

Summary
Idiopathic calcium oxalate (CaOx) stone-formers (ICSFs) differ from patients who make idiopathic calcium phosphate (CaP) stones (IPSFs). ICSFs, but not IPSFs, form their stones as overgrowths on interstitial apatite plaque; the amount of plaque covering papillary surface is positively correlated with urine calcium excretion and inversely with urine volume. The amount of plaque predicts the number of recurrent stones. The initial crystal overgrowth on plaque is CaP, although the stone is mainly composed of CaOx, meaning that lowering supersaturation (SS) for CaOx and CaP is important for CaOx stone prevention. IPSFs, unlike ICSFs, have apatite crystal deposits in inner medullary collecting ducts, which are associated with interstitial scarring. ICSFs and IPSFs have idiopathic hypercalciuria, which is due to decreased tubule calcium reabsorption, but sites of abnormal reabsorption may differ. Decreased reabsorption in proximal tubules (PTs) delivers more calcium to the thick ascending limb (TAL), where increased calcium reabsorption can load the interstitium, leading to plaque formation. The site of abnormal reabsorption in IPSFs may be the TAL, where an associated defect in bicarbonate reabsorption could produce the higher urine pH characteristic of IPSFs. Preventive treatment with fluid intake, protein and sodium restriction, and thiazide will be effective in ICSFs and IPSFs by decreasing urine calcium concentration and CaOx and CaP SS and may also decrease plaque formation by increased PT calcium reabsorption. Citrate may be detrimental for IPSFs if urine pH rises greatly, increasing CaP SS. Future trials should examine the question of appropriate treatment for IPSFs.
Introduction
The common measures of reduced diet sodium and protein, increased fluids, thiazide, potassium citrate, reduced diet purine, and allopurinol have until now been viewed from the perspective of altering urine supersaturations and inhibitors of crystallization. Here, we add to this familiar theme the new work concerning how stones actually form and how the mechanisms that drive their formation actually function. The result is a new level of understanding in the use of accepted treatments that should bring to physicians and their patients a greater confidence and subtlety of management.
The new work has divided calcium stone-formers not only by their clinical appearances but also their renal pathology. We have always distinguished patients who form calcium stones because of systemic disease from idiopathic calcium stone-formers. However, we now know that idiopathic calcium stone-formers whose stones are predominantly calcium oxalate differ markedly from those whose stones are not. This new distinction has real clinical effects and calls for a new trial.
Idiopathic Calcium Oxalate Stone-Formers
Stones Grow over Deposits of Interstitial Apatite (Plaque)
In the most common kind of patient, calcium stones arise from no systemic disease but are, rather, “idiopathic.” Among these, most (1) form stones for which the most abundant crystal is calcium oxalate (CaOx). The kidneys of idiopathic CaOx stone-formers (ICSFs) are normal except for papillary interstitial apatite deposits (2,3) that appear as white clouds (Figure 1A) under the urothelium during ureteroscopy (URS) or percutaneous nephrolithotomy (PERC). The kidney stones grow over these deposits, often called “white plaque,” on the outside of the papilla (Figure 1B). No crystals are seen within the epithelial compartments (4). The deposits evoke no obvious inflammation, and renal papillae are completely normal in appearance except for plaque and overgrowing stones.
Figure 1. Endoscopic and histologic characteristics of interstitial plaque in idiopathic calcium oxalate stone-formers (ICSFs).
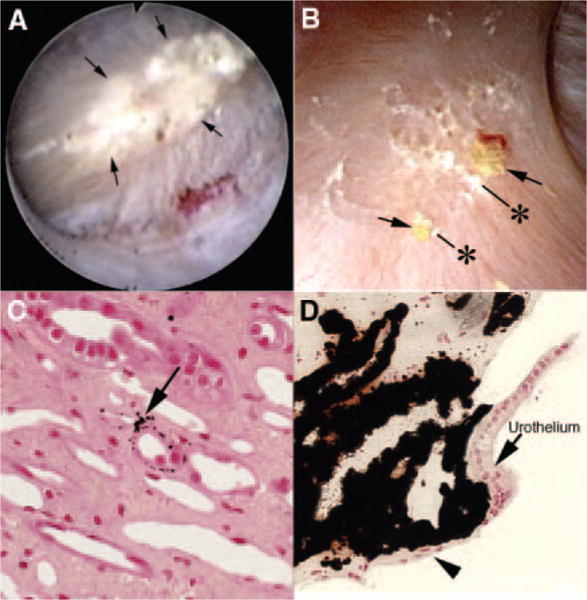
(A) Example of a papilla from an ICSF that was video recorded at the time of the mapping protocol. The papillary tip is covered by multiple sites of an irregular white material (arrows) termed “interstitial” or “Randall’s plaque” that is seen through the urothelial covering. These regions of plaque are sites for stone attachment. (B) Two small stones (asterisks) of approximately 0.5 mm are seen associated with a region of interstitial plaque (arrows). Histologic examination of papillary biopsies taken adjacent to sites of interstitial plaque shows that crystalline material (arrow in C) initially accumulates in the basement membranes of the thin loops of Henle. With time, these small sites of deposits appear to accumulate in the interstitial space as dense islands of mineral proceeding to the basal surface of the urothelial covering of a renal papilla (arrowhead in D). Adapted from references 2,8.
Plaque begins as collections of tiny microspherules that form in the basement membranes of the papillary thin limbs of the loops of Henle (Figure 1C) and spread from there into the interstitium and eventually beneath the urothelium and basement membranes of inner medullary collecting ducts (IMCDs) and terminal ducts of Bellini (BDs), where they appear as white plaque (Figure 1D).
In a prospective study of ICSFs, all stones visualized using PERC were classified as attached or not, and those attached were further classified as attached or not to plaque (5). Most stones were attached, and of those, all were attached to plaque or their attachment site could not be fully verified. Verification required that intraoperative classification during surgery agree with subsequent classification from intraoperative digital video recordings. Despite some difficulties with verification, the results were overwhelmingly in favor of the supposition that attachment is the rule and attachment is virtually always over plaque. In a follow-up study of the unattached stones, most were found to possess apatite surface foci that could have been an attachment to interstitial plaque (6). The main implication of this work is that plaque is essential to stone formation, and if so, plaque abundance and numbers of recurrent stones should correlate; in fact, they do, a finding that strengthens the stone-plaque link (7).
Because CaOx stones in ICSFs grow outside of the nephron and over plaque, the main issues for clinicians concern reduction of overgrowth, given plaque, and prevention of plaque itself. Until recently, we have used our treatments exclusively for the first purpose; we now have reasons to consider both purposes within clinical reach.
