

ABSTRACT
Murine antibody 10H10 raised against human tissue factor is unique in that it blocks the signaling pathway, and thus inhibits angiogenesis and tumor growth without interfering with coagulation. As a potential therapeutic, the antibody was humanized in a two-step procedure. Antigen-binding loops were grafted onto selected human frameworks and the resulting chimeric antibody was subjected to affinity maturation by using phage display libraries. The results of humanization were analyzed from the structural perspective through comparison of the structure of a humanized variant with the parental mouse antibody. This analysis revealed several hot spots in the framework region that appear to affect antigen binding, and therefore should be considered in human germline selection. In addition, some positions in the Vernier zone, e.g., residue 71 in the heavy chain, that are traditionally thought to be crucial appear to tolerate amino acid substitutions without any effect on binding. Several humanized variants were produced using both short and long forms of complementarity-determining region (CDR) H2 following the difference in the Kabat and Martin definitions. Comparison of such pairs indicated consistently higher thermostability of the variants with short CDR H2. Analysis of the binding data in relation to the structures singled out the ImMunoGeneTics information system® germline IGHV1-2*01 as dubious owing to two potentially destabilizing mutations as compared to the other alleles of the same germline and to other human germlines.
KEYWORDS: tissue factor, antibody, humanization, affinity maturation, crystal structure, Vernier zone, germline, induced fit
Introduction
Monoclonal antibodies (mAbs) derived from hybridoma are typically humanized by replacing regions not required for antigen binding with the corresponding human sequence.1 The objective of humanization is to reduce immunogenicity while retaining the affinity of a parental antibody. Quite often, however, grafting of the complementarity-determining regions (CDRs) onto a human framework (FR) yields a molecule with reduced affinity, and additional engineering is required to improve antigen binding. Affinity maturation can be accomplished by a number of methods, including random or directed mutagenesis2 and approaches that reproduce somatic hypermutation in vitro.3
Here, we describe the humanization and affinity maturation of mAb 10H10 obtained from mouse hybridoma4 against human tissue factor (TF), a key initiator of the coagulation cascade. TF is a cell surface transmembrane receptor that binds and activates serine protease factor VII (FVII) and subsequently factor IX (FIX) and factor X (FX), leading to the generation of thrombin. Besides this, the TF-FVII complex induces cell signaling via activation of transmembrane G-protein-coupled protease activated receptors (PARs).5 The signaling pathway is related to tumor progression and metastasis,6 and mAbs blocking the pathway have promising therapeutic potential.
Unlike other anti-TF antibodies, mAb 10H10 specifically blocks activation of PAR2 by the TF-FVII binary complex without inhibiting coagulation.7 10H10 binds the extracellular domain (ECD) of TF with subnanomolar affinity and has demonstrated efficacy in animal models.8 The crystal structure of the 10H10 Fab in complex with the TF ECD revealed the binding epitope, which does not overlap with the binding sites of FVII and FX.9
As part of the therapeutic development of 10H10 mAb, it was humanized using a two-step process called human framework adaption (HFA), which consists of human framework selection and affinity maturation.10 The final optimized mAb contains over 90% human residues within the variable domains and shows an improved binding affinity. To gain an insight into structural aspects of HFA, we determined the crystal structure of the antigen-binding fragment (Fab) of the humanized variant M59 in the unbound form and in complex with TF ECD and compared it with the 10H10 Fab and complex structures that were determined previously.9 The structures revealed a number of hot spots in the FR that are sensitive to amino acid replacements. These results extend the list of residues to be considered during antibody humanization and emphasize the value of structural information.
Results
Humanization of 10H10 mAb
Based on the overall sequence similarity and the length of CDRs, 8 VH and 7 VL human germline genes were selected for the humanization of 10H10 mAb. The VH genes were predominantly from family IGHV-1, whereas all VL genes except one were from family IGKV-2 (ImMunoGeneTics information system® (IMGT) nomenclature).11 Three of the VH variants (IGHV1-69, IGHV1-f and IGHV5-a) were used with both long and short CDR H2 (see Methods for definitions) to produce a total of 11 VH variants (Table 1). All 77 combinations of the humanized VL and VH domains fused to human IgG1 and κ constant domains and a chimeric mouse/human version of 10H10 mAb were expressed in HEK 293 cells. The supernatant from cell cultures was tested for TF binding by enzyme-linked immunosorbent assay (ELISA) (Table 1).
Table 1.
Binding affinities of humanized variants as percentage with respect to the parental mAb 10H10.
| IGHV1-2 | IGHV5-a | IGHV1-46 | IGHV1-3 | IGHV3-74 | IGHV1-69 | IGHV1-18 | IGHV1-f | sIGHV5-a | sIGHV1-69 | sIGHV1-f | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| IGKV4-1 | 0 | 82* | 98* | 93 | 54 | 74 | 90 | 34 | 98* | 90 | 52 |
| IGKV2-40 | 0 | 104* | 68 | 79 | 50 | 63 | 65 | 24 | 101* | 79 | 47 |
| IGKV2-28 | 0 | 97* | 75 | 87 | 50 | 74 | 56 | 43 | 101* | 86 | 49 |
| IGKV2D-29 | 0 | 107* | 111* | 104 | 55 | 84 | 91 | 41 | 100* | 93 | 38 |
| IGKV2-30 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| IGKV2-24 | 0 | 65 | 75 | 63 | 0 | 57 | 64 | 0 | 84 | 65 | 0 |
| IGKV2D-26 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Human germlines are labeled according to the IMGT nomenclature.11 Allele *01 was used in all variants. Heavy-chain germlines with letter “s” indicate variants with short CDR H2. HFA variants marked by an asterisk were selected for further examination.
All VH variants except IGHV1-2 show binding to TF, with affinities in the range of 24–111% as compared with the chimeric 10H10 mAb. Two human VH FRs, IGHV1-f and IGHV3-74, exhibited a 2-fold to 5-fold loss of affinity, whereas a number of variants with IGHV1-3, IGHV1-46 and IGHV5-a retain or exceed the affinity of 10H10. The variants with short CDR H2 appear to be very similar to the corresponding variants with long CDR H2. Out of 7 VL variants, IGKV2-30 and IGKV2D-26 did not show binding with any VH. There is little difference between other VL variants as the pattern follows the more pronounced differences between various VHs, indicating that the heavy chain of 10H10 plays a major role in TF recognition.
