

Abstract
Global climate change includes rising temperatures and increased pCO2 concentrations in the ocean, with potential deleterious impacts on marine organisms. In this case study we conducted a four-week climate change incubation experiment, and tested the independent and combined effects of increased temperature and partial pressure of carbon dioxide (pCO2), on the microbiomes of a foundation species, the giant kelp Macrocystis pyrifera, and the surrounding water column. The water and kelp microbiome responded differently to each of the climate stressors. In the water microbiome, each condition caused an increase in a distinct microbial order, whereas the kelp microbiome exhibited a reduction in the dominant kelp-associated order, Alteromondales. The water column microbiomes were most disrupted by elevated pCO2, with a 7.3 fold increase in Rhizobiales. The kelp microbiome was most influenced by elevated temperature and elevated temperature in combination with elevated pCO2. Kelp growth was negatively associated with elevated temperature, and the kelp microbiome showed a 5.3 fold increase Flavobacteriales and a 2.2 fold increase alginate degrading enzymes and sulfated polysaccharides. In contrast, kelp growth was positively associated with the combination of high temperature and high pCO2 ‘future conditions’, with a 12.5 fold increase in Planctomycetales and 4.8 fold increase in Rhodobacteriales. Therefore, the water and kelp microbiomes acted as distinct communities, where the kelp was stabilizing the microbiome under changing pCO2 conditions, but lost control at high temperature. Under future conditions, a new equilibrium between the kelp and the microbiome was potentially reached, where the kelp grew rapidly and the commensal microbes responded to an increase in mucus production.
Introduction
Kelps form large forests in coastal marine ecosystems that provide habitat for hundreds of species, including benthic and pelagic fishes, invertebrates, vegetation and microbes [1–4]. Microbial communities associated with macroalgae are generally host specific, vary seasonally, and fluctuate based on host condition [5,6]. The host microbiome may represent a majority fraction of the host’s total cellular material and provide the host with additional genomic and metabolic potential [7,8]. Thus, describing and measuring microbial responses to environmental stresses is paramount for evaluating host health.
The marine environment contains over 1800 known species of brown algae, yet the microbial communities or microbiomes have been described in less than 2% [9]. Macroalgal microbiomes have core functions related to vitamin synthesis, biofilm formation, and algae polysaccharide catabolism [9]. Microbiomes benefit the algae by producing micronutrients like B12, enhancing iron uptake, and aiding host immune health by producing antibiotics to restrict colonization of pathogenic bacteria [10–12]. Microbes also negatively impact the algae through disease. While several specific pathogens are described in disease of marine organisms, many pathogens remain unknown and disease to the host occurs because of a shift in the types or functions of the microbes present. Dysbiosis, which is a shift in the microbiome from a stable state to disturbed state where commensal or new colonizers may become opportunists pathogens, occurs when the host system is stressed by abiotic or biotic challenges [13,14]. Determining whether the M. pyrifera microbiome is altered by environmental conditions, such as those associated with global climate change, and whether those alterations affect the function of the host remains an outstanding question.
In the last 150 years global atmospheric concentrations of carbon dioxide have increased 40% to 407 ppm, with an expected increase to 1000 ppm by 2100 leading to a decrease in pH by 0.3–0.4 units [15,16]. Sea surface temperature is predicted to increase by between 1.5 to 4 oC by 2100 [17], while much of the excess carbon dioxide and heat will be absorbed by the ocean. These abiotic fluctuations associated with climate change are expected to be the most extreme in nearshore coastal communities [17], such as those dominated by M. pyrifera. A recent review of over 100 macroalgae revealed that non-calcifying fleshy macroalgae growth and photosynthesis may increase in an elevated pCO2 ocean. Most macroalgae, including M. pyrifera, utilize both CO2 and bicarbonate (HCO3-) as an inorganic carbon source in photosynthesis and therefore an increase in CO2 will reduce use of the less efficient bicarbonate pathway [18]. In contrast, increasing temperature poses risks to macroalgae, especially those living near to their maximum temperature tolerances [19,20]. Enhanced growth and development of the microscopic gametophyte stage of M. pyrifera occurred with elevated pCO2 [21,22], yet growth and photosynthesis of the adult macroscopic sporophyte stage are less affected by fluctuations of pCO2 and more impacted by nutrient availability and elevated temperature [23,24]. M. pyrifera growth and reproduction declines at 18–20 oC [1,25–27]. Elevated temperature causes reductions in frond elongation rates in kelp [28], while gametophytes have maximal photosynthetic rates occurring between 15–20 oC declining at 30 oC [29]. Temperatures above 18 oC have a negative effect on M. pyrifera growth, spore production, germination and recruitment [19,30]. In contrast, the combined effects of elevated temperature and pCO2 together increase M. pyrifera sporophyte growth and photosynthesis [31], but resulted in higher spore mortality and decreased germination [21]. While these ecological and physiological studies have evaluated kelp in relation to changing abiotic conditions [23,27,31–33], few have investigated the microbes, which are a crucial explanatory variable for describing macroalgal health as measured by growth or decay.
As abiotic conditions vary with climate change, microbial metabolic activity and community dynamics are affected. Under elevated temperature regimes, marine microbial heterotroph production and respiration increase [34], and microbial nitrogen uptake and metabolism intensify [35]. Increases in pCO2 was positively correlated with higher abundances of Flavobacteriaceae and Rhodobacteraceae bacteria in sediments, which contribute to higher organic decomposition and a lower abundance of nitrifying bacteria [36]. In a mesocosm experiment where transplanted kelps were grown under elevated pCO2 conditions, shifts occurred in water column microbial community structure, but the kelp microbiome was not assessed. Increases in the relative abundance of Gammaproteobacteria and genes relating to iron acquisition and membrane transport, and decreases in the relative abundance of Flavobacteria were associated with increased pCO2 [37]. While we have shown that microbes in the water column surrounding kelp are affected by changes in pCO2, the effects on the microbial community growing on the kelp surface requires evaluation.
The purpose of our case study was to evaluate the individual and combined effects of elevated temperature and pCO2 associated with climate change on the physiology of the giant kelp, M. pyrifera, its microbiome, and the microbes in the surrounding water column. We hypothesized that the abiotic stressors may affect the microbiomes associated with the kelp and potentially interrupt some of the normal microbiome functions. We demonstrate that the microbiomes of the kelp and water are uniquely affected by changes in pCO2 and temperature, and propose mechanistic links between the microbiome and kelp growth or decay in the context of climate change.
Results
Kelp growth and photosynthetic Carbon uptake was reduced in the elevated temperature treatment, but not affected by increased pCO2, and significantly increased in future conditions (Fig 1A). A total of 19 of the possible 24 microbiomes were generated using whole genome shotgun sequencing from the 12 water column samples (3 replicates per treatment mesocosm) and seven kelp surface samples. Out of the 12 kelp surface samples (3 replicates per treatment mesocosm), only three original samples had at least 100 ng of DNA to make libraries. For the samples with less than 100 ng of DNA, replicates were pooled yielding a total of 7 metagenomic libraries. The microbiomes comprised 14.9 Gb of data, with over 45 million reads and an average of 2.4 million reads per treatment (S1 Table). Over 18 million known proteins were annotated at approximately one million proteins per microbiome. Across the 19 microbiomes, a total of 2,693 genera and 1,097 gene function categories were annotated. Applying the lower cutoff (S1 Fig), the genera were reduced to a total of 361, including 329 genera shared in the water column and kelp surface, consisting of a mean loss of 1.3 ± 0.1% sequences per microbiome. The gene function list was reduced from 1,097 to 718 gene functions, with 689 shared between the water column and kelp surface, consisting of a loss of 3.1 ± 0.178% of sequences per microbiome.
Fig 1. Experimental conditions of kelp mesocosm.
