

Abstract
L-DOPA provides highly effective treatment for Parkinson’s disease, but L-DOPA induced dyskinesia (LID) is a very debilitating response that eventually is presented by a majority of patients. A central issue in understanding the basis of LID is whether it is due to a response to chronic L-DOPA over years of therapy, and/or due to synaptic changes that follow the loss of dopaminergic neurotransmission and then triggered by acute L-DOPA administration. We review recent work that suggests that specific synaptic changes in the D1 dopamine receptor-expressing direct pathway striatal projection neurons due to loss of dopamine in Parkinson’s disease are responsible for LID. Chronic L-DOPA may nevertheless modulate LID through priming mechanisms.
Introduction
Replacement therapy for Parkinson’s disease (PD) with the dopamine (DA) precursor L-DOPA is, in light of the complex nature of the disease, a remarkably successful strategy that substantially improves motor performance in most patients in the early stages after PD diagnosis. The efficacy of the long-term therapy with L-DOPA is reduced with disease duration, however, by the emergence of unwanted effects of which abnormal involuntary movements, or dyskinesia, are the most disabling. L-DOPA induced dyskinesia (LID), which is widely held to be a hypersensitivity to the treatment, depends on two main factors: 1) the severity of the DA denervation, 2) long-term treatment with pulsatile high doses of L-DOPA [1]. In recent years, molecular and genetic tools applied to experimental models of PD have greatly enhanced our understanding of the cellular basis and the neural circuits involved in the expression of LID [2]. There nevertheless remains no effective treatment to overcome LID and it continues to impose a prominent clinical problem.
A central issue for understanding the cause of LID in patients is whether the underlying changes in basal ganglia circuits are in response to chronic L-DOPA treatment, or due to the loss of DA input and then triggered by acute L-DOPA treatment: of course, these two possibilities are not mutually exclusive [3,4]. To date, experimental studies on LID have primarily focused on the effects exerted by L-DOPA. In contrast, the contribution of the DA denervation to LID is less clear, mostly and until recently it has not been possible to induce LID in animal models without administrating DA receptor ligands. It is nevertheless increasingly recognized that DA denervation to the basal ganglia produces multiple pre- and postsynaptic changes associated with hypersensitive responses to L-DOPA [4,5]. These compensatory mechanisms are presumably homeostatic phenomena related to mechanisms by which the basal ganglia circuitry participates in non-associative learning [6] in an attempt to normalize function at glutamatergic and GABAergic synapses in the absence of DA [7]. Here, we will review recent findings that support a fundamental role for DA denervation, in contrast to chronic L-DOPA, as the central cause of acute LID, and particularly new studies that implicate synaptic plasticity in the striatal output direct pathway following DA denervation that could trigger LID.
The initiation of LID depends on DA denervation
It is challenging to separate the relative contribution of chronic and acute use of L-DOPA from disease progression. In the clinic, PD patients typically receive therapy for about 4 to 6 years before LID appears [1]. A further mechanism that complicates analysis is the ability of residual DA axons and sprouting of serotonin axons that sustain DA output and buffer the effects of L-DOPA [8].
Particularly instructive observations are from studies of patients who have severe reduction in DA and did not receive long-term replacement therapy. For example, L-DOPA produces a dramatic and almost immediate development of LID when neurodegeneration of substantia nigra compacta DA neurons is produced acutely by the neurotoxin MPTP [9]. Similarly, LID is observed in patients with genetic defects in DA synthesis, implicating that an absence of endogenous DA as an important factor for LID [10]. A recent study compared LID onset in two groups of patients diagnosed with a comparable stage of PD symptomology: however, one of the groups had a longer history of L-DOPA therapy. Notably, LID developed approximately at the same time in both groups [11], strongly supporting disease progression as a central cause of LID.
In experimental animals, where the degree of DA denervation can be precisely attuned, it is clear that repeated administration is not an absolute requirement to trigger LID; rather, it is required that DA denervation is sufficiently severe [12,13]. In animals with nearly complete destruction of DA neurons, such as in the unilateral 6-OHDA rodent model of PD [14], L-DOPA produces a supersensitive response early in the course of L-DOPA treatment. Indeed, in rodent and monkey PD models with nearly complete ablation of DA by 6-OHDA or MPTP, the first administration of L-DOPA can trigger LID [12].
These observations support the possibility that presentation of LID is due to synaptic changes following DA denervation, with L-DOPA triggering LID expression. Nevertheless, LID responses in animal models can escalate in severity with repeated treatment. The escalation in the behavioral responsiveness to repeated L-DOPA is known as priming, and effects of chronic L-DOPA may contribute to the modulation of clinical LID, even if chronic administration is not required for LID per se.
Striatonigral SPNs and LID
The central pathological feature of the movement disorder aspects of PD is the loss of SN DA neurons that project to the striatum. The replacement of striatal DA in PD by L-DOPA alters the activity of the output neurons of the striatum, which are known as GABAergic spiny projection neurons (SPNs) (Fig 1A). The SPNs are divided into two roughly equal populations on the basis of the expression of either D1 or D2-like of DA receptors and their projections.
