

Abstract
Objective
Tregs need to acquire an effector phenotype to function in inflammatory settings. Whether effector Tregs can limit disease severity in lupus is unknown. IRF4 is an essential controller of effector Tregs and regulates their ability to express IL-10. In non-Tregs, IRF4 activity is modulated by interaction with DEF6 and its homologue SWAP-70. Although mice lacking both DEF6 and SWAP-70 (DKO mice) develop lupus they display normal survival suggesting that, in DKO mice, Tregs can moderate disease development. Here we investigated whether DKO Tregs have an increased capacity to become effector Tregs due to the ability of DEF6 and SWAP-70 to restrain IRF4 activity.
Methods
Tregs were evaluated by FACS. The Blimp1-IL10 axis was assessed by crossing DKO mice with Blimp1-YFP-10BiT dual reporter mice. Deletion of IRF4 in DKO Tregs was achieved by generating Foxp3Cre IRF4fl/fl DKO mice.
Results
The concomitant absence of DEF6 and SWAP-70 leads to increased numbers of Tregs, which acquire an effector phenotype in a cell-intrinsic manner. In addition, DKO Tregs exhibit enhanced expression of the Blimp-1-IL-10 axis. Notably, DKO effector Tregs survive and expand as disease progresses. The accumulation of DKO Tregs was associated with the upregulation of genes controlling autophagy. IRF4 was required for the expansion and function of DKO effector Tregs.
Conclusions
This work uncovers the existence of mechanisms that, by acting on IRF4, can fine-tune the function and survival of effector Tregs in lupus. These studies suggest that the existence of a powerful effector Treg compartment that successfully survives in an unfavorable inflammatory environment could limit disease development.
CD4+ Foxp3+ regulatory T cells (Tregs) are critical for the maintenance of peripheral tolerance [1]. Defects in Treg numbers and/or function have been observed in autoimmune disorders such as Systemic Lupus Erythematosus (SLE), a disease characterized by abnormalities in multiple TH subsets, including TH17 and TFH cells, and by a great degree of clinical heterogeneity [2–5]. Recent studies have demonstrated the existence of specialized Foxp3+ Treg populations, termed effector Tregs, which are recruited to sites of inflammation and are critical for the control of inflammatory responses [6, 7]. Effector Tregs exhibit enhanced expression of key activation markers such as CD44, ICOS, and GITR and produce high-levels of IL-10. Effector Tregs adapt to unique inflammatory environments by acquiring specific migratory and functional properties [6, 7]. For instance, follicular regulatory T (TFR) cells share the expression of CXCR5 with follicular T helper (TFH) cells and localize within the B cell follicles where they control TFH-mediated responses [8, 9]. Coordinated development of TH subsets and the corresponding effector Tregs in response to TCR engagement and environmental cues is essential for the effective restoration of immune homeostasis after an inflammatory response has occurred.
Effector Tregs employ survival mechanisms that are distinct from those utilized by quiescent CD44lo CD62Lhi central Tregs. While survival of central Tregs is predominantly controlled by IL-2, the abundance of effector Tregs is regulated by ICOS [6, 7]. Effector Tregs are furthermore more sensitive than central Tregs to the inhibitory effects of inflammatory mediators like interferons and are short-lived due to lower expression of the antiapoptotic molecule Bcl-2 [10, 11]. The limited life span of effector Tregs, however, could prove counterproductive in chronic inflammatory settings like those encountered in autoimmune diseases. Knowledge of the molecular pathways that enable effector Tregs to function and survive in complex inflammatory milieus could be of great clinical interest for the success of Treg-based therapeutics [12].
At a molecular level, the paired polarization of Treg and TH subsets involves the ability of Tregs to co-express Foxp3 and key transcriptional regulators that are normally associated with the corresponding TH subset [6, 7]. Interferon Regulatory Factor 4 (IRF4) plays a unique role in effector Tregs. Although IRF4 expression in Tregs was initially shown to be critical for the inhibition of TH2 responses, IRF4 has now emerged as an essential controller of all effector Tregs [10, 13]. Fundamental to the function of IRF4 in effector Tregs is its capacity to directly control the expression of Blimp1 and to cooperate with Blimp1 in driving the expression of key effector Treg molecules such as IL-10 [10, 13]. The IRF4-Blimp1 axis is also responsible for the short-lived phenotype of effector Tregs via downregulation of Bcl-2 [10]. IRF4 has also recently been shown to be an essential mediator of TCR-dependent Treg activation in the periphery [14, 15]. Whether under chronic inflammatory conditions, effector Tregs can modulate the function of IRF4 in order to exploit its powerful functional program while lessening its inhibitory impact on their survival is not known.
Amongst IRF4 interacting partners, there is a small family of proteins consisting of two homologous proteins, DEF6 and SWAP-70, which are differentially expressed in TH cells and B cells [16–19]. Both DEF6 and SWAP-70 can interact with IRF4 and selectively restrict its ability to bind to specific subsets of target genes [20, 21]. Studies in TH cells have demonstrated that the function of DEF6 can be controlled in a TCR-dependent manner [22, 23]. Mice lacking both DEF6 and SWAP-70 (Double-knockout=DKO mice) develop a lupus-like disease characterized by the aberrant expansion of TH17 and TFH cells and robust humoral responses ([20] and unpublished observations). Despite displaying many of the hallmarks of lupus including autoantibody production, however, DKO mice do not have a reduced lifespan. Here we demonstrate that Tregs express both DEF6 and SWAP-70 and that the concomitant absence of these two molecules leads to increased numbers of Tregs, which acquire an effector phenotype in a cell-intrinsic manner. DKO effector Tregs accumulate as inflammation progresses due to an enhanced ability to express genes involved in regulating autophagy. We furthermore show that both the function and the expansion of DKO effector Tregs are dependent on IRF4. Thus, DEF6 and SWAP-70 represent important regulatory proteins controlling the access of effector Tregs to gene expression programs, which can maximize their fitness under chronic inflammatory conditions.
Materials and methods
Mice
C57BL/6, B6.SJL (CD45.1), Rag1−/− were obtained from Jackson Laboratory. To generate Def6tr/tr Swap-70−/− (DKO) mice, Def6tr/tr mice were crossed with Swap70−/− mice that had been backcrossed onto the C57BL/6 background as previously described [20]. Foxp3-IRES-mRFP (FIR) mice [24] were obtained from the Jackson Laboratory and crossed with DKO mice to generate FIR-DKO mice. BLIMP-YFP-10BiT double reporter mice were obtained from S. Kaech (Yale University, CT) and have been described previously [25, 26]. BLIMP-YFP-10BiT mice were crossed with DKO mice to generate BLIMP-YFP-10BiT DKO mice. Foxp3-YFP-Cre mice [27] were obtained from A. Rudensky (Memorial Sloan Kettering Cancer Center, NY) and IRF4fl/fl [28] were obtained from U.Klein (Columbia University, NY). Foxp3-YFP-Cre mice were crossed with IRF4fl/fl to produce Foxp3Cre/+ IRF4fl/fl and Foxp3Cre/y IRF4fl/fl mice. These mice were further crossed with DKO mice to produce Foxp3Cre/+ IRF4fl/fl DKO and Foxp3Cre/y IRF4fl/fl DKO mice. All mice used in the experiments were kept under specific pathogen–free conditions. The experimental protocols were approved by the Institutional Animal Care and Use Committee of The Hospital for Special Surgery.
Antibodies and flow cytometry
The following monoclonal antibodies were used for multiparameter flow cytometry: anti-CD4 (RM4-5, Biolegend), anti-CD25 (PC61.5, eBioscience), anti-CCR6 (29-2L17, Biolegend), anti-CD103 (2E7, Biolegend), anti-Ly5.1 (A20, Biolegend), anti-Ly5.2 (104, Biolegend), anti-CTLA4 (UC10-4B9, eBioscience), anti-Ki67 (solA15, eBioscience), anti-Bcl-2 (BCL10C4, Biolegend), anti-ICOS (7E.17G, eBioscience), anti-GITR (DTA-1, eBioscience), anti-Helios (22F6, Biolegend), anti-Neuropilin (FAB566A, R&D systems), anti-PD1 (J43, eBioscience), anti-CXCR5 (2G8, BD Biosciences). The Foxp3 staining kit was used for staining for CTLA-4, Ki67, Helios, Bcl-2 and Bcl6. Stained cells were analyzed on FACS Canto (Becton Dickinson) and were sorted on FACS Aria (Becton Dickinson). Data were analyzed with Flowjo software.
