

Abstract
Introduction
A hormonal role in non-small cell lung cancer (NSCLC) development is well documented. We previously showed that the aromatase inhibitor (AI) anastrozole decreased the development of tobacco carcinogen-induced lung tumors in a murine lung cancer prevention model and that aromatase and estrogen receptor (ER) were expressed in pulmonary inflammatory cells.
Methods
We utilized a tobacco carcinogen-induced lung tumor mouse model [4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone (NNK)] to determine whether an AI combined with non-steroidal anti-inflammatory drugs (NSAIDs) results in greater lung tumor prevention effects than single agent treatment.
Results
Combination of anastrozole (0.1mg/kg/day) with aspirin (25mg/kg/day) following NNK exposure resulted in significantly fewer and smaller lung tumors compared to single agent treatments, accompanied by maximum decreases in circulating β-estradiol (E2) and IL-6, tumor infiltrating macrophages and tumoral Ki67, P-MAPK, P-STAT3 and IL-17A expression. Preneoplasia arising after combination treatment showed the lowest Sox-2 expression, suggesting an inhibitory effect on proliferative capacity in the airways by blocking both E2 and inflammation. Anastrozole combined with ibuprofen instead of aspirin also showed enhanced anti-tumor effects. Moreover, male mice treated with NNK that received E2 in the drinking water showed greater levels of pulmonary macrophages and inflammatory markers compared to control, confirming an E2 effect on inflammation in the microenvironment.
Conclusions
Our results suggest a benefit to joint targeting of the estrogen and inflammatory pathways for NSCLC prevention. Combining AIs with NSAIDs reduces circulating E2, pro-inflammatory cytokines, and macrophage recruitment in the lung microenvironment following tobacco exposure. This strategy could be particularly effective in women who have underlying pulmonary inflammatory diseases.
Keywords: estrogen, NSAIDs, aromatase, lung cancer, prevention
INTRODUCTION
Lung cancer is the number one cause of cancer death worldwide. Despite advances in therapeutic modalities, the 5-year survival for lung cancer patients is a dismal 18% [1], demonstrating the continued need to develop novel therapeutic and preventive approaches for lung cancer. Preclinical and epidemiological studies support a role for estrogenic pathways in driving lung cancer progression [2-10]. Expression of estrogen receptors (ERs), progesterone receptors (PGRs), and/or aromatase, a key enzyme in estrogen biosynthesis, is frequently found in non-small cell lung cancers (NSCLCs) [2-4, 7], and high expression of tissue aromatase is a poor prognostic marker in post-menopausal women with NSCLC [5]. Pre-clinically, β-estradiol (E2) induced proliferation in NSCLC in vitro and in vivo [2, 6, 8] and ERβ was found to be the predominant ER isoform expressed and functional in lung cancer [3, 8]. We and others have demonstrated that the anti-estrogen fulvestrant and aromatase inhibitors (AIs) partially inhibit lung tumor growth [6, 11, 12] and prevent lung tumorigenesis in murine models [13] suggesting that repurposing endocrine therapies developed for ER-positive breast cancer may be effective for prevention or treatment of ER-positive lung cancer.
Estrogen activity on non-tumor cells within the tumor microenvironment is becoming increasingly recognized as playing an important role in tumor progression. We have shown that aromatase is expressed not only in human lung epithelial cells [2, 3] but also in lung macrophages [13], and that estrogen is present locally at sites of pulmonary inflammation [13]. Toniolo et al. recently demonstrated that the pro-tumor M2 macrophage phenotype is promoted by E2 [14], and both macrophages and lymphocytes are known to express ERs [15]. This suggests that inflammation triggered by tobacco exposure in the lungs includes up-regulation of an autocrine estrogen signaling loop in tumor infiltrating immune cells. Estrogen signaling through ER-positive myeloid-derived suppressor cells (MDSCs) has recently been demonstrated in ovarian tumors; the immunosuppressive action of MDSCs induced by E2 may thus contribute to malignant progression [16]. Together these data suggest that inflammatory cells that infiltrate the lungs in response to tobacco carcinogens may be an important source of estrogen synthesis and/or estrogen response, starting early in the carcinogenesis process. This enhanced E2-driven pro-tumorigenic immune phenotype combined with increased local E2 production may stimulate proliferation within the lung epithelial compartment, contributing to lung cancer progression.
