

ABSTRACT
Nicotinamide adenine dinucleotide (NAD) is a key molecule in several cellular processes and is essential for healthy mitochondrial metabolism. We recently reported that mitochondrial dysfunction is among the very first changes to occur within retinal ganglion cells during initiation of glaucoma in DBA/2J mice. Furthermore, we demonstrated that an age-dependent decline of NAD contributes to mitochondrial dysfunction and vulnerability to glaucoma. The decrease in NAD renders retinal ganglion cells vulnerable to a metabolic crisis following periods of high intraocular pressure. Treating mice with the NAD precursor nicotinamide (the amide form of vitamin B3) inhibited many age- and high intraocular pressure- dependent changes with the highest tested dose decreasing the likelihood of developing glaucoma by ∼10-fold. In this communication, we present further evidence of the neuroprotective effects of nicotinamide against glaucoma in mice, including its prevention of optic nerve excavation and axon loss as assessed by histologic analysis and axon counting. We also show analyses of age- and intraocular pressure- dependent changes in transcripts of NAD producing enzymes within retinal ganglion cells and that nicotinamide treatment prevents these transcriptomic changes.
KEYWORDS: Glaucoma, NAD+, nicotinamide, axon degeneration, retinal ganglion cell, optic nerve head cupping
Glaucoma represents a significant economic and health burden. Affecting ∼80 million people, it is a leading cause of irreversible vision loss.1 Glaucoma is characterized by the progressive dysfunction and loss of retinal ganglion cells (RGCs) and their axons, which make up the neural tissue of the optic nerve. Major risk factors for glaucoma include genetics, elevated intraocular pressure (IOP), and age. The DBA/2J (D2) mouse is a widely used model of age-related, hereditary glaucoma, and recapitulates hallmark features of the human disease, including an insult to axons within the optic nerve. In D2 mice, mutant alleles of 2 genes (GpnmbR250X, Tyrp1b) cause a progressive, pigment dispersing, iris disease that results in an age-related, and asynchronous, ocular hypertension (beginning at 6 months of age in our colony). Following a period of IOP elevation, degeneration of the optic nerve occurs from ∼10 months of age onwards and this is almost complete by 12 months of age (∼70% nerves have severe damage).2-6
We recently used RNA-sequencing (RNA-seq) to analyze D2 RGCs at different ages to elucidate the earliest molecular changes that occur in glaucoma.7 We identified mitochondrial dysfunction as one of the first changes within RGCs. These results guided metabolic profiling studies that identified an age-dependent depletion of NAD as a primary driver of RGC vulnerability in glaucoma. Repleting NAD levels, using a diet supplemented in nicotinamide (NAM; an NAD precursor), profoundly protected from glaucoma (∼25% of nerves have severe damage. [550 mg/kg/d nicotinamide added to regular drinking water.]).7 This treatment protected from synapse loss, RGC loss (assessed by soma counts), and optic nerve degeneration. It also protected from declines in RGC electrical activity as assessed by pattern electroretinography (PERG) and loss of anterograde axon transport. Importantly, the treatment prevented mitochondrial changes, and the transcriptional profiles of nicotinamide-treated D2 RGCs match those of no glaucoma controls. As PERG and transcriptional changes are very sensitive measures of dysfunction these data indicate that nicotinamide mediates a remarkably robust protection. We provided further evidence of robust protection from glaucoma in mice with genetically increased NAD levels (increased by the WldS allele), including protection from decreases in both the dendritic field area and branching complexity of RGCs as well as synaptic preservation out to older ages.8,9 To present additional evidence for nicotinamide-mediated protection, we include here results from axon counting and optic nerve head analyses. These data demonstrate that nicotinamide-treated nerves that show no nerve damage are as healthy as non-glaucomatous age-matched controls in terms of their cross sectional area, axon number, and general morphology, without obvious glial changes (Fig. 1). Nicotinamide-treated eyes were also protected from the remodeling and atrophy of the optic nerve head that produces optic nerve cupping, a characteristic feature of human glaucoma (Fig. 2). These findings extend previous studies implicating mitochondria in glaucoma by showing that mitochondrial dysfunction is among the first glaucoma initiating changes within RGCs and that NAD boosting therapy is potently protective.10-13
Figure 1.
