentous fungus Neurospora crassa, Nature, 422, 859–68. [DOI] [PubMed] [Google Scholar]
- 41. Spanu P. D., Abbott J. C., Amselem J., et al. 2010, Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism, Science, 330, 1543–6. [DOI] [PubMed] [Google Scholar]
- 42. Kosiol C., Anisimova M.. 2012, Selection on the protein-coding genome, Methods Mol. Biol., 856, 113–40. [DOI] [PubMed] [Google Scholar]
- 43. Demuth J. P., Hahn M. W.. 2012, The life and death of gene families, Bioessays, 31, 29–39. [DOI] [PubMed] [Google Scholar]
- 44. Powell A. J., Conant G. C., Brown D. E., Carbone I., Dean R. A.. 2008, Altered patterns of gene duplication and differential gene gain and loss in fungal pathogens, BMC Genomics, 9, 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Bie T., Cristianini N., Demuth J. P., Hahn M. W.. 2006, CAFE: a computational tool for the study of gene family evolution, Bioinformatics, 22, 1269–71. [DOI] [PubMed] [Google Scholar]
- 46. Brakhage A. A. 2013, Regulation of fungal secondary metabolism, Nat. Rev. Microbiol., 11, 21–32. [DOI] [PubMed] [Google Scholar]
- 47. Anderson M. R., Nielsen J. B., Klitgaard A., et al. 2013, Accurate prediction of secondary metabolite gene clusters in filamentous fungi, Proc. Natl. Acad. Sci. USA, 110, E99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Khaldi N., Seifuddin F. T., Turner G., et al. 2010, SMURF: genomic mapping of fungal secondary metabolite clusters, Fungal Genet. Biol., 47, 736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Medema M. H., Blin K., Cimermancic P., et al. 2011, antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences, Nucleic Acids Res., 39, W339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Beseli A., Amnuaykanjanasin A., Herrero S., Thomas E., Daub M. E.. 2015, Membrane transporters in self resistance of Cercospora nicotianae to the photo-activated toxin cercosporin, Curr. Genet., 61, 601–20. [DOI] [PubMed] [Google Scholar]
- 51. Vankuyk P. A., Diderich J. A., MacCabe A. P., Hererro O., Ruijter G. J., Visser J.. 2004, Aspergillus niger mstA encodes a high-affinity sugar/H+ symporter which is regulated in response to extracellular pH, Biochem J., 379, 375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang Y., Zhang Z., Zhang X., et al. 2012, CDR4 is the major contributor to azole resistance among four Pdr5p-like ABC transporters in Neurospora crassa, Fungal Biol., 116, 848–54. [DOI] [PubMed] [Google Scholar]
- 53. Brown N. A., Antoniw J., Hammond-Kosack K. E.. 2012, The predicted secretome of the plant pathogenic fungus Fusarium graminearum: a refined comparative analysis, PLoS One, 7, article e33731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Emanuelsson O., Brunak S., von Heijne G., Nielsen H.. 2007, Locating proteins in the cell using TargetP, SignalP and related tools, Nat. Protoc., 2, 953–71. [DOI] [PubMed] [Google Scholar]
- 55. Sonnhammer EL, von Heijne HG, Krogh A A hidden Markov model for predicting transmembrane helices in protein sequences. In: Glasgow, TL, Major, F, Lathrop, D, Sankoff, D, Sensen, C (eds) Proceedings of the Sixth International Conference on Intelligent Systems for Molecular Biology, 1998, p. 175–82. AAAI Press, Menlo Park, CA, USA. [PubMed]
- 56. O'Connell R. J., Thon M. R., Hacquard S., et al. 2012, Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses, Nat. Genet., 44, 1060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Boraston A. B., Bolam D. N., Gilbert H. J., Davies G. J.. 2004, Carbohydrate-binding modules: fine tuning polysaccharide recognition, Biochem. J., 382, 769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Levasseur A., Drula E., Lombard V., Coutinho P. M., Henrissat B.. 2013, Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes, Biotechnol. Biofuels, 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li Z., Yin Z., Fan Y., Xu M., Kang Z., Huang L.. 2015, Candidate effector proteins of the necrotrophic apple canker pathogen Valsa mali can suppress BAX-induced PCD, Front Plant Sci., 6, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yin Z., Liu H., Li Z., et al. 2015, Genome sequence of Valsa canker pathogens uncovers a potential adaptation of colonization of woody bark, New Phytol., 208, 1202–16. [DOI] [PubMed] [Google Scholar]
- 61. Xue M., Yang J., Li Z., et al. 2012, Comparative analysis of the genomes of two field isolates of the rice blast fungus Magnaporthe oryzae, PLoS Genet., 8, article 1002869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Simpson J. T. 2014, Exploring genome characteristics and sequence quality without a reference, Bioinformatics, 30, 1228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Denton J. F., Lugo-Martinez J., Tucker A. E., Schrider D. R., Warren W. C., Hahn M. W.. 2014, Extensive error in the number of genes inferred from draft genome assemblies, PLoS Comput. Biol., 10, article e1003998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang Y., Zhang K., Fang A., et al. 2014, Specific adaptation of Ustilaginoidea virens in occupying host florets revealed by comparative and functional genomics, Nat. Commun., 5, 3849. [DOI] [PubMed] [Google Scholar]
- 65. Baskarathevan J., Jaspers M. V., Jones E. E., Cruickshank R. H., Ridgway H. J.. 2012, Genetic and pathogenic diversity of Neofusicoccum parvum in new Zealand vineyards, Fungal Biol., 116, 276–88. [DOI] [PubMed] [Google Scholar]