Formation of Plaque
Urine Risk Factors
The fraction of papillary surface covered by plaque varies with urine calcium (8), and it varies inversely with urine volume and pH (Figure 2). A multivariate score derived from all three measurements accounts for a high fraction of the variation in plaque abundance between ICSFs and normal controls. To clinicians, this means that high urine volume and control of urine calcium, both prudent means for stone reduction, ought to also benefit plaque formation.
Figure 2. Relationships between plaque abundance and urine findings.
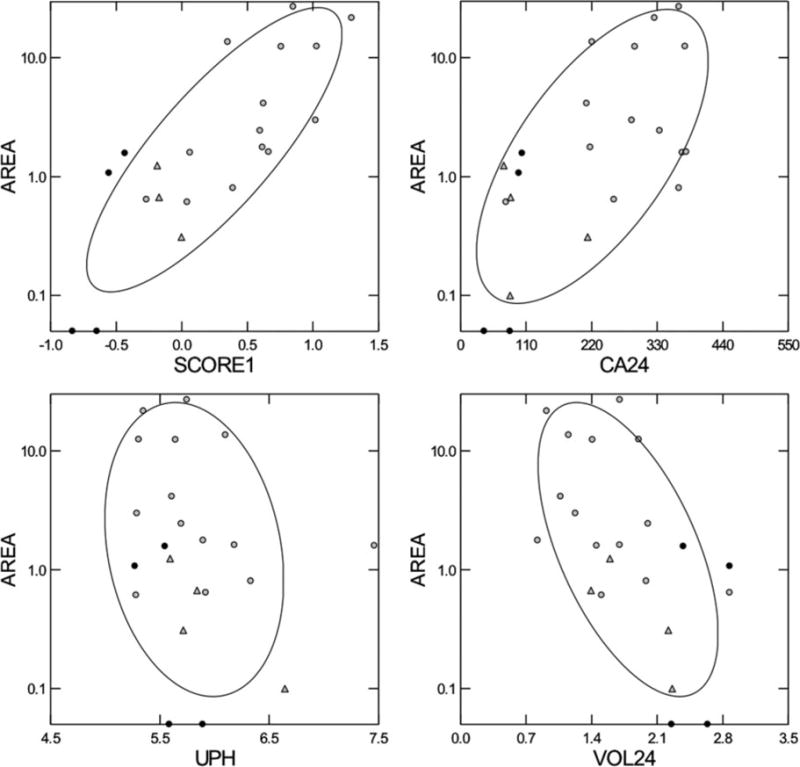
Fraction of papillary surface covered by plaque, as determined from intraoperative digital imaging (y-axis of all four panels), varies with urine calcium (upper right panel), and inversely with urine pH and volume (lower panels). A multivariable score using all three variables accounted for much of the variation in plaque coverage (upper left panel). ○, ICSFs; ● controls; △‚ patients with obesity bypass procedures. Score1, volume calcium pH score; Ca24, urine calcium (mg/day); UPH, urine pH; VOL24, Urine volume (liters/day). Adapted from reference 8.
Physiologic Links between Urine Findings and Plaque
How these urine factors affect plaque requires a deeper look into the renal physiology involved, and such a look increases the nuance and subtlety of clinical management. In the papillae, thin limbs are surrounded by three to four capillaries each (Figure 3, lower panels). The calcium concentration of thin limb fluid exceeds that of blood levels because of water extraction (9), but the epithelium possesses very low calcium permeability and does not transport calcium at all (10). Even so, thin limb fluid is always in contact with its epithelium, so calcium must move into the basement membrane at a higher concentration than blood, especially in the ascending portion that does not permit extensive water movement. However, the outer side of the basement membrane can lose calcium into the surrounding interstitium at a rate that is inversely related to the calcium molarity of the interstitial fluid, and that molarity will inevitably be dominated by the calcium concentration of the blood delivered to the capillaries.
Figure 3. Schematic drawing of outer medulla and papillary tip.
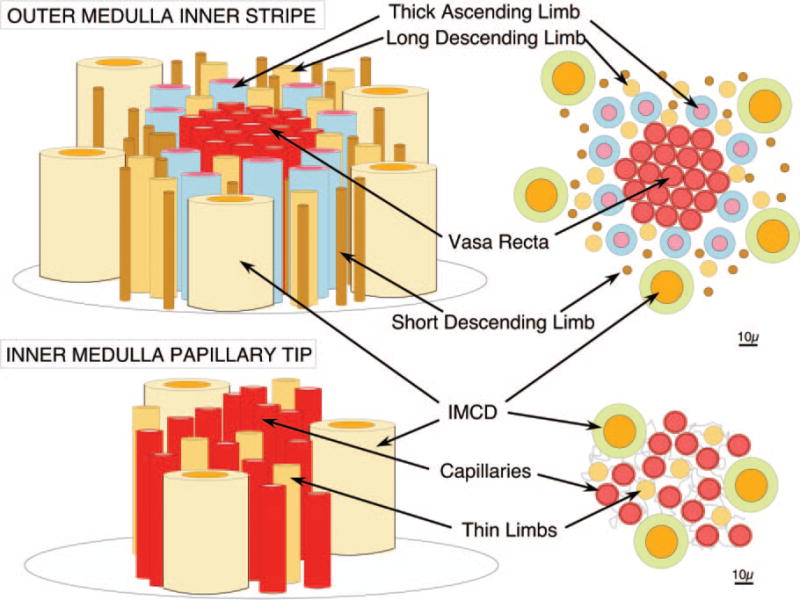
Vasa recta descend in bundles through the inner stripe of the outer medulla (upper panels) surrounded by rings of thick ascending limbs. In the papillary tips, where plaque forms in basement membranes of thin limbs (lower panels), thin limbs are each surrounded by three to four capillaries that derive from the descending vasa recta.
That blood comes down in the descending vasa recta, which form bundles in the inner stripe of the outer medulla that are surrounded by a ring of thick ascending limbs (11) (Figure 3, upper panels). Thick ascending limbs reabsorb calcium without water mainly via a lumen positive transepithelial potential created by the sodium-potassium-chloride cotransporter 2 and the renal outer medullary potassium channel. (10). Delivery of calcium without water must enrich the inner stripe interstitium with calcium, and this must in turn raise the calcium concentration of blood in the descending vasa recta. So, in all conditions, the basement membranes of the papillary thin limbs will be bathed on both sides by fluid with a calcium concentration above that of blood: lumen fluid via water extraction, interstitial fluid via vasa washdown.
This probably accounts for why interstitial plaque forms even in normal people and initiates in this particular site; however, so far we have not explained why plaque would be more abundant in ICSFs than in normal people because the anatomy and functions of thin limbs and thick ascending limbs are essentially alike.
