

Abstract
Huntington’s disease (HD), a dominantly inherited neurodegenerative disease, is defined by its genetic cause, a CAG-repeat expansion in the HTT gene, its motor and psychiatric symptomology and primary loss of striatal medium spiny neurons (MSNs). However, the molecular mechanisms from genetic lesion to disease phenotype remain largely unclear. Mouse models of HD have been created that exhibit phenotypes reminiscent of those in the patient, and specifically, cortico-striatal disconnectivity appears to be a shared pathogenic event shared by HD mouse models and patients. Molecular studies have begun to unveil consistent molecular and cellular pathogenic mechanisms that may account for cortico-striatal miscommunication in various HD mouse models. Systems biological approaches help to illuminate synaptic molecular networks as a nexus for HD cortio-striatal pathogenesis, and may offer new candidate targets to modify the disease.
Introduction
Huntington’s disease is the most common dominantly inherited neurodegenerative disorder with normally middle-age onset of motor, cognitive and psychiatric symptoms and a progressive and lethal disease course [1]. HD pathology is characterized by selective degeneration of striatal medium spiny neurons (MSNs) and, to a lesser extent, cortical pyramidal neurons (CPNs). Currently, there is no effective therapy for HD. The causal mutation in HD is a CAG repeat expansion encoding an elongated polyglutamine (polyQ) repeat in Huntingtin (HTT). Importantly, the length of the repeat is inversely correlated with the age of disease onset [1]. The precise pathogenic mechanisms in HD remains poorly understood, and currently there is no consensus molecular therapeutic target for HD beyond mutant HTT (mHTT) itself. Among the many challenges to understand HD in molecular detail, there is the sheer large size of the HTT protein (>3144 amino acid), the incompletely understood and diverse cellular function of normal HTT, and the nearly ubiquitous expression of mHTT during development and in adult tissues [2]. Thus, detailed analyses of mammalian models of HD (e.g. mouse models), with intact molecular, cellular, and neuronal circuits are crucial to uncover consistent disease mechanisms that facilitate further therapeutic advances. In this review, we will discuss recent studies using mouse genetic models of HD to identify cellular substrates that mHTT targets to initiate the disease and a variety of molecular, physiological, and genetic studies demonstrating the crucial role of cortico-striatal miscommunication, particularly deterioration of synaptic function, in most mouse models of HD. Finally, we will summarize new systems biology studies that highlight the cortico-striatal synapses as a major hub in HD molecular pathogenic networks.
Genetic Mouse Models of Huntington’s Disease
The study of HD molecular pathogenesis in the mammalian brain context often depends on a plethora of genetically engineered mouse models [3]. HD mouse models can be broadly divided into three groups (Figure 1). The first group of models is those expressing a mHTT protein fragment and exhibits more aggressive, disease-like phenotypes [3]. The R6 series of mice (R6/2 and R6/1) is one of the most commonly used “mHTT fragment” models of HD [3]. It uses about 1Kb human HTT promoter to drive the expression of mHTT exon1 with 110 to over 150 CAG repeats, and the model shows early onset of robust motor impairment, brain atrophy, and lethal disease course reminiscent of juvenile HD [3].
Figure 1.

Summary of the design and phenotypes seen in several common murine HD models. Reviewed by [12,13]; References for transcriptomic and proteomic changes, [53,59, Yang lab unpublished]. Abbreviations: KI, Knock In; FL, Full Length; RMP, resting membrane potential; sEPSCs, spontaneous Excitatory Post Synaptic Currents; SynNMDA, synaptic NMDA; exSynNMDA, extrasynaptic NMDA; Ctx, cortex; Str, striatum.
The second group of mouse models are mutant Huntingtin (mHtt) knockin (KI) mice, which have expanded CAG repeats of varying sizes engineered into the endogenous murine Htt locus [3]. These allelic series of mHtt-KI mice have high construct validity and exhibit CAG-length dependent molecular and cellular phenotypes (e.g. transcriptional dysregulation, MSN electrophysiological changes), and for those lines with long CAG repeats (e.g. Q140 or Q175), late-onset behavioral impairment and modest neuronal loss [3].
The third group of mouse models, BACHD and YAC128, expresses full-length human mHTT from human genomic transgenes. These models overexpress human mHTT, and show common disease features such as progressive motor, cognitive and psychiatric-like behavioral deficits, selective forebrain atrophy, and mHtt aggregation in a pattern reminiscent of adult-onset HD [3]. BACHD and YAC128 mice tend to have more robust cortical and striatal atrophy phenotypes compared to HTT-KI mice of comparable polyQ length and age, however they also tend to have less robust nuclear mHTT aggregation and transcriptionopathy. These models are valuable to study pathogenesis elicited by full-length human mHTT.
From a disease modeling perspective, all the existing mouse models of HD have captured varying degrees of progressive, HD-like phenotypes, but they all have imperfect genetic construct and phenotypic validity (Figure 1), and none so far has demonstrated predictive validity that can foretell therapeutic efficacy in patients. Instead of focusing on any single model, we share the view that the field could benefit from studying pathogenic phenotypes and mechanisms that are shared by two or more HD mouse models.
Cortico-striatal miscommunication in HD and HD mouse models
Longitudinal imaging studies of premanifest and manifest HD patients report evidence for progressive disconnection between cortex and striatum [1]. In-depth functional MRI study [1] and a novel graph theoretical analysis of imaging data [4] came to similar conclusions that premanifest and early manifest HD showed a loss of specific cortico-striatal functional network connectivity, which are correlated with clinical progression.
Aberrant cortico-striatal synaptic communication is one of the most consistent findings across HD mouse models (Figure 1) [5,6]. The majority of these models show striatal MSN membrane property changes prior to the onset of behavioral symptoms, including increase in input resistance, depolarizing resting membrane potential, and increased excitability (e.g. rheobase). The synaptic properties of MSN are also altered. In R6/2 and YAC128, there is an early increase in spontaneous excitatory postsynaptic currents (sEPSCs) in MSNs, which is followed by late-onset decreases in sEPSCs [5]. The latter phenotype is shared with other HD mouse models (e.g. BACHD, Q175). Some of these models also showed reduction in evoked synaptic NMDA currents and Long Term Potentiation (LTP) [5,6].
Parallel to the electrophysiological changes is evidence of synaptic morphological degeneration in HD mice. MSN spine loss has been shown in all major HD mouse models (Figure 1) [7–9]. These models also show loss of presynaptic and postsynaptic marker proteins, such as synaptophysin and vGluT1 for presynapse, and PSD-95 and Actn2 for postsynapse (e.g. [5,10,11]). Further, the use of novel reporter mice or neuronal tracers that label axons [12–14] or dendrites [15] also help to show CPN axonal degeneration and MSN dendritic degeneration in mHtt-KI, BACHD and R6/2 mice, offering yet another cellular mechanism for cortico-striatal disconnection in HD mice.