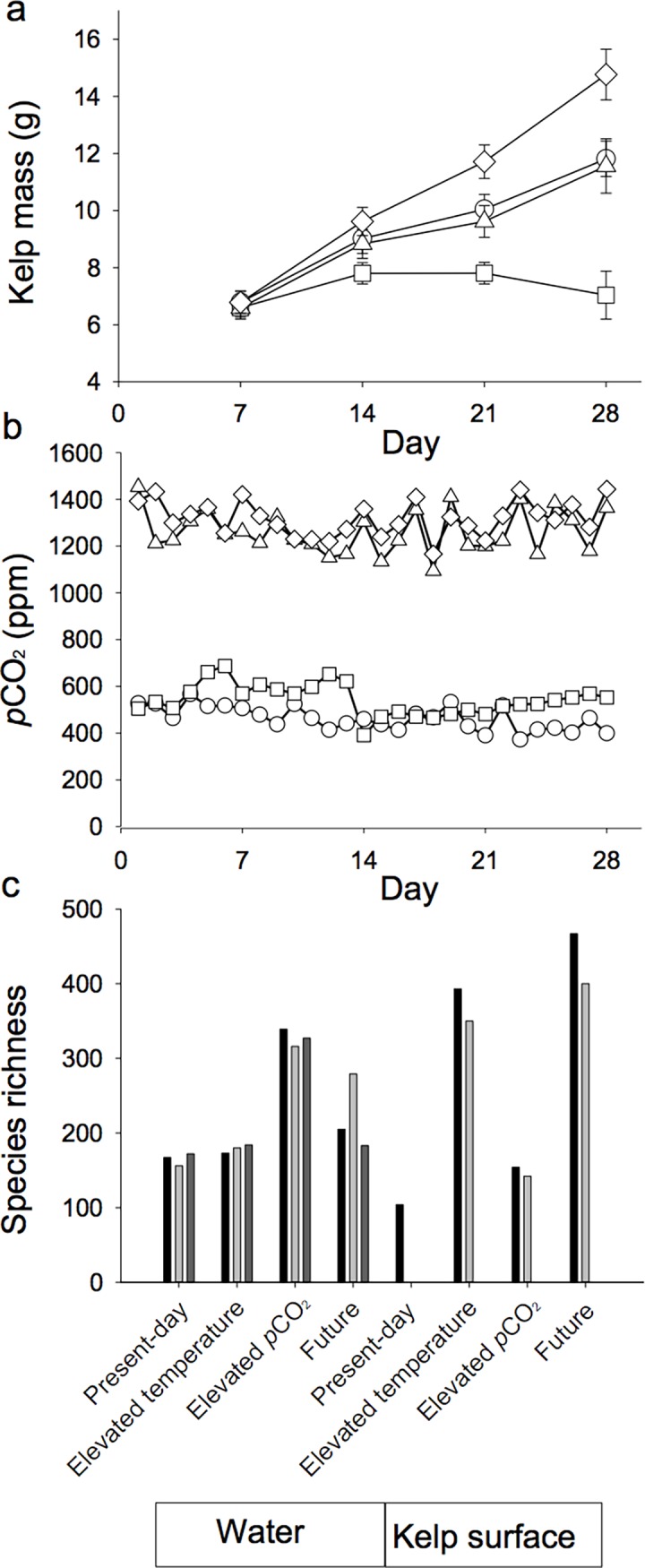
Kelp was grown for four weeks in four different abiotic stressors, present-day 12 oC 500 ppm of CO2 ○, Elevated Temperature 15 oC 500 ppm of CO2 □, Elevated pCO2 12 oC 1300 ppm of CO2 △, and future conditions 15 oC 1300 ppm of CO2 ◇. (a) Growth of kelp over the duration of the experiment. Each week, 18 fronds were sampled and weighed per incubation mesocosm. (b) Elevated pCO2 was maintained between 1,200 and 1,400 ppm of CO2 whereas temperature was held constant at either 12 oC or 15 oC. (c) microbiome species richness. At week 4, microbial communities were sampled from the water and kelp surfaces and sequenced. Alpha diversity, Shannon species richness, was calculated for all microbiome samples using MG-RAST (grey scale represents replicates; three for water and 2 or 1 for kelp surface).
Microbial community analysis
Rarefaction curves indicate microbiomes were sequenced at a depth sufficient to evaluate taxonomical and functional features (S2 Fig). For alpha diversity, water column microbial richness ranged from 165 to 327 species, whereas the kelp surface microbial richness ranged from 104 to 433 species (Fig 1). In the water column microbiomes, the elevated pCO2 mesocosm had a higher species richness of 327 ± 6.6, compared to present day (165 ± 4.7), elevated temperature (179 ± 3.2), and future conditions (222 ± 29.0) (Fdf = 3 = 23.46, P ≤ 0.01), (Tukey HS, P ≤ 0.01). In the kelp surface microbiomes, elevated temperature and future conditions had a species richness of 371 ± 21.5 and 433 ± 33.5 and present day microbiomes had 104 species and elevated pCO2 had 148 ± 6.0 species (Fig 1).
Water and kelp surface microbiomes were impacted differently by global climate stressors
Based on taxonomical composition, the microbiome of the water column was significantly different from the microbiome from the kelp surfaces and microbiomes from each mesocosm clustered (PERMANOVA Pseudo-F = 118.21, P ≤ 0.001) (Table 1A, Fig 2) and explained the largest effect (49.0%) at the genera level. Climate stressors affected each of the microbial communities (Pseudo-F = 16.31, P ≤ 0.001) with a magnitude of effect size of 12.0%, while there was also an interaction between the effect of the climate treatment and microbiome environment (Pseudo-F = 21.65, P ≤ 0.001), magnitude of effect size of 35.0%. This large interaction effect indicates climate stressors altered the water column and kelp surface microbiomes uniquely. The increase in pCO2 had the largest effect on the taxonomic components of water column (23.7% dissimilarity), whereas elevated temperature had the highest effect on the kelp microbiome (51.8% dissimilarity) (Table 1B, Fig 2).
Table 1. (a) PERMANOVA and (b) SIMPER results indicate microbial communities differ taxonomically and functionally between environment (water and kelp surface) and across climate change related stressors.
| a. PERMANOVA | df | Sum of squares | Mean of squares | Pseudo-F | P (perm) | Mag. of effects % |
| Genera | ||||||
| Environment | 1 | 157.73 | 157.73 | 118.21 | < 0.001 | 49 |
| Climate change | 3 | 65.30 | 21.77 | 16.31 | < 0.001 | 12 |
| Interaction | 3 | 86.66 | 28.89 | 21.65 | < 0.001 | 35 |
| Residual | 11 | 14.68 | 1.33 | 4 | ||
| Total | 18 | 324.36 | 100 | |||
| Gene Function | ||||||
| Environment | 1 | 10.91 | 10.91 | 62.29 | < 0.001 | 47 |
| Climate change | 3 | 5.19 | 1.73 | 9.87 | < 0.001 | 13 |
| Interaction | 3 | 5.96 | 1.99 | 11.33 | < 0.001 | 33 |
| Residual | 11 | 1.93 | 0.18 | 7 | ||
| Total | 18 | 23.98 | 100 | |||
| b. SIMPER pairwise analyses | % Dissimilarity | |||||
| Genera | Environment | Water vs. Kelp surface | 54.4 | |||
| Water | Climate change | present-day vs. + temperature | 8.5 | |||
| present-day vs. + pCO2 | 23.7 | |||||
| present-day vs future | 10.0 | |||||
| Kelp surface | Climate change | present-day vs. + temperature | 51.8 | |||
| present-day vs. + pCO2 | 17.0 | |||||
| present-day vs future | 46.4 | |||||
| Gene Function | Environment | 3.9 | ||||
| Water | Climate change | present-day vs. + temperature | 1.1 | |||
| present-day vs. + pCO2 | 1.9 | |||||
| present-day vs future | 1.3 | |||||
| Kelp surface | Climate change | present-day vs. + temperature | 3.3 | |||
| present-day vs. + pCO2 | 1.3 | |||||
| present-day vs future | 3.7 | |||||
Fig 2. Microbial community analysis of water column and kelp surfaces across treatments.

The relatedness of the microbiome from the water (blue) and kelp surface (green) calculated using hierarchical clustering, and nMDS ordination (present-day ○, elevated temperature □, elevated pCO2 △, and future ◇) for (a) genera cluster analysis, (b) Genera nMDS plot, (c) SEED gene function level 3 cluster analysis, (d) SEED Gene Function level 3 nMDS plot.
The distribution of microbial functions across microbiomes followed a similar pattern as the genera analysis with water and kelp microbiomes forming separate clusters (Fig 2). Based on the gene function composition, there was a significant difference between the microbiomes in the water column and kelp surface (Pseudo-F = 62.29, P ≤ 0.001) (Table 1A), with these differences in environments explaining 47.0% of the variation among treatments in the statistical model. Temperature and pCO2 also affected the functional profile of the microbiomes (Pseudo-F = 9.87, P ≤ 0.001), but this was not consistent in the two environments (environment x climate stressor interaction Pseudo-F = 11.33, P ≤ 0.001) (Fig 2C) (Table 1B). The interaction between environment and climate stressors had the second biggest effect in the model (33%). Elevated pCO2 treatment induced the greatest change in the water column microbiome, as the functional profiles showed 1.9% dissimilarity compared with the present day microbiome. The functional profile of the present day kelp surface microbiome was most dissimilar to the profile of the future and elevated temperature treatments (Table 1B).