Idiopathic Hypercalciuria
The crucial difference lies in how calcium is managed in ICSFs as opposed to normal people. Populations of men and women exhibit a wide range of urine calcium excretions from as low as <50 to as high as >500 mg daily, with mean values in the range of 130 to 170 mg daily for both genders (12). ICSFs appear to have been selected from the high tail of this distribution because their mean values are twice that of normal individuals. Like height, calcium excretion is seemingly genetically determined because high values run in families (13) and rodents can be bred for it (14). Such high urine calcium excretion is not a disease, but merely the cause of high stone risk in more hypercalciuric people. ICSFs are said to have idiopathic hypercalciuria (IH), but a better way to think about the matter is that they simply are drawn from one end of the normal genetic endowment of humankind.
In humans and in animals, IH reflects increased tissue vitamin D activation: intestinal calcium absorption and bone mineral mobilization are increased, and a low-calcium diet can cause negative bone mineral balance (13). But of interest here is how more calcium than normal gets into the urine. It could be from increased blood calcium because the calcium from each meal is absorbed at an abnormally brisk pace; it could be from reduced tubule calcium reabsorption. In fact, it is the latter (15). Eating fixed and identical high-calcium and normal diets, ICSFs with IH reduce overall tubule reabsorption with each meal more than matched controls, so that at identical serum calcium levels and calcium-filtered loads they excrete more calcium.
This would not be sufficient to explain higher plaque in ICSFs than normal except we add that, using endogenous lithium clearance as a marker, the proximal tubule participates in the reduction of overall tubule calcium reabsorption (16), and as a consequence, delivery of calcium into the thin limbs is abnormally high (Figure 4). In this figure each point represents 15 clearance periods in ICSFs (black circles) or controls (gray circles) taken throughout a three-meal day in the general clinical research center. Compared with controls, ICSFs deliver far more calcium distally, and urine calcium excretion parallels distal delivery reasonably well.
Figure 4. Relationship between urine calcium excretion (y-axis) and delivery of calcium from proximal tubules (x-axis).
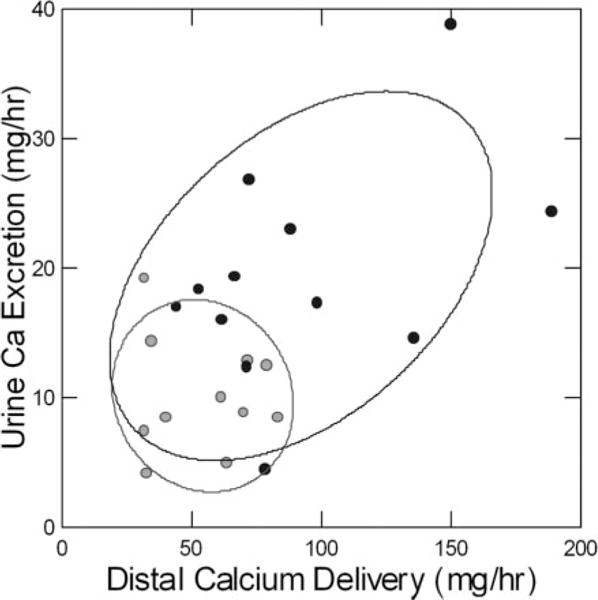
Urine calcium excretion increases with distal delivery from proximal tubules in ICSFs (black circles), whereas values for matched control subjects are clustered in the lower left quadrant (gray circles). Each point is the mean of 15 clearance periods during a three-meal day in the general clinical research center; subjects all ate the same diet. Proximal reabsorption was measured using endogenous lithium clearance. Adapted from reference 45.
The thin limbs do not reabsorb calcium appreciably, so increased distal delivery means increased calcium enters the thick ascending limbs where it is reabsorbed via mainly electrogenic forces. This means that more delivery will generally result in correspondingly more reabsorption, without water, into the interstitium of the outer stripe of the inner medulla. In turn, descending vasa recta will be more preloaded with calcium, and the capillaries of the papillum will enrich the interstitium with calcium to a greater degree, thereby fostering crystal nucleation in thin limb basement membranes. In the broadest sense, one would expect plaque abundance to parallel urine calcium excretion, which it does. Of course, interstitial calcium concentrations must be measured in human tissues if this hypothesis is to be properly tested; such measurements can be made, and we hope that investigators take on this specific test as a research aim. Failure to find increased interstitial calcium would defeat our hypothesis.
Effects of Urine Volume on Plaque
This model also predicts that plaque abundance will be generally inverse to urine volume. Vasopressin, a major agonist of medullary thick ascending limb transport, acting via the V2 receptor, is highest when urine volume is low, and the converse. Moreover, medullary washout from high urine flows will tend to reduce the concentrations of all papillary interstitial solutes, including calcium.
Limitations of This Model
We present this work as in progress and of value because it leads to further tests. For example, lithium clearance is not a direct measurement of proximal tubule reabsorption and may be subject to errors. Other tests (e.g., increase of urine calcium after blockade of the thick ascending limb by furosemide) would be valuable confirmation (17), as would, perhaps, studies of free water clearances. Likewise, Figure 4 makes it clear that not all of our ICSFs display the same degree of proximal tubule alteration, but we cannot presently correlate proximal reabsorption with plaque abundance measurements. This latter comparison would be a critical test of the hypotheses we propose and can be performed in human subjects.
Clinical Measures That May Reduce Plaque Formation
Because plaque cannot be quantified in patients except during surgery, randomized controlled trials (RCTs) for plaque prevention are not as yet practical, but from what we have already presented clinicians can derive some reasonably sound directions that have no risk and high potential benefit.
Reduced diet sodium and protein, long mentioned for moderation of hypercalciuria (18), may increase proximal tubule reabsorption. The first acts via reduction of extracellular fluid volume. The second reduces endogenous acid production from oxidation of sulfur on methionine and cystine; acid loads reduce proximal reabsorption (19). Thiazide-type diuretics are a mainstay in stone prevention (20) because they lower urine calcium excretion. In rodents, and presumably humans as well, they increase proximal reabsorption by reducing extracellular fluid volume (21). High water intake, an obvious stone prevention supported by RCTs (22), will reduce vasopressin, and therefore medullary thick ascending limb reabsorption (23), and foster medullary solute washout. One might consider once-a-night nocturia for patients whose stones recur despite all other measures because urine volume is lowest and vasopressin is presumably highest overnight.
After only one stone, we advise only modest treatment efforts (24), but one might consider that plaque needs to form before ICSFs can make stones and that stone recurrence tends to parallel plaque abundance (7). Given this point, high fluids and reduced diet protein and sodium would be reasonable as soon as possible in the natural history of stone disease, meaning with the first stone.