Genetic evidence for the critical role of cortico-striatal neuronal interactions in HD pathogenesis
It has been known from HD neuropathology that both striatal and cortical neurons are vulnerable to atrophy and degeneration, but a key question is where in the brain mHTT targets to drive disease pathogenesis. To address such a question, a series of mouse models selectively expressing mHTT (or its toxic fragments) in MSNs and CPNs have been developed [16]. Initial studies with fragment models showed evidence for both cell-autonomous and non-cell-autonomous mHtt fragment toxicities in the disease [17–19], but the precise origin for pathological cell-cell interactions was unclear.
BACHD mice are a Cre/LoxP conditional BAC transgenic mouse model designed to interrogate the cell type(s) in which full-length mHTT expression is critical to aspects of the disease [29]. By flanking mHTT-exon1 with loxP sites, the expression of full-length mHTT in this model can be switched off in cells expressing Cre. We crossed BACHD with several Cre mouse lines to genetically reduce mHTT in the striatal MSNs (BACHD/Rgs9-Cre), CPNs (BACHD/Emx1-Cre), or both [20]. Interestingly, the removal of mHTT in the CPNs, but not in MSNs, partially improves motor phenotypes and fully rescues psychiatric-like deficits, but does not improve brain atrophy. Impressively, genetic removal of mHTT in both MSNs and CPNs, while preserving mHTT expression elsewhere in the brain, results in robust rescue of all the disease phenotypes including brain atrophy in this model.
Another interesting finding in this study is the synaptic phenotypic changes in these models. BACHD striatum showed loss of both presynaptic marker (synaptophysin) and postsynaptic markers (PSD-95 and Actn2). As expected, the presynaptic marker loss can be reversed with cortical mHTT reduction. However, surprisingly, the postsynaptic marker loss can be partially restored by either cortical or striatal mHTT reduction, suggesting possible synergy of both cell-autonomous and non-cell-autonomous mechanisms. Consistent with the latter idea, the evoked synaptic NMDA defects in BACHD MSNs is partially reversible by mHTT reduction in either CPNs or MSNs [10]. This latter study is further supported by in vivo recording demonstrating normalization of striatal neuronal activities and behavioral phenotypes with cortical reduction of mHTT [21]. Together, these findings demonstrated distinct but interacting roles of mHTT in cortical and striatal neurons in HD pathogenesis, and suggest both cell-autonomous and non-cell-autonomous mHTT toxicities are important for striatal synaptic dysfunction and synapse loss.
Molecular mechanisms linked to cortico-striatal miscommunication in HD mice
As a molecular genetic disease, a key unanswered question in HD is what is the cascade of molecular events that originated from Htt CAG repeat expansion and leads to the progressive dysfunction and degeneration of the vulnerable neurons? Here we will briefly review selected literature on the known molecular mechanisms that likely contribute to cortico-striatal miscommunications in HD mouse models.
Mutant Htt impairs vesicular transport and multiple presynaptic functions
An evolutionarily conserved function of HTT is vesicular transport [2]. Htt participates in multiple molecular complexes (e.g. Hap1/Dynein/Dynactin/Kinesin, Hap40/Rab5, and Optineurin/Myosin) involved in vesicle and organelle trafficking. Cargos of HTT in fast axonal transport include synaptic precursor vesicles, BDNF-containing vesicles, and APP-positive vesicles (reviewed by [2]). PolyQ-expanded Htt impairs the kinetics of its cargo transport, and in some cases, cargo release (e.g. BDNF) (Figure 2).
Figure 2.
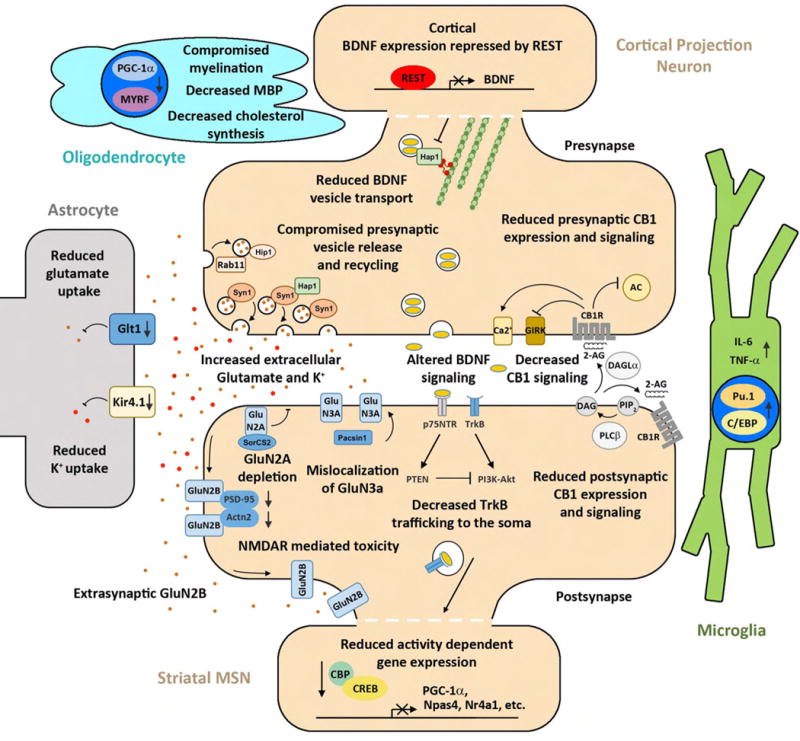
Disconnectivity at the cortico-striatal synapse is mediated by presynaptic and postsynaptic changes in gene expression, vesicle trafficking and release, Glutamate-NMDAR signaling, BDNF signaling, and CB1 signaling as well as glial cell dysfunction impinging on these pathways.
Htt may also directly affect presynaptic vesicular function, including neurotransmitter release. Mutant Htt binds to synaptic vesicles through interaction with Hap1 [22], which is involved in synaptic vesicle exocytosis by interacting with synapsin-1 [23]. Consistent with this mechanism, targeted expression of mHtt-exon1 to the presynaptic terminal interferes with presynaptic glutamate release in the striatum, and produces disease phenotypes in mice [24].
Hip1 is another Htt interactor that appears to have important presynaptic function [25]. The c. elegans homolog of Hip1 (Hip1r) regulates synaptic localization of synaptobrevin, a protein crucial for vesicle fusion. Interestingly, Hip1 null mice have defects in synaptic vesicle recycling. Moreover in the worm model, Hip1r and other presynaptic endocytic proteins are modifiers of mHTT fragment toxicities in neurons [25].