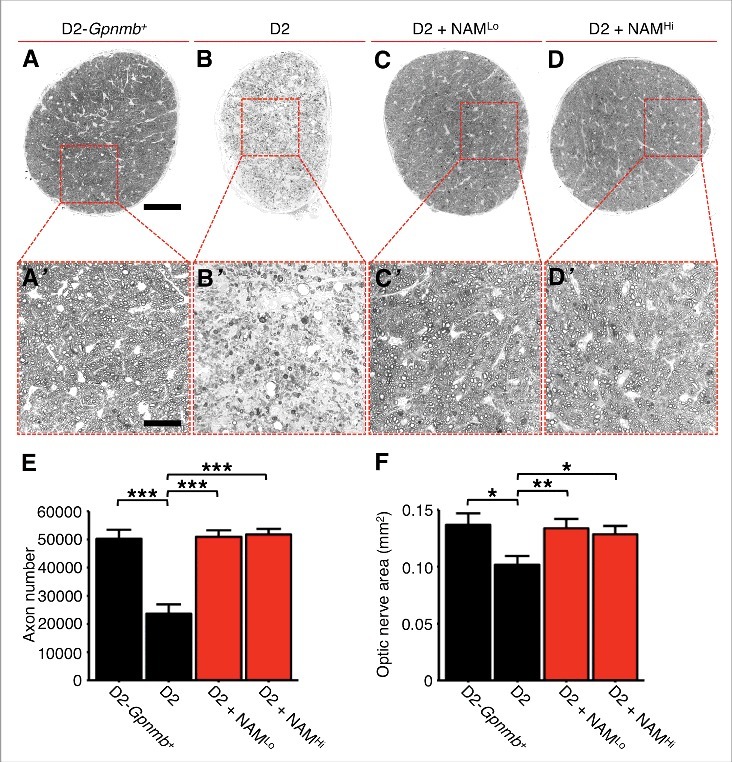
NAM prevents optic nerve atrophy and axon loss in glaucoma. Optic nerves from control (D2-Gpnmb+; A), DBA/2J (D2; B), and treated (D2 + NAMLo, D2 + NAMHi; (C) and D) mice were sectioned and stained with PPD, which darkly stains the axoplasm of dead or dying axons. Surviving axons were counted using AxonJ (E) and optic nerve cross sectional area measured (F). There is a significant decrease in total axon number and optic nerve area in glaucomatous D2 eyes compared with controls. Nerves from NAM treated mice are indistinguishable from no glaucoma controls. Nerves were assessed at 12 months of age. NAMLo and NAMHi refer to our low and high dose of NAM.7 Scale bar = 100 μm (A) and 30 μm (insets). * P < 0.05, * P < 0.01, * P < 0.001.
Figure 2.
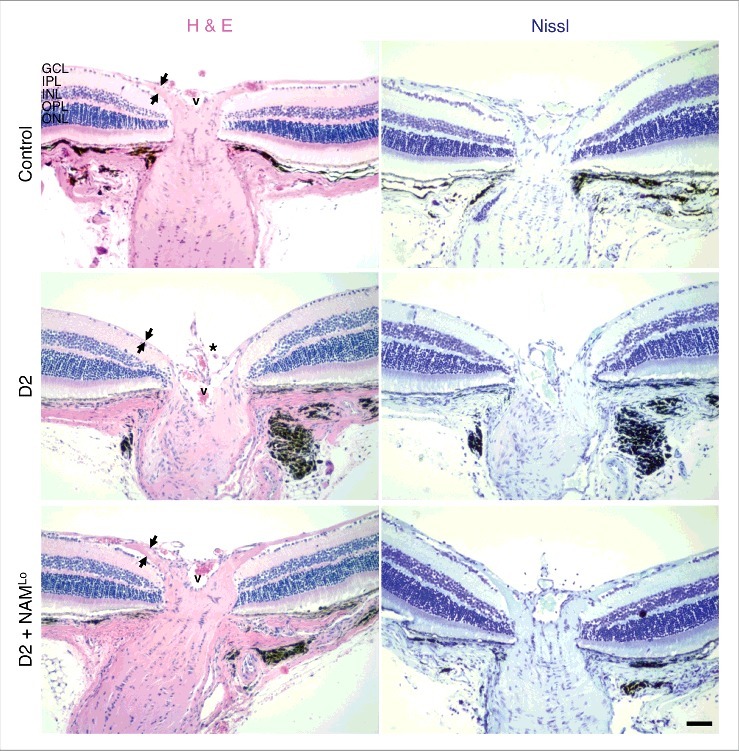
NAM prevents optic nerve cupping in glaucoma. The presence of optic nerve cupping was assessed using haematoxylin and eosin staining (H & E) and cresyl violet staining (Nissl). In control eyes (D2-Gpnmb+) there is a robust ganglion cell layer and nerve fiber layer (which contains axons from the retinal ganglion cells, denoted by the distance between the black arrows). In D2 eyes that have undergone glaucomatous neurodegeneration there is evident optic nerve cupping (asterisk), and a loss of ganglion cell layer cells and nerve fiber layer thickness. Cupping and RGC loss were absent in eyes of NAM treated mice (D2 + NAMLo), thus NAM treatment protected from all assessed signs of glaucoma. Eyes were assessed at 12 months of age. V corresponds to the central retinal vessel. GCL = ganglion cell layer, IPL = inner plexiform layer, INL = inner nuclear layer, OPL = outer plexiform layer, ONL = outer nuclear layer. Scale bar = 50 μm.