Even at the first stone, and often during the course of the disease, surgeons will have access to at least a coarse-grained visual assessment of plaque abundance via URS or PERC. We see every advantage in asking surgeons to share their best impression with us. Given a lot of plaque, stone prevention efforts may best be pushed as far as possible and patients made aware of a higher underlying risk of recurrence. This latter may increase their desire to maintain prevention treatments, especially those pertaining to diet and fluid intake.
How Stones Grow on Plaque
At the attachment site of a 1-mm CaOx stone (Figure 5) the urothelium over a plaque deposit was disrupted, and the plaque surface was exposed to urine (25). The exposed surface was sealed by an inner boundary of organic material of urine origin, containing Tamm–Horsfall protein and osteopontin as well as other molecules. Apatite crystals nucleated in the layer, were covered by more organic matrix, new nucleation occurred, and ultimately alternating crystal and organic layers formed a laminated ribbon. The ribbon terminated when crystallization spread out into the urinary space forming the stone base. In this transmission electron micrograph, plaque is at the lower left, white arrowheads show the inner boundary layer, and crystals are shown at the stone base by an asterisk and double arrows. The inset shows the details of the ribbon and its crystals at higher magnification.
Figure 5. Transmission electron microscopic image of an attachment site of small calcium oxalate (CaOx) stone.
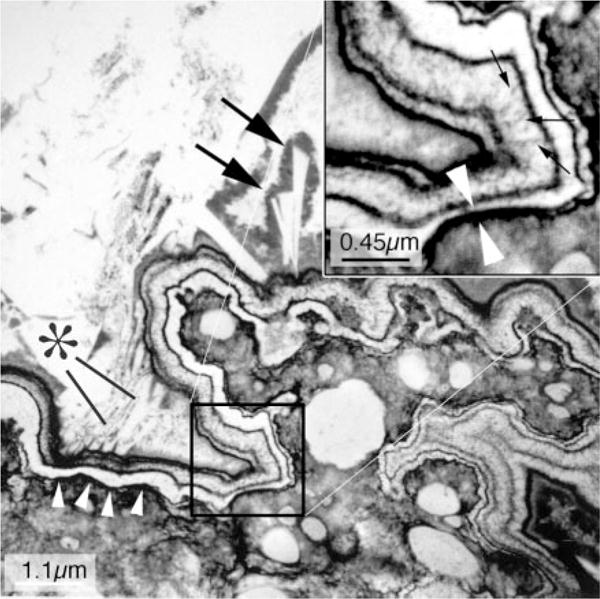
This high-magnification micrograph shows the ultrastructural features of the plaque-stone interface. The upper left hand corner of the micrograph shows the stone material in the urinary space, whereas the lower right corner shows a region of interstitial plaque in the renal papilla. The plaque boundary denoted by a row of four arrowheads has the appearance of a multilayered ribbon. When the portion of this ribbon marked by a square is magnified (see inset at upper right), this ribbon-like structure is seen to have nine separate layers in which five thin black organic lamina alternate with four white lamina. In the thickest of the white lamina, one can see tiny spicules that run perpendicular to the surface and have the appearance of multiple voids that contain tightly packed crystals (small arrows in inset). The two white arrowheads seen in the insert mark the closest black lamina to the renal tissue. An asterisk marks a series of small crystals, whereas two large arrows mark a set of larger crystals that appear to be inserted in the outermost layer (urinary side) of the ribbon. All crystalline material appears to be covered by black matrix material. Adapted from reference 25.
The crucial steps in the process, in addition to formation of plaque itself, are disruption of the urothelium, about which we know nothing at the present time, and the initial apatite nucleation, which drives the formation of the new stone. This nucleation depends entirely upon urine calcium phosphate (CaP) supersaturation (SS), meaning that even in CaOx stone-formers, CaP SS is a critical clinical variable to control as best one can.
In ICSFs, CaP SS far exceeds normal (Figure 6, lower right panel), to an even greater extent than for CaOx SS (Figure 6, upper right panel). Normal people do not, on average, achieve CaP SS values >1, meaning that the initial apatite nucleation might be rare indeed, whereas values in ICSFs are >1 virtually all of the time (26). The highest values of CaP SS are late afternoon and overnight for ICSFs, when the SS of normal people declines modestly. The high CaP SS is driven by high urine calcium molarity (Figure 6, upper left panel), itself arising from IH without any corresponding increase of urine volume (Figure 6, lower middle panel). Differences in urine pH are minimal between ICSFs and normal individuals (Figure 6, lower left panel).
Figure 6. Urine calcium phosphate (CaP) and CaOx supersaturation (SS) throughout a three-meal day in ICSFs (gray bars) and normal controls (black bars).
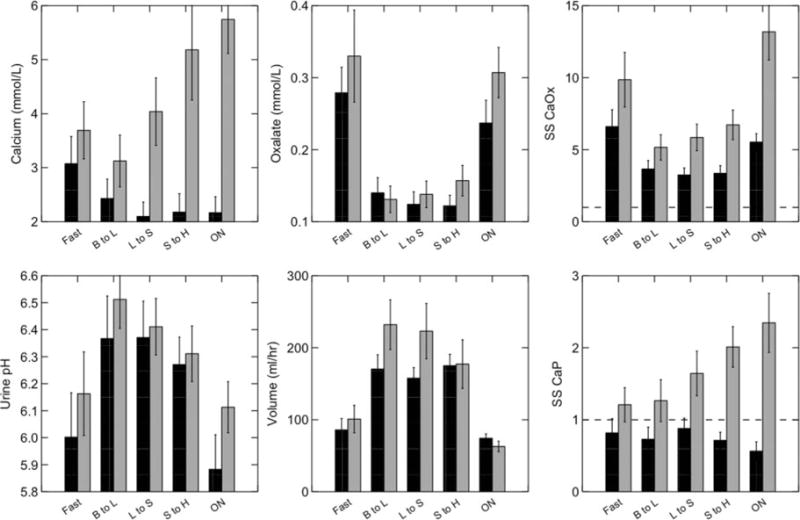
Subjects all ate identical diets and were studied using 15 urine collections during fasting (Fast), from breakfast to lunch (B to L), lunch to supper (L to S), supper to home (S to H), and overnight (ON). CaP SS (lower right panel) of ICSFs exceeded normal controls in the last three periods of the day, and normal controls never achieved an average SS >1 (horizontal dashed line). The main cause of the high CaP SS was urine calcium molarity (upper left panel), which itself arose from hypercalciuria without an accompanying increase of urine volume (lower middle panel). Urine pH values did not differ significantly (lower left panel). Urine CaOx SS of ICSFs exceeded that of normal controls (upper right panel), mainly because of higher calcium molarity; urine oxalate molarity did not differ (upper middle panel). Adapted from reference 46.