- 66. Ullah S. 2016, dbPAF: an integrative database of protein phosphorylation in animals and fungi, Sci. Rep. 6, article e23534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schamber A., Leroch M., Diwo J., Mendgen K., Hahn M.. 2010. The role of mitogen activated protein (MAP) kinase signalling components and the Ste12 transcription factor in germination and pathogenicity of Botrytis cinerea, Mol. Plant Pathol. 11, 105–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kramer B., Thines E., Foster A. J.. 2009, MAP kinase signalling pathway components and targets conserved between the distantly related plant pathogenic fungi Mycosphaerella graminicola and Magnaporthe grisea, Fungal Genet. Biol. 46, 667–81. [DOI] [PubMed] [Google Scholar]
- 69. Alkan N., Espeso E. A., Prusky D.. 2013, Virulence regulation of phytopathogenic fungi by pH, Antioxid. Redox Signal, 19, 1012–25. [DOI] [PubMed] [Google Scholar]
- 70. Siewers V., Viaud M., Jimenez-Teja D., et al. 2005, Functional analysis of the cytochrome P450 monooxygenase gene bcbot1 of Botrytis cinerea indicates that botrydial is a strain-specific virulence factor, Mol. Plant Microbe Interact., 18, 137–49. [DOI] [PubMed] [Google Scholar]
- 71. Nierman W. C., Pain A., Anderson M. J., et al. 2005, Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus, Nature, 438, 1092–3. [DOI] [PubMed] [Google Scholar]
- 72. Luini E., Fleurat-Lessard P., Rousseau L., Roblin G., Berjeaud J.-M.. 2010, Inhibitory effects of polypeptides secreted by the grapevine pathogens Phaeomoniella chlamydospora and Phaeoacremonium aleophilum on plant cell activities, Physiol. Mol. Plant Pathol. 74, 403–11. [Google Scholar]
- 73. Bazzone A., Madej M. G., Kaback H. R., Fendler K.. 2016, pH regulation of electrogenic sugar/H+ symport in MFS sugar permeases, PLoS ONE, 1111, article e0156392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gaur M., Puri N., Manoharlal R., et al. 2008, MFS transportome of the human pathogenic yeast Candida albicans, BMC Genomics, 99, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Marshall R., Kombrink A., Motteram J., et al. 2011, Analysis of two in planta expressed LysM effector homologs from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat, Plant Physiol., 156, 756–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Badel J. L., Piquerez S. J. M., Greenshields D., et al. 2013, In planta effector competition assays detect Hyaloperonospora arabidopsidis effectors that contribute to virulence and localize to different plant subcellular compartments, Mol. Plant Microbe Interact., 26, 745–57. [DOI] [PubMed] [Google Scholar]
- 77. Sharma S., Sharma S., Hirabuchi A., et al. 2013, Deployment of the Burkholderia glumae type III secretion system as an efficient tool for translocating pathogen effectors to monocot cells, Plant J., 74, 701–12. [DOI] [PubMed] [Google Scholar]
- 78. Condon B. J., Leng Y., Wu D., et al. 2013, Comparative genome structure, secondary metabolite, and effector coding capacity across Cochliobolus pathogens, PLoS Genet., 9, article e1003233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Suzuki H., MacDonald J., Syed K., et al. 2012, Comparative genomics of the white-rot fungi, Phanerochaete carnosa and P. chrysosporium, to elucidate the genetic basis of the distinct wood types they colonize, BMC Genomics, 13, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Floudas D., Binder M., Riley R., et al. 2012, The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes, Science, 336, 1715–19. [DOI] [PubMed] [Google Scholar]
- 81. ten Have A., Mulder W., Visser J., van Kan J. A. L.. 1998, The endopolygalacturonase gene Bcpg1 is required for full virulence of Botrytis cinerea, Mol. Plant Microbe Interact., 11, 1009–16. [DOI] [PubMed] [Google Scholar]
- 82. Zhao Z., Liu H., Wang C., Xu J. -R.. 2013, Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi, BMC Genomics, 14, 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Riley R., Salamov A. A., Brown D. W., et al. 2014, Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi, Proc. Natl. Acad. Sci. USA, 111, 9923–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu P., Stajich J. E.. 2015, Characterization of the carbohydrate binding module 18 gene family in the amphibian pathogen Batrachochytrium dendrobatidis, Fungal Genet. Biol., 77, 31–9. [DOI] [PubMed] [Google Scholar]
- 85. Xin X.-F., Nomura K., Aung K., et al. 2016, Bacteria establish an aqueous living space in plants crucial for virulence. Nature, 539, 524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole genome shotgun project for Lasiodiplodia theobromae CSS-01S has been deposited at DDBJ/ENA/GenBank under the accession code MDYX00000000. The version described in this paper is version MDYX01000000. Whole genome sequences of the other five isolates of Lasiodiplodia theobromae (HBQJZ-02S), Botryosphaeria dothidea (Sxg01s, LNXCEYGS-1), Neofusicoccum parvum (CDZ1M1s3, Gxg-01s) has been deposited at GenBank under the accession code SRP108461. All Total mRNA and Polysome-bound mRNA sequencing data generated in this study for Lasiodiplodia theobromae CSS-01S have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm. nih.gov/sra/) under accession number SRP107819.