Figure 1. Modulation of SPN neuronal plasticity occurs at multiple synaptic sites.
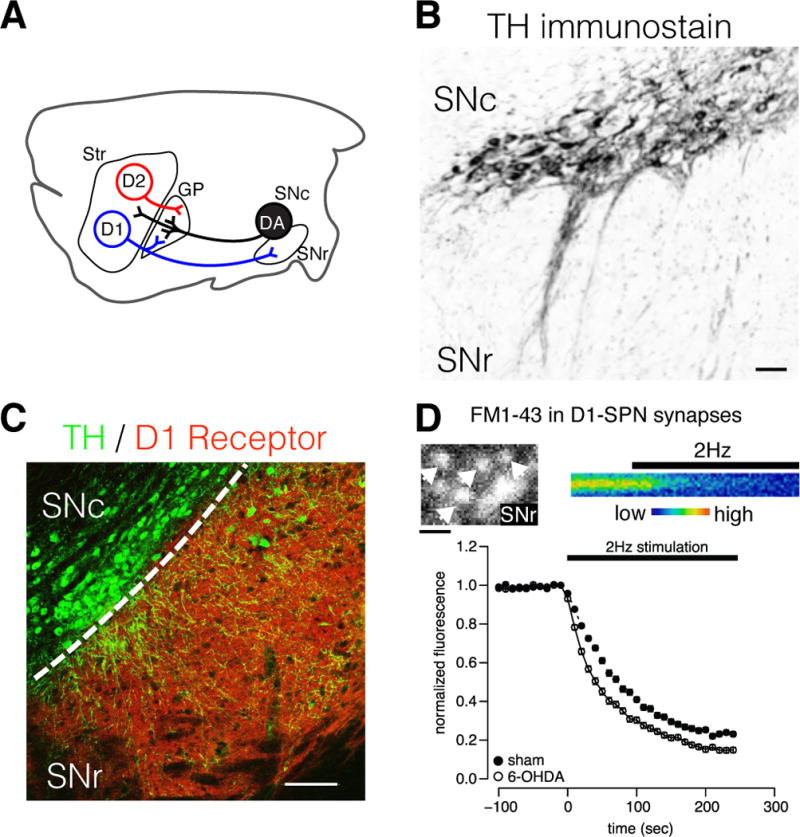
A) Simplified schematic of striatofugal SPN and ascending DA projections in the rodent brain. DA neurons of the SNc can influence neuronal plasticity in distinct microcircuits via post- and presynaptic DA receptors in the striatum, GP and SNr. B) Immunolabel for tyrosine hydroxylase (TH) in mouse ventral midbrain showing DA neurons of the SNc and their dendritic processes extending into the SNr. Somatodendritically released DA presumably influences motor functions by local actions within the midbrain. Scale = 50 μm. C) Immunolabel for TH (green) and D1R (red) in a mouse midbrain section. D1R are preferentially located on the terminals of D1-SPNs, and thus DA neurons directly influence local GABA release from the striatonigral direct pathway. Scale = 100 μm. D) Labeling of D1-SPNs synapses with the fluorescent vesicular probe FM1–43 to study presynaptic plasticity produced by DA denervation. Upper panels show 2-photon images of FM1-43-labeled D1-SPN synaptic boutons in the SNr (left), and a time-series of destaining from a single fluorescent bouton during stimulation of D1-SPN projections (right). Loss of fluorescence represents the fusion of GABA-containing vesicles during electrically evoked synaptic activity and the rate of decay reveals the probability of neurotransmitter release. Bottom panel shows the normalized FM1-43 fluorescence as a function of time from >100 D1-SPN synaptic boutons in brain slice preparations from DA intact (sham) and denervated parkinsonian mice (6-OHDA). The rate of vesicular fusion is greatly increased in DA denervated animals, indicating that increased GABA release during synaptic activity provides a potential mechanism for LID. Scale = 3 μm.
The SPNs of the striatonigral direct pathway are characterized by a expression of DA D1 receptors, and their long-range axons form monosynaptic contacts with the two major output structures of the basal ganglia, the substantia nigra reticulata (SNr) and the globus pallidus internal segment (GPi). In contrast, SPNs of the striatopallidal indirect pathway are enriched in DA D2 receptor expression and control the activity of the basal ganglia output nuclei indirectly through a polysynaptic circuit involving the globus pallidus external segment (GPe) and the subthalamic nucleus (STN). Activation of DA receptors in the striatum promotes motor activity by activating D1-SPN and inhibiting D2-SPNs, which drives increased GABA release from synapses within SNr and GPi. This removes the tonic inhibition provided by these inhibitory output nuclei on thalamocortical and brainstem projections that control of motor initiation [15].