The third endocytic trafficking partner of Htt that may contribute to synaptic pathology is Rab11. Rab11 levels and activity critically depend on the presence of normal Htt, and cortical neurons from Q140 KI mice showed defective Rab11-dependent endocytosis [26]. Interestingly, in a Drosophila model overexpressing mHtt fragments, the presynaptic vesicle deficits, synaptic transmission, and neurodegeneration can be ameliorated by Rab11 overexpression [27,28].
The NMDA receptors at the striatal synapses: neuroprotection vs. neurotoxicity
Several mechanisms have been proposed to implicate the progressive changes in NMDA currents in HD striatal MSNs in disease pathogenesis (Figure 2) [5,6,29]. One set of studies using primarily the YAC transgenic HD models (e.g. YAC128) showed the dysfunction of the GluN2B subunit of the NMDA receptor, particularly its extrasynaptic mislocalization, is likely an early and important pathogenic event [30]. Synaptic NMDA currents and downstream nuclear signaling has been shown to be neuroprotective and extrasynaptic NMDA signaling can induce alternative cell death pathways [29] (Figure 2). Consistent with this model, mHtt appears to alter this synaptic/extrasynaptic NMDA signaling balance by reducing the activity-driven expression or function of Creb, Tcp1, and Pgc-1α, and such deficits can be diminished by low dose memantine [30,31].
An alternative explanation of NMDA receptor mediated toxicity in HD striatal neurons is the selective mis-trafficking of the GluN3A subunit of the NMDA receptor [7]. Mutant Htt binds to and disrupts Pacsin1-GluN3A interactions, leading to the reduction of Pacsin1-mediated recycling of GluN3A [7]. Excessive GluN3A at the striatal postsynapse results in synapse loss in YAC128 which can be prevented by genetic deletion of GlueN3A in these HD mice.
A recently published study suggest mHtt may affect the dendritic and synaptic targeting of GluN2A [32]. GluN2A is significantly reduced on the membrane of Q175 as well as R6/2 mice. The mechanism is due to Htt interaction with and mislocalization of SorCS2, which has an important role in trafficking GluN2A. Haploinsufficiency of SorCS2 resulted in exacerbated motor phenotypes in Q175 mice, suggesting a protective role of SorCS2-NR2A interaction [32].
Currently, it remains unclear what the relative contributions of GluN2A, GluN2B and GluN3A are to HD synatopathogenesis. The concept of neuroprotective synaptic NMDA signaling is an attractive one as it may account for the cortico-striatal pathological interactions in HD mice [10]. Since both cortical and striatal mHTT reduction can partially restore the postsynaptic NMDA currents and PSD-95 and Actn2 levels (both involved in NMDA receptor localization), it is tempting to postulate that disruption of these latter synaptic proteins may play a role in striatal synaptic NMDA dysfunction in HD mice.
Trans-synaptic trophic factor signaling: presynaptic BDNF vs postsynatpic TrkB signaling
Another attractive mechanism that may account for cortico-striatal pathological interactions in HD is the BDNF secretion from cortical neurons and its signaling in the striatum (Figure 2). Striatal MSNs do not synthesize their own BDNF, and instead rely on activity-dependent release of BDNF synthesized in cortical neurons to maintain MSN transcriptional signatures and pro-survival program [33,34]. In the cortex, mHtt affects BDNF levels through several mechanisms. It can reduce BDNF transcription due to elevated nuclear levels of REST, which is normally retained in the cytoplasm by wildtype HTT (but less so by mHTT). Reduction of BDNF mRNA or protein has been described in a several HD mouse models including R6/2, mHtt-KI, BACHD, and YAC128 [35–39]. However, a recent study showed the levels of BDNF is unaltered in two such models [40], which warrants further studies to resolve such discrepancy. The functional significance of BDNF discrepancy has been shown by genetic restoration of BDNF expression or administration of TrkB agonist, which can reduce disease phenotypes in HD mouse models [9,37]. Beyond BDNF synthesis, axonal transport and release of BDNF is also defective in HD (see above). Using a BACHD cortico-striatal primary neuronal co-culture system, a recent study showed BDNF trafficking deficits in cortical neurons is responsible for reduced BNDF release and striatal MSN atrophy, and both deficits can be ameliorated with improvement of chaperonin-mediated proteostasis [41].
Besides the cortical BDNF deficits, emerging evidence also showed impaired BNDF signaling through the TrkB receptor in MSN may also contribute to the disease. Htt plays a key role in the trafficking of TrkB vesicles in MSN dendrites [42]. Mutant Htt alters the attachment of TrkB vesicles to the microtubules, leading to decreased signaling in mHtt-KI neurons [42]. Another study using both BACHD and Q175 KI mice shows consistent defects in BDNF-dependent LTP in the synapse of the indirect-pathway MSNs (D2-MSNs), a subpopulation of MSNs that are most vulnerable in HD [40]. The mechanism for reduced LTP is due to aberrant activation of p75NTR and PTEN, which in turn suppress the PI3K-Akt axis that are critical for LTP induction. Together, these studies provide convincing evidence that mHTT can impair both local and long distance signaling of TrkB receptor signaling in HD MSNs, hence may synergize with the presynaptic BDNF deficits to elicit cortico-striatal pathological interactions in HD.
Endocannibinoid signaling: Where is CB1 critical in HD?
Endocannabinoids (EcB) are lipid signals that mediate retrograde trans-synaptic suppression via binding to the endocannabinoid receptor CB1 [43]. EcB molecules synthesized in the striatal MSN synapses can bind to CB1 in the cortico-striatal glutamatergic synapses to elicit long term depression (LTD) [44]. CB1 is broadly expressed in the brain, and the striatal down-regulation of CB1 receptor is a transcriptional signature of HD (Figure 2) [45]. One key question is whether presynaptic or postsynaptic CB1 is crucial to the disease? A series of studies reveals fascinating roles for CB1 in both CPNs and MSNs in HD models. The first study surprisingly showed that deletion of CB1 in glutamatergic neurons in the forebrain (e.g. CPNs), but not those in GABAergic neurons (e.g. MSNs), exacerbate motor phenotypes and striatal pathology in R6/2 mice [46]. This result suggests presynaptic CB1 is likely important to mediate neuroprotection in this HD model. In the striatum, CB1 expression could also be relevant to the disease. Genetic re-expression of CB1 in R6/2 striatum resulted in rescue of both presynaptic and postsynaptic marker loss, MSN synapse loss, electrophysiological deficits, and striatal atrophy [47,48]. However, these mice did not show behavioral improvement, suggesting the rescue is only partial [48]. Interestingly, chronic CB1 agonist administration seems to improve both motor impairment and reduce MSN marker loss [49]. Together, the studies thus far suggest that CB1 functions in both CPNs and MSNs to provide neuroprotection, and reduction of CB1 in the cortico-striatal axis could be a key to synaptic miscommunication in HD.
