

Abstract
Barrier dysfunction has been implicated in the pathophysiology of eosinophilic esophagitis (EoE). TGF-β1, a potent pleiotropic molecule, is increased in EoE, however, no study has evaluated its influence on esophageal epithelial barrier. We hypothesized that TGF-β1 regulates barrier dysfunction in EoE. We aimed to determine the role of TGF-β1 in epithelial barrier in models of EoE. To examine the impact of TGF-β1 on esophageal barrier, immortalized human esophageal epithelial (EPC2-hTERT) cells were exposed to TGF-β1 during the 3-dimensional air liquid interface (3D-ALI) model in vitro. TGF-β1 exposure diminished EPC2-hTERT barrier function as measured by transepithelial electrical resistance (TEER) and 3kDa FITC dextran paracellular flux (FITC Flux) and H&E assessment revealed prominent cellular separation. In analysis of epithelial barrier molecules, TGF-β1 led to the specific reduction in expression of the tight-junction molecule, claudin-7 and this was prevented by TGF-β receptor I inhibitor. shRNA mediated claudin-7 knockdown diminished epithelial barrier function, while claudin-7 overexpression resulted in protection from TGF-β1-mediated barrier dysfunction. In analysis of pediatric EoE biopsies claudin-7 expression was decreased, altered localization was observed by immunofluorescence analysis and the TGF-β1 downstream transcription factor phosphorylated SMAD2/3 (pSMAD2/3) was increased. Our data suggest that TGF-β1 participates in esophageal epithelial barrier dysfunction through claudin-7 dysregulation.
Keywords: Eosinophilic Esophagitis, Tight Junctions, Claudin-7, pSMAD2/3, Epithelial Barrier
INTRODUCTION
Eosinophilic Esophagitis (EoE) is a chronic, immune/antigen mediated, inflammatory disease characterized by esophageal dysfunction and dense esophageal eosinophilia1. The esophageal epithelial architecture of patients with EoE is disrupted, characterized by dilated intercellular spaces and prominent basal cell hyperplasia2. A growing body of evidence implicates altered esophageal epithelial barrier in the pathophysiology of EoE. For instance, studies have shown decreased cell adhesion molecules such as desmoglein and epithelial barrier molecules such as filaggrin in EoE3–5. In addition, others have demonstrated barrier dysfunction in EoE patients at the electrophysiological level, suggesting impaired esophageal mucosa integrity6. Our previous work also demonstrated altered ultrastructural features with decreased desmosomes in active EoE7. To date, molecular mechanisms focused on the impact of type-2 cytokines, particularly IL-13, on regulating molecules associated with esophageal epithelial barrier3,4.
An increasing body of evidence suggests that transforming growth factor-β1 (TGF-β1), a pleiotropic cytokine that plays a critical role in immunoregulation, fibrosis and remodeling, participates in the pathogenesis of EoE. TGF-β1 signaling is increased in subjects with EoE and TGF-β1 itself is produced by a number of different cells including eosinophils, epithelial cells, and mast cells8–11. Although TGF-β1 protein has been difficult to quantify in esophageal tissue affected by EoE, biopsy specimens from subjects with EoE have demonstrated increased TGF-β1 mRNA and phosphorylated SMAD2/3 (pSMAD2/3) transcription factor protein immunostaining, a downstream nuclear signaling molecule in the TGF-β1 signaling pathway8,12,13. Studies have also shown that TGF-β1 plays a critical role in esophageal remodeling, participates in esophageal smooth muscle contraction, stimulates fibroblast secretion of extracellular matrix, and contributes to esophageal epithelial to mesenchymal transitions (EMT) in vitro10,14–18 and in vivo8,19,20. In vitro models have shown that TGF-β1 alters epithelial barrier function in other epithelial cell types, including columnar epithelium of the lung and pseudostratified epithelium of the epididymis and vas defrens21–23, however, no study has evaluated the influence of TGF-β1 on stratified squamous epithelial barriers including the esophageal epithelium. Therefore, we sought to determine the role of TGF-β1 on epithelial barrier function in the pathogenesis of EoE. We hypothesized that TGF-β1 regulates epithelial barrier dysfunction in EoE.
METHODS
Human Esophageal Epithelial Cell Line in Air Liquid Interface Culture System
Immortalized human esophageal epithelial (EPC2-hTERT)24 cells (kind gift from Drs. Anil Rustgi and Hiro Nakagawa, University of Pennsylvania), claudin-7 knockdown cells, or claudin-7 overexpressing cells were used for cell culture studies. To generate a human CLDN7 expression construct, full length claudin-7 open reading frame (ORF) (NM_001307) was amplified from plasmid mEmerald-claudin7-C-12 (Plasmid 54041; Addgene, Cambridge, MA) by polymerase chain reaction (PCR) using sense primer 5′-AAAGCTAGCAATGGCCAATTCGGGCCTGCAGTT-3 (incorporating a Kozak translation initiation sequence and 5′ NheI restriction site) and antisense primer 5′-TTTATGTTTCAGGTTCAGGGGGAGGTGTGGGAGG-3′. CLDN7 ORF was digested to generate NheI-CLDN7-XbaI for subcloning into lentiviral pLKO.1-puro plasmid with CMV promoter (Addgene, Cambridge, MA) linearized by NheI/XbaI restriction digest. Digested CLDN7 and linearized pLKO.1 vector were purified (WizardPrep Column, Promega, Madison, WI) for ligation using T4 DNA ligase (New England Biolabs, Ipswich, MA). Constructs were verified by sequencing (Molecular Biology Service Center, University Colorado) and packaged into lentiviral particles using 293TN cells (SBI, Palo Alto, CA) and MISSION Lentiviral Packaging mix as previously described (Sigma Aldrich, St. Louis, MO).
To develop claudin-7 knockdown cells, EPC2-hTERT cells were transduced with short hairpin ribonucleic acid (shRNA) lentiviral constructs targeting claudin-7 and a scrambled shRNA as a control (MISSION TRC, Functional Genomics Core, University of Colorado, Boulder) as previously described25. To develop claudin-7 overexpressing cells, EPC2-TERT cells were transduced with the above generated lentiviral construct containing pLKO-claudin-7 overexpressing constructs (pLKO.1-Puro claudin-7 ORF) as previously described25. Forty eight hours after transduction, cells were selected for stable integration using puromycin (0.3μg/ml, Sigma Aldrich, St. Louis, MO).
The 3-dimensional air liquid interface (3D-ALI) culture system was used, as previously described4,26. In brief, EPC2-hTERT cells, claudin-7 knockdown cells, or claudin-7 overexpressing cells were submerged in keratinocyte serum-free media (KSFM) (Thermo Fischer, Waltham, MA) and grown to confluence on 0.4 μm pore-size permeable transwells (Corning, Corning, NY). Once the cells reached confluence, on day 2, they were placed in high calcium KSFM ([Ca2+]=1.8 mM) for an additional 5 days to induce terminal differentiation. To induce stratification, media was removed from the upper chamber in order to expose the cells to air liquid interface for a 4 day period. Treatment with recombinant human TGF-β1 (R&D Systems, Minneapolis, MN, 10 ng/ml) in the basal chamber occurred at the start of 3D-ALI exposure and during the process of differentiation and stratification on days 7 and 9. Cells were collected for analysis of RNA or protein expression and for histologic analysis as previously described4.
Esophageal Epithelial Barrier Measurements
Functional assays of barrier were performed on the 3D-ALI culture system using transepithelial electrical resistance (TEER) with a commercial Ohmmeter (WPI, Sarasota, FL) as previously described25. In vitro measurements for TEER were assessed on day 11 of cells grown at 3D-ALI in TGF-β1 treated cells or on claudin-7 knock down cells.
Paracellular 3kDa FITC Dextran flux assays were also completed on TGF-β1 treated EPC2-hTERT and claudin-7 knock down cells that were grown at 3D-ALI on 0.4 μm permeable polyester membrane inserts on day 11 of culture. Following washing, FITC-Dextran (62.5 μg/ml, 3kDa, Molecular Probes) was placed into the apical chamber. Samples were harvested from the basal chamber every 30 minutes for 90 minutes after to determine rate of flux. Fluorescent spectrophotometry was performed to analyze samples.