A commonly model postulates that LID, as a hyperkinetic response, is concomitant to an abnormal activity in the basal ganglia output system, triggered by an over-stimulated striatonigral direct pathway [2]. In support of this model, the ablation of D1-SPNs decreases LID in mice [16]. A similar decrease occurs when D1-SPNs excitability is decreased through chemogenetic approaches [17,18]. Consistently, reduction of GABA synthesis by conditional inactivation of GAD67 in D1-SPN abolished LID [19], confirming that LID is dependent on GABA neurotransmission from D1-SPNs (Fig. 2).
Figure 2. Model of how DA neurons control motor actions by engaging the striatonigral direct pathway circuit via D1R in the striatum and SNr.
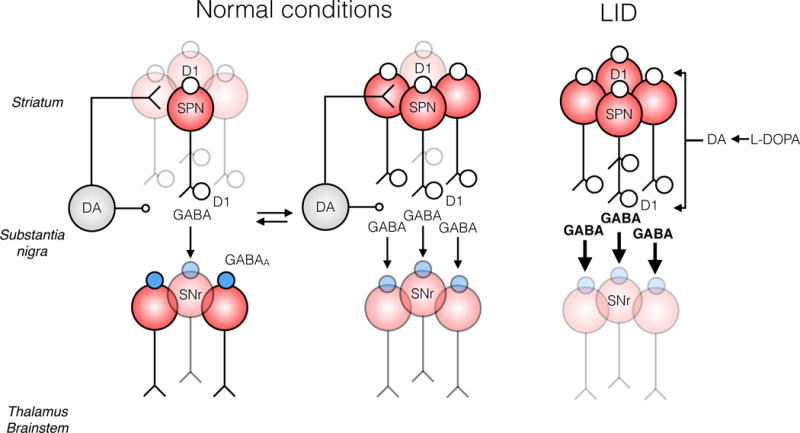
In normal conditions, midbrain DA neurons display various firing rates with episodes of bursts and tonic activity interspersed with pauses. Subsequent changes in extracellular DA will engage different subsets of D1-SPNs in the striatum. D1Rs located in the SNr can directly filter D1-SPN GABA output to provide for action selection, represented here as inhibition of different SNr projection neurons. In PD, DA neurons degenerate and homeostatic plasticity develops which leads to increased intrinsic excitability and evoked GABA release from D1-SPN synapses in response DA replacement with L-DOPA. Increased output from D1-SPNs and loss of filtering by presynaptic receptors, for instance by GABAB receptors (not shown), will result in surges of GABA efflux in the SNr. The lack of selection of GABA output from D1-SPNs will trigger hyperkinetic responses when extracellular DA is produced by L-DOPA administration, such as LID.
In studies using mice which fluorescent biomarkers, LID following DA denervation correlates with the hyperactivation of G-protein signaling downstream of D1Rs and the changes are confined to D1-SPNs [20]. Importantly, the D1R-related responses are evident by the first administration of L-DOPA and remain elevated during chronic treatment, specifically in the most dyskinetic animals [21]. DA denervation further leads to increased intrinsic excitability of D1-SPNs [22–25]. Thus, consistent with LID behavioral studies, homeostatic mechanisms established following DA denervation state appear to provide the alterations in striatal network activity that underlie LID without a requirement for chronic L-DOPA.
Expression of experimental dyskinesia without L-DOPA
As the above reports indicate that chronic L-DOPA is not required for the development of subsequent sensitized behavioral responses in DA denervated anmials, it is helpful to examine whether these changes in synaptic networks cause a hypersensitized response without L-DOPA administration. This has recently become possible by adapting optogenetic [26] and chemogenetic [27,28] techniques [29], which provide means to clearly distinguish effects of DA denervation can be clearly distinguished from effects produced by D1R stimulation during priming.
To examine if DA denervation alone can trigger a sensitized motor response predictive of LID when D1-SPNs are activated, we expressed the light-activated cation channel channelrhodopsin (ChR2) in D1-SPNs of mice which displayed hemiparkinsonism induced by unilateral injections of 6-OHDA [30]. Activation of the D1-SPN synapses within the SNr with blue light produced a vigorous rotational response specifically in DA denervated animals [30]. This recapitulated the response to L-DOPA to unilaterally DA lesioned rodents. Remarkably, a subset of the DA denervated mice also produced clear AIMs, in particular stereotypical orofacial and contralateral limb movements (unpublished observations), when the D1-SPN synapses were activated by light. These results suggest that LID rely on DA denervation and activation of the direct pathway D1-SPNs, but not chronic L-DOPA.
Our results demonstrating that optogenetic activation of D1-SPNs triggers LID-like responses in L-DOPA naïve parkinsonian animals were recently reproduced by other labs in mice [31] and rats [32]. In those studies, while optogenetic stimulation was again capable of producing LID-like responses on its own, administration of L-DOPA produced an additive effect and increased the intensity of the behavioral response. Nevertheless, in animals that received prior L-DOPA treatment, the activation of D1-SPNs produced stronger LID-like responses [31,32]. Thus, as in these behavioral studies, while chronic L-DOPA appears to not be required for LIDs, it may potentiate the responses.
A caveat of the use of optogenetic activation in these studies is that the technique almost certainly interferes with the endogenous firing activity of D1-SPNs during LID [33]. The implication of this manipulation is unknown. A valuable approach may be to use optogenetic inhibition of D1-SPNs, although to our knowledge this has not yet been done.