One must think it odd that urine volume is not higher in ICSFs than normal people given that the apical collecting duct calcium receptor (CaSR) should sense the rising calcium molarity and reduce water reabsorption; however, the facts are simply that this does not occur. For example, overnight, calcium molarity among ICSFs reaches near 6 mM whereas normal is 2 mM, but urine volumes are identical. The CaSR is probably protective, but only at higher levels of urine calcium excretion. The human CaSR studied in vitro is half stimulated at calcium ion molarities of 6 at the pH and ionic strength of urine, but in human urine a total calcium molarity of 6 means only approximately 3 mM calcium ion. The remaining urine calcium is bound in complexes that help create SS, such as with phosphate and oxalate. So the lack of effect in our patients is not actually divergent from what one might expect.
Clinical Measures That May Reduce Stone Overgrowth on Plaque
Reduction of CaP SS may be as important as reduction of CaOx SS even in ICSFs. High urine volume should help, as might all measures to reduce urine calcium, including reduced diet sodium and protein and thiazide. Overnight is so remarkably a time of high CaP SS, 1× nocturia could be important in refractory patients. In other words, means of treatment are the same as for plaque.
However, potassium citrate is complex, if of probable value. Alkalis reduce urine calcium excretion and increase proximal tubule reabsorption via reduced diet net acid load (27), and two RCTs document benefits for calcium stone-formers with hypocitraturia, among whom ICSFs would statistically have predominated (20). Citrate is an excellent inhibitor of apatite nucleation and growth (28,29); however, citrate can raise urine pH and CaP SS, so we believe it is prudent to dose enough to reduce urine ammonia by one half to two thirds; more will almost certainly raise pH. For example, the initial dose in millimoles per day could be one half to two thirds of the urine ammonia, in millimoles per day, as derived from standard commercially available 24-hour urine kidney stone risk panels.
Urine oxalate is far from trivial in any ICSF, so even in this CaP-oriented discussion we note that even with fixed and identical diets oxalate excretion is higher than normal in ICSFs, and CaOx SS is also higher, especially overnight (30). Moreover, ICSFs not infrequently exhibit renal oxalate secretion compared with normal control subjects eating the same diet (31), suggesting new mechanisms by which CaOx crystallization might be promoted. Lowering CaOx SS should reduce bulk nucleation and growth of CaOx on the new CaP stone nidus and is therefore very important. Urine oxalate excretion rates >45 mg/d often arise from low-calcium or high-oxalate diets that can be corrected. Values >65 to 70 mg/d may reflect primary hyperoxaluria or enteric hyperoxaluria (20). No RCTs support reduction of urine oxalate as a treatment for ICSFs.
Finally, hyperuricosuria may reduce CaP and CaOx solubility via salting out and therefore raise the propensity for both nucleations (32–34). This is perhaps why a single RCT with allopurinol was so positive (35). Reduced diet purine should do as well, but no trial has been done.
RCTs Accord with the Model of Plaque Formation and Overgrowth
In an RCT of male ICSFs, a low sodium and protein diet was more effective in stone reduction than a low-calcium diet, which would not increase proximal tubule reabsorption (18). All three fully powered, 3-year thiazide trials (36–38) have been highly positive, as have the two RCTs with potassium citrate in hypocitraturic calcium stone-formers (39,40) and the one RCT with allopurinol in hyperuricosuric calcium stone-formers. The thiazide, citrate, and allopurinol RCTs concerned idiopathic calcium stone-formers, wherein ICSFs must have predominated, but some CaP stone-formers were surely also present.
The ICSFs Have a Specific Disease
Because IH can be linked to plaque via the known anatomy and physiology of the kidney, because their stones grow on plaque through a well defined series of nucleations, and because mechanisms and origins of IH itself also are becoming well known, the stone disease of IH can be understood from its etiology, through its pathogenesis to its eventual tissue expression. Given that the final disease, stones, can be so well explained in mechanistic terms, ICSFs appear to satisfy reasonable criteria of a cohesive disease of mineral metabolism. We note in leaving this subject that a generation of researches has established the remarkable abilities of urine to inhibit nucleation and growth of calcium crystals (41); abnormalities of inhibition may well be part of the ICSF phenotype, but evidence for this is scanty at best, and much more must be done before the matter has clinical relevance.
Idiopathic CaP Stone-Formers
Idiopathic CaP stone-formers (IPSFs) whose stones contain >50% CaP differ so radically from ICSFs that their stones might well come from an entirely different etiology and pathogenesis. However much they may resemble ICSFs on superficial analysis, they do not have that disease. Instead, their kidneys are involved in a far more destructive local process. IPSFs could form stones of apatite or brushite (calcium monohydrogen phosphate); to date, we have data only on the latter group, brushite IPSFs, and what follows applies only to them.
IPSFs Plug BDs and IMCDs with Apatite Crystals
During PERC or URS, white plaque is present (Figure 7A, arrow); papillae show areas of scarring (arrowheads) and scattered hugely dilated BDs plugged with apatite crystals that often protrude out of the duct opening (42) and project into the urinary space (asterisk). On biopsy (Figure 7, B through F) affected BDs can be 20-fold dilated; epithelial cells are absent (arrow). Interstitial fibrosis surrounds affected BDs (legend details changes in each panel). Crystal-mediated injury essentially creates a focal papillary tubulointerstitial nephropathy.
Figure 7. Endoscopic and histologic images of crystalline deposition in brushite stone patients.