NAD is a critical cofactor for enzymes in many cellular processes and is a key metabolic modulator during aging.14-18 It acts as an oxidizing agent (as NAD+) and as a reducing agent (as NADH). Cellular NAD levels are maintained through de novo synthesis and by recycling the by-products of NAD catabolism (Fig. 3A). Sufficient NAD can be produced through de novo synthesis from tryptophan in an 8-step pathway. Alternatively NAD can be produced from vitamin B3. In the literature, vitamin B3 is considered to be either nicotinic acid (NA) or nicotinamide (NAM), and more recently nicotinamide riboside (NR). NAM and NR can be converted to NA in the gut by bacteria. There are salvage pathway routes for NAD production through either NAM or NR that recycle NAD from NAD consuming reactions. Specifically, NAM is a major by-product of NAD catabolism and cells are equipped to replenish NAD levels using NAM (Fig. 3A). In fact, NAM is a major precursor of NAD in vivo when available in large doses.19 In the NAM salvage pathway, nicotinamide mononucleotide (NMN) is produced from NAM by the rate-limiting enzyme NAMPT, and in turn NMN is metabolized to NAD by the spatially restricted enzymes NMNAT1, −2, and −3. Alternatively, NR is converted into NAD through either a 2-step reaction through the nicotinamide riboside kinase (NRK; NRK1, −2) pathway, or through a 3-step reaction through phosphorylation to NAM.20 Because declining NAD levels are thought to be a predisposing factor for age–related changes and neurodegeneration,15,20 there is increasing interest in using NAM or NR to increase NAD levels in various human tissues.
Figure 3.
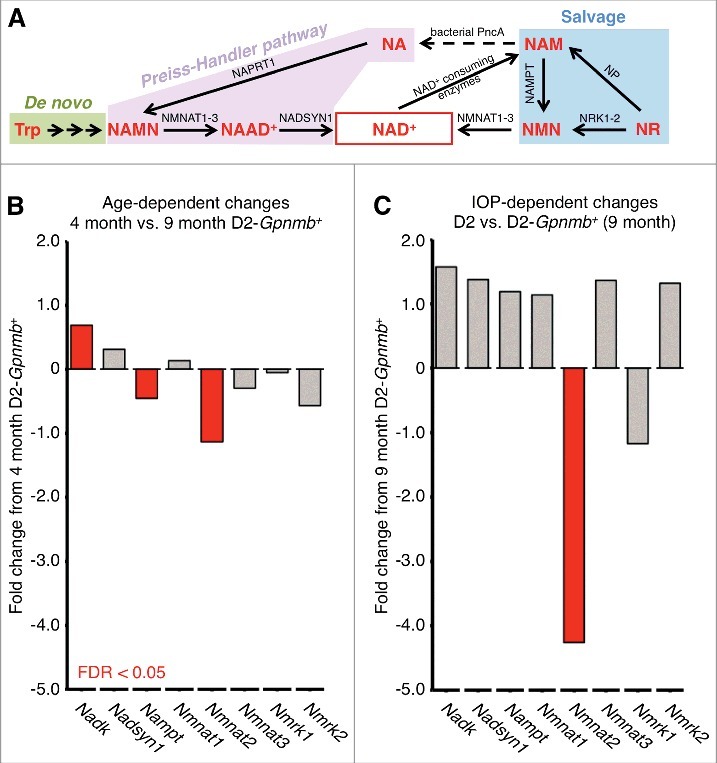
NAD synthesis and NAD relevant genes in RGCs. (A) NAD synthesis. Tryptophan (Trp) is used to form NAD+ de novo from diet in an 8 step pathway with nicotinic acid mononucleotide (NAMN) and nicotinic acid adenine dinucleotide (NAAD+) intermediates. Alternatively NAD+ can be produced through 2 other core pathways; the Preiss-Handler pathway from nicotinic acid (NA), or through the salvage pathway from nicotinamide (NAM). NA is used in the Preiss-Handler pathway to form NAD+ via 2 steps shared with the de novo pathway: NAMN (by nicotinic acid phosphoribosyltransferase; NAPRT1) and NAAD+ (by NAD+ synthetase; NADSYN1). In the salvage pathway, NAM is used to form NAD+ being converted by nicotinamide phosphoribosyltransferase (NAMPT) to nicotinamide mononucleotide (NMN) and subsequently to NAD+ by nicotinamide nucleotide adenylytransferase (NMNAT1, −2, and −3). NAM can also be converted to NA in the gut by bacterial PncA (nicotinamidase) and salvaged into the Preiss-Handler pathway. NAM is available in diet, but can also be produced by NAD+-consuming enzymes. Nicotinamide riboside (NR) can feed into the salvage pathway to form NAD+ by nicotinamide riboside kinases (NRK1, −2; Nmrk1, −2 as mouse genes) via NMN, or via NAM by purine nucleoside phosphorylase (NP). (B) and (C) Retinal ganglion cells exhibit age-dependent changes in NAD+ synthesis-related genes as well as further IOP-dependent declines in Nmnat2, an important NAD producing enzyme linked to axon protection. The decline in NAD is a major age-dependent risk factor for DBA/2J glaucoma. NADK (Nadk gene) is a major NAD-consuming kinase and its upregulation suggests increased NAD consumption / utilization. Differentially expressed genes (FDR < 0.05) are shown in red. Non-differentially expressed genes are shown in gray.