Water column and kelp surface have unique microbiomes
Across all treatment groups, the water and kelp microbiomes were distinctive, as were the communities’ response to climate stressors (Fig 3). In all four treatments, the water microbiomes had higher proportion of Flavobacteriales (26.1% ± 2.7 mean and s.e. percent composition in the microbiome) and Actinomycetales (3.8% ± 1.4), while the kelp surface microbiome was enriched with Alteromonadales (35.4% ± 10.6), Oceanospirillales (6.0% ± 0.9) and Sphingomonadales (3.5% ± 0.5) (Fig 3). Similar differences in taxa between the water column and kelp surface occur in Pt Loma kelp forest (S3 and S4 Figs). The Alteromonadales order accounted for 26.9% of the observed variation between the water and kelp surface microbiomes and the genera Alteromonas, Pseudoalteromonas, Marinobacter and Saccharophagus genera were enriched by an average of 52.2, 11.3, 7.6, and 7.3 fold, respectively, on the kelp surface compared with the water column. The Erythrobacter genus within the Sphingomonadales order was enriched 11.9 fold on the kelp surface (Fig 4A). The water microbiome was enriched with the Flavobacteriales order and had an increased composition of Maribacter, Cellulophaga, Robiginitalea, and Gramella genera at 5.8 fold, 4.9 fold, 7.6 fold, and 3.2 fold, respectively (Fig 4A). The Rhodobacteriadales order was also over represented in the water microbiome (32.2% ± 7.3) compared to the kelp microbiome (19.22% ± 5.35).
Fig 3. Taxonomic differences in community composition of water and kelp surface microbiomes.

Taxonomic compositions of the ten most abundant bacteria orders, in the water column and kelp surface microbiomes from present-day, elevated temperature, elevated pCO2, and future conditions. Rare taxa as labeled in the legend, are microbial orders not in the top ten.
Fig 4. Strongest drivers of microbiome compostional changes in the water and kelp.

The top ten bacterial genera (a) and gene functions (b) differentiating the water column from the kelp surface microbiomes as determined by SIMPER analysis.
Several functions were enriched in the kelp surface microbiome compared to the water microbiomes (Fig 4B). The kelp microbiomes were enriched with genes related to membrane transport, iron acquisition and metabolism, motility and chemotaxis, stress response, and nitrogen metabolism. Within the membrane transport system, the Ton and Tol transport system and general secretion pathways were enriched 2.7 and 8.0 fold on the kelp surface, while iron acquisition was increased 2.5 fold. Within the motility and chemotaxis category; flagellum, flagellar motility and bacteria chemotaxis genes were enriched in the kelp surface microbiomes, 2.5, 3.3, and 3.7 fold, respectively. Three bacterial orders: Rhodobacterales, Flavobacterales, and Alteromonadales, within the water column and kelp surface contained the most flagellar motility genes although the distributions were unique to the water column and kelp surface. Specifically, the water column was mostly comprised of Rhodobacterales (W: 36%; KS: 17%) and Flavobacterales (W: 29%; KS: 1%) while Alteromonadales dominated the kelp surface (W: 10%; KS: 50%). Among the virulence, antibiotics, and toxins category, genes related to cobalt-zinc-cadmium resistance and multidrug resistance efflux pumps were enriched by 1.4 and 1.8 fold respectively on the kelp surface compared with the water microbiome. In both water and kelp surface microbiomes, the Acriflavin resistance protein comprised 63–86% of multidrug efflux pumps. Under nitrogen metabolism, nitrate and nitrite ammonification genes were enriched by 2.4 fold in the kelp surface microbiome and were primarily Nitrite reductase NADPH large subunit EC 1.7.1.4 (mean 20.7%), assimilatory nitrate reductase large subunit EC 1.7.99.4 (mean 19.1%), and the Nitrate ABC transporter, nitrate-binding protein (mean 13.7%). The water microbiome was enriched in cofactors, vitamins, prosthetics group, and sulfur metabolism. Chlorophyll biosynthesis and sulfur oxidation were enriched by 15.3 and 4.4 fold, respectively, in the water column microbiome. The Chlorophyll biosynthesis group had many genes but Protoporphyrin IX Mg-chelatase subunit H EC 6.6.1.1 and Chlorophyllide reductase subunit BchZ EC 1.18 were the two most dominant comprising on average 20% and 8% respectively.
Water microbiomes were most influenced by elevated pCO2
Microbial orders in the water microbiome were differentially affected by changes in environmental conditions (Fig 3) and each condition had a single order that increased in abundance from the present day. Elevated pCO2 showed an increase in the order Rhizobiales, elevated temperature showed an increase in Rhodobacterales, and the combination of pCO2 and temperature showed an increase in Actinomycetales. These changes were also reflected in alterations at the genus level for the various conditions of elevated temperature (Fig 5), elevated pCO2 (Fig 6), and combined elevated temperature and pCO2 (Fig 6). The elevated pCO2 caused the largest effect on the microbiome composition and was 23.7% dissimilar compared with the present day microbiome, whereas elevated temperature was 8.5% dissimilar and future conditions were 10.0% from the present day microbiome (Table 1B). During elevated pCO2, seven genera within the order Rhizobiales increased in abundance including Sinorhizobium (12.6 fold increase), Rhizobium (9.7 fold), Mesorhizobium (12.5 fold), Hoeflea (11.9 fold), and Agrobacterium (8.5 fold) (Fig 6A), whereas the abundances of several genera within the phyla Bacteroidetes and Verrucomicrobia decreased (Fig 6A). Elevated temperature was associated with an increase in the proportion of Rhodobacterales order in the water microbiome, (Fig 3) including increases in the genera Jannaschia (4.4 fold), Rhodobacter (2.3 fold), Roseobacter (1.5 fold), and Ruegeria (1.5 fold) (Fig 5A). Elevated temperature also reduced the proportion of Bacteroidetes and Verrucomicrobia in the water microbiome. The combination of elevated pCO2 and temperature was associated with an increase in the proportion of Actinomycetelales in the water microbiome and a reduction in the proportion of Rhodobacterales. Various genera within this order increased in abundance including: Clavibacter (4.6 fold), unclassified Actinobacteria (4.7 fold) and Leifsonia (4.7 fold). Further, a 49.6 fold increase in the Glaciecola genus within the Alteromonadales order and a 43.4 fold decrease in the Coraliomargarita genus within the Puniceicoccales order occurred in the future mesocosm (Fig 7A).
Fig 5. Effects of temperature on water and kelp surface microbiomes.

Top ten microbial genera and gene functions from the water and kelp surface environments driving the differences between present-day and elevated temperature. Microbial genera in the water column (a) and kelp surface (b) at elevated temperatures were 8.5% and 51.8% dissimilar compared to present day conditions by SIMPER analysis. Microbial gene functions in the water column (c) and kelp surface (d) at elevated temperatures were 1.1% and 3.3% dissimilar compared to present-day conditions.
Fig 6. Effects of pCO2 on water and kelp surface microbiomes.

Top ten microbial genera and gene functions from the water and kelp surface environments driving the differences between present-day and elevated pCO2. Microbial genera in the water column (a) and kelp surface (b) at elevated pCO2 were 23.7% and 17.0% dissimilar compared to present day conditions by SIMPER analysis. Microbial gene functions in the water column (c) and kelp surface (d) at elevated pCO2 were 1.9% and 1.3% dissimilar compared to present-day conditions.
Fig 7. Combined effects of temperature and pCO2 on water and kelp surface microbiomes.

Top ten microbial genera and gene functions from the water and kelp surface environments driving the differences between present-day and future. Microbial genera in the water column (a) and kelp surface (b) at future were 10.0% and 46.4% dissimilar compared to present day conditions by SIMPER analysis. Microbial gene functions in the water column (c) and kelp surface (d) at elevated pCO2 were 1.3% and 3.7% dissimilar compared to present-day conditions.
When comparing water communities based on the gene function composition, elevated pCO2 was the most dissimilar from the present-day at 1.9% dissimilar, followed by future at 1.3% and elevated temperature at 1.1% (Table 1B). The gene functions driving the differences in elevated temperature, elevated pCO2, and future conditions were related to a variety of categories including membrane transport, carbohydrate metabolism, virulence factors and plasmids (Fig 5C, Fig 6C, Fig 7C).
Kelp surface microbiomes were most influenced by elevated temperature and future conditions
Microbial orders in the kelp microbiome were differentially affected by changes in environmental conditions (Fig 3), while each condition showed a general decline in Alteromonadales, increase in Flavobacteriales, Rhodobacterales and Rhizobiales compared to the present day microbiome. The elevated temperature had the largest effect on the kelp microbiome composition, being 51.8% dissimilar compared with the present day microbiome, whereas elevated pCO2 was 17.0% dissimilar, and future conditions were 46.4% dissimilar from the present day microbiome (Table 1B). In elevated temperature, three genera within the order Alteromonadales declined in abundance, Alteromonas (-31.0 fold), Marinobacter (-6.7 fold) and Pseudoaltermonas (-3.7 fold), and two genera, Colwellia (3.7 fold) and Saccharophagus (3.3 fold) increased in abundance. The order Flavobacteriales increased in abundance with increased temperature, including the genera Polaribacter (9.9 fold), Flavobacterium (6.4 fold) and Gramella (4.6 fold) (Fig 5B). Elevated pCO2 caused genera within the order Alteromonadales to decline in abundance, Alteromonas (-1.6 fold), Marinobacter (-1.8 fold) and Pseudoaltermonas (-1.8 fold), and Shewanella (-1.9 fold), but the proportional decline was slight compared with the temperature treatment. The proportion of Rhodobacterales order increased (Fig 3), including increases in the genera Ruegeria (5.1 fold), Roseobacter (4.8 fold), Octadecabacter (11.5 fold) and Hyphomonas (3.1 fold) (Fig 6B). The combination of elevated pCO2 and temperature caused a major decline in the order Alteromonadales, including the genera Alteromonas (-10.3 fold), Pseudoaltermonas (-7.0 fold), Marinobacter (-2.2 fold) and Shewanella (-3.3 fold). In addition, the proportion of Rhodobacterales (27.7 fold), and Planctomycetales increased 29.2 fold in abundance in the future kelp microbiomes (Fig 7B).