TGF-β1 Inhibition
EPC2-hTERT cells were cultured as previously described24. EPC2-hTERT cells were seeded at 125,000 cells per well of a 24-well plate. Twenty-four hours after the cells were plated, they were washed and treated with SB431542, a selective inhibitor of TGF-β type 1 receptor (S4317, Sigma Aldrich, St. Louis, MO, 5μm) for 4 hours. After 4 hours, the cells were treated with recombinant human TGF-β1 (R&D Systems, Minneapolis, MN, 10 ng/ml) in combination with the selective TGF-β type 1 receptor inhibitor (S4317, Sigma Aldrich, St. Louis, MO, 5μm) for 48 hours. Cells were harvested for mRNA analysis using RLT buffer from Qiagen RNeasy kits (Qiagen, Valencia, CA).
RNA Isolation and quantitative RT-PCR
RNA from cells in 3D-ALI and human esophageal biopsies were isolated using RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions and cDNA made using a high-capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Junctional molecule expression was measured by Real-time Reverse Transcription Polymerase Chain Reaction (RT-PCR) using TaqMan gene expression assays with TaqMan probes (Applied Biosystems, Foster City, CA) targeting E-Cadherin, desmoglein-1, desmoglein-2, desmoglein-3, claudin-1, claudin-4, claudin-7, occludin, zonula occluden-1, connective tissue growth factor, smooth muscle α actin, and N Cadherin. Data were normalized to the house keeping gene, 18S, and were calculated for each sample as relative quantity RQ = 2−ΔΔCt, where Ct is the cycle threshold.
Western Blot Analysis
Protein lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer (Sigma Aldrich, St. Louis, MO) and protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Protein electrophoresis was performed using 12% polyacrylamide gels. Blots were probed with the primary antibody overnight and incubated with appropriate HRP labeled secondary antibodies for 1 hour at room temperature. Visualization was performed using a chemiluminescent detection system (SuperSignal West PICO, Thermo Fischer, Waltham, MA). Primary antibodies used include claudin-7 polyclonal rabbit antibody (#34-9100, Invitrogen, Carlsbad, CA), pSMAD2/3 polyclonal rabbit antibody (#8828, Cell Signaling Technology, Danvers, MA), SMAD2/3 monoclonal mouse antibody (#610843, BD Biosciences, San Jose, CA) and β-Actin (#8227, Abcam, Cambridge, MA). Quantification was assessed using comparative densitometry with β-actin as a loading control.
Immunohistochemical & Immunofluorescent Staining
Cells grown at 3D-ALI were fixed with 10% neutral-buffered formalin, processed, and paraffin embedded. Sections were cut into 5 μM sections and stained with hematoxylin and eosin (H&E) (Sigma Aldrich, St. Louis, MO). Immunofluorescent staining was performed on formalin fixed, paraffin-embedded esophageal biopsy samples that were cut into 5 μM sections. Samples were deparaffinized via sequential immersion with xylene followed by graded ethanol immersion and rehydration. Heat induced antigen retrieval in sodium citrate buffer (Vector Laboratories, Burlingame, CA) was used. Sections were blocked in 5% bovine serum albumin in tris-buffered saline for 30 minutes. Primary antibodies claudin-7 (1:100, #51-900, Invitrogen, Carlsbad, CA) or pSMAD2 (1:2000, #3101, Cell Signaling Technology, Danvers, MA) were used and slides were incubated at 4 degrees overnight. Slides were washed and incubated for 30 minutes at room temperature in secondary antibody of Alex Fluor 488 Goat anti-rabbit IgG or Alexa Fluor 555 Goat anti rabbit IgG (Invitrogen, Carlsbad, CA). Slides were washed followed by counterstaining with DAPI and mounted for visualization. Images were acquired using an Olympus IX83 microscope and cellSens v1.14 software.
Characterization of human subject samples
Healthy control subjects underwent upper intestinal endoscopy for indications including abdominal pain, feeding difficulty, poor weight gain, and diarrhea and had normal esophageal histology with a mean eosinophil count of 0 eosinophils per high power field (eos/HPF). The mean age was 8.2±4.1 years in these subjects. Active EoE subjects had symptoms of esophageal dysfunction and ≥15 eos/HPF in whom other causes for eosinophilia had been ruled out according to consensus recommendations1. Symptoms included abdominal pain, dysphagia, feeding difficulty, poor weight gain, and vomiting. Active EoE subjects had a mean eosinophil count of 46.5±34.9 eos/HPF and the mean age was 8.7±5.1 years. Inactive EoE subjects had an established diagnosis of EoE who underwent treatment with topical corticosteroids or dietary elimination with resolution of symptoms and <15 eos/HPF. These subjects had a mean eosinophil count of 1.8±2.9 eos/HPF and the mean age was 7.2±4.0 years. Written informed consent was obtained and all human studies were approved by the Colorado Multi-Institutional Review Board.
Statistical Analysis
Statistical analysis of data was performed by students’ t-test and one-way analysis of variance (ANOVA) where appropriate. A p-value of ≤ 0.05 was considered statistically significant, although in some cases higher levels of significance are noted when applicable (*p≤0.05, **p≤0.01, ***p≤0.001, ****p≤ 0.0001). GraphPad Prism 7 (GraphPad Software, LaJolla, CA) was used to perform statistical analysis and generate figures.
RESULTS
TGF-β1 alters esophageal epithelial barrier
To investigate the functional consequences of transforming growth factor-β1 (TGF-β1) on esophageal epithelia, we measured barrier by transepithelial electrical resistance (TEER) and paracellular permeability by 3kDa FITC Dextran Flux (FITC Flux) in immortalized human esophageal epithelial (EPC2-hTERT) cells grown in a physiologically relevant 3-dimensional air liquid interface (3D-ALI) model in vitro where cells undergo stratification and squamation. We found that TGF-β1 (10 ng/ml) exposure resulted in a significant attenuation of barrier, as measured by TEER, compared to unstimulated control cells (446.6±29.7 vs 323.1±15.6. ohms.cm2, Control vs TGF-β1 10 ng/ml, N=8, p≤0.01) (Figure 1A). We evaluated the epithelial paracellular permeability using 3kDa FITC Flux and found a significant increase in paracellular flux in the epithelium exposed to TGF-β1 (10 ng/ml) when compared to unstimulated cells (1.00±0 vs 1.47±0.15 fold, Control vs TGF-β1 10 ng/ml, N=4, p≤0.05) (Figure 1B). We performed a histological analysis of EPC2-hTERT cells grown in a physiologically relevant 3D-ALI model. H&E stained sections of EPC2-hTERT cells grown in 3D-ALI had normal stratification and differentiation (Figure 1C, top panel). However, TGF-β1 (10 ng/ml) treated cells grown in 3D-ALI had prominent cell separation, particularly in the basal and suprabasal layers (Figure 1C, bottom panel).
Figure 1. TGF-β1 diminishes esophageal epithelial barrier.