Variations in the level or control of NAD producing pathways in the retina may impact vulnerability to glaucoma, as may age-related changes in NAM (NAM is both a product and endogenous inhibitor of NAD catabolizing enzymes). The genes encoding the cellular machinery that drive NAD production from NAM are expressed in retinal ganglion cells (Nampt, Nmnat1, Nmnat2, and Nmnat3) however, transcript abundance of NRK genes (Nmrk1, Nmrk2) that produce NAD from NR are only lowly expressed in retinal ganglion cells.7 Since age-dependent changes in gene expression may modulate glaucoma susceptibility, we have assessed the effects of aging on genes impacting NAD levels. Importantly, there are age-dependent declines in the expression of Nmnat2 and Nampt, and an age-dependent increase in Nadk (whose encoded enzyme converts NAD to NADP) within retinal ganglion cells (Fig. 3B). Nmnat2 is the only NAD-related transcript that we observed to decline in both an age- and IOP- dependent manner (Fig. 3C). All these changes are prevented by nicotinamide treatment.7
The decreased levels of Nampt and Nmnat2 in retinal ganglion cells may be a key feature permitting degeneration in this glaucoma. These decreases are expected to, in turn, decrease the capacity of the salvage pathway in retinal ganglion cells and decrease retinal NAD levels. In our D2 glaucoma experiments we tested viral gene therapy overexpressing the salvage pathway enzyme Nmnat1.7 In this gene therapy experiment the entire gene of interest (Nmnat1) was overexpressed. The nuclear localization signal (NLS) was intact, and so the protein product (NMNAT1) should be localized predominantly at the nucleus within the soma. Poly ADP ribose polymerase (PARP) increases in RGCs during pre-degenerative stages of DBA/2J glaucoma.7 Since PARP depletes nuclear NAD and the NMNAT1 NLS was intact, it seems most likely that NAD-mediated protection is mediated within the soma. However, and possibly arguing against this, a critical insult directly damages RGC axons in glaucoma and keeping cell bodies alive with a BAX mutation does not save the axons.21 Interestingly, NMNAT1 with an intact NLS does not protect axons in transgenic mice arguing that NMNAT1 must reach the axon to be protective.22 cytNmnat1, an NMNAT1 mutant that localizes only to the cytoplasm (and thus axon) provides potent axon protection in transgenic mice.23 Soma, axons, and terminal projections in the brain were protected in our experiments and together, this may argue that NAM, and possibly the virally produced NMNAT1, has protective effects within the axon. It is also possible that overexpression drives some virally produced NMNAT1 (with intact NLS) into axons but this has yet to be empirically tested. If metabolic compromise within the soma is necessary to allow the axons to be insulted, however, it is possible that preserving somal metabolism may also preserve the axons. Thus, our data suggest that maintaining somal health and metabolism can prevent axon degeneration in glaucoma - an axonopathy where it is clear that a critical insult directly attacks axons in the optic nerve head.24 If the axon can be saved through a soma-targeted mechanism, this raises an exciting prospect for other (especially distal) axonopathies where it would not be feasible to treat individual neuronal targets but instead could target a hub of soma or ganglion. However, a protective effect within axons cannot be ruled out. NMNAT2 is emerging as a key NAD producing enzyme within axons.25,26 New evidence shows wide variability of NMNAT2 transcript levels between human post-mortem brains.25 Such variation in human NMNAT2 expression has the potential to modulate neuronal vulnerability to stresses such as elevated IOP in glaucoma. Nmnat3 has also been detected in both somas and axons and overexpression of Nmnat3 protects from axon degeneration in other models of axonopathy and glaucoma.27 Therefore the decreased abundance of Nmnat2 and Nampt transcripts that we observed in D2 retinal ganglion cells may underlie metabolic defects in the axon or throughout the entire cell.