In this study, we evaluated the combined effects of AIs and NSAIDs on lung tumor number/size and explored the link between inflammatory biomarkers, macrophage recruitment in the lung microenvironment and estrogen signaling using a tobacco carcinogen-induced lung tumorigenesis murine model. Epidemiological studies have shown that use of non-steroidal anti-inflammatory drugs (NSAIDs), such as aspirin or ibuprofen, is associated with a decreased risk of lung cancer [17-19]. A recent meta-analysis confirmed a protective effect of aspirin use on lung cancer risk with a reported relative risk of 0.87 [20]. We show that an AI combined with an anti-inflammatory agent prevents tumor development after carcinogen exposure, more so than each single agent treatment, in ovariectomized female mice. These mice are a model of ex-smoking post-menopausal women who could be eligible to use this treatment for chemoprevention. Our results also suggest that inhibiting tissue aromatase activity with an AI and reducing inflammation via an NSAID may be an effective strategy to reduce E2, pro-inflammatory cytokines and macrophage recruitment, magnifying the inhibition of the proliferative and signaling effects of E2. Moreover, we demonstrate that E2 treatment given during carcinogen exposure had an opposite pro-inflammatory effect in murine lungs, including increasing pulmonary macrophage numbers and producing higher inflammatory markers compared to vehicle control. Together our results strongly suggest a combined AI/NSAID prevention strategy could be effective among the tobacco-exposed population, particularly in women who have underlying pulmonary inflammatory diseases such as COPD.
METHODS
Mouse Models
Female ovariectomized FVB/N mice (6 weeks of age) were exposed to the tobacco carcinogen, NNK (3mg per intraperitoneal injection; twice per week) for 4 weeks. Mice received subcutaneous supplementation with 0.1mg androstenedione, the substrate for aromatase, three times per week for the entire course of the experiment. We utilized this post-menopausal model previously to evaluate the anti-tumor effects of anastrozole and fulvestrant [13]. Additionally, this model was chosen because aromatase inhibitors are contra-indicated for use in pre-menopausal women and men. Each of the four treatment groups started with 12-14 mice based on power calculations that assumed at least a 50% reduction in tumor number between treatment groups. Drug treatments were initiated 5 weeks after the final NNK injection to allow for lung preneoplasia development. This model mimics events that are often present in lungs of ex-smokers. Anastrozole (0.1mg/kg p.o; n=12), aspirin (dissolved in water (2.5mg/ml) and administered at 25mg/kg p.o; n=12), the combination of anastrozole with aspirin (n=14) or vehicle control (n=12) was administered 5 times per week for 8 weeks. In a separate experiment, ibuprofen (25mg/kg p.o.) was substituted for aspirin treatment using the same protocol (see animal numbers in Supplemental Table 2). NSAID dosing corresponds to a dose of 150mg daily for humans, using the body surface area normalization method [21]. Additionally, a short-term two-week treatment exposure experiment was performed to evaluate cytokine expression in the lungs and blood prior to tumor formation. We also utilized an E2 exposure mouse model whereby male mice (chosen for this setting to minimize the effects of circulating E2 found in females) received NNK (3mg per intraperitoneal injection; twice per week) for 4 weeks and simultaneously were administered E2 (2μM) or placebo control in the drinking water as described [22]. Mice were sacrificed after 8 weeks of treatment for AI/NSAID studies or 4 week for E2 studies followed by formalin-inflation of lungs under 25cm intra-alveolar pressure and collection of blood. Surface lung tumors were counted under a dissecting microscope and tumor size (defined as surface area, mm2) was measured using Motic Images 2000 software. All animal protocols were approved by the University of Pittsburgh or University of Minnesota Institutional Animal Care and Use Committee and carried out in strict compliance with all guidelines and regulations. No systemic toxicities or differences in body weights were observed with experimental treatment.
β-estradiol ELISA
Blood was collected prior to initiation of experimental treatment from the saphenous vein and at end of treatment by cardiac puncture, processed for serum and stored at -80°C. 17- β-estradiol (pg/ml) was measured in duplicate using the Cayman Estradiol ELISA kit according to the manufacturer’s instructions.
Blood and Lung Tissue Cytokine Analysis
Serum levels of the following markers were measured in duplicate using a customized mouse U-PLEX Biomarker Meso Scale Discovery kit: IL-1β, IL-6, Il-10, IL-12/IL-23p40, IL-17A, IL-23, TNF-α, VEGF-A and IFNγ. Results are reported in pg/ml.