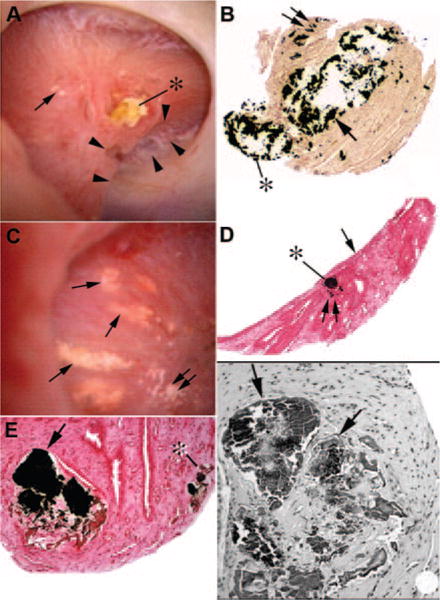
Endoscopic examination of papilla from brushite stone-formers reveals a wide range of pathologies from normal to severe deformities. (A) A papilla with extensive pitting marked by a series of arrowheads and extensive dilation of the distal end of ducts of Bellini (BDs) usually filled with a protruding crystalline plug (asterisk). Small regions of Randall’s plaque (single arrow in A) termed “type 1 crystal pattern” were commonly seen. Histologic examination of papillary biopsies reveals enormously dilated inner medullary collecting ducts (IMCDs) and BDs (arrow in B) with mineral occasionally protruding (asterisk in B) in the urinary space. Sites of interstitial plaque are seen (double arrow in B). Other papilla possessed sites of Randall’s plaque (double arrows in C) and regions of yellow plaque (single arrows in C) termed “type 2 crystal pattern.” Each site of yellow plaque was linear in orientation, and when multiple such areas were near each other they formed a spoke- and wheel-like pattern (C). Histologic examination of a single site of yellow plaque revealed these areas represented IMCDs filled with mineral (asterisk in D) that was positioned just beneath the urothelial covering (single arrow in D). Double arrow in panel D marks a site of Randall’s plaque. Light microscopic examination of Yasue-stained sections revealed that the plugged IMCDs contained large amounts of mineral (arrow in E) that filled the lumens of severely damaged tubules whereas nearby tubules appeared normal. Mineral deposits were also seen in thin loops of Henle (asterisk in E). Tissue sections stained with hemotoxylin and eosin revealed crystalline-filled IMCDs (arrows in F) embedded in a region of interstitial fibrosis. Adapted from reference 42.
The mechanism for plugging and high stone CaP abundance seems simply to be a high urine CaP SS (43). Mean urine CaP SS (Figure 8, left panel) of calcium stone-formers increases smoothly as the fraction of phosphate in stones analyzed increases (Figure 8, x-axes of both panels). This occurs because of rising urine pH (Figure 8, right panel). The reason for the increasing pH is not known. All groups of patients on these plots have equivalent IH. Because IMCD and BD tubule fluid approximates the final urine, CaP SS must be higher at both sites, leading to sporadic and damaging crystallizations.
Figure 8. Urine CaP SS and pH in stone-formers with increasing stone CaP abundance.
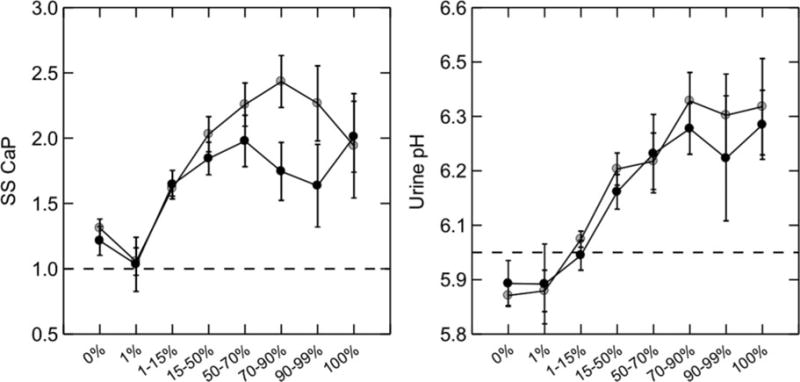
Calcium stone-formers were grouped by percent CaP in all analyzed stones (x-axis). CaP SS (left panel, y-axis) rose progressively with percent CaP in stones; the main reason was a progressive increase in urine pH (right panel, y-axis). Urine calcium and phosphate excretions (not shown) did not vary across stone percentage categories. Women (gray circles) and men (black circles) showed essentially identical behavior. Adapted from reference 43.
The Less Abundant Plaque in IPSFs Raises Important Research Questions
Urine calcium excretion is slightly higher in IPSFs versus ICSFs, and urine volume is approximately the same, but the higher pH is predictive of less plaque. Vas washdown should operate the same way for both groups. However, at least two alternative mechanisms can reduce plaque. Reduced net proton secretion in IMCDs would be expected in IPSFs compared with ICSFs, and this would reduce bicarbonate entry into the papillary interstitium and therefore reduce interstitial fluid pH. Higher pH would foster CaP nucleation in basement membranes of thin loops.
Alternatively, medullary thick ascending limb may have reduced absolute calcium reabsorption in IPSFs compared with ICSFs. Although both exhibit the same urine calcium losses and even reduced overall tubule reabsorptive changes, IPSFs would not load descending vasa recta with calcium to the extent found in ICSFs. Because thick ascending limbs reabsorb approximately 15% of filtered bicarbonate (44), reduced reabsorption would increase distal bicarbonate delivery to IMCDs and BDs, raise the pH there, and foster CaP crystallization. In other words, IH may have several tubule expressions, one centered mainly in proximal tubules and one in thick ascending limbs with perhaps a spectrum in between these extremes. In inbred hypercalciuric rats, proximal tubule and thick ascending limb calcium reabsorptions are abnormally low (17). Comparison of nephron physiology in IPSFs versus ICSFs therefore offers remarkable opportunity for new human discoveries.
Management of IPSFs
The value of reduced diet sodium and protein, and of high fluid intake and thiazide, is the same as in ICSFs— what differs is the role of potassium citrate. Citrate is an inhibitor of CaP nucleation and growth, as mentioned already, but if urine pH and therefore CaP SS rises, stones and plugging can increase. An RCT is badly needed for this exact kind of patient. Given that IPSFs are hardly rare, this trial is long overdue.
One does not have to wait for stone analysis. Any URS or PERC is an opportunity for surgeons to observe plugging, which is not present in ICSFs. The urologists who serve our patients should always be asked if plugging is seen and if even a crude estimate can be given of amounts of plugging and papillary retraction and scarring.
Summary
Information drawn from operative biopsies of the renal papillae in stone-formers is altering our understanding of the ways in which calcium stones form. The importance of plaque in formation of CaOx stones means that methods to reduce plaque formation should become a goal of stone prevention. The role that CaP SS plays in the formation of CaOx and CaP stones means that lowering CaP SS should be a goal for ICSFs and IPSFs. The marked histopathologic differences between ICSFs and IPSFs highlight the need for a trial to evaluate treatments for stone prevention in each group. As our understanding of the pathophysiology of stone formation improves, it will inform our ability to choose appropriate treatments for our patients.
Footnotes
Disclosures
None.