Based on these data, 10 variants were selected for scale up of expression and purification. The affinities of the purified antibodies measured by Biacore confirmed that most bind TF slightly better than 10H10 (Table 2). Interestingly, the IGHV5-a variants with short CDR H2 possess higher affinities in comparison with the long-CDR H2 versions. The humanized mAbs were further characterized for their ability to recognize endogenous TF expressed on human breast cancer cells MDA-MB231. All 10 variants exhibited binding comparable to the parental mAb 10H10 (Table 2).
Table 2.
Binding of recombinant TF ECD and TF-expressing cells by humanized mAbs and their thermal stability.
| KD | Cell binding | Tm | |
|---|---|---|---|
| mAb |
(nM) |
(% to 10H10) |
(°C) |
| 10H10 | 0.76 | 100 | 74.2 |
| H5-a/L4-1 | 0.61 | 107 | 79.4 |
| H5-a/L2-40 | 0.77 | 98 | 75.0 |
| H5-a/L2-28 | 0.37 | 114 | 75.9 |
| H5-a/L2D-29 | 0.55 | 124 | 79.7 |
| sH5-a/L4-1 | 0.20 | 94 | 82.2 |
| sH5-a/L2-40 | 0.40 | 95 | 75.9 |
| sH5-a/L2-28 | 0.47 | 101 | 76.8 |
| sH5-a/L2D-29 | 0.21 | 111 | 81.2 |
| H1-46/L4-1 | 0.66 | 109 | 78.6 |
| H1-46/L2D-29 | 0.41 | 111 | 77.6 |
Thermal stability of the humanized mAbs was measured by the thermofluor method. All 10 selected variants exhibited higher stability compared with chimeric 10H10 (Table 2). The largest increase in Tm, by ∼7-8 °C, was observed for the variants with short CDR H2.
Based on a solubility test (data not shown), variant M59, composed of the IGKV2-40 and IGHV5-a (with short CDR H2) FRs, was selected as the lead candidate. It should be noted that substitution of human FR3 for the mouse FR3 in this variant created a potential N-glycosylation site at position 58 of CDR H2.
Affinity maturation
M59 was subjected to affinity maturation to improve its binding affinity. Two Fab libraries diversifying CDR residues previously shown to be in contact with TF9 were constructed and displayed in the phage coat protein IX system.12 Additionally, the potential glycosylation site in VH was included to find the best replacement for Asn58. In total, 8 positions in each VL and VH, representing all CDRs except CDR L2, were randomized using NGG and NHT codons, where N is any of the four bases and H is either A or T or C. This scheme excludes stop codons and codes for 15 amino acids, excluding Met, Cys, Lys, Gln and Glu. The theoretical size of the resulting library is approximately 3 × 109.
The Fabs composed of the diversified heavy or light chain paired with the opposite parental (M59) chain, were panned with TF ECD and screened for binding. The preferred amino acids selected in each diversified position are listed in Table 3. Variants with an affinity better than that of M59 were converted to IgG1 for cross-chain screening. A number of antibodies exhibited an affinity improvement over M59; however, no additive effect of combining chains was observed. The variant selected for further development, M1587, contained three mutations in VH (Thr31Pro, Ser56Phe and Asn58Thr) and none in VL. It binds TF with an affinity 3 times better than M59 and 5 times better than chimeric 10H10 (Table 4).
Table 3.
Preferred amino acids in diversified positions in combinatorial libraries.
| VH | |
| I30 | all* except FW |
| T31 | APST |
| Y32 | FHY |
| L52 | ILV |
| S54 | PSTV |
| S56 | AFHSWY |
| N58 | all except GPY |
| N100 | NS |
| VL | |
| S30a | all except R |
| S30b | all except D |
| G30c | all except D |
| N30d | all except DHW |
| D91 | D |
| Y92 | all except PRW |
| T93 | all except IPW |
| Y94 | FHILNWY |
means ‘all possible’, i.e. 15 amino acids (ADFGHILNPRSTVWY) coded by NGG+NHT codons.
Table 4.
Kinetic rate constants and affinities of chimeric and humanized mAbs.
| mAb | ka (M−1s−1) ×104 | kd (s−1) ×10−4 | KD (nM) |
|---|---|---|---|
| 10H10 (n=4) | 8.9 (1.1) | 0.68 (0.1) | 0.76 (0.07) |
| M59 (n=6) | 14.2 (0.5) | 0.60 (0.1) | 0.42 (0.06) |
| M1587 (n=2) | 15.7 (0.6) | 0.22 (0.05) | 0.14 (0.04) |
Standard deviations are in parentheses.
Structure of the humanized variant M59
The structure of the humanized variant M59 in the unbound form was determined at 1.6 Å resolution. Pairwise superposition of the VL and VH domains on the 10H10 structure shows that the light chain preserved its conformation better than the heavy chain. The root-mean-square deviation (RMSD) calculated using all Cα atoms is 0.49 Å for VL and 1.77 Å for VH. The rather large RMSD value for VH arises mostly from the difference in the conformation of CDR H3, which folds into the antigen-binding cleft in M59, whereas it points to the outside in 10H10 (Fig. 1). Residues Gly96-Tyr97-Tyr98-Gly99 at the tip of the CDR loop rotate around a hinge formed by the two Gly residues, which results in the main-chain shift of about 10 Å and over 20 Å for the side-chain hydroxyl groups. When residues 96–99 are excluded from the calculation, the RMSD value for the VH domain decreases to 0.89 Å.
Figure 1.