The roles of astrocytes, microglia, and oligodendrocytes in HD neuronal communication defects
Mutant Htt is not exclusively expressed in the neurons, and recent studies have highlighted the role of glia cells, including astrocytes [50], microglia [51], and oligodendrocytes [52], in mediating aspects of HD pathogenesis (Figure 2).
Astrocytes are thought to perform a variety of functions in the healthy brain including but not limited to: maintenance of blood-brain barrier, regulation of synaptic function, and metabolic support for neurons [50,53]. Upon CNS injury, astrocytes become ‘reactive’ and can confer both protective and harmful effects to neurons [53]. In HD, one major hypothesis of mHtt toxicity in astrocytes is the downregulation of glutamate transporter Glt1 (Slc1a2), which is responsible for extracellular glutamate uptake in the forebrain [50]. Such defects are found in HD postmortem brains, and in a majority of HD mouse models. Transgenic expression of mHTT fragments selectively in astrocytes can elicit Glt1 reduction and disease pathology [54]. Downregulation of astrocytic Glt1 is consistent with the model of enhanced excitotoxicity at the striatal MSNs [5]. The β-lactam antibiotic ceftriaxone can upregulate both striatal and cortical Glt1 levels in R6/2 mice, which in turn normalize glutamate uptake and improve behavioral deficits [55].
HD astrocytes may affect MSN function through another mechanism, the reduction of the Kir4.1 potassium channel expressed on the astrocytes [56]. This channel is responsible for extracellular K+ homeostasis. Reduced Kir4.1 levels and currents were found in R6/2 and Q175 mice, and viral-mediated restoration of Kri4.1 channels in the astrocytes can normalize extracellular K+ level, MSN membrane defects, and amelioration multiple disease phenotypes. Subsequent studies showed astrocytic Ca2+ signaling and glutamate transporter defects (likely due to Glt1) in R6/2 and Q175 can be partially improved with Kir4.1 overexpression [57,58].
Besides astrocytes, microglia as the resident innate immune cells in the brain also emerge as a critical player both in normal synaptic homeostasis and in pathogenesis of synaptic degeneration [59]. In HD, mHtt expression causes cell-autonomous activation of microglia via increased expression of myeloid lineage determining transcription factors, which in turn enhance transcription of neuroinflammatory factors and confer non-cell-autonomous toxicity to neurons (Figure 2) [60].
Finally, the oligodendrocyte is a crucial cell type involved in myelination, and its role in HD has been reported but not yet extensively investigated [61]. In mouse models, mHtt affects oligodendrocyte function and elicits reduced myelination through interference of PGC-1α function [62]. Moreover, mHtt can reduce oligodendrocyte-specific gene expression through interference with myelin regulatory factor (MYRF) [52]. Together, these studies underscore the need to further explore oligodendrocyte dysfunction in HD cortico-striatal misscommunication.
Systems biology reveals synapses as a hub for HD molecular pathogenic networks
In neurodegenerative disorders including HD, the gradual dysfunction and eventual degeneration of selected neuronal cells are accompanied by complex molecular changes in both neurons and non-neuronal cells over time. High-throughput sampling of transcriptomes, interactomes, and other types of omics data in disease models and wildtype controls often yield hundreds or thousands of individual molecular changes between genotypes or conditions (e.g. age), which is difficult to interpret and further pursue functional studies. Integrative systems biology approaches, such as Weighted Gene Coexpression Network Analyses (WGCNA), reduce these complex systems into a small number of coherent gene modules based on gene co-expression relationships [63]. A major advantage of such coexpression networks approach, as opposed to the traditional differential expression (DE) or gene enrichment methods, is to provide inter-connectivity relationship (edges) between any two genes (nodes) in the network. Those with the most connected genes in a module are called hub genes [64]. Hub gene status is often meaningful to annotate a module for specific biological factors (e.g. functional pathways, cell types, or genotypes and disease status) [63]. Moreover, hub genes are generally thought to be central to the module biology, hence are considered prime candidates for experimental validation of hypothesis generated by any given module [63]. The latter concept could be particularly helpful in studying the complex biology of HD, as often hundreds to thousands of molecular changes can be seen in any model systems expressing mHtt. To date, such an integrative systems biology approach has been applied to HD in two areas, Htt in vivo interactome [65] and mHtt-driven transcriptomes [39]. Interestingly, both unbiased analyses reveal cortico-striatal synapses appear to be a hub in HD molecular networks.
Huntingtin interactome demonstrates identified modules of interactors at synapses
Through its large protein-protein interaction surfaces, which include dozens of HEAT repeats, Htt and its N-terminal fragments can bind over one thousand interacting partners ex vivo or in vivo [65–68]. Interestingly, a recent meta-analysis of combined Htt interactome datasets, with 1619 interactors, highlighted the enrichment of presynaptic exosome proteins and homeostatic synaptic plasticity proteins among known Htt interactors [69]. To apply systems biology to study the Htt interactome, we used WGCNA to analyze the in vivo spatiotemporal Htt proteomic interactome derived from BACHD and WT mice at 2m and 12m and in three brain tissues, cortex, striatum, and cerebellum (Figure 3) [65]. The study yielded over 740 candidate Htt interactors that are associated with Htt in particular spatiotemporal context. Interestingly, these in vivo Htt interactors are highly enriched with known presynaptic or postsynaptic proteins (Figure 3A, Supplementary Table 1). Using the semi-quantitative peptide counts as input, we defined six WGCNA coexpression modules. Four modules are noteworthy in relationship to cortico-striatal communication. First, the Red Module, which is the most correlated with Htt and contains Htt itself, is enriched with proteins involved in axonal transport, 14-3-3s (synaptic plasticity), chaperonins (e.g. Tcp1), and LTD. Blue module and Yellow modules are enriched with presynaptic and postsynaptic proteins, respectively. Finally, Green module has mostly actin cytoskeletion proteins and synaptic proteins. Importantly, some of the hub genes from these modules are either functionally implicated in HD pathogenesis already, or shown to be novel modifiers of mHtt toxicity in a Drosophila model [65]. Together, this network view of the Htt interactome provides not only different biological contexts for different interactors, but also prioritized molecular candidates (e.g. based on hub-gene status), to study the role of Htt interactors in HD cortico-striatal pathogenesis.
Figure 3.