Conclusions
In conclusion, retinal ganglion cells undergo age-dependent declines in key NAD-salvage pathway transcripts. These declines are expected to contribute to a decline of NAD rendering these cells susceptible to IOP-dependent stresses. Nicotinamide prevents these changes and robustly protects all assessed cell compartments. The mechanisms of how nicotinamide counteracts age- and IOP- dependent changes and whether other NAD precursors have the same effects are important areas for future research. Declining NAD is appearing as a shared mechanism in aging and neurodegeneration.15,16 As such an important avenue in future research will be to test NAD-boosting strategies in other glaucoma models (especially higher order animal models such as non-human primate models) as well as in other models of neurodegenerations. The possibility that a simple dietary supplement can prevent neurodegeneration is an exciting prospect with broad implications for age-related diseases.
Methods
Unless stated, methods used have been previously outlined.7 RNA-seq data are publically available through the Gene Expression Omnibus (accession number GSE90654). For automated axon counts, AxonJ was used to count the entire cross sectional nerve area at a 63x magnification for 10 nerves in each experimental group.28
Funding Statement
The Jackson Laboratory Fellowships (PAW, JMH), EY11721 (SWMJ), the Barbara and Joseph Cohen foundation, the Partridge Foundation, and the Lano Family Foundation (SWMJ). SWMJ is an Investigator of HHMI.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank the Histopathology, Electron Microscopy, and Microscopy services at The Jackson Laboratory, as well as Gareth Howell and members of the John Lab for their scientific support preparing this manuscript.
References
- [1].Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90:262-67. doi: 10.1136/bjo.2005.081224. PMID:16488940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Libby RT, Anderson MG, Pang IH, Robinson ZH, Savinova OV, Cosma IM, Snow A, Wilson LA, Smith RS, Clark AF, et al.. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci. 2005;22:637-48. doi: 10.1017/S0952523805225130. PMID:16332275 [DOI] [PubMed] [Google Scholar]
- [3].Nickells RW. Ganglion cell death in glaucoma: From mice to men. Vet Ophthalmol. 2007;10(Suppl 1):88-94. doi: 10.1111/j.1463-5224.2007.00564.x. PMID:17973839 [DOI] [PubMed] [Google Scholar]
- [4].Fernandes KA, Harder JM, Williams PA, Rausch RL, Kiernan AE, Nair KS, Anderson MG, John SW, Howell GR, Libby RT. Using genetic mouse models to gain insight into glaucoma: Past results and future possibilities. Exp Eye Res. 2015;141:42-56. doi: 10.1016/j.exer.2015.06.019. PMID:26116903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].McKinnon SJ, Schlamp CL, Nickells RW. Mouse models of retinal ganglion cell death and glaucoma. Exp Eye Res. 2009;88:816-24. doi: 10.1016/j.exer.2008.12.002. PMID:19105954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Anderson MG, Smith RS, Hawes NL, Zabaleta A, Chang B, Wiggs JL, John SW. Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nature Genetics. 2002;30:81-85. doi: 10.1038/ng794. PMID:11743578 [DOI] [PubMed] [Google Scholar]
- [7].Williams PA, Harder JM, Foxworth NE, Cochran KE, Philip VM, Porciatti V, Smithies O, John SW. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science. 2017;355:756-60. doi: 10.1126/science.aal0092. PMID:28209901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Harder JM, Braine CE, Williams PA, Zhu X, MacNicoll KH, Sousa GL, Buchanan RA, Smith RS, Libby RT, Howell GR, et al.. Early immune responses are independent of RGC dysfunction in glaucoma with complement component C3 being protective. Proc Natl Acad Sci U S A. 2017;114:E3839-48. doi: 10.1073/pnas.1608769114. PMID:28446616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Williams P, Harder J, Foxworth N, Cardozo B, Cochran K, John S. Nicotinamide and WLDS act together to prevent neurodegeneration in glaucoma. Front Neurosci. 2017;11:232. doi: 10.3389/fnins.2017.00232. PMID:28487632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chrysostomou V, Rezania F, Trounce IA, Crowston JG. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol. 2013;13:12-15. doi: 10.1016/j.coph.2012.09.008. PMID:23069478 [DOI] [PubMed] [Google Scholar]