Real-time RT-PCR
Total RNA was isolated from sorted Treg cells using RNeasy Plus Mini kit (QIAGEN). cDNAs were prepared and analyzed for the expression of the gene of interest by real-time PCR using a SYBR Green PCR master mix kit (Applied Biosystems). The PCR primers for Foxp3, Bag3, and Prdm1 were purchased from QuantiTect Primer Assay (QIAGEN). The expression of each gene was normalized to the expression of β-actin or Cyclophilin A. The primer sequence of Def6 and Swap-70 are as follows: Def6 forward, 5′-CAAGGTTACATGCCCTACCTCAA-3′; Def6 reverse, 5′- CAGTCAGGGTCCAGCACAACT-3′; Swap-70 forward, 5′-TCCTTTCCCATAACCTGTGC-3′; Swap-70 reverse, 5′-ATCTTGTCAAAGTTGTCTTGGAC-3′.
Cell extracts and Western blotting
Whole cell extracts were prepared from FACS-sorted WT and DKO RFP+ CD4+ T cells (Tregs) as described previously [20]. Sorted Tregs were incubated for 1 hr at 37°C with or without 50nM Bafilomycin A1 (B1793; Sigma) for blockade of lysosome function. Primary antibodies used for Western blot analyses were as follows: anti-LC3 A/B (D3U4C, Cell Signaling, 12741), anti-actin (I-19, SantaCruz, SC-1616), anti-SWAP-70 (Q-28, Santa Cruz Biotechnology, SC-81991), anti-LRG47/IRGM1 (A19, Santa Cruz Biotechnology, SC-11075), anti-GBP1-5 (H-300, Santa Cruz Biotechnology, SC-28579), and anti-Tubulin (D66, Sigma, T0198). Anti-DEF6 antibody was generated in rabbit as previously described [18].
Transmission electron microscopy
0.5×106 CD4+ RFP+ (Treg) cells were sorted from FIR-WT and FIR-DKO mice and fixed in 1.5 ml eppendorf tubes by gently overlaying the fixative solution (2.5% gluteraldehyde in 100 mM sodium cacodylate, pH 7.43) and post-fixed in 1% osmium tetroxide in 100 mM sodium cacodylate, pH 7.43, followed by 1% uranyl acetate and further processed at Weill Cornell electron microscopy facility. All grids were viewed on a JEOL JEM 1400 transmission electron microscope at 120 kV. Autophagic vacuoles were identified by visual inspection of the micrographs using previously established criteria.
Bone–marrow Chimeras
To generate mixed bone marrow chimeras, 8×106 T/B-cell-depleted bone marrow cells were injected into Rag1−/− mice, which were lethally irradiated (875 rads). CD45.1+ wild-type bone marrow cells were mixed with an equal number of either CD45.2+ WT or DKO bone marrow cells and injected into Rag1−/− recipients via retro-orbital injection. Recipient mice were analyzed 8–10 wks after the reconstitution.
In vitro Treg suppression assay
FACS-sorted WT or DKO Treg cells were incubated with various ratios of FACS-sorted naive WT CD4+ T cells that were labeled with CFSE (4 μM) in the presence of αCD3 and αCD28 coated dynabeads (Invitrogen). After 72 hours the proliferation of effector CD4+ T cells was measured by CFSE dilution.
Statistics
Comparisons between different groups were done with unpaired two-tailed Student’s t-tests. All P values <0.05 were considered significant. Statistical analysis was performed using Graphpad Prism 5.
RESULTS
Accumulation of effector Tregs in mice lacking DEF6 and SWAP-70
DEF6 and SWAP-70 are the only two members of a unique family of molecules that share a high degree of sequence and structural similarity [16–19]. SWAP-70 has previously been detected in Tregs [29]. To assess whether DEF6 is also present in this compartment we utilized Foxp3-IRES-mRFP (FIR-WT) mice in which expression of RFP marks Foxp3+ cells [24]. In thymocytes, DEF6 was detected in both Treg and non-Treg populations while SWAP-70 was primarily expressed in Tregs (Supplementary Fig. 1A). Analysis of splenic CD4+ T cells confirmed that while naïve TH cells express only DEF6, both DEF6 and SWAP-70 are present in CD4+ Foxp3+ Tregs (Supplementary Fig. 1B–C).
The distinctive pattern of expression of DEF6 and SWAP-70 in Tregs suggested that these molecules might regulate Treg function. To address this possibility we employed mice lacking DEF6 and SWAP-70 (termed Double-Knock-out=DKO) [20]. WT and DKO mice contained similar frequencies of thymic CD4+Foxp3+ Tregs (Supplementary Fig. 2A–B) [30]. DKO mice, however, exhibited an expansion of Tregs in both spleens and lymph nodes as compared to WT mice or to mice lacking only DEF6 or only SWAP-70 (Fig. 1A and Supplementary Fig. 2C–D). This effect could already be observed in young mice and markedly increased with age (Fig. 1B and Supplementary Fig. 2C). Thus, the absence of DEF6 and SWAP-70 leads to an expansion of the peripheral Treg compartment.
Figure 1. Accumulation of effector Tregs in DKO mice.
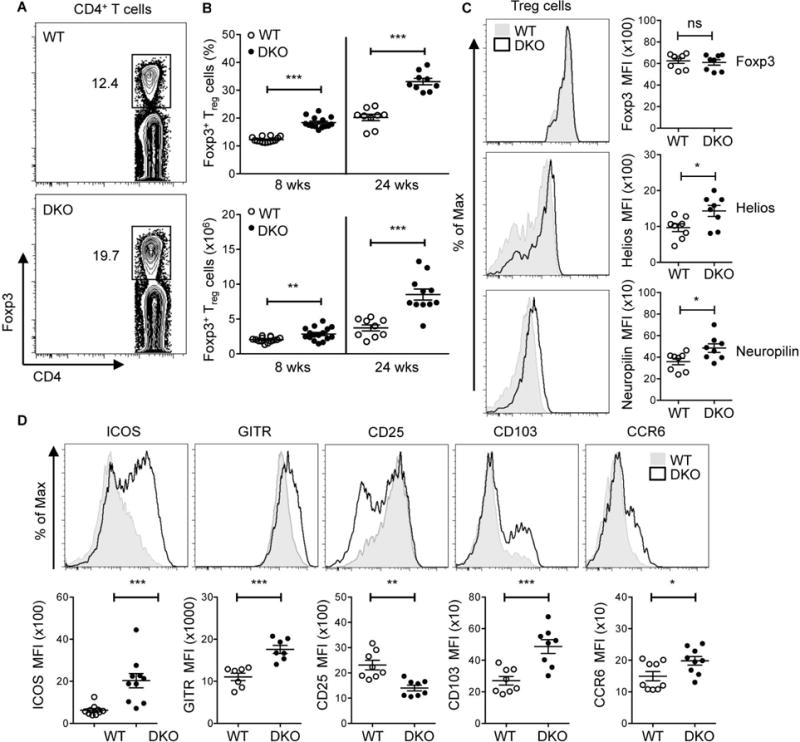
A. Representative flow cytometric analysis of the expression of Foxp3 on CD4+ T cells in WT and DKO mice. B. Percentages (top panel) and cell numbers (bottom panel) of CD4+Foxp3+ (Treg) cells in the spleen of 8-12 wks old mice (termed 8 wks) and >24 wks (termed 24 wks) old WT and DKO mice. Data are pooled from at least five independent experiments (8 wks, n=14 and 24 wks, n=9). C. FACS histograms and Mean Fluorescence Intensity (MFI) showing the expression of Foxp3, Helios, and Neuropilin on CD4+Foxp3+ (Treg) cells in the spleen of 8 wks old WT and DKO mice. Representative plots of at least three independent experiments. D. FACS histograms and Mean Fluorescence Intensity (MFI) of Treg cell-associated molecules (ICOS, GITR, CD25, CD103 and CCR6) on CD4+Foxp3+ (Treg) cells from 8-12 wks old WT and DKO spleens. Data are pooled from at least three independent experiments (n=7-10). Each symbol represents an individual mouse. Error bars represent the mean ± s.e.m., *P<0.05, **P<0.01, ***P<0.001 (unpaired, two-tailed Student’s t-test). ns, not significant.