Immunohistochemistry (IHC)
Lungs were fixed in 10% formalin, paraffin embedded, sliced and mounted on slides. Slides were stained with hematoxylin-eosin (H&E). Sections were evaluated for Ki67, P-MAPK, P-STAT3, F4/80, IL-17A, Sox-2, P-NFκB and VEGF expression in bronchial epithelium, preneoplastic or tumor areas. Sections were incubated with primary antibodies overnight in a humidified chamber at 4°C with the following antibodies: Ki67 (ab833; 1:50; Abcam), P-MAPK (4370; 1:400; Cell Signaling Technology), P-STAT3 (D3A7; 1:400; Cell Signaling Technology), F4/80 (MCA497R; 1:100; Serotec), IL-17A (ab79056; 1:500; Abcam), Sox-2 (ab97959; 1:1000; Abcam), P-NFκB (ab86299; 1:200; Abcam) or VEGF (ab52917; 1:200; Abcam). Positive and negative controls were included for each marker. Bright field images were captured using a Leica DMI 3000B scope and LASv4.7 software (Leica Biosystems). IL-17A, Sox-2, P-NFκB and VEGF staining was graded as low (<30% positive cells per field), moderate (30-60% positive cells per field) or high (>60% positive cells per field). Ki67 cell proliferation index quantitated as the number of positive cells per field divided by the total number of tumor cells per field. For P-MAPK and P-STAT3 quantitation, number of positive cells per field were counted. Tumor infiltrating macrophages (F4/80 staining) was quantitated as the number of positive cells per field.
Statistical Analysis
Poisson regression was used to assess if there was a significant relationship between treatment group and number of tumors per mouse. Mixed effects models were used to determine if there was a significant relationship between treatment group and log-transformed tumor size, excluding mice in which tumors did not form. Group differences in Ki67 proliferation index, macrophage numbers and blood/tumor marker quantitation, were analyzed using Kruskal-Wallis and Mann-Whitney tests. For pre- versus post-treatment blood marker changes, Wilcoxon signed rank tests were used. Group differences in categorized IHC measures were analyzed by Chi square tests of independence. For antigens that showed significance by Chi square test, individual Fisher’s exact tests were performed to determine significant differences for each condition compared to placebo, and for combination treatments compared to single treatments. All statistical tests were two-sided with the threshold for significance defined as p<0.05. Analyses were conducted with GraphPad PRISM (Version 7) or SAS v. 9.4.
RESULTS
The combination of the AI anastrozole with aspirin decreased carcinogen-induced lung tumor formation and tumor size
We have previously shown that lung tumors and infiltrating pulmonary macrophages from the NNK-induced lung tumorigenesis murine model express aromatase and ERβ [13]. Inflammatory responses resulting from tobacco carcinogen exposure could be a key estrogen source driving tumor formation, supporting the idea of estrogen acting as a promoter of lung cancer aggressiveness through the inflammatory process in the lung microenvironment [13]. Therefore, we tested whether or not targeting both the estrogen and inflammatory pathways with anastrozole (0.1mg/kg) plus aspirin (25mg/kg) would have a greater effect on lung tumorigenesis compared to anastrozole or aspirin alone using the NNK lung tumor model.
Number of tumors is summarized for the 4 treatment groups of the anastrozole/aspirin experiment in Table 1 and Fig. 1A. Compared to the vehicle control group, which had an average of 5.8 tumors/animal, the tumor burden was 37% lower in the anastrozole group (3.7 tumors/animal; p=0.02) and 34% lower in the aspirin group (3.8 tumors/animal; p=0.03). When both anastrozole and aspirin were administered together (2.1 tumors/animal), the mean tumor burden was 42% lower than in the anastrozole group (p=0.02), 44% lower than the aspirin group (p=0.01), and 63% lower than for vehicle control (p<0.001).
Table 1.
Descriptive statistics for number of animals, tumors and tumor size by treatment group for anastrozole/aspirin experiment.
| Control | Anastrozole | Aspirin | Combination | |
|---|---|---|---|---|
| Number of Animals | 12 | 12 | 12 | 14 |
| Number of Tumors | ||||
| Total number of tumors | 70 | 44 | 46 | 30 |
| Mean # of tumors per animal (SD) | 5.8 (1.5) | 3.7 (1.5) | 3.8 (0.9) | 2.1 (1.4) |
| Median (min,max) # tumors per animal | 5 (4,8) | 4 (1, 6) | 4 (2, 5) | 2.5 (0, 4) |
| Estimated mean # of tumors per animal (95% CI)* | 5.8 (4.6, 7.4) | 3.7 (2.7, 4.9) | 3.8 (2.9, 5.1) | 2.1 (1.5, 3.1) |
| Tumor Size (mm2)** | ||||
| Mean size (SD) | 0.930 (0.816) | 0.630 (0.400) | 0.720 (0.554) | 0.508 (0.349) |
| Median size (min, max) | 0.631 (0.182, 4.231) | 0.505 (0.152,2.044) | 0.524 (0.208, 2.365) | 0.370 (0.153, 1.871) |
| Estimated mean size (95% CI)*** | 0.728 (0.627, 0.850) | 0.534 (0.443,0.650) | 0.572 (0.476, 0.688) | 0.426 (0.339, 0.535) |
Poisson regression model
Excludes 2 animals in the combination group with 0 tumors
Linear mixed effects regression model of log(tumor size), random intercept
Figure 1.