A second approach is to trigger LID-like responses in DA denervated mice by chemogenetic activation of D1-SPNs [18]. The DREADD technology recapitulates the G-protein signaling aspects of DA receptor activation and can thus provide useful information about the mechanisms underlying DA denervation-induced sensitization. Interestingly, activation of the D1-SPNs with stimulatory DREADDs unmasked similar DA denervation-induced sensitization in the motor response to the optogenetic experiments, and activation of Gs coupled, but not the Gq, DREADD-receptor produced a magnitude of AIMs in L-DOPA naïve mice comparable to L-DOPA treatment [18]. Interestingly, activation of the Gs-DREADD shared the ability of D1R itself to stimulate the cAMP/PKA/DARPP-32 and ERK1/2 signaling cascades, which have been shown to correlate with LID severity in both rodents [21] and primates [34]. This suggests that a set of molecular cascades occur in striatal neurons after acute administration of L-DOPA and, while DA denervation is required for expression of LID, that L-DOPA elicits additional effects that may modulate plasticity and subsequent LID.
A synaptic basis for denervation-induced sensitization
The evidence summarized so far indicates that synaptic plasticity at the direct pathway D1-SPN synapses triggers LID, but not the forms of the plasticity. DA regulates both the somatodendritic excitability of D1-SPNs, but also influences GABA activity within the basal ganglia output nuclei via presynaptic D1R on striatonigral efferents (Fig. 1B–C). The DA within the SNr is released from dendrites and soma of the SNc DA cells that degenerate in PD [35].
D1Rs in the SNr are implicated in the sensitized motor responses to L-DOPA in DA denervated animals [36,37] and LID is associated with increased glutamate and GABA release within the SNr triggered by local D1Rs [38–40]. In addition, the SNr is densely innervated by serotonergic projections from the raphe nucleus. LID has been associated with unregulated synaptic release of DA from serotonergic terminals (see review [8]), which possess the necessary components to convert L-DOPA into DA and to store DA in VMAT2-expressing synaptic vesicles. Thus, the aberrant firing activity of the SNr, which triggers the expression of LID [41,42], could be dependent on both somatodendritic and presynaptic changes in D1-SPN plasticity.
The most direct means to characterize these changes would be to measure GABA release within the SNr, but this is challenging. First, SNr GABA neurons are spontaneously active (>15 Hz), providing a high GABAergic tone within the local synaptic microcircuit that obscures analysis, particularly with methods that measure changes over long time scales such as microdialysis [43]. Second, only synaptic recordings can distinguish the D1-SPN striagonigral and GPe pallidonigral synapses that both release GABA [44]. Third, inhibition of one population of SNr neurons could release others from tonic inhibition via lateral excitation [45]. Finally, SNr neurons are relatively large with a substantial sodium conductance in dendrites [46] and are modulated by postsynaptic D1Rs [47]. This introduces technical challenges in recordings with somatic microelectrodes, which complicates interpretation of currents in the soma as readouts of presynaptic plasticity.
To bypass these issues, we adapted an optical approach, the use of FM1–43 as a marker of synaptic vesicle fusion [48], to measure presynaptic function from individual D1-SPN synapses within the SNr (Fig. 1D). We combined this optical method with patch-clamp recordings from SNr neurons during optogenetic activation of D1-SPN synapses in L-DOPA naïve 6-OHDA lesioned mice. These direct measurements of presynaptic activity revealed a substantially increased probability of release of GABA from individual D1-SPNs in the DA denervated SNr [30]. Increased D1R stimulation of GABA release from D1-SPN synapses also occurred in a genetic model of SNc DA neurodegeneration, the Pitx3 null mice [49], in which ventral tegmental area DA neurons were intact and continued to provide DA input to D1-SPN in the ventral striatum. There was still a global scaling of D1-SPN synapses in the SNr, suggesting that the loss of DA within the midbrain elicits this synaptic plasticity [49].
How might plasticity in D1-SPN synapses trigger the supersensitive motor response to L-DOPA? In our studies of DA denervated mice, we found that the increased release probability at D1-SPN synapses in the SNr was due to a loss of feedback inhibition of GABA release normally provided by metabotropic GABAB receptors [30]. Cell-type-specific mRNA translational profiling using translating ribosome affinity purification (TRAP) illustrated that the responses of striatal neurons to DA denervation and subsequent L-DOPA administration is complex, however, and affect many genes that control the expression of regulators of neuronal excitability and neurotransmitter release, including voltage-gated calcium and potassium channels [50]. Consistently, DA denervation decreases A-type potassium currents in striatal neurons [22]. Notably, the molecular signaling pathways implicated in D1R responses in the D1-SPN are also expressed in D1-SPN terminals within the SNr, although it is unknown how these molecules affect local GABA release (Fig. 3).
Figure 3. Presynaptic signaling mechanisms in D1-SPNs that may be involved in synaptic plasticity triggered by disrupted D1R signaling in PD.