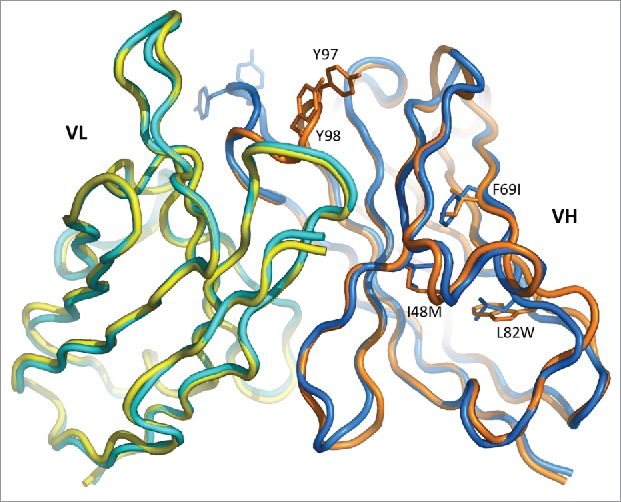
Superposition of the variable domains of 10H10 (blue/cyan) and M59 (orange/yellow). Amino acid substitutions are indicated for residues where deviations are the largest.
Given that the conformation of CDR H3 is maintained in free 10H10, in 10H10:TF complex and in M1587-TF complex (see below), the unique conformation observed in free M59 is probably a crystallographic artefact. Indeed, CDR H3 in that structure makes contacts to the neighboring Fab molecules. Since this unique conformation is incompatible with the TF binding, it may be called unproductive.
Besides CDR H3, significant deviations occur in FR3 of VH (Fig. 1). The likely cause of the rearrangement is the amino acid substitutions at the hydrophobic core of the VH domain, namely Ile48Met, Phe69Ile and, most notably, Leu82Trp. In order to accommodate the bulky indole ring of Trp82, the main chain at residues 82–84 moves outward by almost 3 Å. Although the rearrangements are noticeable in several segments of the VH domain, they seem to be local and do not affect the conformation of CDR H1 and CDR H2.
The mutual orientation of the VL and VH domains measured by the tilt angle has changed by 2.6°, which is within the limits observed for the identical Fabs in different crystal settings.13 Overall, mutations in FRs introduced upon humanization resulted in some conformational rearrangements, particularly in the VH domain. However, the changes apparently did not affect residues critical for antigen binding, as indicated by affinity measurements.
The M59 Fab was expressed in HEK cells and was expected to be glycosylated at Asn58 in CDR H2, owing to the sequence Asn58-Tyr59-Ser60. Although Asn58 is exposed in the structure, no traces of glycosylation are observed in the electron density. Residues 58–60 are part of the β-sheet, which may restrict access to the glycosylation site. Moreover, the amide group of Asn58 forms a hydrogen bond to Asp50, which seems to act as a particularly strong hydrogen acceptor in the presence of Glu35 (Fig. 2). The strength of the interaction may be another factor preventing glycosylation.
Figure 3.
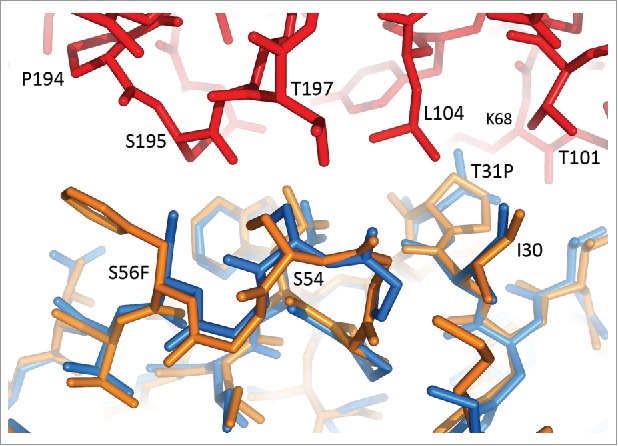
Affinity maturation mutations Thr31Pro and Ser56Phe at the interface between CDR H1, CDR H2 and TF. 10H10-TF and M1587-TF structures are superimposed on the VH domains. 10H10 is shown in orange, M1587 in blue, TF (from the M1587 complex) in red.
Figure 2.
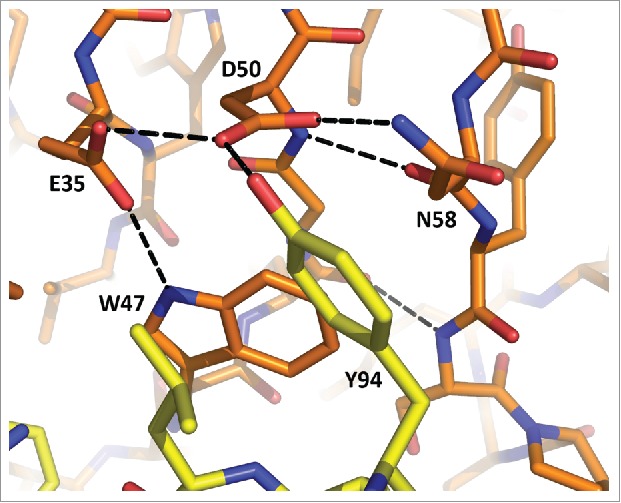
Interactions at the potential site of glycosylation in M59. Heavy chain residues are shown in orange, light chain in yellow. Hydrogen bonds are shown as dashed lines.
Structure of the humanized Fab in complex with TF
The structure of the M1587 Fab in complex with TF ECD was determined at 2.6 Å resolution. As expected, humanization did not change the binding epitope, which had been identified from the 10H10-TF structure.9 Superposition of the two structures on the TF molecule shows that a possible rotation of the variable domains of 10H10 and M1587 does not exceed 2°, indicating that the orientation of Fv relative to TF also has not changed.
In contrast, the elbow angle between the variable domains and the constant domains of the Fab changed significantly, from 119° in 10H10-TF to 156° in M1587-TF. The angle of 119° is the smallest angle observed for the Fabs with κ light chain, whereas 156° is in the middle of the range.14 The elbow angle of 10H10 also varies when comparing the unbound Fab (135°) and the complex (119°). The elbow angle in the unbound M59 Fab is 165°. The wide range of the angles observed in the four structures likely reflects the effect of crystal contacts on the quaternary structure rather than a consequence of humanization.