To assess whether the lack of DEF6 and SWAP-70 alters the phenotype of Tregs we analyzed the expression of key Treg molecules. While Foxp3 levels were similar between WT and DKO Tregs, DKO Tregs expressed high levels of both Helios and Neuropilin-1, markers that are normally associated with thymic-derived Tregs (Fig. 1C). In vitro suppression assays demonstrated similar suppressive activities of WT and DKO Tregs (Supplementary Fig. 3). An evaluation of the expression of ICOS, GITR, surface and intracellular CTLA4, CD103, and CCR6, however, revealed that DKO Tregs in spleens and lymph nodes expressed higher surface levels of these effector Treg markers as compared to WT Tregs (Fig. 1D, Supplementary Fig. 4, and data not shown). Consistent with an effector Treg phenotype, splenic DKO Tregs expressed lower levels of CD25 than WT Tregs (Fig. 1D). Thus the lack of DEF6 and SWAP-70 is accompanied by an expansion of effector Tregs in the periphery, which can be observed even in young mice.
The Blimp1-IL10 gene module is upregulated in DKO Tregs
One of the hallmarks of all effector Tregs is their ability to produce high-levels of IL-10 [6, 7, 31]. IL-10 production by effector Tregs is controlled by IRF4 via its dual ability to upregulate Blimp1 and to directly bind to the IL-10 regulatory regions together with Blimp1 [10]. In line with the finding that DEF6 and SWAP-70 regulate the activity of IRF4 in non-Treg compartments [20, 21], sorted Tregs from DKO mice expressed higher levels of both Blimp1 and IL-10 than WT Tregs (Fig. 2A). To further facilitate the evaluation of the Blimp1-IL-10 axis in vivo we crossed WT and DKO mice to Blimp1-YFP-10BiT dual reporter mice [25]. Blimp1-expressing cells in these mice can be identified by expression of YFP while IL-10 competent cells can be detected by evaluating the surface levels of Thy1.1. Based on the expression of YFP and Thy1.1, Tregs were classified as Blimp1−IL10− (Q1), Blimp1+IL10− (Q2) or Blimp1+IL10+ (Q3). The majority (~85%) of Tregs in WT mice were Blimp1−IL10−, indicative of their naïve phenotype, ~10% were Blimp1+IL-10− and very few cells were Blimp1+IL10+. In contrast, DKO mice exhibited a marked increase in the frequencies of Blimp1+IL10− and Blimp1+IL10+ cells in both spleens and lymph nodes (Fig. 2B–C and data not shown). Thus the Blimp1-IL-10 gene module is highly upregulated in DKO Tregs.
Figure 2. Upregulation of the Blimp1-IL10 axis in DKO Tregs.
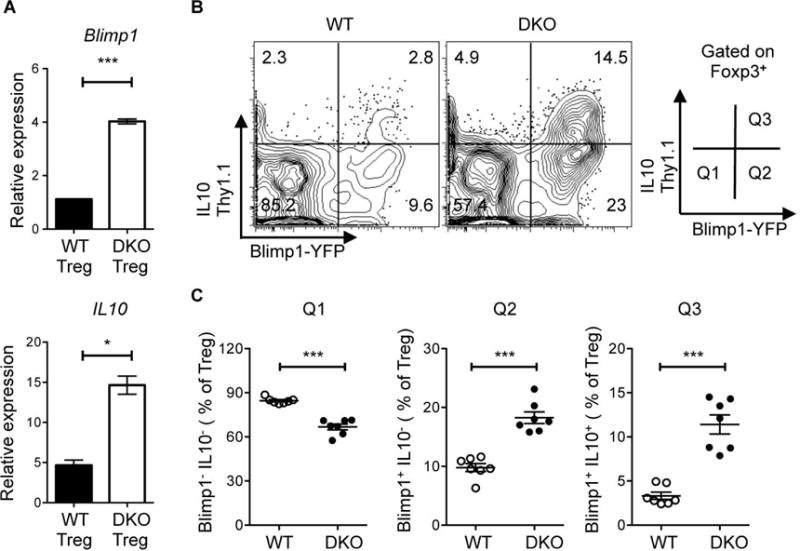
A. Quantitative RT-PCR analysis of the expression of Blimp1 and IL10 mRNA in CD4+Foxp3+ (Treg) cells sorted from 8 wks and 24 wks old Foxp3-IRES-mRFP (FIR)-WT and FIR-DKO mice. Data are normalized relative to the expression of cyclophilin A and are representative of at least two independent experiments. B. Representative flow cytometric analysis and gating strategy of the expression of Blimp1 and IL10 on CD4+ Foxp3+ (Treg) cells in the spleen of WT and DKO Blimp1-YFP and IL10-Thy1.1 double-reporter mice. C. Percentage of WT and DKO Treg cells in Q1 (Blimp1−IL10−), Q2 (Blimp1+IL10−) and Q3 (Blimp1+IL10+). Data in B and C are pooled from at least three independent experiments, each with two to three mice per group. Each symbol represents an individual mouse (n=7). Error bars indicate the mean ± s.e.m., *P<0.05, **P<0.01, ***P<0.001 (unpaired, two-tailed Student’s t-test). ns, not significant.
DKO mice exhibit an altered TFH/TFR balance
Amongst effector Treg subsets, TFR cells play a fundamental role in inhibiting TFH-mediated responses and the TFH/TFR ratio has been proposed to be a critical predictor of the levels of humoral responses [8, 9, 32]. Given that DKO mice spontaneously develop autoantibodies and systemic autoimmunity together with an aberrant accumulation of TFH cells, we directly assessed the effects of the lack of DEF6 and SWAP-70 on the TFR compartment. Splenocytes from WT mice and young unimmunized DKO mice were evaluated for the presence of CD4+CXCR5+PD1+cells. We further subdivided this population based on the expression of Foxp3 or Bcl6, by performing intracellular staining, to confirm their identities as TFH or TFR cells (Fig. 3A). We then assessed the TFH/TFR ratio over time. DKO mice exhibited an increased TFH/TFR ratio, which became particularly striking with age (Fig. 3B). To determine whether the alterations in the TFH/TFR ratio were associated with defective expansion of the TFR compartment, we directly evaluated the number of TFR cells. Unlike the striking expansion exhibited by DKO Tregs that had not acquired a TFR phenotype, the number of DKO TFR cells displayed only a minimal increase with age (Fig. 3C).
Figure 3. DKO mice exhibit an altered TFH and TFR ratio.