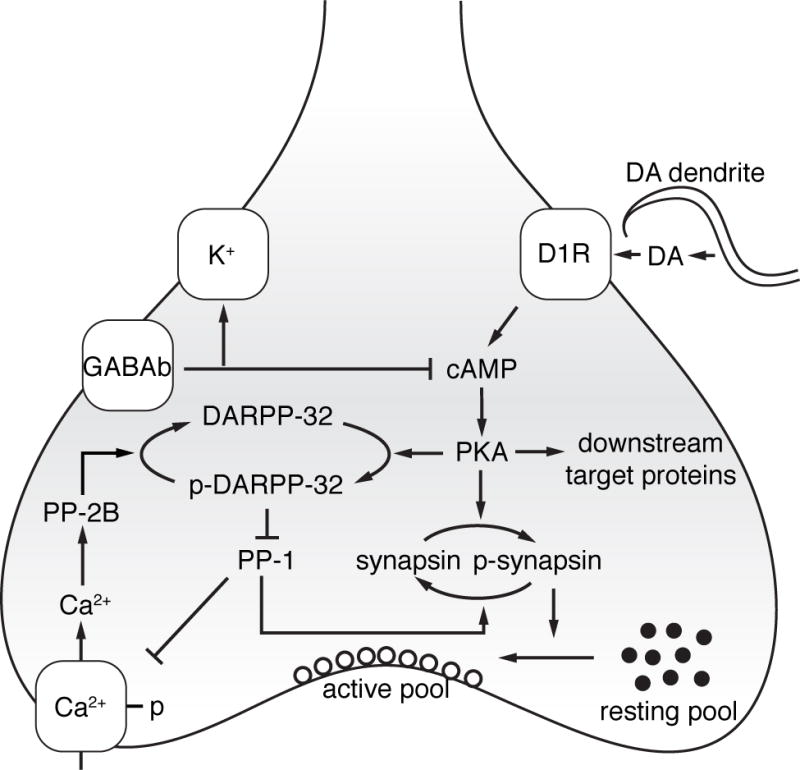
Activation of D1Rs triggers a series of downstream signal transduction pathways, principally driven by the control of the cAMP/PKA/DARPP-32 cascade. Direct control of presynaptic activity by somatodendritic DA release is achieved by regulation of voltage-dependent potassium and calcium channels that together control the probability of GABA release. GABAB receptor activity, which inhibits D1R driven activation of cAMP, is lost in DA denervated animals [30], presumably resulting from compensatory changes in D1R mediated signal transduction. Long-term changes in synaptic function may also involve the regulation of synapsins, a group of phosphoproteins that control the recruitment of synaptic vesicles from the resting pool. Compensatory mechanisms may result in homeostatic plasticity that attempt to overcome the loss of DA and sustain normal synaptic function in prodromal PD. As neurodegeneration progresses, however, the homeostatic plasticity eventually can trigger LID.
Future directions
Together, an array of new findings suggests that synaptic changes established in response to DA denervation are a requirement for LID. The mechanistic effects of pulsatile administration of L-DOPA have neverthless been the major focus of attention. In such studies, responses are likely due to a complex combination of molecular and synaptic events induced by both loss of endogenous DA and subsequent repeated stimulation of DA receptors within the basal ganglia and associated circuitry from exogenously-derived DA.
The chemogenetic and optogenetic experiments reviewed here and the relationship between 1) the degree of DA denervation, 2) the acute supersensitive behavioral response to L-DOPA, and 3) the hyperactivity of D1-SPNs that is evident at the first administration of L-DOPA, suggest, that the denervation triggered the homeostatic plasticity, which is expressed upon D1R activation. The responses to D1R stimulation by D1-SPNs may balance increased intrinsic hyperexcitability of D1-SPNs and altered corticostriatal plasticity. The change in intrinsic excitability appears sufficient to produce LID in experimental animals, in part due to a loss of presynaptic inhibition of D1-SPN synapses in the SNr [30]. Nevertheless, priming seems to require ongoing L-DOPA, and so chronic L-DOPA appears likely to modulate and enhance LIDs in patients (Fig. 2).
There is a considerable potential to reduce the risk of LID by establishing strategies to limit the impact of the homeostatic plasticity triggered by the loss of DA. Future experiments that specifically investigate the mechanisms involved in this form of plasticity thus have great promise for the development of improved pharmacotherapy for PD.
Highlights.
-
*
Denervation of dopamine neurons triggers changes in intrinsic excitability in striatal spiny projection neurons.
-
*
Dyskinesia can be elicited without prior L-DOPA priming in dopamine-denervated animals with optogenetic and chemogenetic approaches.
-
*
Increased GABA output is responsible for sensitized responses to L-DOPA in DA denervated animals.
-
*
Disease progression sets the stage for dyskinesia in Parkinson’s disease with chronic L-DOPA triggering the response.