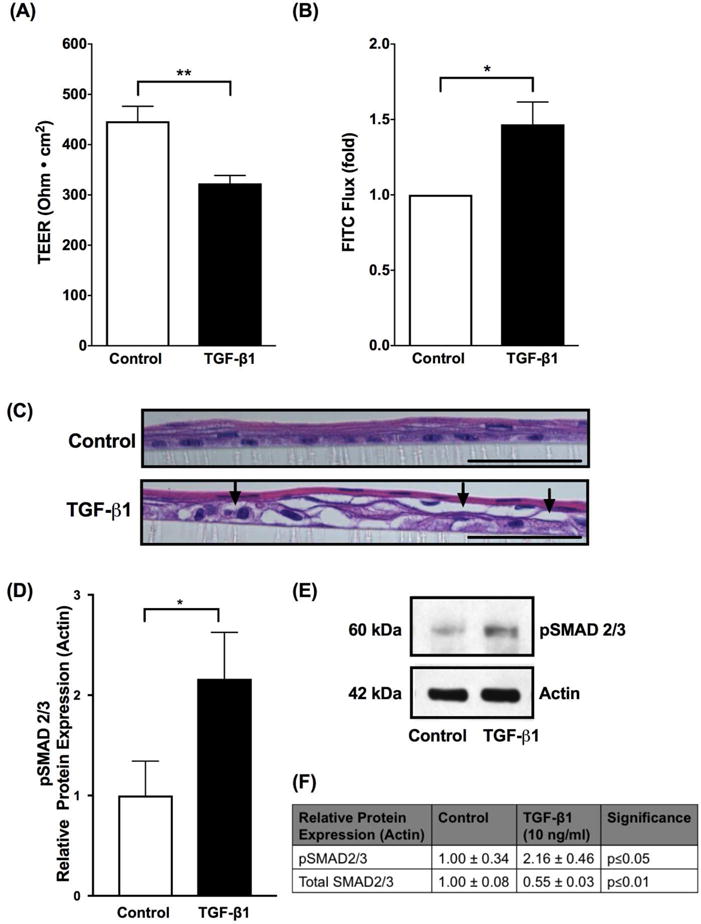
Using immortalized human esophageal epithelial (EPC2-TERT) cells in a stratified squamous 3-dimensional air liquid interface (3D-ALI) culture, transforming growth factor-β1 (TGF-β1) (10 ng/ml) diminished barrier as measured by (A) transepithelial electrical resistance (TEER) and increased permeability as measured by (B) 3kDa FITC dextran paracellular flux (FITC flux) (N=4–8, *p≤0.05, **p≤0.01). (C) H&E stained sections from EPC2-TERT cells grown in 3D-ALI culture and treated with TGF-β1 (10 ng/ml). Arrows indicate prominent cellular separation. Black scale bar represents 50 μm. (D) To confirm that TGF-β1 signaling is increased, we performed western blot densitometry which shows increase in protein expression of phosphorylated SMAD2/3 (pSMAD2/3) when cells are exposed to TGF-β1 (10 ng/ml), indicating that TGF-β1 activates SMAD2/3 via phosphorylation in the in vitro 3-D ALI model (N=4, *p≤0.05). (E) Representative western blot showing increase in protein expression of pSMAD2/3 in vitro when exposed to TGF-β1 (10 ng/ml). (F) Protein expression of pSMAD2/3 and total SMAD2/3 when EPC2-TERT cells exposed to TGF-β1 (10 ng/ml). Data are expressed as mean fold change versus unstimulated controls ± SEM. Treatment with recombinant human TGF-β1 (10 ng/ml) occurred at the start of 3D-ALI exposure and during the process of differentiation and stratification on days 7 and 9.
Because activation of the canonical TGF-β1 pathway leads to phosphorylation of SMAD2/3, we performed western blot of phosphorylated SMAD2/3 (pSMAD2/3) to confirm the effect of TGF-β1 at the signaling level in the 3-D ALI model. We found that TGF-β1 (10 ng/ml) exposure resulted in an increase in pSMAD2/3 protein expression (1.00±0.34 vs 2.16±0.46, Control vs TGF-β1 10 ng/ml, N=4, p≤0.05) and decrease in total SMAD 2/3 protein expression (1.00±0.08 vs 0.55±0.03, Control vs TGF-β1 10 ng/ml, N=4, p≤0.01) compared to unstimulated control cells, as measured by densitometry indicating that TGF-β1 activates SMAD2/3 via phosphorylation in the in vitro 3-D ALI model (Figure 1D–F).
TGF-β1 attenuates expression of tight junction claudin-7 in vitro
In light of the functional barrier changes observed after exposure to TGF-β1, we sought to understand the expression patterns of various epithelial cell-cell junction barrier molecules in 3D-ALI cells following TGF-β1 exposure. We analyzed transmembrane epithelial barrier molecule expression of the epithelial cell-cell junctions including tight junctions, adherens junctions and desmosomes using real time reverse transcription polymerase chain reaction (RT-PCR). These molecules were targeted because they are the transmembrane proteins involved in cell-cell junctions important in epithelial barrier function. In particular, we performed an array analysis using real time RT-PCR to determine which claudins were present in the esophageal epithelium using EPC2-hTERT cells. Consistent with previous literature in the field27–29 we found that claudin 1, 4 and 7 were the predominantly expressed claudins in the esophageal epithelium differentiated at 3D-ALI (Figure 2A). We subsequently found the specific and significantly attenuated expression of claudin-7 (CLDN7) mRNA in 3D-ALI cells exposed to TGF-β1 (10 ng/mL) when compared with unstimulated cells (0.93±0.10 vs 0.42±0.02, Control vs TGF-β1 10 ng/ml, N=3, p≤0.01) (Figure 2B). However, no significant differences were observed in other epithelial barrier molecules including adherens junction components (E-Cadherin), desmosomes (desmoglein-1, desmoglein-2, and desmoglein-3), or tight junction components (claudin-1, claudin-4, occludin, and zonula occluden-1) (Table 1). Additionally, there were no significant differences in mesenchymal genes including connective tissue growth factor or alpha smooth muscle actin. There was however a significant increase in N-Cadherin. With no concomitant decrease in E-Cadherin in our cells following 5 days of culture at 3D-ALI in the presence of TGF-β1, this does not meet the definition of epithelial mesenchymal transition (EMT)30 (Table 1). Western blot analysis of cell lysates showed consistent findings, with a significant decrease in protein expression of claudin-7 relative to unstimulated cells as measured by densitometry (1.00±0.17 vs 0.28±0.12, Control vs TGF-β1 10 ng/ml, N=3, p≤0.05,) (Figure 2C & 2D). To determine if inhibition of the TGF-β1 SMAD pathway would protect against the TGF-β1 mediated attenuation in claudin-7, we exposed esophageal epithelial cells to SB431542, a selective inhibitor of TGF-β type 1 receptor, prior to TGF-β1. We found that when cells were exposed to TGF-β1, there was an expected attenuation in claudin-7, however when cells were exposed to TGF-β type 1 receptor inhibitor (TGF-β RI) prior to TGF-β1, cells were protected from the TGF-β1 mediated claudin-7 attenuation, indicating that the TGF-β1 pathway directly influences the attenuation of claudin-7 (1.00±0 vs 0.08±0.02 vs 1.38±0.17, Control vs TGF-β1 10 ng/ml vs TGF-β type 1 receptor inhibitor 5μm, N=4, p≤0.01) (Figure 2E).
Figure 2. TGF-β1 attenuates expression of Claudin-7.
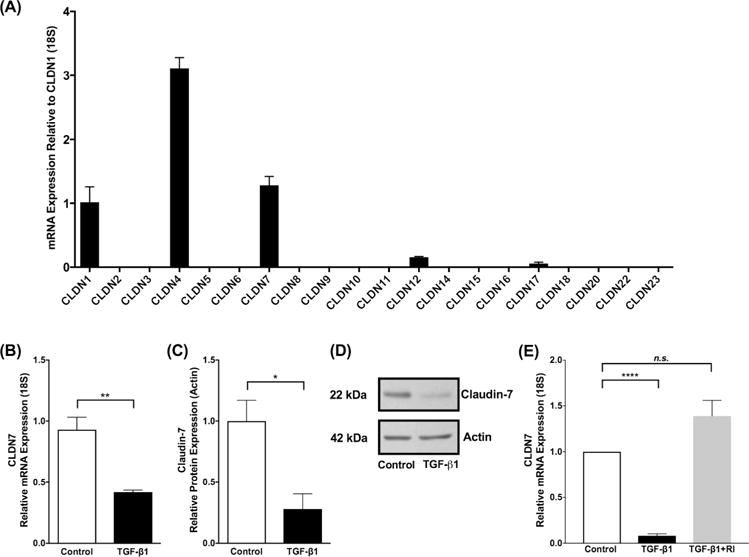
(A) Claudin mRNA expression in immortalized human esophageal epithelial (EPC2-hTERT) cells as mean fold change relative to CLDN1. CLDN 1, 4 and 7 were the predominantly expressed claudin mRNAs in the esophageal epithelium differentiated and stratified in 3D-ALI culture. (B) Transforming growth factor-β1 (TGF-β1) exposure (10 ng/ml) led to no significant effect on expression of selected epithelial barrier molecules, but decreased expression of CLDN7 mRNA and (C) claudin-7 protein in EPC2-hTERT cells at 3D-ALI (N=3, *p≤0.05, **p≤0.01). (C) Representative western blot demonstrates decreased claudin-7 protein in the presence of TGF-β1 (10 ng/ml). (E) EPC2-hTERT cells exposed to TGF-β1 (10 ng/ml) leads to attenuation in claudin-7, however when exposed to TGF-β type 1 receptor inhibitor (TGF-β RI) (5μm) prior to TGF-β1, cells were protected from the attenuation in claudin-7, indicating that the TGF-β1 pathway directly influences the attenuation of claudin-7 (N=4, ****p≤0.0001).