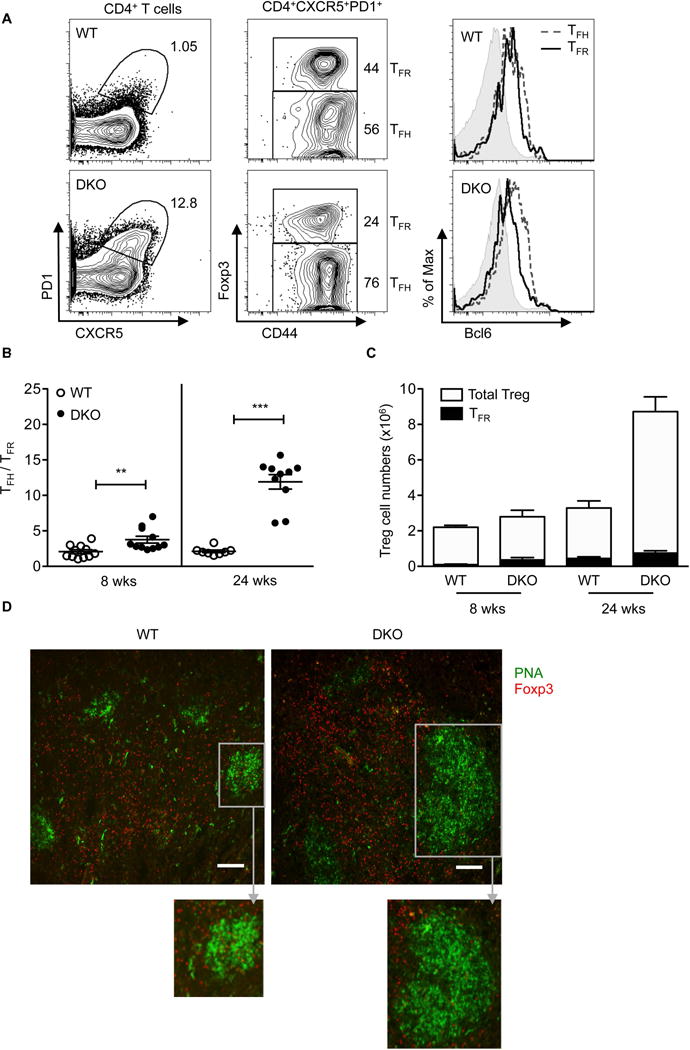
A. Representative FACS plots of PD1 and CXCR5 expression (left panel) on CD4+ T cells in 8 wks old WT and DKO mice. (Middle panel) CD4+CXCR5+PD1+ cells were further fractionated into Foxp3+ (TFR= CD4+CXCR5+PD1+CD44+FOXP3+) and Foxp3− (TFH= CD4+CXCR5+PD1+CD44+FOXP3−) cells. (Right panel) Representative histograms showing Bcl6 expression in TFH (dotted lines), TFR (black solid line), and naïve (gray shaded, CD4+CXCR5−PD1−FOXP3− CD44−) cell populations. Data are representative of at least three independent experiments. B. TFH/TFR ratio in spleens of 8 wks and 24 wks old WT and DKO mice. Data are pooled from at least five independent experiments (8 wks, n=11 and 24 wks, n=8-10). C. Total Treg and TFR cell numbers in 8 and 24 wks old WT and DKO mice. Data are pooled from at least five independent experiments (n=8-12). Each symbol represents an individual mouse. Error bars indicate the mean ± s.e.m., *P<0.05, **P<0.01, ***P<0.001 (unpaired, two-tailed Student’s t-test). ns, not significant. D. Representative immunofluorescence images of splenic sections from 24 wks old WT and DKO mice (two independent experiments, n=2-3). PNA, green: Foxp3, red, scale bar: 100μm. Inset (gray box) represents magnified view of PNA+ germinal center.
To confirm these findings we also performed immunofluorescence imaging of spleens from WT and DKO mice (Fig. 3D). Foxp3+ cells were located both outside and within the small GCs that are detected in older WT mice. In contrast, DKO mice exhibited a marked accumulation of Foxp3+ Tregs outside the GCs but only few Foxp3+ cells localized within the robust GCs that these mice spontaneously develop. Thus, unlike other DKO Tregs, DKO TFR cells do not expand as markedly and fail to accumulate to levels commensurate to those of DKO TFH cells, the corresponding TH subset that they need to restrain.
Expansion of effector Tregs in DKO mice is cell-intrinsic
Accumulation of effector Tregs in DKO mice could be observed even in young mice prior to the development of systemic autoimmunity suggesting that the expansion of DKO Tregs was not simply a response to the inflammation that develops in these mice over time. To directly assess whether accumulation of effector Tregs in DKO mice is due to cell-intrinsic effects, we generated mixed bone marrow chimeras by transferring a mixture of equal numbers of bone marrow cells from CD45.1+ wild-type and CD45.2+ DKO donors into irradiated Rag1−/− recipient mice (Fig. 4A). Control chimeras were also generated by transferring a mixture of equal numbers of CD45.1+ wild-type and CD45.2+ wild-type bone marrow cells (Fig. 4A). Eight weeks after transfer, we evaluated the recipient mice for the presence of Tregs in the spleen and peripheral lymph nodes. CD45.2+CD4+ DKO T cells contained an increased proportion of CD4+Foxp3+ cells as compared to CD45.1+CD4+ WT T cells. CD45.2+CD4+Foxp3+ T cells also demonstrated higher levels of expression of effector Treg markers such as ICOS, GITR, and CCR6 (Fig. 4B). The lack of DEF6 and SWAP-70, therefore, leads to the cell-intrinsic expansion of effector Tregs.
Figure 4. Accumulation of effector DKO Tregs is cell-intrinsic.
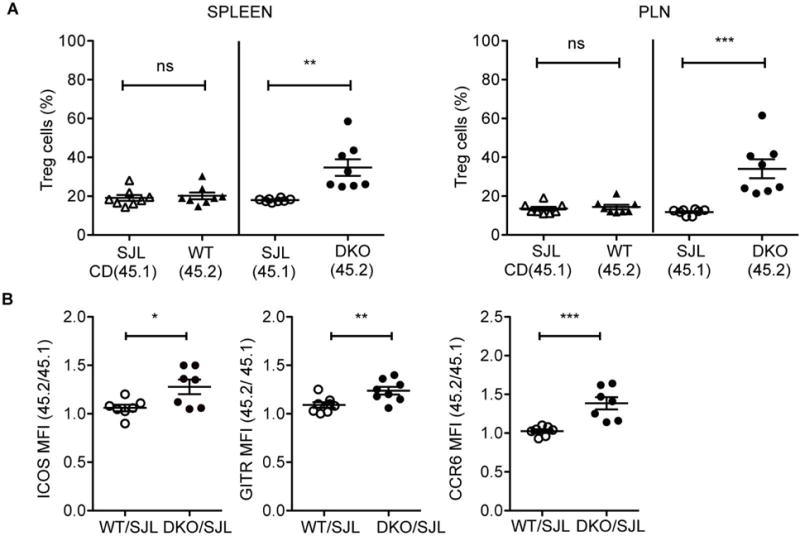
A. Frequency of Foxp3+ (Treg) cells among CD4+ T cells of each genotype in the spleen and peripheral lymph nodes (PLN) of mixed bone marrow (BM) chimeric mice generated by transferring 1:1 ratio of bone marrow cells from 45.1+ (SJL) WT and 45.2+ DKO mice or control 45.1+ (SJL) WT and 45.2+ WT mice. B. Fold changes in the MFI of Treg-associated markers (GITR, ICOS, and CCR6) on 45.2+ compared to 45.1+ Treg cells in the PLNs of BM chimeric mice. Data are pooled from two independent experiments with at least three mice in each group. Each symbol represents an individual mouse (n=7-8). Error bars indicate the mean ± s.e.m., *P<0.05, **P<0.01, ***P<0.001 (unpaired, two-tailed Student’s t-test). ns, not significant.
Accumulation of DKO Tregs is associated with enhanced autophagy
While expression of Blimp1 by Tregs enables them to upregulate a number of important effector molecules, it also endows them with a shortened life-span due to the ability of Blimp1 to downregulate Bcl-2 [10]. The accumulation of Blimp1-expressing effector Tregs in DKO mice was thus surprising leading us to evaluate the expression of Bcl-2 in DKO Tregs. Interestingly, DKO Tregs still exhibited markedly lower Bcl-2 levels than WT Tregs (Fig. 5A). WT and DKO Tregs furthermore did not exhibit any differences in their proliferative capabilities based on the frequencies of CD4+Foxp3+ cells expressing Ki67, a cell cycle-associated antigen expressed in proliferating but not quiescent cells (Fig. 5B). Thus the accumulation of DKO Tregs occurs despite low levels of Bcl-2 and is not due to an increased proliferative capability of these cells.
Figure 5. DKO effector Treg cells exhibit enhanced autophagy.