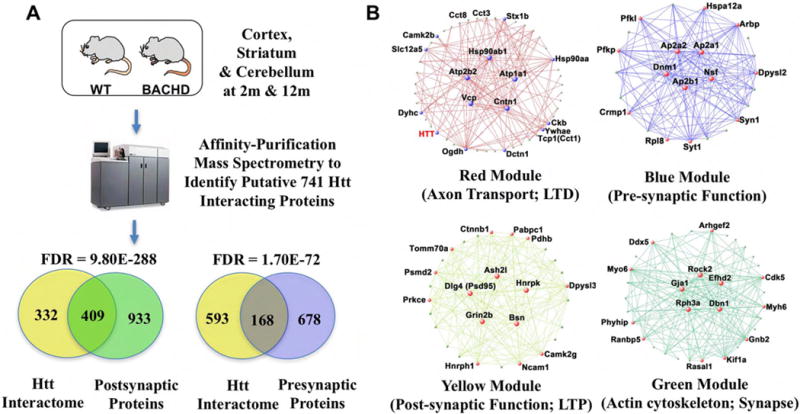
Htt interacting proteins are enriched for presynaptic and postsynaptic functions. (A) Experimental design and Venn diagrams of the overlap between Htt interacting proteins and pre-and post-synaptic function. (B) Weighted Gene Correlation Network Analysis (WGCNA) modules describing Htt interactor networks associated with synaptic function. Adapted from [81].
Huntingtin CAG-length dependent transcriptomic and proteomic networks also impair cortico-striatal synapses
The study by Langfelder et al. [39] used an allelic series of murine Htt-KI mice with increasing CAG repeats to address the question of why mHtt CAG-expansion appears to be inversely correlated with the severity of disease reminiscent of a similar relationship in HD. By densely sampling the transcriptomes and proteomes of this mHtt allelic series of mouse lines at multiple ages and tissues (Figure 4A), the study helps to build a rich resource to study age and CAG-length dependent molecular changes in HD mice. To probe the underlying biological factors driving Htt CAG-length dependent differential gene expression, we applied WGNCA to identify coexpression gene modules that are robustly CAG-length and age dependent. We found 13 modules in the striatum, 5 in the cortex, and none in the liver. Importantly, several of the striatal and cortical CAG-length dependent gene modules are relevant to understanding synaptopathogenesis in HD (Figure 4B). First, striatal M2 module, the most significant CAG-length dependent downregulated module with 1867 genes, is highly enriched with several types of genes: striatal MSN marker genes and D2-MSN marker genes, GPCR & cAMP signaling, postsynaptic density (PSD), LTP, and interestingly, neuronal synaptic activity dependent genes (Figure 4B; supplementary Table 1). Another interesting striatal module is M25, which is down-regulated with Htt CAG expansion and is enriched with genes involved in glutamatergic receptor signaling, synaptic LTP, and D2-MSN genes (Figure 4; Supplementary Table 1). Since the mHtt-KI mice do not show any MSN loss at the age we did RNA-sequencing, the M2 and M25 modules provide the following insights: First, mHtt CAG expansion seems to impair the maintenance of the striatal MSN identity gene expression, particularly those for the vulnerable D2-MSNs. Second, the transcription of synaptic genes is also impaired, especially those involved in LTP. Finally, the synaptic activity driven transcription is reduced, including those implicated in neuroprotection (e.g. Npas4, Nr4a1, etc.) [29]. In the cortex, we found M4 is down-regulated with CAG-length and age, and is enriched with genes in Ca2+ signaling, synapse/PSD, and markers of glutamatergic neurons. Since Ca2+ signaling is important for neuronal activity and neurotransmission, we interpret M4 module could be relevant to study mHtt induced reduction in presynaptic neurotransmission in CPNs. Together, our transcriptomic network analyses provide evidence for mHtt CAG-length dependent impairment in presynaptic and postsynaptic gene expression in CPNs and MSNs, respectively, and evidence for reduced neuronal activity-driven transcription in MSNs.
Figure 4.
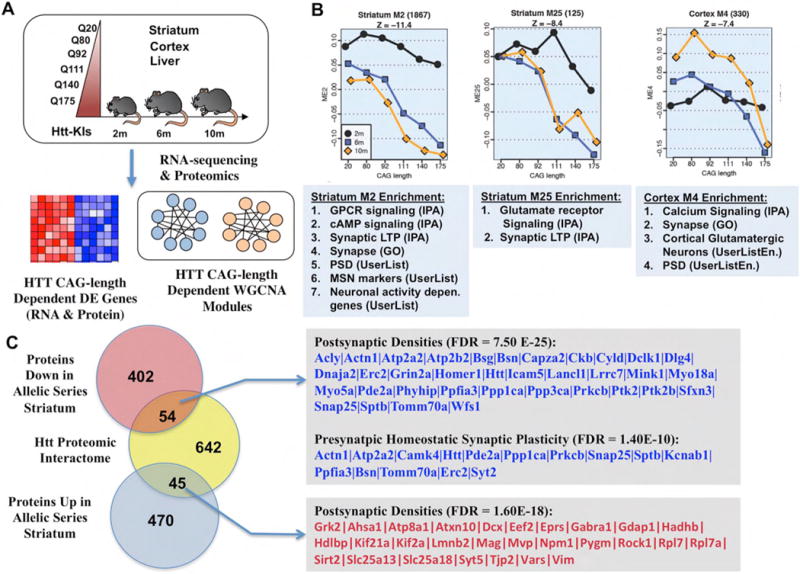
Systems biological analysis of protein and gene interaction networks derived from HD knockin models with varying CAG-repeat lengths (Allelic series), highlights significant changes in both cortical and striatal synapses. (A) Schematic of the experimental design. (B) Three significantly enriched synaptic WGCNA modules, two in the striatum and one in the cortex, showing age and CAG-length dependent down-regulation. (C) Venn diagram and gene lists of significantly enriched synaptic Htt interactors that overlap with genes up- or down-regulated in the striatum of allelic series RNA-seq data. For complete gene lists see Supplemental table. Adapted from [53].
Finally, our study also provides several strategies for further investigation of molecular mechanisms underlying cortico-striatal pathogenesis in HD. First, one can prioritize in an unbiased manner, based on the hub gene status for top CAG-length dependent modules in striatum and cortex, for genetic perturbation to test novel gene function in modifying disease molecular networks. Second, programs such as Ingenuity Pathway Analyses (IPA) can predict potential upstream regulators of a given module. Interestingly, for the striatal CAG-length dependent down-regulated genes, the top upstream regulators are Htt, Creb1, CBP (Crebpp), Crem, and BDNF (Supplementary Table 1). These upstream regulators are consistent with the hypothesis that both BDNF and NMDAR-driven nuclear signaling defects contribute to striatal transcriptionopathy in mHtt-KI mice. Finally, using label-free quantitative proteomics, we validated 790 genes as being dysregulated in a CAG-length dependent manner at both RNA and protein levels mHtt-KI striata [39]. Importantly, these mHtt dysregulated proteins are also highly enriched with synaptic proteins (Supplementary Table 1), and a significant subset of these dysregulated proteins are among candidate Htt interactors (Figure 4C). These latter proteins provide another systems biology generated gene list to investigate synaptopathogenesis in HD.