Table 1. Expression of Tight Junction, Adherens Junction, Desmosome, and Mesenchymal genes in transforming growth factor-β1 (TGF-β1) exposed immortalized human esophageal epithelial (EPC2-hTERT cells).
Data are expressed as mean fold change versus unstimulated controls ± SEM.
| Name | Fold Change ± SEM TGF-β1 (10 ng/ml) | Significance |
|---|---|---|
| Tight Junction | ||
| Claudin-1 | 1.27 ± 0.13 | n.s. |
| Claudin-4 | 0.63 ± 0.12 | n.s. |
| Claudin-7 | 0.45 ± 0.02 | p<0.01 |
| Occludin | 0.91 ± 0.14 | n.s. |
| Zonula Occluden-1 | 0.79 ± 0.16 | n.s. |
| Adherens Junction | ||
| E-Cadherin | 1.04 ± 0.01 | n.s. |
| Desmosome | ||
| Desmoglein-1 | 1.09 ± 0.30 | n.s. |
| Desmoglein-2 | 1.42 ± 0.17 | n.s. |
| Desmoglein-3 | 1.14 ± 0.33 | n.s. |
| Mesenchymal | ||
| Connective Tissue Growth Factor | 0.88 ± 0.33 | n.s. |
| Smooth Muscle α Actin | 1.16 ± 0.23 | n.s. |
| N-Cadherin | 49.69 ± 11.12 | p<0.05 |
Claudin-7 knockdown results in esophageal epithelial barrier dysfunction in vitro
In order to assess whether TGF-β1’s role was mediated, at least in part, through the suppression of CLDN7 expression, we generated CLDN7 knockdown cells using shRNA mediated approach and examined barrier function in these esophageal cells. To first validate the level of CLDN7 suppression in our cells, we examined CLDN7 mRNA and claudin-7 protein expression levels. We found our cells to have 97% reduction in CLDN7 mRNA expression (1.00±0 vs 0.03±0.004, shRNA control vs shRNA CLDN7 KD, N=3, p≤0.0001) as compared to control cells (Figure 3D). This resulted in a 90% reduction in claudin-7 expression at the protein level (1.00±0 vs 0.098±0.091, shRNA control vs shRNA CLDN7 KD, N=3, p≤0.01) as compared to control cells (Figure 3A–C). Specificity for CLDN7 was confirmed by assessing the expression of other related esophageal claudins at the protein level by western blot, including claudin-1 (1.00±0 vs 1.16±0.28, shRNA control vs shRNA CLDN7 KD, N=2, n.s.) and claudin-4 (1.00±0 vs 0.61±0.1, shRNA control vs shRNA CLDN7 KD, N=2, n.s.). No significant effects on these associated molecules were observed (Figure 3A & 3B).
Figure 3. Claudin-7 knockdown validation.
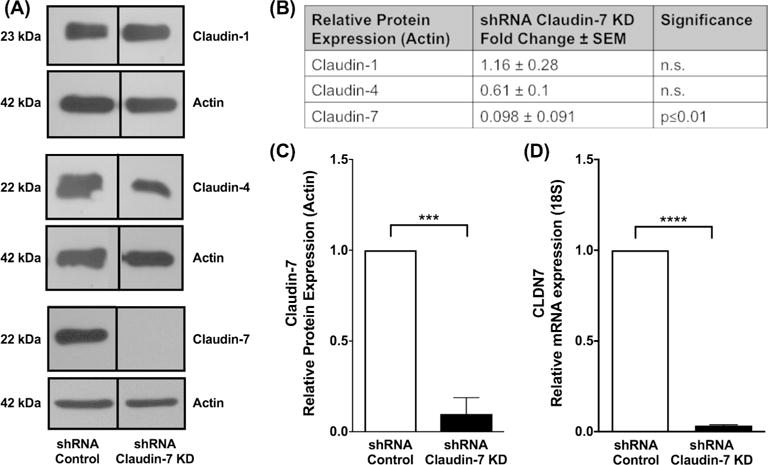
Claudin-7 knockdown cells were generated with shRNA constructs targeted against CLDN7 mRNA to determine the role of claudin-7 in epithelial barrier dysfunction independent of transforming growth factor-β1 (TGF-β1). (A) Western blot to assess specificity for CLDN7 was confirmed by assessing the expression esophageal claudins (claudin-1, claudin-4 and claudin-7) at the protein level by western blot on scrambled shRNA control and claudin-7 knockdown cells. (B) No significant effects on claudin-1 and claudin-4 were observed. Claudin-7 knockdown cells resulted in (C) 90% reduction in claudin-7 protein expression and (D) a 97% reduction in CLDN7 mRNA expression.
Functional barrier assessments of CLDN7 knockdown cells in the 3D-ALI model were performed to evaluate the importance of this specific molecule on esophageal epithelial barrier. We found a significant decrease in TEER (437.4± 19.5 vs 336.0±23.5 ohms.cm2, shRNA control vs shRNA CLDN7 KD, N=12, p≤0.01) and a parallel increase in paracellular flux (1.00±0 vs 1.54±0.31, shRNA control vs shRNA CLDN7 KD, N=11, p≤0.05) when compared to control cells (Figure 4A & 4B). We performed a histological analysis of these cells in the 3D-ALI model. H&E stained sections of CLDN7 knockdown cells grown in 3D-ALI exhibit prominent cell separation, particularly in the basal and suprabasal layers (Figure 4C, bottom panel) when compared to control cells, which displayed normal epithelial stratification (Figure 4C, top panel).
Figure 4. Claudin-7 knockdown results in diminished esophageal epithelial barrier function.

Knockdown of CLDN7 diminished barrier function as measured by (A) transepithelial electrical resistance (TEER) and increased permeability as measured by (B) 3kDa FITC dextran paracellular flux (FITC flux) (N=11, *p≤0.05, **p≤0.01). (C) H&E stained sections from CLDN7 knockdown cells grown in 3-dimensional air liquid interface (3D-ALI) culture. Arrows indicate prominent cellular separation. Black scale bar represents 50 μm. These data suggest that in the setting of attenuated claudin-7, epithelial barrier defects exist. (D) CLDN7 overexpressing cells (pLKO.1-Puro claudin-7 ORF) were generated and functional assessments were done in the presence and absence of TGF-β1 (10 ng/ml) in the 3-D ALI model. There was no significant decrease in TEER and (E) no significant increase in paracellular flux following TGF-β1 exposure. (N=5, n.s.). (F) H&E stained sections from CLDN7 overexpressing cells grown in 3D-ALI culture in the absence (top panel) and presence of TGF-β1 (bottom panel). Black scale bar represents 50 μm.
Additionally, we generated CLDN7 overexpressing cells (pLKO.1-Puro claudin-7 ORF) and compared functional assessments in these esophageal cells in the presence and absence of TGF-β1 () in the 3-D ALI model. Claudin-7 overexpressing cells, pLKO.1-Puro claudin-7 ORF, were validated and had 4.9 fold increase in mRNA expression and 2.5 fold increase in protein expression by western blot compared to control cells, pLKO.1-Puro EV. When pLKO.1-Puro claudin-7 ORF cells were grown in the presence and absence of TGF-β1, there was no significant decrease in TEER (376.2± 46.8 vs 341.0±53.1 ohms.cm2, pLKO.1-Puro claudin-7 ORF without TGF-β1 versus pLKO.1-Puro claudin-7 ORF with TGF-β1 10 ng/ml, N=5, n.s.) or increase in paracellular flux (1.00±0 vs 1.40±0.30, pLKO.1-Puro claudin-7 ORF without TGF-β1 versus pLKO.1-Puro claudin-7 ORF with TGF-β1 10 ng/ml, N=4, n.s.) compared to control cells following TGF-β1 exposure (Figure 4D & 4E). We performed a histological analysis of CLDN7 overexpressing cells in the 3D ALI model. H&E stained sections of these cells grown in 3D-ALI had normal stratification and differentiation (Figure 4F, top panel). CLDN7 overexpressing cells exposed to TGF-β1 (10 ng/ml) in the 3D-ALI model did not exhibit the same degree of cell separation as either EPC-hTERT cells exposed to TGF-β1 or as CLDN7 KD cells (Figure 4F, bottom panel). These results show that overexpression of claudin-7 in the presence of TGF-β1, will partially but not completely, protect the epithelial barrier and indicates that TGF-β1 impairs the barrier in part through a claudin-7 dependent manner. Our findings suggest that in the setting of attenuated claudin-7, even in the absence of other potential effects of TGF-β1, epithelial barrier defects exist. This finding suggests that suppression of claudin-7 in response to TGF-β1 exposure plays an important role in esophageal epithelial barrier integrity in vitro.