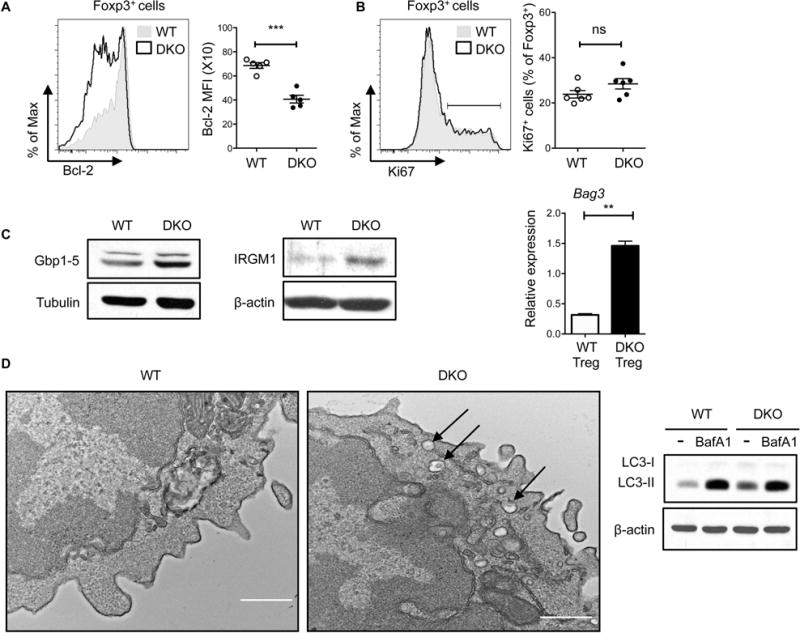
A. Bcl-2 expression in splenic Tregs from 8 wks old WT and DKO mice. Data are pooled from three independent experiments (n=5). B. Ki67 expression on splenic Tregs from 8 wks WT and DKO mice. Data are pooled from at least three independent experiments (n=6). C. Western blot of Gbp1-5 (left panel) and IRGM1 (middle panel) in sorted Tregs from 8 wks old FIR-WT and FIR-DKO mice. (Right panel) Quantitative RT-PCR analysis of Bag3 mRNA in sorted Tregs from FIR-WT and FIR-DKO mice. Data are normalized to the expression of cyclophilin A. Data are representative of at least two independent experiments. D. Electron microscopic images of sorted Tregs from FIR-WT and FIR-DKO mice. Arrows denote double membrane autophagosomes. Images are representative of ten cells (at least five fields were imaged from single cell) from each group of mice. Scale bar, 500nm. (Right panel) Western blot of LC3-I and LC3-II in sorted WT and DKO Tregs treated for 1 hr with DMSO or Bafilomycin A1 (BafA1, 50nM). Data are representative of at least two independent experiments. Each symbol represents an individual mouse. Error bars indicate the mean ± s.e.m., * P<0.05, **P<0.01, ***P<0.001 (unpaired, two-tailed Student’s t-test). ns, not significant.
The ability of DKO Tregs to accumulate over time raised the possibility that these Tregs utilize a distinct set of survival mechanisms in order to expand under inflammatory conditions. To investigate this possibility we sorted RFP+ Treg cells from WT and DKO mice and evaluated their gene expression profile by RNASeq analysis. A total of 114 genes (>2 fold) were differentially expressed between WT and DKO Tregs, 31 of those genes were upregulated in the DKO Tregs. Gene Ontology analysis revealed that several of the differentially expressed genes belong to a group of interferon-induced GTPases [33] that are important in vesicular trafficking, phagocytosis and neutrophil activation (data not shown). Prominent amongst the transcripts were a number of genes known to be involved in the regulation of autophagy, including members of the GBP (Guanylate Binding Proteins) and IRG (Immunity-related GTPase) family and Bag3, a gene implicated in Chaperone-assisted selective autophagy (CASA) [34, 35]. Differential expression of these genes in WT and DKO Tregs was confirmed by Western blotting and/or quantitative real-time RT-PCR (Fig. 5C).
In view of the ability of autophagy to promote the survival of cells under stressful conditions [36] we next considered the possibility that DKO Tregs might upregulate this critical process. TEMs of sorted WT and DKO Tregs indeed exhibited an accumulation of autophagic vacuoles in DKO Tregs (Fig. 5D). Furthermore, evaluation of the levels of LC3 demonstrated that DKO Tregs exhibit higher levels of the lipidated form of LC3 (LC3-II) than WT Tregs (Fig. 5D). The increased expression of LC3-II was due to increased autophagic flux since DKO Tregs exhibited increased LC3-II expression upon exposure to BafilomycinA1 (Fig. 5D). Taken together these studies support the idea that the enhanced ability of DKO Tregs to upregulate a transcriptional program controlling autophagy enables these Tregs to survive in the inflammatory milieu that accompanies the development of lupus.
The function and expansion of DKO effector Tregs are dependent on IRF4
Analysis of overrepresented sequencing motifs using XXMotif [37] in regions surrounding the transcription start site (−1kb to +200bp) of 176 genes induced in DKO Tregs (>1.4 fold) revealed an enrichment of sequence motif that is strikingly similar to IRF1 and IRF4 binding sites reported in the JASPAR database [38]. These motifs were found upstream of Gbp 2,4,6, Igtp, Iigp1 and Irgm2, among others (data not shown). Given the ability of DEF6 and SWAP-70 to interact with IRF4 and considering the enrichment of IRF-binding sites in DKO induced genes we wanted to directly evaluate whether the striking expansion of effector Tregs in DKO mice was dependent on this transcriptional regulator. To assess this possibility we crossed DKO mice with Foxp3Cre IRF4fl/fl mice to generate DKO mice selectively lacking IRF4 in Tregs. We first assessed the effects of the loss of IRF4 in heterozygous Foxp3Cre/+ IRF4fl/fl DKO female mice where, because of X chromosome inactivation, Tregs that lack IRF4 (Foxp3Cre−YFP+) can be directly compared to Tregs expressing IRF4 (Foxp3Cre−YFP−) within the same inflammatory milieu. Foxp3Cre/+ IRF4fl/fl female mice were used as controls. Appropriate deletion of IRF4 within the Foxp3Cre−YFP+ DKO compartment was confirmed by intracellular staining (Fig. 6A). An examination of YFP+Foxp3+ versus YFP−Foxp3+ populations in spleens and peripheral lymph nodes revealed a dramatic reduction in the ratio of YFP+ to YFP− Treg cells in DKO mice (Fig. 6B and data not shown). In addition, as compared to YFP−Foxp3+ cells, the remaining YFP+Foxp3+ DKO cells expressed much lower levels of effector Treg markers such as ICOS, GITR, CD103, and CCR6 (Fig. 6C). Thus, both the expansion and the activation of the DKO effector Tregs require IRF4.
Figure 6. Expansion of DKO effector Tregs is dependent on IRF4.
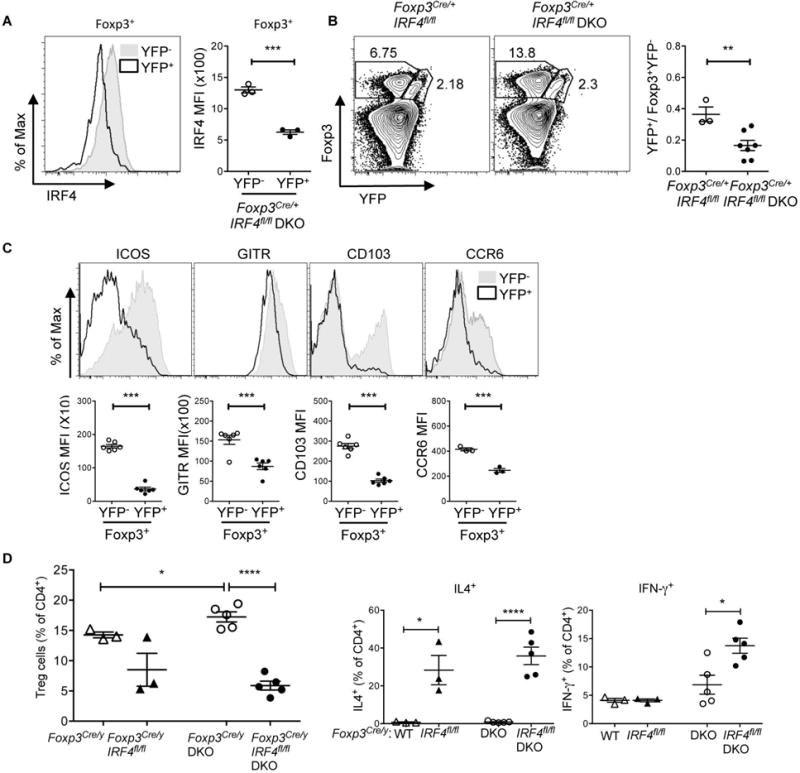
A. Representative FACS histograms and MFI (right panel) of IRF4 on Foxp3+YFP− and Foxp3+YFP+ Treg cells in the spleen of 8 wks old Foxp3Cre/+ IRF4fl/fl DKO female mice. Data are representative of two independent experiments (n=3). B. Flow cytometry plots of Foxp3 and YFP on CD4+ T cells and ratios of Foxp3+YFP+ versus Foxp3+YFP− Treg cells in spleen of Foxp3Cre/+ IRF4fl/fl and Foxp3Cre/+ IRF4fl/fl DKO mice. Data are pooled from at least two independent experiments (n=3-7). C. FACS histograms and MFI of Treg cell-associated molecules (ICOS, GITR, CD103 and CCR6) on Foxp3+YFP− and Foxp3+YFP+ Treg cells in the spleens of Foxp3Cre/+ IRF4fl/fl DKO mice. Data are pooled from at least two independent experiments (n=3-6). C. Percentages of CD4+Foxp3+ (Treg) cells in the spleens of (5-12 wks old) Foxp3Cre/y, Foxp3Cre/y IRF4fl/fl, Foxp3Cre/y DKO and Foxp3Cre/y IRF4fl/fl DKO male mice. Data are pooled from at least three independent experiments (n=4-6). D. Percentages showing the intracellular expression of IL-4, and IFN-γ in splenocytes from the indicated mice. Data are pooled from at least two independent experiments (n=4-5). Each symbol represents an individual mouse. Error bars indicate the mean ± s.e.m., * P<0.05, **P<0.01, ***P<0.001 (unpaired, two-tailed Student’s t-test). ns, not significant.