The evaluation of the functional profiles of the kelp microbiome showed that the future conditions were most dissimilar to the present day conditions (3.7% dissimilar), followed by elevated temperature (3.3%) and elevated pCO2 (1.3%) (Table 1B). The gene functions driving the differences in the elevated temperature regime were a 2.0 fold decrease in Ton and Tol transport systems, 1.9 fold decrease in iron acquisition, and 4.9 fold increase in r1t-like streptococcal phages, 2.2 fold increase in alginate metabolism, and 4.4 fold increase in sulfatases (Fig 5D). For elevated pCO2, Ton and Tol transport systems and iron acquisition were reduced, while a 2.8 fold and 6.0 fold increase in respiratory complex I and CO dehydrogenase contributed to functional differences (Fig 6D). The enrichment in CO dehydrogenase genes associated with increased CO2 on the kelp surface (Figs 6D and 7D), primarily belonged to the Ruegeria and Roseobacter genera which were also elevated at the taxonomic level (Figs 6B and 7B). In the future conditions incubation mesocosms, Ton and Tol transport systems and iron acquisition were both reduced 2.7 and 2.5 fold while respiratory complex I and CO dehydrogenase were enriched 4.4 and 8.8 fold (Fig 7D). The Alteromonadales order contains the majority of the genes (mean 57%) annotated to Ton and Tol transport system with the TonB-dependent receptor being the most prominent gene (mean 50%). Alginate metabolism genes included alginate lyases (mean 47%), phosphomannomutase (mean 16%), mannose-6-phosphate isomerase (7%), poly(beta-D-mannuronate) lyase (15%), and poly(beta-D-mannuronate) O-acetylase (8%) suggesting the primary polysaccharides being alginate and mannose. During elevated temperature, the alginate lyase genes were enriched compared to all other metagenomes (mean 63% vs. 40%) while the phosphomannomutase genes were lower (mean 9% vs. 19%). Furthermore, the bacteria associated with alginate lyase genes on the kelp surface were primarly Pseudoalteromonas (mean 23%) and Saccharophagus (mean 26%) with Saccharophagus becoming enriched in the elevated temperature to (38%).
Discussion
Macroalgae development, growth and decay are facilitated by bacteria and bacterial physiology is tied to surrounding abiotic conditions, such as temperature, pH and nutrient availability. Our case study experiment tested the independent and combined effects of elevated temperature and pCO2 on the microbiomes of the M. pyrifera kelp surface and surrounding water column. Microbiomes of the water column and kelp surface were unique, and were affected differently by global climate change stressors. The water column microbiome was altered most by increased pCO2, while the kelp surface microbiome was altered most by increased temperature and future conditions. The disturbances of the kelp microbiome in elevated temperature correlated with decreased kelp growth, while the future condition microbiome associated with prolific kelp growth. Combined effects of future conditions were significant on the kelp surface microbiome indicating the importance of multifactorial experimental designs addressing multiple variables to evaluate ecological consequences of climate change stressors on host organisms. Although our overall replication of study conditions were limited due to technical constraints, our intra-tank replicates were consistent and provide a foundation for future targeted studies both in situ and in the lab.
Climate stressors induced changes to water microbiomes
The water microbiome showed a distinct increase in single bacterial orders with each climate change condition with elevated pCO2 being the most influential. These microbiomes had the highest alpha diversity and a 7.3 fold increase in Rhizobiales. The Rhizobiales order assimilates dissolved inorganic carbon (e.g. CO2) in coastal environments [38] and could be utilizing excess pCO2 alongside kelp. In addition, species within the Rhizobiales order have important roles in both denitrification [39] along with nitrogen fixation in soil, and are attached to organic particles in the deep ocean and degrade xenobiotic and refractory compound [40,41]. The functional analysis identified an increase in the proportion of sequences similar to denitrification and 4-hydroxphenylacetic acid catabolic pathways which can stimulate algal growth [42]. An increase in denitrification may be a response to the increase in some Rhizobiales and nitrogen fixation. Nitrogen fixation on M. pyrifera surfaces is up to ten times higher than other macroalgae and is associated with decomposition [43]. An alternative hypothesis to increased denitrification would be explained by an increase in overall NO3 and NO2 availability in the ecosystem which also could be a result of bacterial metabolism.
M. pyrifera microbiome was dominated by Alteromonadales and unique functions
The Alteromonadales order was a major component of the M. pyrifera microbial community, similar to in situ sampling of kelp at Pt Loma (S3 and S4 Figs), and declined in abundance in the altered climate condition treatments. Alteromonadales, while important in the Point Loma kelp forest, were not described in the M. pyrifera microbiome from Monterey Bay [44] indicating that the frond surface microbiome may vary regionally or alternatively may be attributed to the difference in metagenomic sampling and sequencing methods. Within the Alteromonadales order, Alteromonas, Cytophaga, Pseudomonas and Pseudoalteromonas genera contain enzymes like agarases, alginate lyases, cellulases, pectinases, and fucoidanases which catabolize kelp polysaccharides [45] with some causing disease in other species of brown algae [46,47]. However, in this study Alteromonadales were enriched on the kelp surfaces during the present day treatment where the kelp was thriving, and was reduced by 51.0% in the elevated temperature treatment where the kelp showed reduced growth. Future work should focus on the strain ecology and metabolic functions of Alteromonadales microbes throughout the life history, geographic range, and seasonality of M. pyrifera. The kelp surface microbiome was enriched with genes related to nutrient acquisition, motility, and nitrogen metabolism. Genes related to Ton and Tol transport systems, which are important for gram negative marine bacteria to obtain extracellular dissolved organic matter, vitamins and iron from the environment [48,49]. Nitrate and nitrite ammonification related genes were enriched on the kelp surface microbiome suggesting a possible mutualistic mechanism for nitrogen cycling between kelp and bacteria. Other microbes on the kelp surface had an enrichment of genes related to environmental sensing and motility, including bacterial chemotaxis and flagellum, which may be associated with biofilm formation [50] or macroalgae colonization [51]. Furthermore, although we were able to identify candidate gene families associated with algae degradation across all treatments, future studies should strive to link transcriptional or protein profiles of the bacteria to demonstrate functional activity.
Increased temperature and an altered microbiome reduced kelp growth
Host macroalgae susceptibility to bacterial pathogenesis is increased during elevated temperature regimes and is related to reduced chemical defense responses [5,19]. We found that elevated temperature was associated with a unique shift in the kelp surface microbiome which corresponded with reduced M. pyrifera growth.The kelp surface microbiome showed a loss of Alteromondales and increase in Flavobacterales indicating that Flavobacterales may be negatively associated with kelp growth. Flavobacteriales strains, including Flavobacterium sp. LAD-1 [52], Kordia algicida OT-1 [53], Gramella forsetii KT0803 [54], and Cellulophaga lytica DMS 7489 [55] are algicidal bacteria which contribute to disease in red and brown macroalgae. Similarly, our study identified Polaribacter, Flavobacterium, and Gramella enriched on the kelp surface in the elevated temperature condition and these genera have alginate lyase enzymes which assist in degrading brown algae [55–58]. The functional analysis corroborated the taxonomic change with an increase in alginate metabolism and sulfatases. An increase in Colwellia and Saccharophagus genera, which contain enzymes that degrade algae polysaccharides [55,59] occurred at higher temperature. Saccharophagus degradans 2–40, for example, has numerous enzymes that degrade marine plant cellulose [60] and five agarase enzymes [61]. Similarly in the brown macroalgae, Fucus vesiculosus, increased temperature was associated with an increase in Flavobacteriales and reduced kelp growth, while CO2 had no effect [62], suggesting a common mechanism across brown macroalgae.