The VL/VH tilt angle in the set of the four structures is within 1.5° from the average value, indicating no major rearrangement of the domains either after humanization or upon antigen binding. The structures of the humanized Fabs in the bound and unbound forms show no major deviations. The RMSD is 0.36 Å for all Cα atoms of VL and 0.35 Å for VH (excluding the tip of the CDR H3 loop, residues 96 to 99). In the complex with TF, CDR H3 of the humanized mAb M1587 adopts a ‘productive’ conformation virtually identical to that observed in 10H10.
Comparison of the 10H10-TF and M1587-TF complexes allowed us to describe the affinity maturation results in structural terms. The mAbs differ by only three amino acids within the CDRs. The Asn58Thr mutation, introduced to knock out a potential glycosylation site, is ∼8 Å from the nearest TF residue and is unlikely to affect its binding. Two other mutations, Thr31Pro in CDR H1 and Ser56Phe in CDR H2, improve hydrophobic interactions with TF (Fig. 4). Pro31 fits into a hydrophobic pocket between Thr101, Leu104 and Lys68 of TF. Phe56 increases the interface with residues 194–195 of TF by covering ∼30 Å2 more of TF surface. These mutations account for a 3-fold improvement in the KD value even though they represent only a small fraction of the mAb-TF interface.
Figure 4.
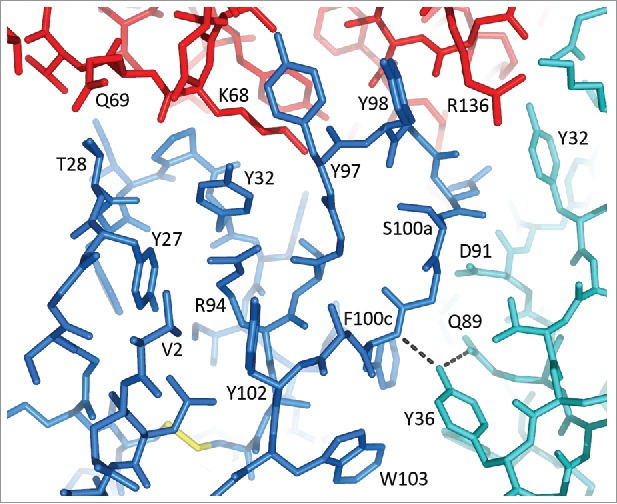
CDR H1 and CDR H3 in the structure of M1587-TF complex. Colors: VH in blue, VL in cyan, TF in red. Hydrogen bonds of Tyr36(VL) are shown as dashed lines.
Discussion
Humanization of murine antibodies by grafting their CDRs into human antibody FRs has been successfully used for decades. Numerous reports highlight various aspects of the humanization process, but usually they present a successful path without paying too much attention to the pitfalls. Here, we discuss, based on the crystal structure, why certain FRs selected for the humanization matrix did not work. The matrix consisted of 7 VLs and 11 VHs. The binding data (Table 1) indicate that only about half of all variants retain their ability to bind TF at the levels comparable to the parental mAb 10H10. A number of variants completely lost binding, although their expression was in the normal range. In particular, VL germlines IGKV2D-26*01 and IGKV2-30*01 and VH germline IGHV1-2*01 showed no binding regardless of the pairing germline.
Three VH germlines (IGHV1-69, IGHV1-f and IGHV5-a) were repeated twice in the matrix to include variants with a shorter CDR H2. In those variants, residues 59–65 were considered part of FR3 and were replaced by the corresponding human germline sequence. The binding data show that in each long/short pair virtually no difference exists between the long and the short variants (Table 1). Apparently the use of a shorter version of CDR H2 does not affect CDR grafting in a significant way, but may yield a more ‘human’ antibody. Remarkably, the variants with short CDR H2 have consistently higher Tm values than the corresponding variants with long CDR H2 (Table 2). The current data set is too small to make conclusions about the importance of residues 59–65 for the stability of the variable domain. It would be interesting to see if the trend exists in other antibodies.
All variants with the IGHV1-2*01 germline failed, whereas other germlines from the same family (IGHV-1) fared much better. One may think that substitution of Arg for Ala at position 71 may have an adverse effect as this position is considered important for the canonical conformation of CDR H2.15 However, germlines IGHV1-3, IGHV1-46 and IGHV3-74, which also possess Arg71, produced good binders, suggesting that this position is not always essential. This finding agrees with the observation that replacement of Lys71 by Val or Ala in VH of anti-lysozyme mAb D1.3 caused no structural changes.16
The sequence and the conformation of CDR H2 in 10H10 correspond to cluster H2-10-3 (classification of North et al.17) characterized by Pro52a-Gly53 at the turn of the β-hairpin. Pro52a occupies a pocket between the 70s loop and CDR H1 that may be otherwise occupied by Arg71. Although the conformation of CDR H2 in 10H10 is consistent with the presence of Ala at position 71, cluster H2-10-3 represents only about 10% of all possible conformations.17 The majority of mAbs with Ala71 adopt canonical structure H2-10-1, which is very similar to H2-10-3 with the exception of a peptide flip at 52a-53. Arg at position 71 is typically associated with canonical cluster H2-10-2 with a different CDR conformation. Apparently, the conformational change in CDR H2 of 10H10 inevitably caused by the Ala71Arg replacement is well tolerated in terms of interaction with TF.
Inspection of the sequences revealed a likely reason for the failure of IGHV1-2*01. This germline, according to the IMGT database, has Val at position 88, while all other germlines in the humanization matrix have Ala, as it is in mouse 10H10 mAb. In fact, Ala88 is conserved in all human germlines. It is also conserved in the other four alleles of IGHV1-2. Moreover, it is Ala88 in IGHV1-2*01 according to the UniProtKB entry HV102_HUMAN for this germline. The difference in one nucleotide (GTC instead of GCC) translates into the Ala88Val mutation according to IMGT. However, the question remains whether this allele is functional or even exists at all. From the structural perspective, Ala88 resides at the bottom of the VH domain in a tightly packed environment. Val at this position would inevitably cause a major rearrangement in the interior β-sheet. Therefore, the Ala88Val mutation would most likely result in a misfolded VH domain. Among more than 2,000 Fab structures in the Protein Data Bank (PDB), there is no structure with Val88. Equally unique is Ser at position 69 in IGHV1-2*01. This residue occupies a niche in a hydrophobic core between two β-sheets. Usually it is Ile, and in some instances Met, as is the case for alleles 2, 3, 4 and 5 of IGHV1-2 (also Met in the UniProtKB entry HV102_HUMAN). In conclusion, IMGT germline IGHV1-2*01 looks questionable and probably should be avoided as an acceptor FR.