Claudin-7 expression is attenuated in pediatric subjects with active EoE
We next sought to evaluate the clinical relevance of these in vitro findings. Pediatric subjects were characterized by disease activity based on clinical symptoms and histopathology. We evaluated esophageal biopsies from active eosinophilic esophagitis (EoE) subjects (N=7), inactive EoE subjects (N=6) and healthy controls (N=10).
Consistent with our in vitro findings, we found that CLDN7 mRNA expression was significantly decreased in subjects with active EoE compared to control subjects. In addition, following successful treatment of EoE, in inactive EoE subjects, CLDN7 mRNA expression was restored to similar levels as in our control subjects (1.00±0.10 vs 0.30±0.10 vs 0.97±0.13, control subjects vs active EoE subjects vs inactive EoE, N=6–10 per group, p≤0.001) (Figure 5). We next performed immunofluorescent staining for claudin-7 protein in esophageal biopsies in order to characterize its expression pattern and localization. Control and inactive EoE subjects showed a crisp continuous linear pattern outlining the cell membrane in the suprabasal layer of the epithelium with a honeycomb appearance. However, claudin-7 immunofluorescence in active EoE subjects showed a diffuse granular perimembraneous pattern outlining the cell membrane in addition to increased staining present within in the cytoplasm of the cells. This was particularly the case in cells of the suprabasal layers. (Figure 6 A–C).
Figure 5. Claudin-7 expression is decreased in pediatric subjects with EoE.
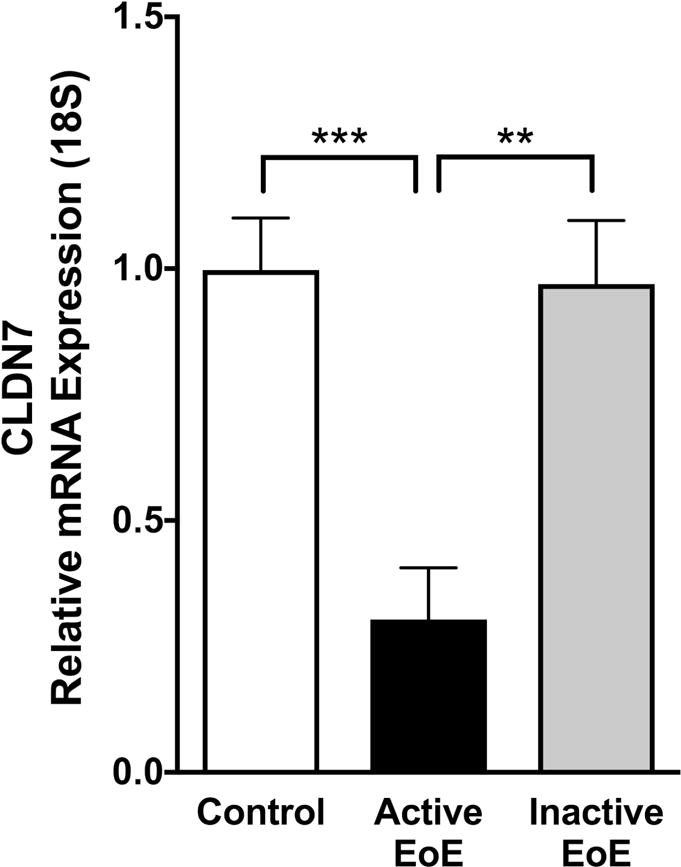
CLDN7 mRNA is decreased in active eosinophilic esophagitis (EoE) (N=7) compared to control subjects (N=10). Levels returned to near normal following successful clinical treatment (inactive EoE, N=6) (**p≤0.01, ***p≤0.001).
Figure 6. Claudin-7 immunofluorescence in pediatric subjects.
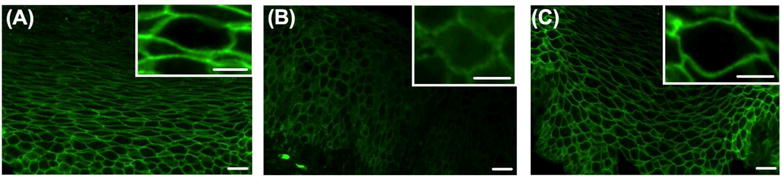
Immunofluorescent staining of claudin-7 in human esophageal biopsies from pediatric (A) control (B) active eosinophilic esophagitis (EoE) and (C) inactive EoE subjects. Immunofluorescence of (B) active EoE shows a diffuse granular perimembraneous pattern outlining the cell membrane with staining present within in the cytoplasm of the cells in the suprabasal layer when compared to (A) control and (C) inactive EoE subjects which show a continuous linear pattern outlining the cell membrane in the suprabasal layer of the epithelium with a honeycomb appearance. Images at 400× magnification. White scale bar represents 20 μm. White scale bar in inset image represents 2 μm.
TGF-β1 signaling increased in EoE
Because activation of the canonical TGF-β1 pathway leads to phosphorylation of SMAD2/3 and nuclear translocation of SMAD complexes that affect gene transcription31, we evaluated phosphorylated SMAD2/3 (pSMAD2/3) in active EoE subjects compared to controls and inactive EoE subjects. pSMAD2 staining was present in both the lamina propria and the epithelium of subjects with active EoE, similar to previously published findings8. Future studies better powered to focus on the functional effects of lamina propria pSMAD2/3 should be pursued. However, for our focus on TGF-β1’s effect on the epithelium, we found increased nuclear pSMAD2 staining in esophageal epithelial cells in active EoE subjects compared to control and inactive EoE (500±54 vs 695±61 vs 504±35 positively stained epithelial cells per high power field, N=3 × 3 fields of 40× view, p≤0.05) (Figure 7A–D) indicating increased TGF-β1 signaling in the epithelium of subjects with active EoE. In addition, semi-quantitative analysis of pSMAD2/3 protein expression by western blot was consistent with these findings, with increased expression in active EoE when compared to control and inactive EoE subjects as measured by densitometry (1.17±0.20 vs 3.17±0.64 vs 1.88±0.47, control subjects vs active EoE subjects vs inactive EoE subjects, N=6 per group, p≤0.05) (Figure 7E–F). To determine whether the regulation of SMAD2/3 is at the activation or expression level, total SMAD2/3 was also assessed by western blot in a cohort of these samples. There was no significant difference in total SMAD2/3 protein expression observed between control, active and inactive EoE subjects (1.00±0.41 vs 2.06±0.70 vs 2.92±1.21, normal subjects vs active EoE subjects vs inactive EoE subjects, N=3–4 per group, n.s.), indicating that there is an increase in the activation of SMAD2/3 by phosphorylation in active EoE subjects (Figure 7G).
Figure 7. pSMAD2/3 in pediatric subjects.