Conclusions
The miscommunication between cortex and striatum is a key pathological event found in HD patients and recapitulated in multiple HD mouse models. Genetic and molecular analyses in HD mice support distinct but interacting roles of mHTT in cortical and striatal neurons, as well as glia, in eliciting synaptic communication deficits and overall disease phenotypes. Detailed molecular studies begin to yield diverse molecular and cellular mechanisms that may contribute to cortico-striatal pathogenesis in HD. Finally, systems biology offers a new avenue to obtain a more global view of mHtt-induced molecular pathogenic networks, which so far also highlight synaptic gene networks in HD mice. Together, these approaches are converging to depict a more coherent molecular picture on how mHtt may elicit cortico-striatal miscommunication in HD mouse brains, which in turn may provide new disease modifying targets for therapeutic development.
Supplementary Material
Highlights.
Diverse mouse models of HD have important phenotypic and molecular similarities
Disconnectivity of the cortico-striatal synapse drives the disease progression
Distinct molecular mechanisms and glia cells play roles in cortico-striatal miscommunication
Systems analysis reveal cortico-striatal synapses as key hubs in HD molecular pathogenic networks
Acknowledgments
We thank Peter Langfelder and Fuying Gao for help in bioinformatics, and members of the Yang lab for thoughtful discussions and support of this review. The authors apologize to researchers whose work could not be cited due to space constraints. This work was supported by grants from the NIH (R01-NS084298 and U01-MH106008), Hereditary Disease Foundation (HDF), and CHDI Foundation, Inc. X.W.Y. is also supported by Leslie Gehry Brenner Prize from HDF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Permanent Address: 695 Charles E Young Drive South, Los Angeles, CA 90095-1761
Reference and recommended reading
- 1.Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10:204–216. doi: 10.1038/nrneurol.2014.24. [DOI] [PubMed] [Google Scholar]
- 2.Saudou F, Humbert S. The Biology of Huntingtin. Neuron. 2016;89:910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Crook ZR, Housman D. Huntington’s disease: can mice lead the way to treatment? Neuron. 2011;69:423–435. doi: 10.1016/j.neuron.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McColgan P, Seunarine KK, Razi A, Cole JH, Gregory S, Durr A, Roos RA, Stout JC, Landwehrmeyer B, Scahill RI, et al. Selective vulnerability of Rich Club brain regions is an organizational principle of structural connectivity loss in Huntington’s disease. Brain. 2015;138:3327–3344. doi: 10.1093/brain/awv259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raymond LA, André VM, Cepeda C, Gladding CM, Milnerwood AJ, Levine MS. Pathophysiology of Huntington’s disease: time-dependent alterations in synaptic and receptor function. Neuroscience. 2011;198:252–273. doi: 10.1016/j.neuroscience.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plotkin JL, Surmeier DJ. Corticostriatal synaptic adaptations in Huntington’s disease. Curr Opin Neurobiol. 2015;33:53–62. doi: 10.1016/j.conb.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marco S, Giralt A, Petrovic MM, Pouladi MA, Martínez-Turrillas R, Martínez-Hernández J, Kaltenbach LS, Torres-Peraza J, Graham RK, Watanabe M, et al. Suppressing aberrant GluN3A expression rescues synaptic and behavioral impairments in Huntington’s disease models. Nat Med. 2013;19:1030–1038. doi: 10.1038/nm.3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J, Ryskamp DA, Liang X, Egorova P, Zakharova O, Hung G, Bezprozvanny I. Enhanced Store-Operated Calcium Entry Leads to Striatal Synaptic Loss in a Huntington’s Disease Mouse Model. J Neurosci. 2016;36:125–141. doi: 10.1523/JNEUROSCI.1038-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simmons DA, Belichenko NP, Yang T, Condon C, Monbureau M, Shamloo M, Jing D, Massa SM, Longo FM. A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington’s disease. J Neurosci. 2013;33:18712–18727. doi: 10.1523/JNEUROSCI.1310-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang N, Gray M, Lu XH, Cantle JP, Holley SM, Greiner E, Gu X, Shirasaki D, Cepeda C, Li Y, et al. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington’s disease. Nat Med. 2014;20:536–541. doi: 10.1038/nm.3514. This study uses conditional deletion of the BACHD transgene in cortical, striatal, or both tissues to demonstrate cortical, but not striatal, reduction of mHtt can partially ameliroate motor and psychiatric symptoms. Combined deletion of mHTT in both the cortex and striatum rescued behavioral deficits and neurodegenerative pathology in BACHD. Importantly, both mHTT in both CPNs and MSNs appear to contribute to reduction of striatal synaptic markers and evoked synaptic NDMA currents, suggesting a crucial role of both cortical and striatal neurons in striatal synaptopathogenesis in HD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng YP, Wong T, Bricker-Anthony C, Deng B, Reiner A. Loss of corticostriatal and thalamostriatal synaptic terminals precedes striatal projection neuron pathology in heterozygous Q140 Huntington’s disease mice. Neurobiol Dis. 2013;60:89–107. doi: 10.1016/j.nbd.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marangoni M, Adalbert R, Janeckova L, Patrick J, Kohli J, Coleman MP, Conforti L. Age-related axonal swellings precede other neuropathological hallmarks in a knock-in mouse model of Huntington’s disease. Neurobiol Aging. 2014;35:2382–2393. doi: 10.1016/j.neurobiolaging.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 13.Gatto RG, Chu Y, Ye AQ, Price SD, Tavassoli E, Buenaventura A, Brady ST, Magin RL, Kordower JH, Morfini GA. Analysis of YFP(J16)-R6/2 reporter mice and postmortem brains reveals early pathology and increased vulnerability of callosal axons in Huntington’s disease. Hum Mol Genet. 2015;24:5285–5298. doi: 10.1093/hmg/ddv248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hintiryan H, Foster NN, Bowman I, Bay M, Song MY, Gou L, Yamashita S, Bienkowski MS, Zingg B, Zhu M, et al. The mouse cortico-striatal projectome. Nat Neurosci. 2016;19:1100–1114. doi: 10.1038/nn.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu XH, Yang XW. Genetically-directed Sparse Neuronal Labeling in BAC Transgenic Mice through Mononucleotide Repeat Frameshift. Sci Rep. 2017;7:43915. doi: 10.1038/srep43915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee CY, Cantle JP, Yang XW. Genetic manipulations of mutant huntingtin in mice: new insights into Huntington’s disease pathogenesis. FEBS J. 2013;280:4382–4394. doi: 10.1111/febs.