Acknowledgments
Our research in Parkinson’s disease is supported by the Parkinson’s, Michael J. Fox, William F. Richter, and JPB Foundations (DLS). DLS is a NARSAD Brain and Behavior Distinguished Investigator. AB is also supported by the Swedish Research Council (2009–563 and 2016–03129) and Sweden-America Foundation. OL is supported by T32 NIGMS GM007367.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
Authors disclose no conflict of interest.
References
- 1.Aquino CC, Fox SH. Clinical spectrum of levodopa-induced complications. Mov Disord. 2015;30:80–89. doi: 10.1002/mds.26125. [DOI] [PubMed] [Google Scholar]
- 2.Bastide MF, Meissner WG, Picconi B, Fasano S, Fernagut PO, Feyder M, Francardo V, Alcacer C, Ding Y, Brambilla R, et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog Neurobiol. 2015;132:96–168. doi: 10.1016/j.pneurobio.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9:665–677. doi: 10.1038/nrn2471. [DOI] [PubMed] [Google Scholar]
- 4.Nadjar A, Gerfen CR, Bezard E. Priming for l-dopa-induced dyskinesia in Parkinson’s disease: a feature inherent to the treatment or the disease? Prog Neurobiol. 2009;87:1–9. doi: 10.1016/j.pneurobio.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iravani MM, McCreary AC, Jenner P. Striatal plasticity in Parkinson’s disease and L-dopa induced dyskinesia. Parkinsonism Relat Disord. 2012;18(Suppl 1):S123–125. doi: 10.1016/S1353-8020(11)70038-4. [DOI] [PubMed] [Google Scholar]
- 6.Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Villalba RM, Smith Y. Loss and remodeling of striatal dendritic spines in Parkinson’s disease: from homeostasis to maladaptive plasticity? J Neural Transm (Vienna) 2017 doi: 10.1007/s00702-017-1735-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mosharov EV, Borgkvist A, Sulzer D. Presynaptic effects of levodopa and their possible role in dyskinesia. Mov Disord. 2015;30:45–53. doi: 10.1002/mds.26103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langston JW, Ballard P. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): implications for treatment and the pathogenesis of Parkinson’s disease. Can J Neurol Sci. 1984;11:160–165. doi: 10.1017/s0317167100046333. [DOI] [PubMed] [Google Scholar]
- 10.Pons R, Syrengelas D, Youroukos S, Orfanou I, Dinopoulos A, Cormand B, Ormazabal A, Garzia-Cazorla A, Serrano M, Artuch R. Levodopa-induced dyskinesias in tyrosine hydroxylase deficiency. Mov Disord. 2013;28:1058–1063. doi: 10.1002/mds.25382. [DOI] [PubMed] [Google Scholar]
- 11*.Cilia R, Akpalu A, Sarfo FS, Cham M, Amboni M, Cereda E, Fabbri M, Adjei P, Akassi J, Bonetti A, et al. The modern pre-levodopa era of Parkinson’s disease: insights into motor complications from sub-Saharan Africa. Brain. 2014;137:2731–2742. doi: 10.1093/brain/awu195. This elegant study on patients with Parkinson’s disease provides evidence suggesting that disease progression and high dose of L-DOPA are the two major risk factors for LID. The study compared the onset of motor complications in two patient groups with simliar stages of PD: one from Italy and one from Ghana. The two groups developed LID approximately at the same time, altough L-DOPA therapy was introduced later in Ghana. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Putterman DB, Munhall AC, Kozell LB, Belknap JK, Johnson SW. Evaluation of levodopa dose and magnitude of dopamine depletion as risk factors for levodopa-induced dyskinesia in a rat model of Parkinson’s disease. J Pharmacol Exp Ther. 2007;323:277–284. doi: 10.1124/jpet.107.126219. [DOI] [PubMed] [Google Scholar]
- 13.Shan L, Diaz O, Zhang Y, Ladenheim B, Cadet JL, Chiang YH, Olson L, Hoffer BJ, Backman CM. L-Dopa induced dyskinesias in Parkinsonian mice: Disease severity or L-Dopa history. Brain Res. 2015;1618:261–269. doi: 10.1016/j.brainres.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cenci MA. Dopamine dysregulation of movement control in L-DOPA-induced dyskinesia. Trends Neurosci. 2007;30:236–243. doi: 10.1016/j.tins.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Revy D, Jaouen F, Salin P, Melon C, Chabbert D, Tafi E, Concetta L, Langa F, Amalric M, Kerkerian-Le Goff L, et al. Cellular and behavioral outcomes of dorsal striatonigral neuron ablation: new insights into striatal functions. Neuropsychopharmacology. 2014;39:2662–2672. doi: 10.1038/npp.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engeln M, Bastide MF, Toulme E, Dehay B, Bourdenx M, Doudnikoff E, Li Q, Gross CE, Boue-Grabot E, Pisani A, et al. Selective Inactivation of Striatal FosB/DeltaFosB-Expressing Neurons Alleviates L-DOPA-Induced Dyskinesia. Biol Psychiatry. 2016;79:354–361. doi: 10.1016/j.biopsych.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 18*.Alcacer C, Andreoli L, Sebastianutto I, Jakobsson J, Fieblinger T, Cenci MA. Chemogenetic stimulation of striatal projection neurons modulates responses to Parkinson’s disease therapy. J Clin Invest. 