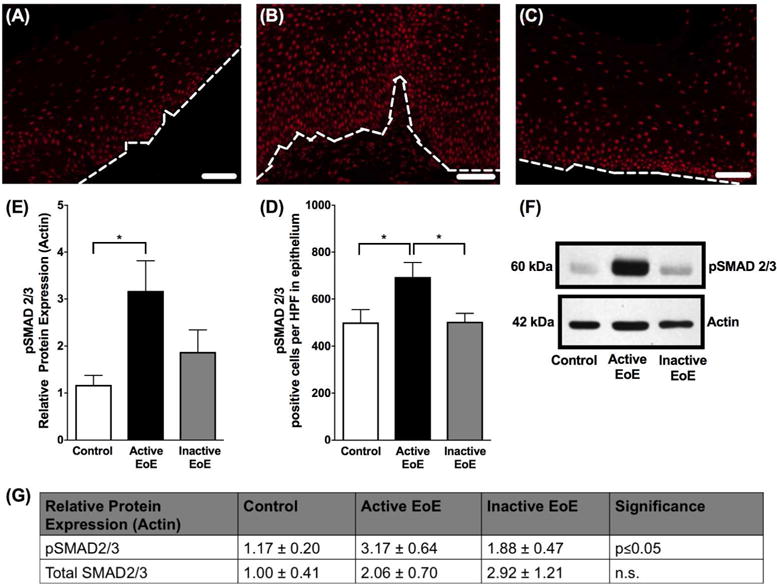
Immunofluorescent staining of phosphorylated SMAD2/3 (pSMAD2/3) in human esophageal biopsies from (A) control (B) active eosinophilic esophagitis (EoE) and (C) inactive EoE subjects. A C at 200× magnification, white scale bar represents 50 μm. White dotted line represents basal lamina. (D) Quantification shows increased positive cells per high power field of pSMAD2/3 staining in active EoE compared to control and inactive EoE. (E) Western blot densitometry shows increase in protein expression of pSMAD2/3 in active EoE vs control and inactive EoE. (F) Western blot showing increase in protein expression of pSMAD2/3 in active EoE vs control and inactive EoE. (G) Relative protein expression of pSMAD2/3 and total SMAD2/3 in active EoE vs control and inactive EoE.
DISCUSSION
In this study, we demonstrate that transforming growth factor-β1 (TGF-β1) leads to functional esophageal epithelial barrier defects in vitro. We show that esophageal epithelial cells cultured in 3-dimensional air liquid interface (3D-ALI) stratified squamous cultures exposed to TGF-β1 led to a specific attenuation of claudin-7 expression and that shRNA-mediated knockdown results in barrier defects, indicating a role for claudin-7 in maintaining epithelial barrier integrity. Notably, we demonstrate that claudin-7 expression is decreased and phosphorylated SMAD2/3 (pSMAD2/3) is increased in pediatric subjects with active EoE when compared to control and inactive EoE subjects. The results herein suggest that the down regulation of claudin-7 by TGF-β1 plays a role in epithelial barrier dysfunction in eosinophilic esophagitis (EoE).
Our data shows that in the presence of TGF-β1, transepithelial electrical resistance (TEER) decreases and 3 kDa FITC Dextran Flux (FITC flux) increases, suggesting that TGF-β1 leads to an impaired epithelial barrier. Recent clinical studies implicate epithelial barrier dysfunction in the pathogenesis of EoE, including dilated intercellular spaces and decreased electrical impedance in subjects with active EoE5,32,33. Additionally, in translational studies, decreased expression of junctional cell cell adhesion complex molecules, such as desmoglein and barrier forming filaggrin, further suggests an impaired epithelial barrier in EoE3,4,7,34. Some of these studies have focused on the impact of the type-2 cytokine IL-13 as a mediator of the negative regulation of epithelial barrier molecules associated with esophageal epithelium3,4. Our study adds to the growing body of evidence that altered epithelial barrier plays a role in the pathogenesis of EoE and indeed is the first study to examine the importance of claudin-7 in esophageal epithelial barrier function. Other studies have shown important roles for this molecule in the simple columnar epithelium of the colon and the cuboidal epithelium of the salivary glands35,36. In vitro models examining the role of TGF-β1 and impaired epithelial barrier function has been evaluated in other epithelia types, including the columnar epithelia of the lung, pseudostratified epithelium of the epididymis and vas defrens, however this has not been previously examined the in stratified epithelium or indeed the esophagus21–23. Our study is the first to show the effect of TGF-β1 on altered epithelial barrier function and specifically the key role of claudin-7 in the esophagus. Previous EoE studies have shown that TGF-β1 plays a role in esophageal remodeling, participates in esophageal smooth muscle contraction, stimulates fibroblast secretion of extracellular matrix, and contributes to esophageal epithelial to mesenchymal transition (EMT)8,10,14–20. Our findings suggest that it is possible that chronic TGF-β1 mediated altered esophageal epithelial barrier or increased esophageal epithelial permeability may allow passage of allergens that enhances antigen presentation an immune activation and contributes in a previously unconsidered way to the chronicity of disease in EoE patients, though this would require further examination outside the remit of the current studies.
The findings in our study demonstrate that claudin-7 is down-regulated both at the transcriptional and translational/post-translational level in the presence of TGF-β1. The claudin family of proteins are transmembrane proteins found in tight junctions that fall into 2 functional categories: barrier-forming claudins that serve to tighten the barrier or pore forming claudins that increase permeability37. Tight junctions serve as a barrier to paracellular permeability by forming a seal between epithelial cells and are important to the integrity of the epithelial barrier37. Claudin-7 is a barrier-forming claudin found in the epithelium of a variety of tissues37. Several previous studies have shown that claudin-1 and claudin-4, which are both barrier-forming claudins, are affected in in vitro models of gastroesophageal reflux disease (GERD) in conjunction with increased permeability and decreased transepithelial electrical resistance, indicating a disrupted epithelial barrier27,28. In addition, the esophageal epithelium in subjects with GERD has significantly decreased claudin-1 and claudin-4 expression29. After exposure to TGF-β1 in vitro, we did not see a significant decrease in claudin-1 or claudin-4, which was previously shown in subjects with GERD. Instead, we observed a significant and specific decrease in claudin-7, indicating that this is specific to TGF-β1 signaling. In addition, in pediatric subjects with active EoE, we found that claudin-7 expression was significantly decreased as compared to control subjects, suggesting that the attenuation in claudin-7 may be specific to TGF-β1 signaling in EoE and is important for esophageal barrier function.
In pediatric subjects with active EoE, we show that pSMAD2/3, is increased when compared to normal subjects and inactive EoE subjects, suggesting increased TGF-β1 signaling activity in the esophageal epithelium in active EoE. This is consistent with previous studies that have shown increased TGF-β1 and pSMAD2/3 in active EoE with the cellular source of TGF-β1 being eosinophils, mast cells and epithelial cells8–11. In the canonical SMAD dependent TGF-β1 signaling pathway, binding of TGF-β1 to TGF-β receptors I and II allows TGF-β receptor II to trans phosphorylate TGF-β receptor I leading to the phosphorylation of SMAD 2 and 331. pSMAD2/3 complexes with the common SMAD4 and trans-locates into the nucleus31. SNAI1, a transcription repressor, in complex with SMAD in the canonical TGF-β1 signaling pathway, has been shown to target the gene promoter of several tight junction molecules, specifically claudin-3 and E-cadherin in breast epithelial cells38. In other systems, SNAI1 plays a role in transcriptional repression of claudin-739–41. Other regulators of claudin-7 include hepatocyte nuclear factor 4alpha 36. Therefore, a potential mechanism for the attenuation of claudin-7 is through the TGF-β1 signaling pathway with induction of the SMAD/SNAI1 transcriptional repressor complex leading to the transcriptional repression of claudin-7 with resulting epithelial barrier dysfunction. Specifically, in vitro lung injury models revealed that TGF-β1 reduced levels of claudin-521. In vitro models of the vas deferens, which is a columnar epithelium, have shown that TGF-β1 disrupts epithelial barrier with attenuation in claudin-723. Our study shows a specific attenuation of claudin-7 and decreased barrier function in esophageal epithelial cells after exposure to TGF-β1. We show that in the esophagus, which is a stratified squamous epithelium, subjects with active EoE have a reduction in claudin-7 and alterations in the localization of claudin-7. We also demonstrate that knockdown of claudin-7 in esophageal epithelial cells resulted in a significant decrease in barrier function as measured by decreased TEER and increased paracellular permeability as measured by FITC flux implicating the importance of this molecule in esophageal epithelial barrier function, and the lack of redundancy with other claudin molecules expressed in the esophagus. Overall, our data suggests that claudin-7 plays a key role in maintaining the esophageal epithelial barrier function.