Two VH germlines selected for humanization (IGHV3-74 and IGHV1-f) produced variants with the affinity significantly lower than that of 10H10. Germline IGHV1-f is particularly interesting because it is one of the best candidates based on sequence similarity, with 65% identity overall and 70% identity in the FR. IGHV1-f is unique to have Thr at position 94, just before CDR H3. The position is occupied by Arg in 10H10 and in all other VH variants, where Arg94 is packed between the phenol groups of Tyr27, Tyr32 and Tyr102. Substitution of Thr for Arg94 would create a void that may prompt CDR H1 and CDR H3 to shift towards each other. Both CDRs form numerous contacts to TF, so that even a slight rearrangement may affect binding, which probably happens in the IGHV1-f variants.
Germline IGHV3-74 was included in the humanization matrix for diversity, but failed to produce good binders. A possible reason may be Leu at position 78 in FR3. This residue sits in the hydrophobic core of the VH domain opposite Ile34 of CDR H1. Selection of IGHV3-74 introduces Leu instead of Ala, which occurs in the parental antibody and is typical for family IGHV-1. This is a clear example of the impact a residue from the Vernier zone18 may have on binding. An obvious choice would be to select a germline from family IGHV-1 or IGHV-5, both of which have Ala78. If this was not possible, one would have to compensate for the replacement of residue 78 by a corresponding mutation at position 34.
Two VL FRs out of 7 show no binding in any VH combination. There is only one position where IGKV2-30 and IGKV2D-26 deviate from the other germlines. The residue in position 36, following CDR L1, plays a key role in mediating interactions between CDR L3 and CDR H3. In 10H10, Tyr36 forms hydrogen bonds to Gln89 of CDR L3 and the amino group of Phe100c in CDR H3, thus stabilizing the CDR conformations (Fig. 4). Apparently, the hydroxyl group of Tyr36 is indispensable in this role because substitution of Phe for Tyr results in the complete loss of binding in all variants with IGKV2-30 and IGKV2D-26. Variants with IGKV2-24, which has Leu36, are only marginally better.
There is little difference between the best variants in terms of the binding affinity, although they represent different families of human germlines in both VH and VL. This indicates that amino acid variations at many FR positions, especially in FR1, are well tolerated by the antibody. One example is residue 71 in VH, which could be replaced without any adverse effect. In fact, some of the best variants based on IGHV1-46 and IGHV1-3 contain the Ala71Arg mutation, which is usually considered detrimental. This is particularly surprising given that residue 71 potentially affects the conformation of CDR H2, which, in 10H10, is very much involved in the interactions with TF.
In conclusion, antibody humanization by CDR grafting onto human FR is usually based on the sequence similarity, but structural considerations in germline selection should not be underestimated. Humanization of anti-TF mAb 10H10 revealed several positions in the FR that may guide the germline selection. Residue 94 immediately preceding CDR H3 (typically Arg) is particularly interesting since it is often directly involved in antigen binding. However, this position is not included in CDR H3 in any of the common conventions and therefore not transferred to human FR as part of the non-human CDR. Each humanization project reveals a number of sensitive positions in both VH and VL that need to be preserved either by germline selection or by back mutations. To keep that number to a reasonable minimum, analysis of 3D structures obtained either experimentally or by modeling may prove to be useful.
Materials and methods
Protein expression and purification
The human TF ECD expression construct contains residues 1–219 of the mature form, which correspond to residues 33–251 of the full-length sequence (UniProtKB entry TF_HUMAN), and a hexahistidine tag at the C-terminus. The construct was cloned into the mammalian expression vector, which was transiently expressed in HEK 293 cells. The protein was purified by affinity (Ni-NTA) and size exclusion (Superdex 200) chromatography and was used in selection and characterization of the anti-TF antibodies.
For crystallization, the His-tagged TF ECD (residues 1–213) was expressed in Escherichia coli BL21Star (DE3) cells and purified using affinity (Ni-NTA) and ion exchange chromatography as described.9
MAbs were constructed by fusing the variable domains with the human IgG1 and kappa constant domains and were transiently expressed in HEK 293 cells. Cell supernatants were collected 96 h following transfection, clarified by centrifugation, filtered using a 0.2 μ PES membrane (Corning), concentrated 10-fold using an LV Centramate (Pall) device, diluted 10-fold with phosphate-buffered saline (PBS) and loaded onto a HiTrap MabSelect Sure Protein A column (GE Healthcare) equilibrated with PBS pH 7. The protein was eluted with 10 column volumes of 0.1 M Na acetate pH 3. Fractions of 2 mL containing mAbs were neutralized immediately in the collector tubes filled with 0.4 mL 2.0 M Tris pH 7. The fractions were pooled and dialyzed against PBS pH 7 overnight at 4 °C.
The Fabs of mAb M59 and mAb M1587 were constructed by fusing the variable domains of the mAbs with the human IgG1 and kappa constant domains. A hexahistidine tag was attached to the C-terminus of the heavy chain. The Fabs were transiently expressed in HEK 293 cells and purified using affinity and size exclusion chromatography. The supernatant was loaded onto a 5-mL HisTrap column (GE Healthcare) in 50 mM Na phosphate pH 7.1, 350 mM NaCl and eluted with a 0 to 80% gradient of 100 mM imidazole, 150 mM NaCl, 50 mM Na phosphate pH 7.1. The fractions containing Fabs were pooled and dialyzed into 20 mM Tris pH 7.4, 50 mM NaCl. The Fabs were further purified using a HiLoad 26/60 Superdex 200 column (GE Healthcare).