Kelp grew prolifically in future conditions with marked changes in the microbiome
Elevated temperature combined with elevated pCO2 were surprisingly associated with increased M. pyrifera growth [23] and were related to increased microbial diversity. Microbes within the Planctomycetales and Rhodobacterales orders were both enriched on the kelp surface, similar to other studies of microbes associated with macroalgae (Lachnit et al., 2011; Friedrich 2012). Planctomycetales, produce sulfatases enabling catabolism of sulfated polysaccharides and are associated with a variety of hosts, including, marine sponges, corals, ascidians, and macroalgae biofilms [63–65]. Sulfatase genes were increased in the kelp microbiome under future conditions and were primarily comprised of aryl sulfatase, choline-sulfatase, sulfatase, and sulfatase modifying factor 1 precursor all of which were present in the Planctomycetales microbes. Two Planctomycetales genera, Planctomyces and Rhodopirellula, enriched on the kelp surface, are macroalgae symbionts with resistance to antimicrobial compounds produced by macroalgae [66]. A strain Rhodopirellula baltica SH1, contains 110 different sulfatase genes [64]. Within the Rhodobacterales, the Roseobacter genera were enriched on the kelp surface under future conditions. One species, Dinorosebacter shibae DFL12, synthesizes B1 and B12 vitamins for the algae host [67]. We suggest that in future conditions, a novel symbiotic relationship where Planctomycetales and Rhodobacterales are able to thrive on the kelp surface utilizing M. pyrifera sulfated polysaccharides as an energy source, while resisting M. pyrifera induced antimicrobial barrage and outcompeting deleterious Flavobacteriales that were enriched in the increased temperature treatment. In the elevated CO2 tanks, there was an increase of carbon monoxide dehydrogenase, which is a bidirectional catalyst of carbon dioxide, which suggests the response of the kelp microbiome to presence of increased CO2.
Kelp forests located in lower latitudes, like southern California, where upper temperature tolerances (above 18 oC) are reached three to four months out of the year, may be at risk for shorter life cycles, increased seasonal senescence, and increased disease prevalence in an ocean with rising sea surface temperatures unless increasing CO2 is sufficient to mitigate this effect. Here we demonstrate that the water and kelp surface microbiomes respond differently to climate stressors. Water microbiomes were most influenced by increased pCO2 conditions and primarily associated with an increase in Rhizobiales. Kelp surface microbiomes on the other hand, dominated by Alteromonadales, were perturbed by increased temperature, corresponding with increased Flavobacterales, alginate metabolism, and kelp degradation. While kelp microbiomes were largely not disturbed by increases in pCO2, perturbations in the water column microbes due to pCO2 could have an impact in kelp communities by changing the available kelp surface colonizers. The results from this initial case study are preliminary observations which need to be followed up with more highly replicated mesocosms or in situ experiments to verify the findings. Alternatively, environmental sampling across seasonal variances of temperature and pCO2 could also be valuable for understanding the microbial influence in this kelp forest ecosystem. Future conditions increased kelp growth and carbon monoxide dehydrogenase corresponding with increased Planctomycetales, Rhodobacterales and sulfatase genes. Our study demonstrates that kelp growth, as a result of changing climate conditions, is directly tied to the host microbiome.
Materials and methods
Macrocystis pyrifera fronds were collected from 72 randomly selected sporophytes from the Point Loma kelp forest, San Diego, California, USA (32°39'59.56"N, 117°14'50.62"W) at 12–15 m deep in August 2012. Fronds containing meristems (shoots or stems) and laminae (blades) were immediately placed in a cooler filled with seawater and transported to San Diego State Coastal Marine Institute Laboratory Point Loma, CA. M. pyrifera was collected in accordance with U.S. Fisheries and Wildlife Service Permit #SC-13075. At the lab the fronds were trimmed to 7 g and placed in one of four 90 L acrylic incubation mesocosms (n = 18 fronds per mesocosm), each containing recirculating seawater collected from the kelp forest. For four weeks, temperature and pCO2 levels in each mesocosm were maintained at the following conditions: “present day” at 12 oC and 500 pCO2, “elevated temperature” at 15 oC and 500 ppm pCO2, “elevated pCO2” at 12 oC and 1300 ppm pCO2, and “future” at 15 oC and 1300 ppm pCO2 to reflect predicted temperature and CO2 increases in nearshore temperate marine environments over the next 100 years [17,68]. Four weeks was selected to provide sufficient time for the kelp to grow and the microbes to change. A 12 hour—light/dark cycle was maintained at 15–20 μmol photons m-2 s-1 irradiance to simulate light levels experienced in the kelp forest while nutrient levels were maintained by supplemental addition of Proline algae fertilizer to ensure nitrate levels remained above 1–2 uM (Brown, 2014). Each of the 18 pseudo-replicate kelp fronds were resampled from the incubation mesocosms each week, and weighed to monitor growth, while the microbiomes on the kelp fronds and in the water column were collected at the end of the experiment. Measurements of pCO2 were taken on a daily basis.
At the end of four weeks, replicate microbiomes were collected from the seawater and the kelp frond surfaces incubated in each mesocosm. To sample the water column microbiome, 45 liters of seawater were passed through a 100 μm Nitex filter to exclude zooplankton, larger phytoplankton and organic debris, and then concentrated to 500 mL using tangential flow filtration (TFF) (100 kDa pore, GE Healthcare Life Sciences, Pittsburg, PA). The concentrate was passed through three replicate 0.22 μm Sterivex filters to collect the microbial fraction, (including Bacteria, Archaea, and micro-eukaryotes) [69]. Sterivex filters were sealed with parafilm and stored at -20°C until DNA extraction. Microbes were collected from kelp frond surfaces using a closed circuit two-way 50 mL syringe referred to as a ‘supersucker’ [70]. While pressing the close circuit syringe against the kelp fronds, sterile, 0.02 μm filtered seawater was passed over the surface, dislodging surface microbes from the algal surface and re-circulating the microbes laden water back into the syringe. The microbes and water from the kelp was dispensed through a 0.22 μm Sterivex. The sterivex was sealed with parafilm and immediately stored at -20°C for transport back to a -80°C freezer at SDSU. The sampling process was repeated on three kelp fronds per incubation treatment.
The gDNA was extracted from the Sterivex filters using a modified Macherey Nagel column protocol as described previously [37]. To remove RNA from the eluent, samples were incubated at 37 oC for one hour with RNase A and reprocessed through a DNeasy blood extraction kit (Qiagen, Valencia, CA). Samples were quantified using a Qubit (Thermofisher, Waltham, WA). For kelp surface biological replicates, the gDNA content was low for 9 of the 12 samples (less than 100 ng), thus samples from the two lowest gDNA replicates were pooled into one for each mesocosm (elevated temperature, elevated CO2, and future conditions). All three replicates had to be pooled for the present-day mesocosm, to meet requirements for library preparation.
A total of 19 microbiomes, whole genome sequencing, samples from the water column (n = 12) and kelp surfaces (n = 7) of the four incubation mesocosms were prepared into libraries using 100 ng of gDNA (Illumina PCR Nano prep kit) and quantified using the KAPA qPCR (Kapa Biosystems, Wilmington, MA) and Bioanalyzer (Agilent, Santa Clara, CA) on the high sensitivity assays. Nextera was not chosen due to known biases in fragmentation. Libraries were sequenced over two MiSeq runs using the Illumina V3 2 x 300 sequencing kits (S1 Table). All FASTQ files generated were quality filtered using PRINSEQ [71] and MG-RAST [72] to remove artificial replicate [73], while low quality sequence reads were trimmed to contain less than 5 bases of a Q-score ≤ 15 [74]. The metagenome-microbiomes were uploaded to MG-RAST version 3.5 [75]. Annotations of microbial organisms were generated in MG-RAST at the order and genera level using the Representative Hit Classification setting (e-value = 1 x 10−5, minimum percent identity cutoff of 65%, and minimum alignment length cutoff of 50 bp) against the M5 non-redundant database. To identify gene functions associated with the microbes, relative abundances of protein encoded gene annotations were generated at the SEED functional level 3 or gene pathways [69], using the Hierarchical Classification setting with cut-off described above. Rarefaction curves for each microbiome were generated based on ORF detection. Species richness of taxonomy was calculated at the genera level, whereby means of water column replicates were compared using One Way ANOVA followed by a Tukey HSD test with significance of P < 0.05. Kelp microbiome species richness was compared qualitatively.
When conducting a homology based approach to obtain relative abundances of organisms or functions of the microbes from DNA sequences, there is lower confidence in the identification of taxa in the rare tail of the distribution. One of the reasons for this is that metagenomics experiments rarely sample all of the microbes in a given sample, thus one will always have to deal with subsampling error. Although methods exist to quantify this including rarefaction curves and Good’s coverage of singletons [76], there can also be other sources of error including contamination events during sample processing [77] and bioinformatic errors caused by sequencing or analysis with incomplete databases [78]. Here, we quantitatively determined the lower cutoff for our analysis of the genera and functions, and apply this cutoff to the downstream analyses. Consequently, the analysis was conducted on 2,693 genera and 1,007 gene functions across the 12 microbiomes from the water column samples. We then applied these cutoff values to the kelp associated microbiomes since we did not have the added replicates for calculating CV values. First, the relative abundances of each 2,693 genera or 1,007 gene functions were calculated (abundance of category) / total reads x 100%) and binned into seven abundance bins (S2 Table). Second, for each genera and gene function that was identified, the mean, standard deviation, and coefficient of variation (CV) were calculated across the three replicates from each water column treatment. Within each bin, the mean coefficient of variation and standard error for each treatment was calculated and plotted (S1 Fig). The graphs show an elbow formation when the mean CV falls below 20% at the relative abundance bin of 0.01 suggesting an appropriate cutoff level. Thus if a microbiome has 1,000,000 reads passing QC, the acceptable minimum reads of a genera or function category would be 100 reads (total hits/100 X correction factor or 1,000,000 /100 x 0.01 = 100 hits). The calculation was applied and truncated the genera and gene function categories across all microbiomes to 361 genera and 718 gene functions. The same cut-off was applied to the kelp microbiomes.