There are limitations to our study. First, our findings of esophageal epithelial barrier dysfunction are from in vitro studies and those with EoE patient biopsies. Future functional studies evaluating mucosal integrity by intraluminal impedance in conjunction with evaluating cell adhesion molecules, such as claudin-7, and activation of the TGF-β1 signaling pathway are further needed. Second, earlier studies illustrated a role for TGF-β1 in EMT20. Unique to original studies on submerged cells the focus of this study was to determine the effects of TGF-β1 specifically on epithelial barrier in cells grown in an organotypic fashion. Although cells exposed to TGF-β1 in this 3D-ALI environment increased N-cadherin expression, it did not decrease E-cadherin expression. Loss of E-cadherin is a key event in the EMT cascade. By definition, Kalluri and Weinberg classify EMT as a process in which an epithelial cell completely loses its polarized, barrier forming identity in turn for a mesenchymal state that functionally includes enhanced migration and invasiveness, increased apoptotic resistance, and the greatly increased production of extra-cellular matrix components42. Our studies, found a specific and significant decrease in CLDN7, an increase in N-cadherin and no change in E-cadherin immediately in response to TGF-β1 exposure. In future studies, longer-term exposure to TGF-β1 at ALI culture may lead to a functional EMT-like process that will critically involve the loss of epithelial molecular and functional phenotypes. EMT plays an important role in wound healing following inflammation and injury. Pathological fibrosis can arise from excessive wound healing, a process in which TGF-β1 has been shown to play a key role 31. In EoE, studies have shown that TGF-β1 plays a role in esophageal remodeling, esophageal smooth muscle contraction, EMT, and fibroblast secretion of extracellular matrix in vitro10,14–18 and in vivo8,19,20. Thus, TGF-β1 may not only play a role in EoE as it relates to EMT and fibrosis, but also in epithelial barrier dysfunction.
In summary, our findings indicate a pathogenic role for TGF-β1 in esophageal barrier dysfunction. TGF-β1, which is elevated in EoE, leads to alterations in epithelial barrier function, specifically through diminishing claudin-7 expression. With the growing body of clinical evidence suggesting a role of altered barrier in EoE specifically, and in atopic diseases in general, our findings support a potential role for TGF-β1 in this process. The advent of TGF-β1 inhibiting therapeutics presents an opportunity to consider this novel therapeutic approach that could alter both fibrotic as well as barrier alterations in EoE.
Acknowledgments
We are grateful to our patients and families who consented to be in this study. We thank the physicians, nurses, and research assistants who contributed to this work by helping to recruit subjects and collect samples. We thank the members of all the participating laboratories for insightful discussions and critical comments. The immortalized human esophageal epithelial cells (EPC2-hTERT) were a kind gift from Drs. Anil Rustgi and Hiro Nakagawa, University of Pennsylvania.
Supported by: This study was supported by grants from the United States National Institutes of Health (NIH) 2T32DK067009-11 (Nguyen N), 1K24DK100303 (Furuta GT), K01-DK106315 (Masterson JC), the American Partnership for Eosinophilic Disorders (Masterson JC). The study is also funded by U54 AI117804, which is part of the Rare Disease Clinical Research Network (RDCRN), an initiative of the Office of Rare Disease Research (ORDR), NCATS, and is funded through collaboration between NCATS, NIAID and NIDDK, as well as the patient advocacy groups American Partnership for Eosinophilic Disorders (APFED), CURED and the Eosinophilic Family Coalition (EFC), which have collectively resulted in the Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR). (Furuta GT). Manuscript contents are the authors’ sole responsibility and do not necessarily represent official NIH views. The study sponsors played no role in the study design in the collection, analysis, and interpretation of data.
Abbreviations
- 3D-ALI
3-dimensional air liquid interface
- ANOVA
Analysis of variance
- Ca2+
Calcium
- CLDN7
Claudin-7
- EMT
Epithelial Mesenchymal Transition
- EoE
Eosinophilic Esophagitis
- Eos/HPF
eosinophils per high power field
- EPC2-hTERT
immortalized human esophageal epithelial
- FITC Flux
3kDa FITC dextran paracellular flux
- GERD
Gastroesophageal Reflux Disease
- H&E
hematoxylin and eosin
- KSFM
Keratinocyte serum-free media
- ORF
Open reading frame
- pSMAD2/3
phosphorylated SMAD2/3
- RIPA
radioimmunoprecipitation assay
- RT-PCR
Reverse Transcription Polymerase Chain Reaction
- shRNA
Short hairpin ribonucleic acid
- TEER
Transepithelial electrical resistance
- TGF-β1
Transforming growth factor-β1
- TGF-β RI
TGF-β type 1 receptor inhibitor
Footnotes
AUTHOR CONTRIBUTIONS
Study concept and design (NN, SDF, SPC, GTF, JCM); acquisition of data (NN, SDF, KAB, JAH, KEC, DAK); analysis and interpretation of data (NN, SDF, LEG, SPC, GTF, JCM); drafting of the manuscript (NN, GTF, JCM); critical revision of the manuscript for important intellectual content (NN, SDF, SPC, GTF, JCM); statistical analysis (NN, SDF, JCM); obtained funding (NN, GTF, JCM); study supervision (NN, GTF, JCM).
DISCLOSURES
The authors declare no conflicts of interest.
Conflicts of Interest: None.
References
- 1.Liacouras CA, Furuta GT, Hirano I, et al. Eosinophilic esophagitis: Updated consensus recommendations for children and adults. J Allergy Clin Immunol. 2011;128:3–20 e6. doi: 10.1016/j.jaci.2011.02.040. [DOI] [PubMed] [Google Scholar]
- 2.Collins MH. Histopathologic features of eosinophilic esophagitis and eosinophilic gastrointestinal diseases. Gastroenterol Clin North Am. 2014;43:257–68. doi: 10.1016/j.gtc.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 3.Blanchard C, Stucke EM, Burwinkel K, et al. Coordinate interaction between IL-13 and epithelial differentiation cluster genes in eosinophilic esophagitis. J Immunol. 2010;184:4033–41. doi: 10.4049/jimmunol.0903069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sherrill JD, Kc K, Wu D, et al. Desmoglein-1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis. Mucosal immunology. 2014;7:718–29. doi: 10.1038/mi.2013.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katzka DA, Tadi R, Smyrk TC, et al. Effects of topical steroids on tight junction proteins and spongiosis in esophageal epithelia of patients with eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2014;12:1824–9 e1. doi: 10.1016/j.cgh.2014.02.039. [DOI] [PubMed] [Google Scholar]
- 6.van Rhijn BD, Kessing BF, Smout AJ, Bredenoord AJ. Oesophageal baseline impedance values are decreased in patients with eosinophilic oesophagitis. United European gastroenterology journal. 2013;1:242–8. doi: 10.1177/2050640613496411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Capocelli KE, Fernando SD, Menard-Katcher C, Furuta GT, Masterson JC, Wartchow EP. Ultrastructural features of eosinophilic oesophagitis: impact of treatment on desmosomes. J Clin Pathol. 2015;68:51–6. doi: 10.1136/jclinpath-2014-202586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aceves SS, Newbury RO, Dohil R, Bastian JF, Broide DH. Esophageal remodeling in pediatric eosinophilic esophagitis. J Allergy Clin Immunol. 2007;119:206–12. doi: 10.1016/j.jaci.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 9.Straumann A, Conus S, Grzonka P, et al. Anti-interleukin-5 antibody treatment (mepolizumab) in active eosinophilic oesophagitis: a randomised, placebo controlled, double blind trial. Gut. 2010;59:21–30. doi: 10.1136/gut.2009.178558. [DOI] [PubMed] [Google Scholar]
- 10.Aceves SS, Chen D, Newbury RO, Dohil R, Bastian JF, Broide DH. Mast cells infiltrate the esophageal smooth muscle in patients with eosinophilic esophagitis, express TGF-beta1, and increase esophageal smooth muscle contraction. J Allergy Clin Immunol. 2010;126:1198–204 e4. doi: 10.1016/j.jaci.2010.08.050. [DOI] [PubMed] [Google Scholar]