- [11].Lee S, Van Bergen NJ, Kong GY, Chrysostomou V, Waugh HS, O'Neill EC, Crowston JG, Trounce IA. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp Eye Res. 2011;93:204-12. doi: 10.1016/j.exer.2010.07.015. PMID:20691180 [DOI] [PubMed] [Google Scholar]
- [12].Chen SD, Wang L, Zhang XL. Neuroprotection in glaucoma: Present and future. Chin Med J (Engl). 2013;126:1567-77. PMID:23595396 [PubMed] [Google Scholar]
- [13].Inman DM, Harun-Or-Rashid M. Metabolic vulnerability in the neurodegenerative disease glaucoma. Front Neurosci. 2017;11:146. doi: 10.3389/fnins.2017.00146. PMID:28424571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D'Amico D, Ropelle ER, Lutolf MP, Aebersold R, et al.. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436-43. doi: 10.1126/science.aaf2693. PMID:27127236 [DOI] [PubMed] [Google Scholar]
- [15].Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350:1208-13. doi: 10.1126/science.aac4854. PMID:26785480 [DOI] [PubMed] [Google Scholar]
- [16].Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464-71. doi: 10.1016/j.tcb.2014.04.002. PMID:24786309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, et al.. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624-38. doi: 10.1016/j.cell.2013.11.037. PMID:24360282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Cantó C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, et al.. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430-41. doi: 10.1016/j.cell.2013.06.016. PMID:23870130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ijichi H, Ichiyama A, Hayaishi O. Studies on the biosynthesis of nicotinamide adenine dinucleotide. 3. Comparative in vivo studies on nicotinic acid, nicotinamide, and quinolinic acid as precursors of nicotinamide adenine dinucleotide. J Biol Chem. 1966;241:3701-07. PMID:4288134 [PubMed] [Google Scholar]
- [20].Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115-30. doi: 10.1146/annurev.nutr.28.061807.155443. PMID:18429699 [DOI] [PubMed] [Google Scholar]
- [21].Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, John SW. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005;1:17-26. doi: 10.1371/journal.pgen.0010004. PMID:16103918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, et al.. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 2007;14:116-27. doi: 10.1038/sj.cdd.4401944. PMID:16645633 [DOI] [PubMed] [Google Scholar]
- [23].Sasaki Y, Vohra BP, Baloh RH, Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J Neurosci. 2009;29:6526-34. doi: 10.1523/JNEUROSCI.1429-09.2009. PMID:19458223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, et al.. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179:1523-37. doi: 10.1083/jcb.200706181. PMID:18158332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ali YO, Allen HM, Yu L, Li-Kroeger D, Bakhshizadehmahmoudi D, Hatcher A, McCabe C, Xu J, Bjorklund N, Taglialatela G, Bennett DA, et al.. NMNAT2:HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 2016;14:e1002472. doi: 10.1371/journal.pbio.1002472. PMID:27254664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. PMID:20126265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kitaoka Y, Munemasa Y, Kojima K, Hirano A, Ueno S, Takagi H. Axonal protection by Nmnat3 overexpression with involvement of autophagy in optic nerve degeneration. Cell Death Dis. 2013;4:e860. doi: 10.1038/cddis.2013.391. PMID:24136224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zarei K, Scheetz TE, Christopher M, Miller K, Hedberg-Buenz A, Tandon A, Anderson MG, Fingert JH, Abràmoff MD. Automated axon counting in rodent optic nerve sections with AxonJ. Sci Rep. 2016;6:26559. doi: 10.1038/srep26559. PMID:27226405 [DOI] [PMC free article] [PubMed] [Google Scholar]