An examination of Foxp3Cre/y IRF4fl/fl male DKO mice, which lack IRF4 in all Tregs, confirmed that the frequencies of CD4+Foxp3+ T cells were dramatically reduced as compared to Foxp3Cre/y male DKO mice (Fig. 6D). Similarly to what had been observed in the YFP+ compartment of Foxp3Cre/+ IRF4fl/fl DKO female mice, the remaining Tregs expressed lower levels of ICOS (data not shown). In line with a strong requirement for the presence of IRF4 in DKO Tregs to limit the inflammatory potential of DKO TH cells, Foxp3Cre/yI RF4fl/fl DKO mice exhibited a shortened survival and displayed an expansion of cytokine producing TH cells, which, compared to Foxp3Cre/y IRF4fl/fl mice, encompassed increases not only in IL-4 but also in IFN-γ production (Fig. 6D and data not shown). Thus, effector Tregs in DKO mice are critical for dampening the function of CD4+ Foxp3− DKO T cells.
DISCUSSION
Acquisition of specialized effector programs in the periphery is critical for the ability of Tregs to restore homeostasis in response to a wide range of inflammatory conditions [6, 7, 31]. Despite their phenotypic diversity, all effector Tregs rely on one critical transcription factor, IRF4 [10]. Here we demonstrate that the activity of IRF4 in Tregs can be modulated by DEF6 and SWAP-70, the only two members of a unique family of immunoregulators. Both DEF6 and SWAP-70 contain a number of signaling motifs that could be targeted by TCR signaling in Tregs [18, 39–43]. These molecules could thus serve as a useful relay system whereby a Treg, by regulating the interaction of these molecules with IRF4, can precisely tailor IRF4 activation to the degree of TCR engagement. By ensuring that IRF4 activity is commensurate to the extent and the chronicity of the inflammatory insult such a system can help fine-tune effector Treg function and survival during the development of lupus and help limit disease severity.
Our ability to selectively delete IRF4 in Tregs in a spontaneous model of lupus enabled us to specifically assess the role of IRF4 in the regulation of effector Tregs in this disease. Particularly useful was the analysis of Foxp3Cre−YFP/+ IRF4fl/fl DKO female mice where we could directly compare IRF4 sufficient and deficient Tregs exposed to the same inflammatory milieu. These studies revealed a marked loss of key effector Treg markers by IRF4 deficient Tregs supporting the notion that IRF4 is indeed a crucial regulator of the effector Treg program. Deletion of IRF4 in all Tregs in Foxp3Cre/y IRF4fl/fl DKO male mice confirmed that its presence in Tregs was necessary to dampen the inflammatory function of TH cells in these mice. The shortened survival of Foxp3Cre/y IRF4fl/fl DKO mice was accompanied by increased production of both IL-4 and IFN-γ indicating a more complex inflammatory environment than that exhibited by Foxp3Cre/y IRF4fl/fl mice whose limited lifespan was primarily marked by exaggerated TH2 inflammatory responses as previously reported [13].
In addition to acquiring an effector phenotype, DKO Tregs also exhibited a cell-intrinsic ability to expand leading to a remarkable accumulation of effector Tregs over time. This was surprising in view of the known ability of the IRF4-Blimp1 axis to impart effector Tregs with a shortened life span [10]. The expansion of effector Tregs in DKO mice was IRF4-dependent and was marked by the upregulation of autophagy, a process known to facilitate cell survival under stressful conditions [44]. Thus, by restricting the ability of IRF4 to upregulate autophagy-dependent pathways, DEF6 and SWAP-70 can regulate the conversion of short-lived effector Tregs to more potent and longer-lived regulators. Consistent with a direct role for IRF4 in controlling this program, DKO Tregs upregulate a number of genes involved in the control of autophagy including members of the IRG and GBP family, which have previously been shown to be regulated by IRFs [45–47]. Intriguingly increased autophagy has previously been linked to the ability of autoreactive lupus T cells to survive [48] suggesting that, in lupus, this pathway can be exploited for both pathogenic and protective responses.
Unlike the remarkable accumulation of most effector Tregs, DKO mice exhibited only a minimal expansion of TFR cells resulting in an imbalanced TFH/TFR ratio over time. It is possible that the high levels of IL-21 produced by DKO TFH cells may contribute to this effect since IL-21 has been reported to decrease the frequencies and numbers of TFR cells [49]. Thus although the shared utilization of transcriptional modules should ensure the parallel differentiation of TH subsets and the corresponding effector Treg subset [6, 7, 31], the unique characteristics of local inflammatory milieus can overwhelm these regulatory systems and create selective imbalances in specific TH/Treg subset pairs. These complexities are likely to be highly relevant to the pathophysiology of human SLE and are well exemplified by the DKO lupus mouse model where the simultaneous defects in TFR cells can fuel the aberrant autoantibody production while the marked expansion of non-TFR effector Tregs could help limit tissue damage. These findings thus support the idea that the cellular and molecular networks that mediate tissue-injury do not fully overlap with those driving systemic autoimmunity and that these two components of lupus pathophysiology could be differentially targeted. Interestingly, pharmacologic inhibition of CaMK4 (calcium/calmodulin-dependent protein kinase IV) was recently shown to favor the recruitment of Tregs to the kidneys in the MRL/lpr lupus mouse model resulting in decreased kidney damage [50] suggesting that therapeutic interventions aimed at promoting the expansion/recruitment of effector Tregs into tissues could be effective at ameliorating end-organ damage.
Our findings carry potentially important clinical implications. Indeed, these studies suggest that despite the presence of autoimmune features such as dysregulated humoral responses, the existence of a powerful effector Treg compartment that successfully survives in an unfavorable inflammatory environment can effectively limit disease development. Such a scenario is highly relevant to diseases such as SLE whose clinical heterogeneity could be linked, at least in part, to the presence/absence of long-lived effector Tregs. The identification of the molecular pathways that endow effector Tregs with increased potency and survival may also provide valuable information for the development of appropriate therapies aimed at targeting Tregs for the treatment of SLE and, possibly, for a broader range of clinical settings including cancer and transplantation where manipulations of effector Tregs could also be beneficial.