To identify whether perturbations in abiotic climate conditions affected the structure of the microbiomes, the relative abundances of reads in the 361 genera and 718 gene functions were square root transformed and compared among treatments using hierarchical cluster analyses and non-metric multidimensional scaling (nMDS) based on Euclidean distances in the PRIMER V6 software (PRIMER-E Ltd., Devon, UK) [79]. The experiment had two microbiome environments, water or kelp surface, and each of these were exposed to the four environmental, abiotic conditions: present-day, elevated temperature, elevated pCO2, and future conditions. To determine if these factors independently or synergistically influenced the microbiomes, the genera and gene function read abundances were independently compared using a two factor permutational multivariate analysis of variance (PERMANOVA) based on Euclidean Distance similarities [80,81] and the relative importance of each factor was determined by calculating their magnitude of effects using variance components analyses (Graham and Edwards, 2001).
To evaluate whether abiotic factors altered the microbiomes in the water and kelp surface environments at a finer scale, microbiomes were each compared to the present-day condition using a SIMPER analysis [79]. The SIMPER analyses were used to quantitatively determine how dissimilar treatment microbiomes were to the present-day control. In addition, the SIMPER analysis was used to determine the relative importance of each of the 361 genera and 718 gene functions to the overall microbiome group dissimilarity between the environmental types and abiotic stressors. All level 3 gene functions from the Cluster Based Subsystem reported from the SIMPER analysis were described based on the more specific gene function ontological classification (level 4). To describe the effects of a treatment (elevated temperature, CO2, or combined) within an environment (water or kelp), genera and gene function feature tables were compared to present-day condition using STAMP [82]. Pairwise comparisons were made using the Two-sided, white’s non parametric t-test [83], with confidence intervals of 95% and a 5% FDR with Benjamini-Hochberg multiple test correction [84].
Supporting information
The coefficient of variance as a function of relative abundances of (a) genera and (b) gene function Level 3 categories in three biological replicates across four environmental conditions (present-day ○, elevated temperature □, elevated pCO2 △, and future ◇) indicates a minimum cutoff of 0.01% relative abundance.
(TIFF)
Rarefaction curve of (a) water column and (b) kelp surface microbiomes across the four environmental conditions (present-day ‘black’ ○, elevated temperature ‘red’ □, elevated pCO2 ‘green’△, and future ‘purple’ ◇. Diversity index was calculated based off of open reading frames, protein annotations from MG-RAST.
(TIFF)
(TIF)
(TIF)
(DOCX)
(DOCX)
Acknowledgments
We would like to thank the support of the National Science Foundation, with Dinsdale being supported by grants from the Division of Undergraduate Education #1323809 and the Division of Molecular and Cellular Biology # 1330800. At the time of the research, Todd P Michael was an employee at Ibis Biosciences, an Abbott Laboratories company, and he was involved as a committee member on Minich’s Masters Thesis committee and contributed intellectually to the manuscript by interpretation of the results. This commercial affiliation does not alter our adherence to all PLOS ONE policies on sharing data and materials. The funders had no role in the study design, data collection, analysis, decision to publish, or preparation of the manuscript.
Data Availability
All SRA data is available here: https://www.ncbi.nlm.nih.gov/sra/SRP132751 SRA accession SRP132751.
Funding Statement
The authors would like to thank the support of the National Science Foundation, with Elizabeth Dinsdale being supported by grants from the Division of Undergraduate Education #1323809 and the Division of Molecular and Cellular Biology # 1330800. The funders had no role in the study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Dayton PK. Ecology of Kelp Communities. Annu Rev Ecol Syst. 1985;16: 215–245. [Google Scholar]
- 2.Fredriksen S. Food web studies in a Norwegian kelp forest based on stable isotope (δ13C and δ15N) analysis. Mar Ecol Prog Ser. 2003;260: 71–81. doi: 10.3354/meps260071 [Google Scholar]
- 3.Mazure HGF, Field JG. Density and ecological importance of bacteria on kelp fronds in an upwelling region. J Exp Mar Biol Ecol. 1980;43: 173–182. doi: 10.1016/0022-0981(80)90024-6 [Google Scholar]
- 4.Steneck RS, Graham MH, Bourque BJ, Corbett D, Erlandson JM, Estes JA, et al. Kelp forest ecosystems: biodiversity, stability, resilience and future. Environ Conserv. 2002;29: 436–459. doi: 10.1017/S0376892902000322 [Google Scholar]
- 5.Campbell AH, Harder T, Nielsen S, Kjelleberg S, Steinberg PD. Climate change and disease: bleaching of a chemically defended seaweed. Glob Change Biol. 2011;17: 2958–2970. doi: 10.1111/j.1365-2486.2011.02456.x [Google Scholar]
- 6.Lachnit T, Meske D, Wahl M, Harder T, Schmitz R. Epibacterial community patterns on marine macroalgae are host-specific but temporally variable. Environ Microbiol. 2011;13: 655–665. doi: 10.1111/j.1462-2920.2010.02371.x [DOI] [PubMed] [Google Scholar]
- 7.Gill SR, Pop M, DeBoy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic Analysis of the Human Distal Gut Microbiome. Science. 2006;312: 1355–1359. doi: 10.1126/science.1124234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sender R, Fuchs S, Milo R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell. 2016;164: 337–340. doi: 10.1016/j.cell.2016.01.013 [DOI] [PubMed] [Google Scholar]
- 9.Singh RP, Reddy CRK. Unraveling the Functions of the Macroalgal Microbiome. Front Microbiol. 2016;6 doi: 10.3389/fmicb.2015.01488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Croft MT, Lawrence AD, Raux-Deery E, Warren MJ, Smith AG. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature. 2005;438: 90–93. doi: 10.1038/nature04056 [DOI] [PubMed] [Google Scholar]
- 11.Kanagasabhapathy M, Sasaki H, Haldar S, Yamasaki S, Nagata S. Antibacterial activities of marine epibiotic bacteria isolated from brown algae of Japan. Ann Microbiol. 2006;56: 167–173. doi: 10.1007/BF03175000 [Google Scholar]
- 12.Keshtacher-Liebso E, Hadar Y, Chen Y. Oligotrophic Bacteria Enhance Algal Growth under Iron-Deficient Conditions. Appl Environ Microbiol. 1995;61: 2439–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christian N, Whitaker BK, Clay K. Microbiomes: unifying animal and plant systems through the lens of community ecology theory. Front Microbiol. 2015;6 doi: 10.3389/fmicb.2015.00869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut. 2004;53: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caldeira K, Wickett ME. Ocean model predictions of chemistry changes from carbon dioxide emissions to the atmosphere and ocean. J Geophys Res Oceans. 2005;110: C09S04 doi: 10.1029/2004JC002671 [Google Scholar]
- 16.Orr JC, Fabry VJ, Aumont O, Bopp L, Doney SC, Feely RA, et al. Anthropogenic ocean acidification over the twenty-first century and its impact on calcifying organisms. Nature. 2005;437: 681–686. doi: 10.1038/nature04095 [DOI] [PubMed] [Google Scholar]
- 17.Pachauri RK, Allen MR, Barros VR, Broome J, Cramer W, Christ R, et al. Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change [Internet]. Pachauri RK, Meyer L, editors. Geneva, Switzerland: IPCC; 2014. Available: http://epic.awi.de/37530/
- 18.Axelsson L, Mercado J, Figueroa F. Utilization of HCO3 − at high pH by the brown macroalga Laminaria saccharina. Eur J Phycol. 2000;35: 53–59. doi: 10.1080/09670260010001735621 [Google Scholar]
- 19.Harley CDG, Anderson KM, Demes KW, Jorve JP, Kordas RL, Coyle TA, et al. Effects of Climate Change on Global Seaweed Communities. J Phycol. 2012;48: 1064–1078. doi: 10.1111/j.1529-8817.2012.01224.x [DOI] [PubMed] [Google Scholar]
- 20.Koch M, Bowes G, Ross C, Zhang X-H. Climate change and ocean acidification effects on seagrasses and marine macroalgae. Glob Change Biol. 2013;19: 103–132. doi: 10.1111/j.1365-2486.2012.02791.x [DOI] [PubMed] [Google Scholar]