Human framework selection
The Chothia numbering scheme of antibody residues is used throughout the manuscript.19 For the purpose of HFA, the CDRs were assigned using the Kabat definition20 with the exception of CDR H1, which was extended to also include residues as defined by Chothia and Lesk.19 Thus, the CDRs include the following residues: 24–34 for CDR L1, 50–56 for CDR L2, 89–97 for CDR L3, 26–35 for CDR H1, 50–65 for CDR H2 and 95–102 for CDR H3. In some HFA variants, a shorter version of CDR H2 (50-58) was used by excluding 7 residues on the C-terminal side. The CDR assignments with the long CDR H1 and short CDR H2, used in this study, correspond to the Martin definition.21
Human FRs, defined as the regions in the variable domains not included in the CDRs, were selected from the repertoire of functional human germline genes IGKV, IGKJ, IGHV and IGHJ, alleles *01, using the IMGT database.11 Initial selection of human sequences was based on sequence similarity to the entire length of the mouse variable region. The selected germlines were ranked taking into account the length of the CDRs. Sequences of the parental mAb and humanized variants are given in Supplemental Materials.
ELISA assay
Binding of the mAbs to human TF was performed in the ELISA format using chemiluminescent detection. For primary screening of crude supernatants, samples were normalized to 50 ng/mL in FreeStyle 293 HEK media (Gibco) and assayed at a single concentration. About 5 ng of mAb per well was used to bind 10 ng/well of the His-tagged TF ECD.
The 96-well MaxiSorp plates were coated with 100 μL of 4 μg/mL goat anti-human IgG Fc antibody (Jackson ImmunoResearch Laboratories, Cat. No. 109-005-008) diluted in carbonate-bicarbonate buffer pH 9.4, kept at 4 °C overnight, washed 3 times with PBS buffer with 0.05% Tween-20 and blocked with 1% bovine serum albumin (BSA) in 10 mM PBS for 1 hour followed by a washing as before. Samples were diluted to 50 ng/mL in Assay Buffer (1% BSA in PBS with 0.05% Tween-20), 100 μL was added to the assay plate and incubated at room temperature for 1 hour. The plates were washed three times, 100 μL of human His-tagged TF ECD at 100 ng/mL was added in each well and incubated at room temperature for 2 hours. After washing, 100 μL of Qiagen anti-His antibody conjugated to horseradish peroxidase (HRP) at 1:2000 dilution in Assay Buffer was added in each well and incubated at room temperature for 1 hour. After the final wash, 100 μL of the Chemiluminescence Substrate (Roche) at 1:100 dilution in Assay Buffer was added to the plates. After 10 minutes the plates were read on a Perkin Elmer Envision Reader.
Affinity measurements
Affinity measurements using surface plasmon resonance (SPR) were performed with a Biacore 3000 optical biosensor. The biosensor surface was prepared by coupling anti-IgG Fc antibody mixture of anti-mouse mAb (Jackson cat#315-005-046) and anti-human mAb (Jackson cat#109-005-098) to the carboxymethylated dextran surface of a CM-5 chip (Biacore) using the manufacturer instructions for amine-coupling chemistry. Approximately 19,000 RU (response units) of mAb were immobilized in each of 4 flow cells. The kinetic experiments were performed at 25 °C in running PBS buffer containing 0.005% P20, 3 mM EDTA and 100 μg/mL BSA. Serial dilutions of TF ECD from 100 nM to 0.4 nM were prepared in running buffer. About 200 RU of mAb were captured on flow cell 2 to 4 of the sensor chip. Flow cell 1 was used as reference surface. Capture of mAb was followed by 3 minutes of injection of antigen at 50 μL/min (association phase), followed by 10 minutes of buffer flow (dissociation phase). The chip surface was regenerated by two pulses of 18 sec injections of 100 mM H3PO4 at 50 μL/min.
The collected data were processed using BIAevaluation software, version 3.2 (Biacore). First, double reference subtraction of the data was performed by subtracting the curves generated by buffer injection from the reference-subtracted curves for analyte injections. Then, kinetic analysis of the data was performed using 1:1 binding model with global fit.
Cell binding
To detect mAb binding to endogenous human TF, adherent MDA-MB-231 cells were rinsed in a culture flask with PBS, lifted with Versene and counted to seed 200,000 cells per well in a polystyrene V-bottom plate. The pellets were prepared in an Allegra X-15R centrifuge at 450 g for 3 min at 4 °C, resuspended in fluorescence-activated cell sorting (FACS) Buffer (1% fetal bovine serum in PBS) and plated 200,000 cells per well in 200 μL. The cells were pelleted at 450 g for 3 minutes at 4 °C. The supernatants were discarded and 100 μL/well of test or control mAbs was added to designated wells followed by incubation on ice or at 4 °C for 1 hour. The cells were then pelleted at 450 g for 3 min at 4 °C and the supernatants were discarded. The cells were resuspended in 200 μL/well FACS buffer and were pelleted at 450 g for 3 min at 4 °C. The supernatants were discarded and 100 μL/well of the secondary antibody was added to designated wells and incubated on ice for 1 hour. The cells were then pelleted at 450 g for 3 min at 4 °C. The supernatants were discarded and the cells were washed 2 times in FACS buffer before resuspending them in 200 μL/well FACS Buffer. The cells were then pelleted at 450 g for 3 min at 4 °C. The supernatant was discarded and the cells were resuspended in 100 μL/well CytoFix Buffer. The reactions were analyzed by flow cytometry (BD FACSArray). The FlowJo Software was used for FACS data analysis by gating the main population of cells in the unstained control well and applying the gate to the whole data set.
Thermofluor assay
Thermofluor analysis was performed using the ProteoStat Thermal Shift Stability Assay (Enzo Life Sciences). This assay measures thermostability using a fluorescent dye that binds to hydrophobic patches exposed as the protein unfolds. The dye solution contained 20 μL of the 40 mM 8-anilinonaphthalene-1-sulfonic acid (ANS), 2.8 μL 10% Tween and 1.98 mL PBS. Antibody concentration was normalized to 0.5 mg/mL. Fluorescence was measured using a LightCycler 480 (Roche) at 480 nm excitation and 610 nm emission. Samples were heated continuously from 50 °C to 95 °C at 0.5 °C/sec.