References
- 1.Parks JH, Coe FL. Evidence for durable kidney stone prevention over several decades. BJU Int. 2009;103:1238–1246. doi: 10.1111/j.1464-410X.2008.08170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evan AP, Lingeman JE, Coe FL, Parks JH, Bledsoe SB, Shao Y, Sommer AJ, Paterson RF, Kuo RL, Grynpas M. Randall’s plaque of patients with nephrolithiasis begins in basement membranes of thin loops of Henle. J Clin Invest. 2003;111:607–616. doi: 10.1172/JCI17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evan A, Lingeman J, Coe FL, Worcester E. Randall’s plaque: Pathogenesis and role in calcium oxalate nephrolithiasis. Kidney Int. 2006;69:1313–1318. doi: 10.1038/sj.ki.5000238. [DOI] [PubMed] [Google Scholar]
- 4.Evan AP, Coe FL, Gillen D, Lingeman JE, Bledsoe S, Worcester EM. Renal intratubular crystals and hyaluronan staining occur in stone formers with bypass surgery but not with idiopathic calcium oxalate stones. Anat Rec (Hoboken) 2008;291:325–334. doi: 10.1002/ar.20656. [DOI] [PubMed] [Google Scholar]
- 5.Miller NL, Gillen DL, Williams JC, Jr, Evan AP, Bledsoe SB, Coe FL, Worcester EM, Matlaga BR, Munch LC, Lingeman JE. A formal test of the hypothesis that idiopathic calcium oxalate stones grow on Randall’s plaque. BJU Int. 2009;103:966–971. doi: 10.1111/j.1464-410X.2008.08193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller NL, Williams JC, Jr, Evan AP, Bledsoe SB, Coe FL, Worcester EM, Munch LC, Handa SE, Lingeman JE. In idiopathic calcium oxalate stone-formers, unattached stones show evidence of having originated as attached stones on Randall’s plaque. BJU Int. 2010;105:242–245. doi: 10.1111/j.1464-410X.2009.08637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim SC, Coe FL, Tinmouth WW, Kuo RL, Paterson RF, Parks JH, Munch LC, Evan AP, Lingeman JE. Stone formation is proportional to papillary surface coverage by Randall’s plaque. J Urol. 2005;173:117–119. doi: 10.1097/01.ju.0000147270.68481.ce. [DOI] [PubMed] [Google Scholar]
- 8.Kuo RL, Lingeman JE, Evan AP, Paterson RF, Parks JH, Bledsoe SB, Munch LC, Coe FL. Urine calcium and volume predict coverage of renal papilla by Randall’s plaque. Kidney Int. 2003;64:2150–2154. doi: 10.1046/j.1523-1755.2003.00316.x. [DOI] [PubMed] [Google Scholar]
- 9.Asplin JR, Mandel NS, Coe FL. Evidence of calcium phosphate supersaturation in the loop of Henle. Am J Physiol. 1996;270:F604–F613. doi: 10.1152/ajprenal.1996.270.4.F604. [DOI] [PubMed] [Google Scholar]
- 10.Friedman PA. Renal Calcium Metabolism. In: Alpern R, Hebert SC, editors. The Kidney: Physiology and Pathohysiology. 4th. Burlington, MA: Academic Press; 2008. pp. 1851–1890. [Google Scholar]
- 11.Jamison RL, Kriz W. Urinary Concentrating Mechanism: Structure and function. 1st. New York: Oxford University Press; 1982. [Google Scholar]
- 12.Coe FL, Parks JH, Evan AP, Worcester E. Pathogenesis and Treatment of Nephrolithiasis. In: Alpern R, Hebert SC, editors. Seldin and Giebisch’s The Kidney. 4th. Amsterdam: Elsevier Academic Press; 2008. pp. 1945–1977. [Google Scholar]
- 13.Worcester EM, Coe FL. New insights into the pathogenesis of idiopathic hypercalciuria. Semin Nephrol. 2008;28:120–132. doi: 10.1016/j.semnephrol.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bushinsky DA, Asplin JR, Grynpas MD, Evan AP, Parker WR, Alexander KM, Coe FL. Calcium oxalate stone formation in genetic hypercalciuric stone-forming rats. Kidney Int. 2002;61:975–987. doi: 10.1046/j.1523-1755.2002.00190.x. [DOI] [PubMed] [Google Scholar]
- 15.Worcester EM, Gillen DL, Evan AP, Wright K, Trumbore L, Nakagawa Y, Coe FL. Evidence that postprandial reduction of renal calcium reabsorption mediates hypercalciuria of patients with calcium nephrolithiasis. Am J Physiol Renal Physiol. 2007;292:F66–F75. doi: 10.1152/ajprenal.00115.2006. [DOI] [PubMed] [Google Scholar]
- 16.Worcester EM, Coe FL, Evan AP, Bergsland KJ, Parks JH, Willis LR, Clark DL, Gillen DL. Evidence for increased postprandial distal nephron calcium delivery in hypercalciuric stone-forming patients. Am J Physiol Renal Physiol. 2008;295:F1286–F1294. doi: 10.1152/ajprenal.90404.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuruoka S, Bushinsky DA, Schwartz GJ. Defective renal calcium reabsorption in genetic hypercalciuric rats. Kidney Int. 1997;51:1540–1547. doi: 10.1038/ki.1997.212. [DOI] [PubMed] [Google Scholar]
- 18.Borghi L, Schianchi T, Meschi T, Guerra A, Allegri F, Maggiore U, Novarini A. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med. 2002;346:77–84. doi: 10.1056/NEJMoa010369. [DOI] [PubMed] [Google Scholar]
- 19.Wang T, Egbert AL, Jr, Aronson PS, Giebisch G. Effect of metabolic acidosis on NaCl transport in the proximal tubule. Am J Physiol. 1998;274:F1015–F1019. doi: 10.1152/ajprenal.1998.274.6.F1015. [DOI] [PubMed] [Google Scholar]
- 20.Coe FL, Evan A, Worcester E. Kidney stone disease. J Clin Invest. 2005;115:2598–2608. doi: 10.1172/JCI26662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115:1651–1658. doi: 10.1172/JCI24134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water and recurrences of idiopathic calcium nephrolithiasis: A 5-year randomized prospective study. J Urology. 1996;155:839–843. [PubMed] [Google Scholar]
- 23.Gamba G. The nanopeptide hormone vasopressin is a new player in the modulation of renal Na(+)-Cl(−) cotransporter activity. Kidney Int. 2010;78:127–129. doi: 10.1038/ki.2010.147. [DOI] [PubMed] [Google Scholar]
- 24.Worcester EM, Coe FL. Clinical practice. Calcium kidney stones. N Engl J Med. 2010;363:954–963. doi: 10.1056/NEJMcp1001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evan AP, Coe FL, Lingeman JE, Shao Y, Sommer AJ, Bledsoe SB, Anderson JC, Worcester EM. Mechanism of formation of human calcium oxalate renal stones on Randall’s plaque. Anat Rec (Hoboken) 2007;290:1315–1323. doi: 10.1002/ar.20580. [DOI] [PubMed] [Google Scholar]