- 11.Straumann A, Conus S, Degen L, et al. Budesonide is effective in adolescent and adult patients with active eosinophilic esophagitis. Gastroenterology. 2010;139:1526–37. 37 e1. doi: 10.1053/j.gastro.2010.07.048. [DOI] [PubMed] [Google Scholar]
- 12.Lucendo AJ, Arias A, De Rezende LC, et al. Subepithelial collagen deposition, profibrogenic cytokine gene expression, and changes after prolonged fluticasone propionate treatment in adult eosinophilic esophagitis: a prospective study. J Allergy Clin Immunol. 2011;128:1037–46. doi: 10.1016/j.jaci.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Mishra A, Wang M, Pemmaraju VR, et al. Esophageal remodeling develops as a consequence of tissue specific IL-5-induced eosinophilia. Gastroenterology. 2008;134:204–14. doi: 10.1053/j.gastro.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beppu LY, Anilkumar AA, Newbury RO, Dohil R, Broide DH, Aceves SS. TGF-beta1-induced phospholamban expression alters esophageal smooth muscle cell contraction in patients with eosinophilic esophagitis. J Allergy Clin Immunol. 2014;134:1100–7 e4. doi: 10.1016/j.jaci.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muir AB, Dods K, Henry SJ, et al. Eosinophilic Esophagitis-Associated Chemical and Mechanical Microenvironment Shapes Esophageal Fibroblast Behavior. J Pediatr Gastroenterol Nutr. 2016;63:200–9. doi: 10.1097/MPG.0000000000001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muir AB, Dods K, Noah Y, et al. Esophageal epithelial cells acquire functional characteristics of activated myofibroblasts after undergoing an epithelial to mesenchymal transition. Exp Cell Res. 2015;330:102–10. doi: 10.1016/j.yexcr.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muir AB, Lim DM, Benitez AJ, et al. Esophageal epithelial and mesenchymal cross-talk leads to features of epithelial to mesenchymal transition in vitro. Exp Cell Res. 2013;319:850–9. doi: 10.1016/j.yexcr.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rieder F, Nonevski I, Ma J, et al. T-helper 2 cytokines, transforming growth factor beta1, and eosinophil products induce fibrogenesis and alter muscle motility in patients with eosinophilic esophagitis. Gastroenterology. 2014;146:1266–77. e1–9. doi: 10.1053/j.gastro.2014.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho JY, Doshi A, Rosenthal P, et al. Smad3-deficient mice have reduced esophageal fibrosis and angiogenesis in a model of egg induced eosinophilic esophagitis. J Pediatr Gastroenterol Nutr. 2014;59:10–6. doi: 10.1097/MPG.0000000000000343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kagalwalla AF, Akhtar N, Woodruff SA, et al. Eosinophilic esophagitis: epithelial mesenchymal transition contributes to esophageal remodeling and reverses with treatment. J Allergy Clin Immunol. 2012;129:1387–96 e7. doi: 10.1016/j.jaci.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohta H, Chiba S, Ebina M, Furuse M, Nukiwa T. Altered expression of tight junction molecules in alveolar septa in lung injury and fibrosis. American journal of physiology Lung cellular and molecular physiology. 2012;302:L193–205. doi: 10.1152/ajplung.00349.2010. [DOI] [PubMed] [Google Scholar]
- 22.Stammler A, Muller D, Tabuchi Y, Konrad L, Middendorff R. TGFbetas modulate permeability of the blood-epididymis barrier in an in vitro model. PLoS One. 2013;8:e80611. doi: 10.1371/journal.pone.0080611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierucci-Alves F, Yi S, Schultz BD. Transforming growth factor beta 1 induces tight junction disruptions and loss of transepithelial resistance across porcine vas deferens epithelial cells. Biology of reproduction. 2012;86:36. doi: 10.1095/biolreprod.111.092262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harada H, Nakagawa H, Oyama K, et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res. 2003;1:729–38. [PubMed] [Google Scholar]
- 25.Saeedi BJ, Kao DJ, Kitzenberg DA, et al. HIF dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Molecular biology of the cell. 2015;26:2252–62. doi: 10.1091/mbc.E14-07-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kc K, Rothenberg ME, Sherrill JD. In vitro model for studying esophageal epithelial differentiation and allergic inflammatory responses identifies keratin involvement in eosinophilic esophagitis. PLoS One. 2015;10:e0127755. doi: 10.1371/journal.pone.0127755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Oshima T, Tomita T, et al. Acidic bile salts modulate the squamous epithelial barrier function by modulating tight junction proteins. Am J Physiol Gastrointest Liver Physiol. 2011;301:G203–9. doi: 10.1152/ajpgi.00096.2011. [DOI] [PubMed] [Google Scholar]
- 28.Oshima T, Koseki J, Chen X, Matsumoto T, Miwa H. Acid modulates the squamous epithelial barrier function by modulating the localization of claudins in the superficial layers. Lab Invest. 2012;92:22–31. doi: 10.1038/labinvest.2011.139. [DOI] [PubMed] [Google Scholar]
- 29.Bjorkman EV, Edebo A, Oltean M, Casselbrant A. Esophageal barrier function and tight junction expression in healthy subjects and patients with gastroesophageal reflux disease: functionality of esophageal mucosa exposed to bile salt and trypsin in vitro. Scand J Gastroenterol. 2013;48:1118–26. doi: 10.3109/00365521.2013.828772. [DOI] [PubMed] [Google Scholar]
- 30.Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119:1417–9. doi: 10.1172/JCI39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nature reviews Drug discovery. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Rhijn BD, Weijenborg PW, Verheij J, et al. Proton pump inhibitors partially restore mucosal integrity in patients with proton pump inhibitor-responsive esophageal eosinophilia but not eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2014;12:1815–23 e2. doi: 10.1016/j.cgh.2014.02.037. [DOI] [PubMed] [Google Scholar]
- 33.Ravelli A, Villanacci V, Cadei M, Fuoti M, Gennati G, Salemme M. Dilated intercellular spaces in eosinophilic esophagitis. J Pediatr Gastroenterol Nutr. 2014;59:589–93. doi: 10.1097/MPG.0000000000000491. [DOI] [PubMed] [Google Scholar]
- 34.Simon D, Radonjic Hosli S, Straumann A, Yousefi S, Simon HU. Active eosinophilic esophagitis is characterized by epithelial barrier defects and eosinophil extracellular trap formation. Allergy. 2015;70:443–52. doi: 10.1111/all.12570. [DOI] [PubMed] [Google Scholar]
- 35.Abe A, Takano K, Kojima T, et al. Interferon-gamma increased epithelial barrier function via upregulating claudin-7 expression in human submandibular gland duct epithelium. Journal of molecular histology. 2016;47:353–63. doi: 10.1007/s10735-016-9667-2. [DOI] [PubMed] [Google Scholar]
- 36.Farkas AE, Hilgarth RS, Capaldo CT, et al. HNF4alpha regulates claudin-7 protein expression during intestinal epithelial differentiation. Am J Pathol. 2015;185:2206–18. doi: 10.1016/j.ajpath.2015.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gunzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiological reviews. 2013;93:525–69. doi: 10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vincent T, Neve EP, Johnson JR, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial mesenchymal transition. Nature cell biology. 2009;11:943–50. doi: 10.1038/ncb1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu Y, Yu L, Yang M, et al. The effects of shRNA-mediated gene silencing of transcription factor SNAI1 on the biological phenotypes of breast cancer cell line MCF-7. Molecular and cellular biochemistry. 2014;388:113–21. doi: 10.1007/s11010-013-1903-4. [DOI] [PubMed] [Google Scholar]
- 40.Mittal MK, Myers JN, Bailey CK, Misra S, Chaudhuri G. Mode of action of the retrogene product SNAI1P, a SNAIL homolog, in human breast cancer cells. Molecular biology reports. 2010;37:1221–7. doi: 10.1007/s11033-009-9492-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pereira F, Barbachano A, Silva J, et al. KDM6B/JMJD3 histone demethylase is induced by vitamin D and modulates its effects in colon cancer cells. Hum Mol Genet. 2011;20:4655–65. doi: 10.1093/hmg/ddr399. [DOI] [PubMed] [Google Scholar]
- 42.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]