Phage display
Fab libraries displayed on phage coat protein IX were panned against biotinylated human TF ECD. Phage was produced by helper phage infection. Binders were retrieved by addition of streptavidin beads to form a bead/antigen/phage complex. After the final wash, phage was rescued by infection of exponentially growing Escherichia coli TG-1 cells. Phage was again produced and subjected to additional rounds of panning.
The pIX gene was excised by NheI/SpeI digestion from the selected clones to allow for secreted soluble Fab production. After religation, the DNA was transformed into TG-1 cells and grown on LB/Agar plates overnight. The cultures were used for (1) colony PCR and sequencing of the V-regions, and (2) soluble Fab production. The soluble Fabs were captured onto plates by a polyclonal anti-human Fd antibody. After appropriate washing and blocking, biotinylated TF ECD was added at 0.2 nM and was detected by HRP-conjugated streptavidin and chemiluminescence. The variants were ranked with respect to the binding of the parent M59 Fab, which was present as a control in all plates.
Crystallization
M1587-TF complex was prepared by mixing 4.8 mg Fab with 2.8 mg of TF ECD at an approximate molar ratio of 1:1.2 (excess TF). The mixture was incubated for 4 days at 4°C and concentrated to ∼700 μL. The complex was then purified by SEC on a Superdex 200 (10/300) column (GE Healthcare) equilibrated in 20 mM HEPES pH 7.5, 0.1 M NaCl. The shift in the elution profile indicated complex formation. Fractions corresponding to the main peak were pooled, concentrated to 8.7 mg/mL and used for crystallization.
Crystallization of both the complex and free Fab was carried out by the vapor-diffusion method in sitting drops at 20 °C using an Oryx4 robot (Douglas Instruments). Equal volumes of protein and reservoir solutions were dispensed in Corning plates 3550. Initial screening was performed with the Hampton Crystal Screen HT and in-house screens.22 Needle-like crystals appeared in various conditions and were used to make seeds for microseed matrix screening (MMS).22,23 Diffraction quality crystals of the complex were obtained from 0.1 M Na acetate pH 4.5, 18% PEG 3350, 0.2 M ammonium acetate. The optimized conditions for M59 Fab were 0.1 M MES buffer pH 6.5, 16% PEG 3350, 0.2 M Na formate. Crystal data are given in Table 5.
Table 5.
X-ray data and refinement statistics.
| M59 Fab | TF+M1587 Fab | |
|---|---|---|
| X-Ray Data | ||
| Space group | P212121 | P21212 |
| Unit cell (Å) | 43.38, 80.65, 112.16 | 68.10, 175.56, 63.31 |
| Molecules/asymmetric unit | 1 | 1 |
| Vm (Å3/Da)/solvent (%) | 2.02/39 | 2.61/53 |
| Resolution (Å) | 30–1.64 (1.68–1.64) | 30–2.6 (2.67–2.60) |
| Number of measured reflections | 402,169 (5781) | 165,595 (10363) |
| Number of unique reflections | 45,702 (2034) | 23,936 (1678) |
| Completeness (%) | 93.1(56.4) | 99.5 (94.7) |
| Multiplicity | 8.8 (2.8) | 6.9 (6.2) |
| Rsym (I) | 0.040 (0.179) | 0.088 (0.403) |
| Mean I/σ(I) | 38.0 (5.7) | 16.0 (4.7) |
| B factor from Wilson plot (Å2) | 22.3 | 41.1 |
| Refinement | ||
| Resolution (Å) | 20–1.64 | 20–2.6 |
| Total number of atoms | 3710 | 5016 |
| Number of water molecules | 355 | 107 |
| Rcryst (%) | 18.7 | 18.6 |
| Rfree (%) | 20.4 | 23.4 |
| RMSD bond lengths (Å) | 0.006 | 0.007 |
| RMSD bond angles (°) | 1.2 | 1.1 |
| Mean B-factor from model (Å2) | 18.7 | 42.9 |
| Ramachandran plot, most favored (%) | 92.8 | 91.4 |
| Ramachandran plot, disallowed (%) | 0.3 | 0.2 |
Values for highest resolution shell are in parentheses.
X-ray structure determination
For X-ray data collection, one crystal each of the Fab and the complex was soaked for a few seconds in the corresponding cryoprotectant. Mother liquor supplemented with either 25% PEG 400 (for the Fab crystal) or with 20% glycerol (for the complex). The crystals were flash cooled in liquid nitrogen at 100 K. Diffraction data for the Fab were collected using a MicroMax-007HF microfocus X-ray generator equipped with an Osmic VariMax confocal optics, Saturn 944 CCD detector, and an X-stream 2000 cryocooling system (Rigaku). Diffraction data for the Fab were collected at the IMCA beamline 17-ID equipped with an ADSC Quantum-210 CCD detector at the Advanced Photon Source (Argonne, IL). The X-ray data were processed with XDS.24
The structures were determined by molecular replacement with Phaser25 and refined using Refmac26 and Coot.27 The structure of 10H10 Fab (PDB entry 4M7K) and the TF ECD structure (PDB entry 1UJ3) were used as search models. X-ray data and refinement statistics are given in Table 5. Ramachandran statistics was calculated with Procheck.28 The solvent-accessible surface area was calculated with Areaimol from the CCP4 suite.29 Figures were prepared with PyMol (Schrödinger).
Accession numbers
The coordinates and structure factors have been deposited in the PDB under accession codes 5W05 for M59 Fab and 5W06 for M1587-TF complex.