- 26.Bergsland KJ, Coe FL, Gillen DL, Worcester EM. A test of the hypothesis that the collecting duct calcium-sensing receptor limits rise of urine calcium molarity in hypercalciuric calcium kidney stone formers. Am J Physiol Renal Physiol. 2009;297:F1017–F1023. doi: 10.1152/ajprenal.00223.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemann J, Jr, Pleuss JA, Hornick L, Hoffman RG. Dietary NaCl-restriction prevents the calciuria of KCl-deprivation and blunts the calciuria of KHCO3- deprivation in healthy adults. Kidney Int. 1995;47:899–906. doi: 10.1038/ki.1995.134. [DOI] [PubMed] [Google Scholar]
- 28.Johnsson M, Richardson CF, Sallis JD, Nancollas GH. Adsorption and mineralization effects of citrate and phosphocitrate on hydroxyapatite. Calcif Tissue Int. 1991;49:134–137. doi: 10.1007/BF02565136. [DOI] [PubMed] [Google Scholar]
- 29.Berg C, Tiselius HG. The effects of citrate on hydroxyapatite induced calcium oxalate crystallization and on the formation of calcium phosphate crystals. Urol Res. 1989;17:167–172. doi: 10.1007/BF00256245. [DOI] [PubMed] [Google Scholar]
- 30.Bergsland KJ, Coe FL, Gillen DL, Worcester EM. A test of the hypothesis that the collecting duct calcium-sensing receptor limits rise of urine calcium molarity in hypercalciuric calcium kidney stone formers. Am J Physiol Renal Physiol. 2009;297:F1017–F1023. doi: 10.1152/ajprenal.00223.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergsland KJ, Zisman AL, Asplin JR, Worcester EM, Coe FL. Evidence for net renal tubule oxalate secretion in patients with calcium kidney stones. Am J Physiol Renal Physiol. 2011;300:F311–F318. doi: 10.1152/ajprenal.00411.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grover PK, Ryall RL, Marshall VR. Effect of urate on calcium oxalate crystallization in human urine: Evidence for a promotory role of hyperuricosuria in urolithiasis. Clin Sci. 1990;79:9–15. doi: 10.1042/cs0790009. [DOI] [PubMed] [Google Scholar]
- 33.Grover PK, Ryall RL, Potezny N, Marshall VR. The effect of decreasing the concentration of urinary urate on the crystallization of calcium oxalate in undiluted human urine. J Urol. 1990;143:1057–1061. doi: 10.1016/s0022-5347(17)40183-2. [DOI] [PubMed] [Google Scholar]
- 34.Grover PK, Ryall RG, Marshall VR. Calcium oxalate crystallization in urine: Role of urate and glycosaminoglycans. Kidney Int. 1992;41:149–154. doi: 10.1038/ki.1992.20. [DOI] [PubMed] [Google Scholar]
- 35.Ettinger B, Tang A, Citron JT, Livermore B, Williams T. Randomized trial of allopurinol in the prevention of calcium oxalate calculi. N Engl J Med. 1986;315:1386–1389. doi: 10.1056/NEJM198611273152204. [DOI] [PubMed] [Google Scholar]
- 36.Laerum E, Larsen S. Thiazide prophylaxis of urolithiasis: A double-blind study in general practice. Acta Med Scand. 1984;215:383–389. [PubMed] [Google Scholar]
- 37.Borghi L, Meschi T, Guerra A, Novarini A. Randomized prospective study of a nonthiazide diuretic, indapamide, in preventing calcium stone recurrences. J Cardiovasc Pharmacol. 1993;22(Suppl 6):S78–S86. [PubMed] [Google Scholar]
- 38.Ettinger B, Citron JT, Livermore B, Dolman LI. Chlorthalidone reduces calcium oxalate calculous recurrence but magnesium hydroxide does not. J Urol. 1988;139:679–684. doi: 10.1016/s0022-5347(17)42599-7. [DOI] [PubMed] [Google Scholar]
- 39.Ettinger B, Pak CY, Citron JT, Thomas C, Adams-Huet B, Van-gessel A. Potassium-magnesium citrate is an effective prophylaxis against recurrent calcium oxalate nephrolithiasis. J Urol. 1997;158:2069–2073. doi: 10.1016/s0022-5347(01)68155-2. [DOI] [PubMed] [Google Scholar]
- 40.Barcelo P, Wuhl O, Servitge E, Rousaud A, Pak CY. Randomized double-blind study of potassium citrate in idiopathic hypocitraturic calcium nephrolithiasis. J Urol. 1993;150:1761–1764. doi: 10.1016/s0022-5347(17)35888-3. [DOI] [PubMed] [Google Scholar]
- 41.Kumar V, Lieske JC. Protein regulation of intrarenal crystallization. Curr Opin Nephrol Hypertens. 2006;15:374–380. doi: 10.1097/01.mnh.0000232877.12599.f4. [DOI] [PubMed] [Google Scholar]
- 42.Evan AP, Lingeman JE, Coe FL, Shao Y, Parks JH, Bledsoe SB, Phillips CL, Bonsib S, Worcester EM, Sommer AJ, Kim SC, Tinmouth WW, Grynpas M. Crystal-associated nephropathy in patients with brushite nephrolithiasis. Kidney Int. 2005;67:576–591. doi: 10.1111/j.1523-1755.2005.67114.x. [DOI] [PubMed] [Google Scholar]
- 43.Parks JH, Worcester EM, Coe FL, Evan AP, Lingeman JE. Clinical implications of abundant calcium phosphate in routinely analyzed kidney stones. Kidney Int. 2004;66:777–785. doi: 10.1111/j.1523-1755.2004.00803.x. [DOI] [PubMed] [Google Scholar]
- 44.Kwon TH, Nielsen J, Kim YH, Knepper MA, Frøkiaer J, Nielsen S. Regulation of sodium transporters in the thick ascending limb of rat kidney: Response to angiotensin II. Am J Physiol Renal Physiol. 2003;285:F152–F165. doi: 10.1152/ajprenal.00307.2002. [DOI] [PubMed] [Google Scholar]
- 45.Worcester EM, Coe FL, Evan AP, Bergsland KJ, Parks JH, Willis LR, Clark DL, Gillen DL. Evidence for increased postprandial distal nephron calcium delivery in hypercalciuric stone-forming patients. Am J Physiol Renal Physiol. 2008;295:F1286–F1294. doi: 10.1152/ajprenal.90404.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bergsland KJ, Coe FL, Gillen DL, Worcester EM. A test of the hypothesis that the collecting duct calcium-sensing receptor limits rise of urine calcium molarity in hypercalciuric calcium kidney stone formers. Am J Physiol Renal Physiol. 2009;297:F1017–F1023. doi: 10.1152/ajprenal.00223.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]