Supplementary Material
A. Quantitative RT-PCR analysis of the expression of Def6, SWAP-70, and Foxp3 mRNA in thymic CD4+CD8−Foxp3+ (Treg), CD4+CD8−Foxp3− (CD4SP), CD8+CD4− (CD8SP), and CD4+CD8+ (DP) cells sorted from 4-wks old FIR-WT mice. Data are normalized relative to the expression of β-actin and are representative of four independent experiments. B. Quantitative RT-PCR analysis of the expression of Def6, SWAP-70, and Foxp3 mRNA in splenic CD4+CD44−62L+ (TH) and CD4+ Foxp3+ (Treg) cells sorted from 4-wks old FIR-WT mice. Data are normalized relative to the expression of β-actin and are representative of three independent experiments. Error bars represent mean ± s.e.m., *P<0.05, **P<0.01, ***P<0.001 (unpaired, two-tailed Student’s t-test). ns, not significant. C. Western blot analysis of the levels of DEF6, SWAP-70 in CD4+CD62L+RFP− (TH) and CD4+RFP+ (Treg) cells sorted from the spleen of 8 wks old FIR-WT and FIR-DKO mice. β-actin was used as a loading control. Data are representative of two independent experiments.
A. Flow cytometry analysis showing the expression of Foxp3 and CD25 on CD4+CD8− (CD4SP) thymocytes in 4-6 wks old WT and DKO mice. B. Percentages of CD4+Foxp3+CD25+ thymocytes in in WT and DKO mice. Data are pooled from two independent experiments (n=6). C. Percentages of Foxp3+ (Treg) cell among CD4+ T cells in the pooled peripheral lymph nodes (PLN) of 8 wks and 24 wks old WT and DKO mice (n=9-11). D. Percentages of Foxp3+ (Treg) cell among CD4+ T cells in the spleen of 8wks old WT, Def6tr/tr, S70−/− and DKO mice (n=6-10).
A. Proliferation of naïve WT T cells stimulated with magnetic beads covalently coupled to αCD3 and αCD28 in the presence of increasing ratios of Tregs derived from either WT or DKO mice was assessed by CFSE dilution. Data are representative of two independent experiments.
A. Representative FACS histograms and Mean Fluorescence Intensity (MFI) of CTLA4 on CD4+Foxp3+ (Treg) cells from 8-18 wks old WT and DKO spleens. CTLA4 was stained for both intracellular (i) and surface (s) expression. Data are pooled from two independent experiments (n=4-5). Each symbol represents an individual mouse. Error bars represent the mean ± s.e.m., **P<0.01, (unpaired, two-tailed Student’s t-test). ns, not significant.
Acknowledgments
Funding sources: The research was supported by an NIH T32 Rheumatology Training grant (UC), the Peter Jay Sharp Foundation, the Alliance for Lupus Research, the David Z. Rosensweig Genomics Research Center, the American Heart Association (YC), and the National Institutes of Health.
Footnotes
Competing Financial Interests: The authors declare no competing financial interests.
References
- 1.Rudensky AY. Regulatory T cells and Foxp3. Immunological reviews. 2011;241(1):260–268. doi: 10.1111/j.1600-065X.2011.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lourenco EV, La Cava A. Natural regulatory T cells in autoimmunity. Autoimmunity. 2011;44(1):33–42. doi: 10.3109/08916931003782155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sawla P, Hossain A, Hahn BH, Singh RP. Regulatory T cells in systemic lupus erythematosus (SLE); role of peptide tolerance. Autoimmunity reviews. 2012;11(9):611–614. doi: 10.1016/j.autrev.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Crispin JC, Kyttaris VC, Terhorst C, Tsokos GC. T cells as therapeutic targets in SLE. Nature reviews Rheumatology. 2010;6(6):317–325. doi: 10.1038/nrrheum.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nature reviews Rheumatology. 2012;8(6):337–347. doi: 10.1038/nrrheum.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gratz IK, Campbell DJ. Organ-specific and memory treg cells: specificity, development, function, and maintenance. Frontiers in immunology. 2014;5:333. doi: 10.3389/fimmu.2014.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol. 2014;14(3):154–165. doi: 10.1038/nri3605. [DOI] [PubMed] [Google Scholar]
- 8.Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, Srivastava M, Divekar DP, Beaton L, Hogan JJ, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nature medicine. 2011;17(8):975–982. doi: 10.1038/nm.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, Wang YH, Lim H, Reynolds JM, Zhou XH, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nature medicine. 2011;17(8):983–988. doi: 10.1038/nm.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M, Belz GT, Smyth GK, Busslinger M, Nutt SL, et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nature immunology. 2011;12(4):304–311. doi: 10.1038/ni.2006. [DOI] [PubMed] [Google Scholar]
- 11.Srivastava S, Koch MA, Pepper M, Campbell DJ. Type I interferons directly inhibit regulatory T cells to allow optimal antiviral T cell responses during acute LCMV infection. The Journal of experimental medicine. 2014;211(5):961–974. doi: 10.1084/jem.20131556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bluestone JA, Trotta E, Xu D. The therapeutic potential of regulatory T cells for the treatment of autoimmune disease. Expert opinion on therapeutic targets. 2015:1–13. doi: 10.1517/14728222.2015.1037282. [DOI] [PubMed] [Google Scholar]
- 13.Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, Corcoran L, Treuting P, Klein U, Rudensky AY. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458(7236):351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nature immunology. 2014;15(11):1070–1078. doi: 10.1038/ni.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, Ohkura N, Morikawa H, Poeck H, Schallenberg S, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. 2014;41(5):722–736. doi: 10.1016/j.immuni.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 16.Borggrefe T, Wabl M, Akhmedov AT, Jessberger R. A B-cell-specific DNA recombination complex. The Journal of biological chemistry. 1998;273(27):17025–17035. doi: 10.1074/jbc.273.27.17025. [DOI] [PubMed] [Google Scholar]
- 17.Hotfilder M, Baxendale S, Cross MA, Sablitzky F. Def-2, -3, -6 and -8, novel mouse genes differentially expressed in the haemopoietic system. British journal of haematology. 1999;106(2):335–344. doi: 10.1046/j.1365-2141.1999.01551.x. [DOI] [PubMed] [Google Scholar]
- 18.Gupta S, Lee A, Hu C, Fanzo J, Goldberg I, Cattoretti G, Pernis AB. Molecular cloning of IBP, a SWAP-70 homologous GEF, which is highly expressed in the immune system. Human immunology. 2003;64(4):389–401. doi: 10.1016/s0198-8859(03)00024-7. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka Y, Bi K, Kitamura R, Hong S, Altman Y, Matsumoto A, Tabata H, Lebedeva S, Bushway PJ, Altman A. SWAP-70-like adapter of T cells, an adapter protein that regulates early TCR-initiated signaling in Th2 lineage cells. Immunity. 2003;18(3):403–414. doi: 10.1016/s1074-7613(03)00054-2. [DOI] [PubMed] [Google Scholar]
- 20.Biswas PS, Gupta S, Stirzaker RA, Kumar V, Jessberger R, Lu TT, Bhagat G, Pernis AB. Dual regulation of IRF4 function in T and B cells is required for the coordination of T-B cell interactions and the prevention of autoimmunity. The Journal of experimental medicine. 2012;209(3):581–596. doi: 10.1084/jem.20111195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Q, Yang W, Gupta S, Biswas P, Smith P, Bhagat G, Pernis AB. IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity. 2008;29(6):899–911. doi: 10.1016/j.immuni.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biswas PS, Bhagat G, Pernis AB. IRF4 and its regulators: evolving insights into the pathogenesis of inflammatory arthritis? Immunological reviews. 2010;233(1):79–96. doi: 10.1111/j.0105-2896.2009.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becart S, Altman A. SWAP-70-like adapter of T cells: a novel Lck-regulated guanine nucleotide exchange factor coordinating actin cytoskeleton reorganization and Ca2+ signaling in T cells. Immunological reviews. 2009;232(1):319–333. doi: 10.1111/j.1600-065X.2009.00839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan YY, Flavell RA. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(14):5126–5131. doi: 10.1073/pnas.0501701102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parish IA, Marshall HD, Staron MM, Lang PA, Brustle A, Chen JH, Cui W, Tsui YC, Perry C, Laidlaw BJ, et al. Chronic viral infection promotes sustained Th1-derived immunoregulatory IL-10 via BLIMP-1. J Clin Invest. 2014;124(8):3455–3468. doi: 10.1172/JCI66108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, Weaver CT. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3- precursor cells in the absence of interleukin 10. Nature immunology. 2007;8(9):931–941. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- 27.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28(4):546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 28.Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, Ludwig T, Rajewsky K, Dalla-Favera R. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nature immunology. 2006;7(7):773–782. doi: 10.1038/ni1357. [DOI] [PubMed] [Google Scholar]