- 21.Gaitán-Espitia JD, Hancock JR, Padilla-Gamiño JL, Rivest EB, Blanchette CA, Reed DC, et al. Interactive effects of elevated temperature and pCO2 on early-life-history stages of the giant kelp Macrocystis pyrifera. J Exp Mar Biol Ecol. 2014;457: 51–58. doi: 10.1016/j.jembe.2014.03.018 [Google Scholar]
- 22.Roleda MY, Morris JN, McGraw CM, Hurd CL. Ocean acidification and seaweed reproduction: increased CO2 ameliorates the negative effect of lowered pH on meiospore germination in the giant kelp Macrocystis pyrifera (Laminariales, Phaeophyceae). Glob Change Biol. 2012;18: 854–864. doi: 10.1111/j.1365-2486.2011.02594.x [Google Scholar]
- 23.Brown MB. The effects of rising ocean temperature and pco2 on the physiology and growth of giant kelp, macrocystis pyrifera, and grazing by purple urchins, strongylocentrotus purpuratus [Internet]. M.S., San Diego State University. 2013. Available: http://search.proquest.com/docview/1497967254/abstract
- 24.Subiabre F, Andrea P. The effects of ocean acidification on photosynthesis, growth, and carbon and nitrogen metabolism of Macrocystis pyrifera [Internet]. Thesis, University of Otago. 2016. Available: https://ourarchive.otago.ac.nz/handle/10523/6227
- 25.Hay CH. The distribution of Macrocystis (Phaeophyta: Laminariales) as a biological indicator of cool sea surface temperature, with special reference to New Zealand waters. J R Soc N Z. 1990;20: 313–336. doi: 10.1080/03036758.1990.10426716 [Google Scholar]
- 26.Rothäusler E, Gómez I, Karsten U, Tala F, Thiel M. Physiological acclimation of floating Macrocystis pyrifera to temperature and irradiance ensures long-term persistence at the sea surface at mid-latitudes. J Exp Mar Biol Ecol. 2011;405: 33–41. doi: 10.1016/j.jembe.2011.05.018 [Google Scholar]
- 27.Schiel DR, Foster MS. The Biology and Ecology of Giant Kelp Forests. Univ of California Press; 2015. [Google Scholar]
- 28.Liu F, Pang SJ. Performances of growth, photochemical efficiency, and stress tolerance of young sporophytes from seven populations of Saccharina japonica (Phaeophyta) under short-term heat stress. J Appl Phycol. 2009;22: 221–229. doi: 10.1007/s10811-009-9445-6 [Google Scholar]
- 29.Fain SR, Murray SN. Effects of Light and Temperature on Net Photosynthesis and Dark Respiration of Gametophytes and Embryonic Sporophytes of Macrocystis Pyrifera1. J Phycol. 1982;18: 92–98. doi: 10.1111/j.1529-8817.1982.tb03161.x [Google Scholar]
- 30.Buschmann AH, Vásquez JA, Osorio P, Reyes E, Filún L, Hernández-González MC, et al. The effect of water movement, temperature and salinity on abundance and reproductive patterns of Macrocystis spp. (Phaeophyta) at different latitudes in Chile. Mar Biol. 2004;145: 849–862. doi: 10.1007/s00227-004-1393-8 [Google Scholar]
- 31.Brown MB, Edwards MS, Kim KY. Effects of climate change on the physiology of giant kelp, Macrocystis pyrifera, and grazing by purple urchin, Strongylocentrotus purpuratus. ALGAE. 2014;29: 203–215. doi: 10.4490/algae.2014.29.3.203 [Google Scholar]
- 32.Behera S, Singh R, Arora R, Sharma NK, Shukla M, Kumar S. Scope of Algae as Third Generation Biofuels. Front Bioeng Biotechnol. 2015;2 doi: 10.3389/fbioe.2014.00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graham MH, Vásquez JA, Buschmann AH. Global ecology of the giant kelp Macrocystis: from ecotypes to ecosystems. Oceanogr Mar Biol Annu Rev. 2007; Available: http://www.vliz.be/en/imis?refid=111466 [Google Scholar]
- 34.Sarmento H, Montoya JM, Vázquez-Domínguez E, Vaqué D, Gasol JM. Warming effects on marine microbial food web processes: how far can we go when it comes to predictions? Philos Trans R Soc B Biol Sci. 2010;365: 2137–2149. doi: 10.1098/rstb.2010.0045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lomas MW, Glibert PM, Shiah F-K, Smith EM. Microbial processes and temperature in Chesapeake Bay: current relationships and potential impacts of regional warming. Glob Change Biol. 2002;8: 51–70. doi: 10.1046/j.1365-2486.2002.00454.x [Google Scholar]
- 36.Raulf FF, Fabricius K, Uthicke S, de Beer D, Abed RMM, Ramette A. Changes in microbial communities in coastal sediments along natural CO2 gradients at a volcanic vent in Papua New Guinea. Environ Microbiol. 2015; n/a-n/a. doi: 10.1111/1462-2920.12729 [DOI] [PubMed] [Google Scholar]
- 37.Minich J, Dinsdale EA. Experimental Metagenomics: Influence of Pulses of Carbon Dioxide on Kelp Forest Microbial Ecology In: Nelson KE, editor. Encyclopedia of Metagenomics. Springer New York; 2013. pp. 1–8. Available: http://link.springer.com/referenceworkentry/10.1007/978-1-4614-6418-1_137-3 [Google Scholar]
- 38.DeLorenzo S, Bräuer SL, Edgmont CA, Herfort L, Tebo BM, Zuber P. Ubiquitous Dissolved Inorganic Carbon Assimilation by Marine Bacteria in the Pacific Northwest Coastal Ocean as Determined by Stable Isotope Probing. PLOS ONE. 2012;7: e46695 doi: 10.1371/journal.pone.0046695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Falk S, Liu B, Braker G. Isolation, genetic and functional characterization of novel soil nirK-type denitrifiers. Syst Appl Microbiol. 2010;33: 337–347. doi: 10.1016/j.syapm.2010.06.004 [DOI] [PubMed] [Google Scholar]
- 40.Eloe EA, Shulse CN, Fadrosh DW, Williamson SJ, Allen EE, Bartlett DH. Compositional differences in particle-associated and free-living microbial assemblages from an extreme deep-ocean environment. Environ Microbiol Rep. 2011;3: 449–458. doi: 10.1111/j.1758-2229.2010.00223.x [DOI] [PubMed] [Google Scholar]
- 41.Martín-Cuadrado A-B, López-García P, Alba J-C, Moreira D, Monticelli L, Strittmatter A, et al. Metagenomics of the deep Mediterranean, a warm bathypelagic habitat. PloS One. 2007;2: e914 doi: 10.1371/journal.pone.0000914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu X, Chen L, Zhang W. Chemicals to enhance microalgal growth and accumulation of high-value bioproducts. Front Microbiol. 2015;6 doi: 10.3389/fmicb.2015.00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamersley MR, Sohm JA, Burns JA, Capone DG. Nitrogen fixation associated with the decomposition of the giant kelp Macrocystis pyrifera. Aquat Bot. 2015;125: 57–63. doi: 10.1016/j.aquabot.2015.05.003 [Google Scholar]
- 44.Michelou VK, Caporaso JG, Knight R, Palumbi SR. The Ecology of Microbial Communities Associated with Macrocystis pyrifera. PLoS ONE. 2013;8: e67480 doi: 10.1371/journal.pone.0067480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goecke F, Labes A, Wiese J, Imhoff J. Chemical interactions between marine macroalgae and bacteria. Mar Ecol Prog Ser. 2010;409: 267–299. doi: 10.3354/meps08607 [Google Scholar]
- 46.Sawabe T, Makino H, Tatsumi M, Nakano K, Tajima K, Iqbal MM, et al. Pseudoalteromonas bacteriolytica sp. nov., a marine bacterium that is the causative agent of red spot disease of Laminaria japonica. Int J Syst Bacteriol. 1998;48: 769–774. doi: 10.1099/00207713-48-3-769 [DOI] [PubMed] [Google Scholar]
- 47.Vairappan CS, Suzuki M, Motomura T, Ichimura T. Pathogenic bacteria associated with lesions and thallus bleaching symptoms in the Japanese kelp Laminaria religiosa Miyabe (Laminariales, Phaeophyceae). Hydrobiologia. 2001;445: 183–191. doi: 10.1023/A:1017517832302 [Google Scholar]
- 48.Danese I, Haine V, Delrue R-M, Tibor A, Lestrate P, Stevaux O, et al. The Ton System, an ABC Transporter, and a Universally Conserved GTPase Are Involved in Iron Utilization by Brucella melitensis 16M. Infect Immun. 2004;72: 5783–5790. doi: 10.1128/IAI.72.10.5783-5790.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang K, Jiao N, Liu K, Zhang Y, Li S. Distribution and Functions of TonB-Dependent Transporters in Marine Bacteria and Environments: Implications for Dissolved Organic Matter Utilization. PLoS ONE. 2012;7: e41204 doi: 10.1371/journal.pone.0041204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol. 1998;30: 295–304. doi: 10.1046/j.1365-2958.1998.01062.x [DOI] [PubMed] [Google Scholar]
- 51.Burke C, Steinberg P, Rusch D, Kjelleberg S, Thomas T. Bacterial community assembly based on functional genes rather than species. Proc Natl Acad Sci. 2011;108: 14288–14293. doi: 10.1073/pnas.1101591108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sunairi M, Tsuchiya H, Tsuchiya T, Omura Y, Koyanagi Y, Ozawa M, et al. Isolation of a bacterium that causes anaaki disease of the red algae Porphyra yezoensis. J Appl Bacteriol. 1995;79: 225–229. doi: 10.1111/j.1365-2672.1995.tb00939.x [Google Scholar]