Abbreviations
- BSA
bovine serum albumin
- CHES
N-cyclohexyl-2-aminoethanesulfonic acid
- CCD
charge coupled device
- CDR
complementarity determining region
- ECD
extracellular domain
- EDTA
ethylenediaminetetraacetic acid
- FR
framework region
- FVII
coagulation factor VII
- FX
coagulation factor X
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HRP
horseradish peroxidase
- mAb
monoclonal antibody
- PAR
G-protein-coupled protease activated receptor
- PBS
phosphate-buffered saline
- PDB
Protein Data Bank
- PEG
polyethylene glycol
- RMSD
root-mean-square deviation
- SPR
surface plasmon resonance
- TF
tissue factor
- VH
variable domain of the heavy chain
- VL
variable domain of the light chain
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 1986;321:522–525. doi: 10.1038/321522a0. [DOI] [PubMed] [Google Scholar]
- 2.Wark KL, Hudson PJ. Latest technologies for the enhancement of antibody affinity. Adv Drug Deliv Rev. 2006;58:657–670. doi: 10.1016/j.addr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 3.King DJ, Bowers PM, Kehry MR, Horlick RA. Mammalian cell display and somatic hypermutation in vitro for human antibody discovery. Curr Drug Discov Technol. 2014;11:56–64. doi: 10.2174/15701638113109990037. [DOI] [PubMed] [Google Scholar]
- 4.Edgington TS, Morrissey JH. Hybridomas producing monoclonal antibodies reactive with human tissue-factor glycoprotein heavy chain. US patent 5223427. 1993. [Google Scholar]
- 5.Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci USA. 2000;97:5255–5260. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruf W, Disse J, Carneiro-Lobo TC, Yokota N, Schaffner F. Tissue factor and cell signalling in cancer progression and thrombosis. J Thromb Haemost. 2011;9:306–315. doi: 10.1111/j.1538-7836.2011.04318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahamed J, Versteeg HH, Kerver M, Chen VM, Mueller BM, Hogg PJ, Ruf W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci USA. 2006;103:13932–13937. doi: 10.1073/pnas.0606411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Versteeg HH, Schaffner F, Kerver M, Petersen HH, Ahamed J, Felding-Habermann B, Takada Y, Mueller BM, Ruf W. Inhibition of tissue factor signaling suppresses tumor growth. Blood. 2008;111:190–199. doi: 10.1182/blood-2007-07-101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teplyakov A, Obmolova G, Malia TJ, Wu B, Zhao Y, Taudte S, Anderson GM, Gilliland GL. Crystal structure of tissue factor in complex with antibody 10H10 reveals the signaling epitope. Cell Signal. 2017;36:139–144. doi: 10.1016/j.cellsig.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Fransson J, Teplyakov A, Raghunathan G, Chi E, Cordier W, Dinh T, Feng Y, Giles-Komar J, Gilliland G, Lollo B, Malia TJ, Nishioka W, Obmolova G, Zhao S, Zhao Y, Swanson RV, Almagro JC. Human framework adaptation of a mouse anti-human IL-13 antibody. J Mol Biol. 2010;398:214–231. doi: 10.1016/j.jmb.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Lefranc MP, Giudicelli V, Kaas Q, Duprat E, Jabado-Michaloud J, Scaviner D, Ginestoux C, Clément O, Chaume D, Lefranc G. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2005;33:D593–D597. doi: 10.1093/nar/gki065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tornetta M, Baker S, Whitaker B, Lu J, Chen Q, Pisors E, Shi L, Luo J, Sweet R, Tsui P. Antibody Fab display and selection through fusion to the pIX coat protein of filamentous phage. J Immunol Methods. 2010;360:39–46. doi: 10.1016/j.jim.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 13.Teplyakov A, Luo J, Obmolova G, Malia TJ, Sweet R, Stanfield RL, Kodangattil S, Almagro JC, Gilliland GL. Antibody modeling assessment II. Structures and models. Proteins. 2014;82:1563–1582. [DOI] [PubMed] [Google Scholar]
- 14.Stanfield RL, Zemla A, Wilson IA, Rupp B. Antibody elbow angles are influenced by their light chain class. J Mol Biol. 2006;357:1566–1574. doi: 10.1016/j.jmb.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 15.Tramontano A, Chothia C, Lesk AM. Framework residue 71 is a major determinant of the position and conformation of the second hypervariable region in the VH domains of immunoglobulins. J Mol Biol. 1990;215:175–182. doi: 10.1016/S0022-2836(05)80102-0. [DOI] [PubMed] [Google Scholar]
- 16.Holmes MA, Buss TN, Foote J. Structural effects of framework mutations on a humanized anti-lysozyme antibody. J Immunol. 2001;167:296–301. doi: 10.4049/jimmunol.167.1.296. [DOI] [PubMed] [Google Scholar]
- 17.North B, Lehmann A, Dunbrack RL. A new clustering of antibody CDR loop conformations. J Mol Biol. 2011;406:228–256. doi: 10.1016/j.jmb.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foote J, Winter G. Antibody framework residues affecting the conformation of the hypervariable loops. J Mol Biol. 1992;224:487–499. doi: 10.1016/0022-2836(92)91010-M. [DOI] [PubMed] [Google Scholar]
- 19.Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 1987;196:901–917. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- 20.Kabat EA, Wu TT, Perry HM, Gottesmann KS, Foeller C. Sequences of Proteins of Immunological Interest, 5th edit., NIH Publication no 91–3242, U.S: Department of Health and Human Services, 1991. [Google Scholar]
- 21.Martin ACR. Protein Sequence and Structure Analysis of Antibody Variable Domains. In: Kontermann R, Dübel S, (Eds.), Antibody engineering. Berlin: Springer; 2014 page 33–51. [Google Scholar]
- 22.Obmolova G, Malia TJ, Teplyakov A, Sweet R, Gilliland GL. Promoting crystallization of antibody-antigen complexes via microseed matrix screening. Acta Crystallogr. 2010;D66:927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D'Arcy A, Villard F, Marsh M. An automated microseed matrix-screening method for protein crystallization. Acta Crystallogr. 2007;D63:550–554. [DOI] [PubMed] [Google Scholar]
- 24.Kabsch W. XDS. Acta Crystallogr. 2010;D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: Model building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 28.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 29.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al.. Overview of the CCP4 suite and current developments. Acta Crystallogr. 2011;D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]