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu X, Li C, Wei W, Lo V, Gong S, Li SH, Iwasato T, Itohara S, Li XJ, Mody I, et al. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46:433–444. doi: 10.1016/j.neuron.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 18.Gu X, André VM, Cepeda C, Li SH, Li XJ, Levine MS, Yang XW. Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington’s disease. Mol Neurodegener. 2007;2:8. doi: 10.1186/1750-1326-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown TB, Bogush AI, Ehrlich ME. Neocortical expression of mutant huntingtin is not required for alterations in striatal gene expression or motor dysfunction in a transgenic mouse. Hum Mol Genet. 2008;17:3095–3104. doi: 10.1093/hmg/ddn206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng GG, Wei W, Yang XW, Zhang YB, Xu W, Gong NB, Lü Y, Wang FF. New coumarins from the roots of Angelica dahurica var. formosana cv. Chuanbaizhi and their inhibition on NO production in LPS-activated RAW264.7 cells. Fitoterapia. 2015;101:194–200. doi: 10.1016/j.fitote.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 21.Estrada-Sánchez AM, Burroughs CL, Cavaliere S, Barton SJ, Chen S, Yang XW, Rebec GV. Cortical efferents lacking mutant huntingtin improve striatal neuronal activity and behavior in a conditional mouse model of Huntington’s disease. J Neurosci. 2015;35:4440–4451. doi: 10.1523/JNEUROSCI.2812-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Wyman T, Yu ZX, Li SH, Li XJ. Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum Mol Genet. 2003;12:2021–2030. doi: 10.1093/hmg/ddg218. [DOI] [PubMed] [Google Scholar]
- 23.Mackenzie KD, Lumsden AL, Guo F, Duffield MD, Chataway T, Lim Y, Zhou XF, Keating DJ. Huntingtin-associated protein-1 is a synapsin I-binding protein regulating synaptic vesicle exocytosis and synapsin I trafficking. J Neurochem. 2016;138:710–721. doi: 10.1111/jnc.13703. [DOI] [PubMed] [Google Scholar]
- 24.Xu Q, Huang S, Song M, Wang CE, Yan S, Liu X, Gaertig MA, Yu SP, Li H, Li S, et al. Synaptic mutant huntingtin inhibits synapsin-1 phosphorylation and causes neurological symptoms. J Cell Biol. 2013;202:1123–1138. doi: 10.1083/jcb.201303146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parker JA, Metzler M, Georgiou J, Mage M, Roder JC, Rose AM, Hayden MR, Néri C. Huntingtin-interacting protein 1 influences worm and mouse presynaptic function and protects Caenorhabditis elegans neurons against mutant polyglutamine toxicity. J Neurosci. 2007;27:11056–11064. doi: 10.1523/JNEUROSCI.1941-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Sapp E, Chase K, Comer-Tierney LA, Masso N, Alexander J, Reeves P, Kegel KB, Valencia A, Esteves M, et al. Disruption of Rab11 activity in a knock-in mouse model of Huntington’s disease. Neurobiol Dis. 2009;36:374–383. doi: 10.1016/j.nbd.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richards P, Didszun C, Campesan S, Simpson A, Horley B, Young KW, Glynn P, Cain K, Kyriacou CP, Giorgini F, et al. Dendritic spine loss and neurodegeneration is rescued by Rab11 in models of Huntington’s disease. Cell Death Differ. 2011;18:191–200. doi: 10.1038/cdd.2010.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinert JR, Campesan S, Richards P, Kyriacou CP, Forsythe ID, Giorgini F. Rab11 rescues synaptic dysfunction and behavioural deficits in a Drosophila model of Huntington’s disease. Hum Mol Genet. 2012;21:2912–2922. doi: 10.1093/hmg/dds117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron. 2010;65:178–190. doi: 10.1016/j.neuron.2010.01.008. The authors characterize, in detail, the role of extrasynaptic NMDAR in pathogenic HD mouse model phenotypes and the rescue of these phenotypes with extrasynaptic NMDAR inhibition with low-dose memantine. [DOI] [PubMed] [Google Scholar]
- 31.Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma Q, Yang J, Milner TA, Vonsattel JG, Palko ME, Tessarollo L, Hempstead BL. SorCS2-mediated NR2A trafficking regulates motor deficits in Huntington’s disease. JCI Insight. 2017;2 doi: 10.1172/jci.insight.88995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strand AD, Baquet ZC, Aragaki AK, Holmans P, Yang L, Cleren C, Beal MF, Jones L, Kooperberg C, Olson JM, et al. Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J Neurosci. 2007;27:11758–11768. doi: 10.1523/JNEUROSCI.2461-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuccato C, Belyaev N, Conforti P, Ooi L, Tartari M, Papadimou E, MacDonald M, Fossale E, Zeitlin S, Buckley N, et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J Neurosci. 2007;27:6972–6983. doi: 10.1523/JNEUROSCI.4278-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie Y, Hayden MR, Xu B. BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J Neurosci. 2010;30:14708–14718. doi: 10.1523/JNEUROSCI.1637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conforti P, Mas Monteys A, Zuccato C, Buckley NJ, Davidson B, Cattaneo E. In vivo delivery of DN:REST improves transcriptional changes of REST-regulated genes in HD mice. Gene Ther. 2013;20:678–685. doi: 10.1038/gt.2012.84. [DOI] [PubMed] [Google Scholar]
- 39.Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci. 2016 doi: 10.1038/nn.4256. The authors applied Weighted Coexpression Network Analsyes (WGCNA) to analyze RNA-seq and proteomic datasets from an allelic series of HD knockin mice with increasing CAG length. The study defined CAG-length dependent coexpression modules in the striatum and cortex of murine Htt-KI mice. These modules shed light on molecular networks driven by mHtt CAG repeat expansion, including multiple pathological RNA and protein networks highly enriched with cortico-striatal synaptic genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plotkin JL, Day M, Peterson JD, Xie Z, Kress GJ, Rafalovich I, Kondapalli J, Gertler TS, Flajolet M, Greengard P, et al. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron. 2014;83:178–188. doi: 10.1016/j.neuron.2014.05.032. This study shows defects of TrkB signaling and LTP in striatal D2-MSNs in BACHD and Q175 mice, and the mechanism is aberrant P75 neurotrophic receptor signaling through PTEN that inhibits TrkB-PI3K-Akt signaling pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao X, Chen XQ, Han E, Hu Y, Paik P, Ding Z, Overman J, Lau AL, Shahmoradian SH, Chiu W, et al. TRiC subunits enhance BDNF axonal transport and rescue striatal atrophy in Huntington’s disease. Proc Natl Acad Sci U S A. 2016;113:E5655–5664. doi: 10.1073/pnas.1603020113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liot G, Zala D, Pla P, Mottet G, Piel M, Saudou F. Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J Neurosci. 2013;33:6298–6309. doi: 10.1523/JNEUROSCI.2033-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- 45.Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, Becanovic K, Pouladi MA, Sathasivam K, Cha JH, et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet. 2007;16:1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- 46.Chiarlone A, Bellocchio L, Blázquez C, Resel E, Soria-Gómez E, Cannich A, Ferrero JJ, Sagredo O, Benito C, Romero J, et al. A restricted population of CB1 cannabinoid receptors with neuroprotective activity. Proc Natl Acad Sci U S A. 2014;111:8257–8262. doi: 10.1073/pnas.1400988111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blázquez C, Chiarlone A, Bellocchio L, Resel E, Pruunsild P, García-Rincón D, Sendtner M, Timmusk T, Lutz B, Galve-Roperh I, et al. The CB cannabinoid receptor signals striatal neuroprotection via a PI3K/Akt/mTORC1/BDNF pathway. Cell Death Differ. 2015;22:1618–1629. doi: 10.1038/cdd.2015.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naydenov AV, Sepers MD, Swinney K, Raymond LA, Palmiter RD, Stella N. Genetic rescue of CB1 receptors on medium spiny neurons prevents loss of excitatory striatal synapses but not motor impairment in HD mice. Neurobiol Dis. 2014;71:140–150. doi: 10.1016/j.nbd.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pietropaolo S, Bellocchio L, Ruiz-Calvo A, Cabanas M, Du Z, Guzmán M, Garret M, Cho YH. Chronic cannabinoid receptor stimulation selectively prevents motor impairments in a mouse model of Huntington’s disease. Neuropharmacology. 2015;89:368–374. doi: 10.1016/j.neuropharm.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 50.Khakh BS, Beaumont V, Cachope R, Munoz-Sanjuan I, Goldman SA, Grantyn R. Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci. 2017;40:422–437. doi: 10.1016/j.tins.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crotti A, Glass CK. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015;36:364–373. doi: 10.1016/j.it.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang B, Wei W, Wang G, Gaertig MA, Feng Y, Wang W, Li XJ, Li S. Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron. 2015;85:1212–1226. doi: 10.1016/j.neuron.2015.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liddelow SA, Barres BA. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity. 2017;46:957–967. doi: 10.1016/j.immuni.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 54.Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, Li S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci U S A. 2009;106:22480–22485. doi: 10.1073/pnas.0911503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller BR, Dorner JL, Shou M, Sari Y, Barton SJ, Sengelaub DR, Kennedy RT, Rebec GV. Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington’s disease phenotype in the R6/2 mouse. Neuroscience. 2008;153:329–337. doi: 10.1016/j.neuroscience.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci. 2014;17:694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang R, Diaz-Castro B, Looger LL, Khakh BS. Dysfunctional Calcium and Glutamate Signaling in Striatal Astrocytes from Huntington’s Disease Model Mice. J Neurosci. 2016;36:3453–3470. doi: 10.1523/JNEUROSCI.3693-15.2016. This study demonstrates reduction of Kir4.1 expression in R6/2 and Q175 astrocytes is responsible for elevated striatal extracellular K+ and increased MSN excitability, and restoration of Kir4.1 expression in astrocytes normalize extracellular K+ levels, MSN membrane properties, and multiple disease phenotypes in R6/2 mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dvorzhak A, Vagner T, Kirmse K, Grantyn R. Functional Indicators of Glutamate Transport in Single Striatal Astrocytes and the Influence of Kir4.1 in Normal and Huntington Mice. J Neurosci. 2016;36:4959–4975. doi: 10.1523/JNEUROSCI.0316-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016;36:128–134. doi: 10.1016/j.conb.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crotti A, Benner C, Kerman BE, Gosselin D, Lagier-Tourenne C, Zuccato C, Cattaneo E, Gage FH, Cleveland DW, Glass CK. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. 2014;17:513–521. doi: 10.1038/nn.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Myers RH, Vonsattel JP, Paskevich PA, Kiely DK, Stevens TJ, Cupples LA, Richardson EP, Bird ED. Decreased neuronal and increased oligodendroglial densities in Huntington’s disease caudate nucleus. J Neuropathol Exp Neurol. 1991;50:729–742. doi: 10.1097/00005072-199111000-00005. [DOI] [PubMed] [Google Scholar]
- 62.Xiang Z, Valenza M, Cui L, Leoni V, Jeong HK, Brilli E, Zhang J, Peng Q, Duan W, Reeves SA, et al. Peroxisome-proliferator-activated receptor gamma coactivator 1 α contributes to dysmyelination in experimental models of Huntington’s disease. J Neurosci. 2011;31:9544–9553. doi: 10.1523/JNEUROSCI.1291-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parikshak NN, Gandal MJ, Geschwind DH. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet. 2015;16:441–458. doi: 10.1038/nrg3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- 65.Shirasaki DI, Greiner ER, Al-Ramahi I, Gray M, Boontheung P, Geschwind DH, Botas J, Coppola G, Horvath S, Loo JA, et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. 2012;75:41–57. doi: 10.1016/j.neuron.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, Strand A, Torcassi C, Savage J, Hurlburt A, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007;3:e82. doi: 10.1371/journal.pgen.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Culver BP, Savas JN, Park SK, Choi JH, Zheng S, Zeitlin SO, Yates JR, Tanese N. Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J Biol Chem. 2012;287:21599–21614. doi: 10.1074/jbc.M112.359307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stroedicke M, Bounab Y, Strempel N, Klockmeier K, Yigit S, Friedrich RP, Chaurasia G, Li S, Hesse F, Riechers SP, et al. Systematic interaction network filtering identifies CRMP1 as a novel suppressor of huntingtin misfolding and neurotoxicity. Genome Res. 2015;25:701–713. doi: 10.1101/gr.182444.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang JKT, Langfelder P, Horvath S, Palazzolo MJ. Exosomes and Homeostatic Synaptic Plasticity Are Linked to Each other and to Huntington’s, Parkinson’s, and Other Neurodegenerative Diseases by Database-Enabled Analyses of Comprehensively Curated Datasets. Front Neurosci. 2017;11:149. doi: 10.3389/fnins.2017.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.