2017;127:720–734. doi: 10.1172/JCI90132. The authors use chemogenetic stimulation of direct and indirect SPNs in hemiparkinsonian mice to show that D1-SPN activation with the prodrug clozapine-N-oxide triggers behavioral sensitization in L-DOPA naive animals and potentiates LID when administered with L-DOPA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang K, Chammas C, Soghomonian JJ. Loss of glutamic acid decarboxylase (Gad67) in striatal neurons expressing the Drdr1a dopamine receptor prevents L-DOPA-induced dyskinesia in 6-hydroxydopamine-lesioned mice. Neuroscience. 2015;303:586–594. doi: 10.1016/j.neuroscience.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 20.Feyder M, Bonito-Oliva A, Fisone G. L-DOPA-Induced Dyskinesia and Abnormal Signaling in Striatal Medium Spiny Neurons: Focus on Dopamine D1 Receptor-Mediated Transmission. Front Behav Neurosci. 2011;5:71. doi: 10.3389/fnbeh.2011.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA, Herve D, Greengard P, Fisone G. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci. 2007;27:6995–7005. doi: 10.1523/JNEUROSCI.0852-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Azdad K, Chavez M, Don Bischop P, Wetzelaer P, Marescau B, De Deyn PP, Gall D, Schiffmann SN. Homeostatic plasticity of striatal neurons intrinsic excitability following dopamine depletion. PLoS One. 2009;4:e6908. doi: 10.1371/journal.pone.0006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warre R, Thiele S, Talwar S, Kamal M, Johnston TH, Wang S, Lam D, Lo C, Khademullah CS, Perera G, et al. Altered function of glutamatergic cortico-striatal synapses causes output pathway abnormalities in a chronic model of parkinsonism. Neurobiol Dis. 2011;41:591–604. doi: 10.1016/j.nbd.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 24.Fieblinger T, Graves SM, Sebel LE, Alcacer C, Plotkin JL, Gertler TS, Chan CS, Heiman M, Greengard P, Cenci MA, et al. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat Commun. 2014;5:5316. doi: 10.1038/ncomms6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suarez LM, Solis O, Aguado C, Lujan R, Moratalla R. L-DOPA Oppositely Regulates Synaptic Strength and Spine Morphology in D1 and D2 Striatal Projection Neurons in Dyskinesia. Cereb Cortex. 2016;26:4253–4264. doi: 10.1093/cercor/bhw263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roth BL. DREADDs for Neuroscientists. Neuron. 2016;89:683–694. doi: 10.1016/j.neuron.2016.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferguson SM, Eskenazi D, Ishikawa M, Wanat MJ, Phillips PE, Dong Y, Roth BL, Neumaier JF. Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat Neurosci. 2011;14:22–24. doi: 10.1038/nn.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossi MA, Calakos N, Yin HH. Spotlight on movement disorders: What optogenetics has to offer. Mov Disord. 2015;30:624–631. doi: 10.1002/mds.26184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30*.Borgkvist A, Avegno EM, Wong MY, Kheirbek MA, Sonders MS, Hen R, Sulzer D. Loss of Striatonigral GABAergic Presynaptic Inhibition Enables Motor Sensitization in Parkinsonian Mice. Neuron. 2015;87:976–988. doi: 10.1016/j.neuron.2015.08.022. The authors show that optogenetic activation of D1-SPN synapses in the SNr triggers behavioral sensitization in L-DOPA naive hemiparkisonian mice. This increased responsiveness of the D1-SPN was associated with increased GABA release due to loss of GABAB-mediated inhibition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31*.Perez XA, Zhang D, Bordia T, Quik M. Striatal D1 medium spiny neuron activation induces dyskinesias in parkinsonian mice. Mov Disord. 2017;32:538–548. doi: 10.1002/mds.26955. In this study, the authors optogenetically activate D1-SPN in the striatum of hemiparkinsonian mice. Activation of D1-SPN caused abnormal involuntary movements in L-DOPA naive mice and the severity was increased by L-DOPA priming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hernandez FL, Castela I, Ruiz-DeDiego I, Obeso JA, Moratalla R. Striatal activation by optogenetics induces dyskinesias in the 6-hydroxydopamine rat model of Parkinson disease. Mov Disord. 2017;32:530–537. doi: 10.1002/mds.26947. [DOI] [PubMed] [Google Scholar]
- 33.Alberico SL, Kim YC, Lence T, Narayanan NS. Axial levodopa-induced dyskinesias and neuronal activity in the dorsal striatum. Neuroscience. 2017;343:240–249. doi: 10.1016/j.neuroscience.2016.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Santini E, Sgambato-Faure V, Li Q, Savasta M, Dovero S, Fisone G, Bezard E. Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in L-DOPA-induced dyskinesia. PLoS One. 2010;5:e12322. doi: 10.1371/journal.pone.0012322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geffen LB, Jessell TM, Cuello AC, Iversen LL. Release of dopamine from dendrites in rat substantia nigra. Nature. 1976;260:258–260. doi: 10.1038/260258a0. [DOI] [PubMed] [Google Scholar]