- 53.Lee HS, Kang SG, Kwon KK, Lee J-H, Kim S-J. Genome Sequence of the Algicidal Bacterium Kordia algicida OT-1. J Bacteriol. 2011; JB.05241-11 doi: 10.1128/JB.05241-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kabisch A, Otto A, König S, Becher D, Albrecht D, Schüler M, et al. Functional characterization of polysaccharide utilization loci in the marine Bacteroidetes “Gramella forsetii” KT0803. ISME J. 2014;8: 1492–1502. doi: 10.1038/ismej.2014.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martin M, Barbeyron T, Martin R, Portetelle D, Michel G, Vandenbol M. The Cultivable Surface Microbiota of the Brown Alga Ascophyllum nodosum is Enriched in Macroalgal-Polysaccharide-Degrading Bacteria. Front Microbiol. 2015;6 doi: 10.3389/fmicb.2015.01487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Descamps V, Colin S, Lahaye M, Jam M, Richard C, Potin P, et al. Isolation and Culture of a Marine Bacterium Degrading the Sulfated Fucans from Marine Brown Algae. Mar Biotechnol. 2006;8: 27–39. doi: 10.1007/s10126-005-5107-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong S, Yang J, Zhang X-Y, Shi M, Song X-Y, Chen X-L, et al. Cultivable Alginate Lyase-Excreting Bacteria Associated with the Arctic Brown Alga Laminaria. Mar Drugs. 2012;10: 2481–2491. doi: 10.3390/md10112481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uchida M, Nakayama A, Abe S. Distribution and Characterization of Bacteria Capable of Decomposing Brown Algae Fronds in Waters Associated with Laminaria Vegetation. Fish Sci. 1995;61: 117–120. doi: 10.2331/fishsci.61.117 [Google Scholar]
- 59.Kim HT, Chung JH, Wang D, Lee J, Woo HC, Choi I-G, et al. Depolymerization of alginate into a monomeric sugar acid using Alg17C, an exo-oligoalginate lyase cloned from Saccharophagus degradans 2–40. Appl Microbiol Biotechnol. 2012;93: 2233–2239. doi: 10.1007/s00253-012-3882-x [DOI] [PubMed] [Google Scholar]
- 60.Taylor LE, Henrissat B, Coutinho PM, Ekborg NA, Hutcheson SW, Weiner RM. Complete Cellulase System in the Marine Bacterium Saccharophagus degradans Strain 2-40T. J Bacteriol. 2006;188: 3849–3861. doi: 10.1128/JB.01348-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ekborg NA, Taylor LE, Longmire AG, Henrissat B, Weiner RM, Hutcheson SW. Genomic and Proteomic Analyses of the Agarolytic System Expressed by Saccharophagus degradans 2–40. Appl Environ Microbiol. 2006;72: 3396–3405. doi: 10.1128/AEM.72.5.3396-3405.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mensch B, Neulinger SC, Graiff A, Pansch A, Künzel S, Fischer MA, et al. Restructuring of Epibacterial Communities on Fucus vesiculosus forma mytili in Response to Elevated pCO2 and Increased Temperature Levels. Front Microbiol. 2016;7 doi: 10.3389/fmicb.2016.00434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bengtsson MM, Øvreås L. Planctomycetes dominate biofilms on surfaces of the kelp Laminaria hyperborea. BMC Microbiol. 2010;10: 261 doi: 10.1186/1471-2180-10-261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Glöckner FO, Kube M, Bauer M, Teeling H, Lombardot T, Ludwig W, et al. Complete genome sequence of the marine planctomycete Pirellula sp. strain 1. Proc Natl Acad Sci. 2003;100: 8298–8303. doi: 10.1073/pnas.1431443100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lage OM, Bondoso J. Planctomycetes and macroalgae, a striking association. Front Microbiol. 2014;5 doi: 10.3389/fmicb.2014.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lage OM, Bondoso J. Planctomycetes diversity associated with macroalgae. FEMS Microbiol Ecol. 2011;78: 366–375. doi: 10.1111/j.1574-6941.2011.01168.x [DOI] [PubMed] [Google Scholar]
- 67.Wagner-Döbler I, Ballhausen B, Berger M, Brinkhoff T, Buchholz I, Bunk B, et al. The complete genome sequence of the algal symbiont Dinoroseobacter shibae: a hitchhiker’s guide to life in the sea. ISME J. 2009;4: 61–77. doi: 10.1038/ismej.2009.94 [DOI] [PubMed] [Google Scholar]
- 68.Solomon S, Qin D, Manning M, Marquis M, Averyt K, others. Technical Summary in Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, 2007 24 National Research Council. Am Clim Choices. 2011; [Google Scholar]
- 69.Dinsdale EA, Edwards RA, Hall D, Angly F, Breitbart M, Brulc JM, et al. Functional metagenomic profiling of nine biomes. Nature. 2008;452: 629–632. doi: 10.1038/nature06810 [DOI] [PubMed] [Google Scholar]
- 70.Haas AF, Knowles B, Lim YW, McDole Somera T, Kelly LW, Hatay M, et al. Unraveling the Unseen Players in the Ocean—A Field Guide to Water Chemistry and Marine Microbiology. J Vis Exp. 2014; doi: 10.3791/52131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27: 863–864. doi: 10.1093/bioinformatics/btr026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Glass EM, Meyer F. The Metagenomics RAST Server: A Public Resource for the Automatic Phylogenetic and Functional Analysis of Metagenomes In: Bruijn FJ de, editor. Handbook of Molecular Microbial Ecology I. John Wiley & Sons, Inc.; 2011. pp. 325–331. Available: http://onlinelibrary.wiley.com/doi/10.1002/9781118010518.ch37/summary [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gomez-Alvarez V, Teal TK, Schmidt TM. Systematic artifacts in metagenomes from complex microbial communities. ISME J. 2009;3: 1314–1317. doi: 10.1038/ismej.2009.72 [DOI] [PubMed] [Google Scholar]
- 74.Cox MP, Peterson DA, Biggs PJ. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;11: 485 doi: 10.1186/1471-2105-11-485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, et al. The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9: 386 doi: 10.1186/1471-2105-9-386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smith EP, van Belle G. Nonparametric Estimation of Species Richness. Biometrics. 1984;40: 119–129. doi: 10.2307/2530750 [Google Scholar]
- 77.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12: 87 doi: 10.1186/s12915-014-0087-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hoff KJ. The effect of sequencing errors on metagenomic gene prediction. BMC Genomics. 2009;10: 520 doi: 10.1186/1471-2164-10-520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clarke KR. Non-parametric multivariate analyses of changes in community structure. Aust J Ecol. 1993;18: 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x [Google Scholar]
- 80.Graham MH, Edwards MS. Statistical significance versus fit: estimating the importance of individual factors in ecological analysis of variance. Oikos. 2001;93: 505–513. doi: 10.1034/j.1600-0706.2001.930317.x [Google Scholar]
- 81.McArdle BH, Anderson MJ. Fitting Multivariate Models to Community Data: A Comment on Distance-Based Redundancy Analysis. Ecology. 2001;82: 290–297. doi: 10.1890/0012-9658(2001)082[0290:FMMTCD]2.0.CO;2 [Google Scholar]
- 82.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30: 3123–3124. doi: 10.1093/bioinformatics/btu494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.White JR, Nagarajan N, Pop M. Statistical Methods for Detecting Differentially Abundant Features in Clinical Metagenomic Samples. PLOS Comput Biol. 2009;5: e1000352 doi: 10.1371/journal.pcbi.1000352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B Methodol. 1995;57: 289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The coefficient of variance as a function of relative abundances of (a) genera and (b) gene function Level 3 categories in three biological replicates across four environmental conditions (present-day ○, elevated temperature □, elevated pCO2 △, and future ◇) indicates a minimum cutoff of 0.01% relative abundance.
(TIFF)
Rarefaction curve of (a) water column and (b) kelp surface microbiomes across the four environmental conditions (present-day ‘black’ ○, elevated temperature ‘red’ □, elevated pCO2 ‘green’△, and future ‘purple’ ◇. Diversity index was calculated based off of open reading frames, protein annotations from MG-RAST.
(TIFF)
(TIF)
(TIF)
(DOCX)
(DOCX)
Data Availability Statement
All SRA data is available here: https://www.ncbi.nlm.nih.gov/sra/SRP132751 SRA accession SRP132751.