- 29.Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, Xiong H, Dolpady J, Frey AB, Ruocco MG, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. The Journal of experimental medicine. 2012;209(10):1723–1742. S1721. doi: 10.1084/jem.20120914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tai X, Erman B, Alag A, Mu J, Kimura M, Katz G, Guinter T, McCaughtry T, Etzensperger R, Feigenbaum L, et al. Foxp3 transcription factor is proapoptotic and lethal to developing regulatory T cells unless counterbalanced by cytokine survival signals. Immunity. 2013;38(6):1116–1128. doi: 10.1016/j.immuni.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cretney E, Kallies A, Nutt SL. Differentiation and function of Foxp3(+) effector regulatory T cells. Trends in immunology. 2013;34(2):74–80. doi: 10.1016/j.it.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nature immunology. 2013;14(2):152–161. doi: 10.1038/ni.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim BH, Shenoy AR, Kumar P, Bradfield CJ, MacMicking JD. IFN-inducible GTPases in host cell defense. Cell host & microbe. 2012;12(4):432–444. doi: 10.1016/j.chom.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gamerdinger M, Carra S, Behl C. Emerging roles of molecular chaperones and co-chaperones in selective autophagy: focus on BAG proteins. Journal of molecular medicine. 2011;89(12):1175–1182. doi: 10.1007/s00109-011-0795-6. [DOI] [PubMed] [Google Scholar]
- 35.Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol. 2012;24(1):21–31. doi: 10.1016/j.coi.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Molecular cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartmann H, Guthohrlein EW, Siebert M, Luehr S, Soding J. P-value-based regulatory motif discovery using positional weight matrices. Genome research. 2013;23(1):181–194. doi: 10.1101/gr.139881.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004;5(4):276–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- 39.Gupta S, Fanzo JC, Hu C, Cox D, Jang SY, Lee AE, Greenberg S, Pernis AB. T cell receptor engagement leads to the recruitment of IBP, a novel guanine nucleotide exchange factor, to the immunological synapse. The Journal of biological chemistry. 2003;278(44):43541–43549. doi: 10.1074/jbc.M308960200. [DOI] [PubMed] [Google Scholar]
- 40.Fos C, Becart S, Canonigo Balancio AJ, Boehning D, Altman A. Association of the EF-hand and PH domains of the guanine nucleotide exchange factor SLAT with IP(3) receptor 1 promotes Ca(2)(+) signaling in T cells. Science signaling. 2014;7(345):ra93. doi: 10.1126/scisignal.2005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Becart S, Balancio AJ, Charvet C, Feau S, Sedwick CE, Altman A. Tyrosine-phosphorylation-dependent translocation of the SLAT protein to the immunological synapse is required for NFAT transcription factor activation. Immunity. 2008;29(5):704–719. doi: 10.1016/j.immuni.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hey F, Czyzewicz N, Jones P, Sablitzky F. DEF6, a novel substrate for the Tec kinase ITK, contains a glutamine-rich aggregation-prone region and forms cytoplasmic granules that co-localize with P-bodies. The Journal of biological chemistry. 2012;287(37):31073–31084. doi: 10.1074/jbc.M112.346767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shinohara M, Terada Y, Iwamatsu A, Shinohara A, Mochizuki N, Higuchi M, Gotoh Y, Ihara S, Nagata S, Itoh H, et al. SWAP-70 is a guanine-nucleotide-exchange factor that mediates signalling of membrane ruffling. Nature. 2002;416(6882):759–763. doi: 10.1038/416759a. [DOI] [PubMed] [Google Scholar]
- 44.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marquis JF, Kapoustina O, Langlais D, Ruddy R, Dufour CR, Kim BH, MacMicking JD, Giguere V, Gros P. Interferon regulatory factor 8 regulates pathways for antigen presentation in myeloid cells and during tuberculosis. PLoS genetics. 2011;7(6):e1002097. doi: 10.1371/journal.pgen.1002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Yao ZQ, Moorman JP, Xu Y, Ning S. Gene expression profiling identifies IRF4-associated molecular signatures in hematological malignancies. PloS one. 2014;9(9):e106788. doi: 10.1371/journal.pone.0106788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Briken V, Ruffner H, Schultz U, Schwarz A, Reis LF, Strehlow I, Decker T, Staeheli P. Interferon regulatory factor 1 is required for mouse Gbp gene activation by gamma interferon. Mol Cell Biol. 1995;15(2):975–982. doi: 10.1128/mcb.15.2.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caza TN, Talaber G, Perl A. Metabolic regulation of organelle homeostasis in lupus T cells. Clinical immunology. 2012;144(3):200–213. doi: 10.1016/j.clim.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ding Y, Li J, Yang P, Luo B, Wu Q, Zajac AJ, Wildner O, Hsu HC, Mountz JD. Interleukin-21 promotes germinal center reaction by skewing the follicular regulatory T cell to follicular helper T cell balance in autoimmune BXD2 mice. Arthritis & rheumatology. 2014;66(9):2601–2612. doi: 10.1002/art.38735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koga T, Mizui M, Yoshida N, Otomo K, Lieberman LA, Crispin JC, Tsokos GC. KN-93, an inhibitor of calcium/calmodulin-dependent protein kinase IV, promotes generation and function of Foxp3(+) regulatory T cells in MRL/lpr mice. Autoimmunity. 2014;47(7):445–450. doi: 10.3109/08916934.2014.915954. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Quantitative RT-PCR analysis of the expression of Def6, SWAP-70, and Foxp3 mRNA in thymic CD4+CD8−Foxp3+ (Treg), CD4+CD8−Foxp3− (CD4SP), CD8+CD4− (CD8SP), and CD4+CD8+ (DP) cells sorted from 4-wks old FIR-WT mice. Data are normalized relative to the expression of β-actin and are representative of four independent experiments. B. Quantitative RT-PCR analysis of the expression of Def6, SWAP-70, and Foxp3 mRNA in splenic CD4+CD44−62L+ (TH) and CD4+ Foxp3+ (Treg) cells sorted from 4-wks old FIR-WT mice. Data are normalized relative to the expression of β-actin and are representative of three independent experiments. Error bars represent mean ± s.e.m., *P<0.05, **P<0.01, ***P<0.001 (unpaired, two-tailed Student’s t-test). ns, not significant. C. Western blot analysis of the levels of DEF6, SWAP-70 in CD4+CD62L+RFP− (TH) and CD4+RFP+ (Treg) cells sorted from the spleen of 8 wks old FIR-WT and FIR-DKO mice. β-actin was used as a loading control. Data are representative of two independent experiments.
A. Flow cytometry analysis showing the expression of Foxp3 and CD25 on CD4+CD8− (CD4SP) thymocytes in 4-6 wks old WT and DKO mice. B. Percentages of CD4+Foxp3+CD25+ thymocytes in in WT and DKO mice. Data are pooled from two independent experiments (n=6). C. Percentages of Foxp3+ (Treg) cell among CD4+ T cells in the pooled peripheral lymph nodes (PLN) of 8 wks and 24 wks old WT and DKO mice (n=9-11). D. Percentages of Foxp3+ (Treg) cell among CD4+ T cells in the spleen of 8wks old WT, Def6tr/tr, S70−/− and DKO mice (n=6-10).
A. Proliferation of naïve WT T cells stimulated with magnetic beads covalently coupled to αCD3 and αCD28 in the presence of increasing ratios of Tregs derived from either WT or DKO mice was assessed by CFSE dilution. Data are representative of two independent experiments.
A. Representative FACS histograms and Mean Fluorescence Intensity (MFI) of CTLA4 on CD4+Foxp3+ (Treg) cells from 8-18 wks old WT and DKO spleens. CTLA4 was stained for both intracellular (i) and surface (s) expression. Data are pooled from two independent experiments (n=4-5). Each symbol represents an individual mouse. Error bars represent the mean ± s.e.m., **P<0.01, (unpaired, two-tailed Student’s t-test). ns, not significant.