- 36*.Kozlowski MR, Sawyer S, Marshall JF. Behavioural effects and supersensitivity following nigral dopamine receptor stimulation. Nature. 1980;287:52–54. doi: 10.1038/287052a0. In this seminal report, the authors show, for the first time, that L-DOPA triggers behavioral sensitization in DA lesioned rats due to actions on D1R expressed within the SNr. In addition, this study provides the first evidence showing that the plasticity develops in response to prolonged reductions in DA signaling. [DOI] [PubMed] [Google Scholar]
- 37.Robertson GS, Robertson HA. Evidence that L-dopa-induced rotational behavior is dependent on both striatal and nigral mechanisms. J Neurosci. 1989;9:3326–3331. doi: 10.1523/JNEUROSCI.09-09-03326.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rangel-Barajas C, Silva I, Lopez-Santiago LM, Aceves J, Erlij D, Floran B. L-DOPA-induced dyskinesia in hemiparkinsonian rats is associated with up-regulation of adenylyl cyclase type V/VI and increased GABA release in the substantia nigra reticulata. Neurobiol Dis. 2011;41:51–61. doi: 10.1016/j.nbd.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 39.Mango D, Bonito-Oliva A, Ledonne A, Nistico R, Castelli V, Giorgi M, Sancesario G, Fisone G, Berretta N, Mercuri NB. Phosphodiesterase 10A controls D1-mediated facilitation of GABA release from striato-nigral projections under normal and dopamine-depleted conditions. Neuropharmacology. 2014;76(Pt A):127–136. doi: 10.1016/j.neuropharm.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 40.Mela F, Marti M, Bido S, Cenci MA, Morari M. In vivo evidence for a differential contribution of striatal and nigral D1 and D2 receptors to L-DOPA induced dyskinesia and the accompanying surge of nigral amino acid levels. Neurobiol Dis. 2012;45:573–582. doi: 10.1016/j.nbd.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 41.Meissner W, Ravenscroft P, Reese R, Harnack D, Morgenstern R, Kupsch A, Klitgaard H, Bioulac B, Gross CE, Bezard E, et al. Increased slow oscillatory activity in substantia nigra pars reticulata triggers abnormal involuntary movements in the 6-OHDA-lesioned rat in the presence of excessive extracellular striatal dopamine. Neurobiol Dis. 2006;22:586–598. doi: 10.1016/j.nbd.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 42.Aristieta A, Ruiz-Ortega JA, Miguelez C, Morera-Herreras T, Ugedo L. Chronic L-DOPA administration increases the firing rate but does not reverse enhanced slow frequency oscillatory activity and synchronization in substantia nigra pars reticulata neurons from 6-hydroxydopamine-lesioned rats. Neurobiol Dis. 2016;89:88–100. doi: 10.1016/j.nbd.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 43.Timmerman W, Westerink BH. Extracellular gamma-aminobutyric acid in the substantia nigra reticulata measured by microdialysis in awake rats: effects of various stimulants. Neurosci Lett. 1995;197:21–24. doi: 10.1016/0304-3940(95)11888-4. [DOI] [PubMed] [Google Scholar]
- 44.Connelly WM, Schulz JM, Lees G, Reynolds JN. Differential short-term plasticity at convergent inhibitory synapses to the substantia nigra pars reticulata. J Neurosci. 2010;30:14854–14861. doi: 10.1523/JNEUROSCI.3895-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown J, Pan WX, Dudman JT. The inhibitory microcircuit of the substantia nigra provides feedback gain control of the basal ganglia output. Elife. 2014;3:e02397. doi: 10.7554/eLife.02397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Atherton JF, Bevan MD. Ionic mechanisms underlying autonomous action potential generation in the somata and dendrites of GABAergic substantia nigra pars reticulata neurons in vitro. J Neurosci. 2005;25:8272–8281. doi: 10.1523/JNEUROSCI.1475-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou FW, Jin Y, Matta SG, Xu M, Zhou FM. An ultra-short dopamine pathway regulates basal ganglia output. J Neurosci. 2009;29:10424–10435. doi: 10.1523/JNEUROSCI.4402-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- 49.Ding S, Li L, Zhou FM. Nigral dopamine loss induces a global upregulation of presynaptic dopamine D1 receptor facilitation of the striatonigral GABAergic output. J Neurophysiol. 2015;113:1697–1711. doi: 10.1152/jn.00752.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heiman M, Heilbut A, Francardo V, Kulicke R, Fenster RJ, Kolaczyk ED, Mesirov JP, Surmeier DJ, Cenci MA, Greengard P. Molecular adaptations of striatal spiny projection neurons during levodopa-induced dyskinesia. Proc Natl Acad Sci U S A. 2014;111:4578–4583. doi: 10.1073/pnas.1401819111. [DOI] [PMC free article] [PubMed] [Google Scholar]