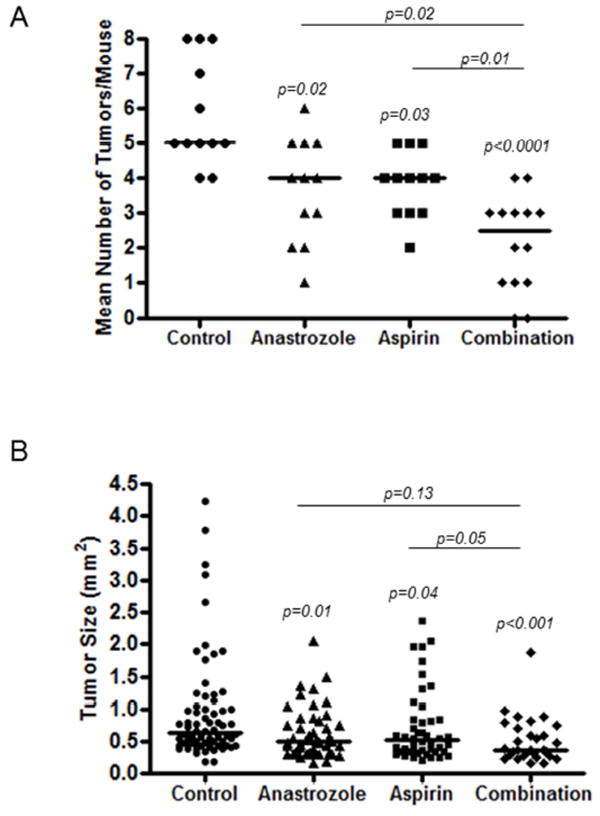
A, Number of tumors per mouse by treatment group; control, anastrozole, aspirin or combination. Each point represents a single mouse. Line represents the median value. P-values are shown above each group as assessed by Poisson regression. B, Tumor size (mm2) for each individual tumor by treatment group; control, anastrozole, aspirin or combination. Each point represents a single tumor. Line represents the median value. P-values are shown above each group as analyzed by linear mixed effects regression for log-transformed size.
We also observed a significant association between treatment group and lung tumor size (Table 1 and Fig. 1B). Tumors ranged in size from 0.15mm2 to 4.23mm2, with the largest tumor in the vehicle control group and no tumor larger than 1.87mm2 in the combination group. Tumors in the anastrozole and aspirin alone groups were 27% (p=0.01) and 21% (p=0.04) smaller, respectively, compared to those in the vehicle control group. Tumors in the combination treatment group were on average 20% smaller than in the anastrozole group (p=0.13), 26% smaller than the aspirin group (p=0.05) and 41% smaller than in the vehicle control group (p<0.001).
Circulating serum β-estradiol was lower with AI and aspirin treatment
Anastrozole is known to lower estrogen levels in postmenopausal women. In addition, studies analyzing sex hormone concentrations in postmenopausal women reported that regular users of aspirin and other analgesics have lower circulating estrogen levels versus nonusers, which could be a potential mechanism to lower their risk of hormone-associated cancers [23, 24]. We therefore investigated whether or not circulating β-estradiol (E2) was decreased by NSAID use in our animal model and whether or not adding NSAIDs to anastrozole had a further effect on E2 reduction. We collected serum prior to the initiation of experimental treatments and again at the end of the experiment to measure changes in E2 levels in each mouse by treatment. We observed 26% (p=0.03) and 23% (p=0.04) average decreases in circulating E2 after 8 weeks of anastrozole and aspirin treatment, respectively, compared to pre-treatment levels (Fig. 2A). With combination treatment, a 45% decrease (p=0.002) in E2 was observed (Fig. 2A). No change in pre- versus post-treatment E2 levels in control treated mice were observed.
Figure 2.
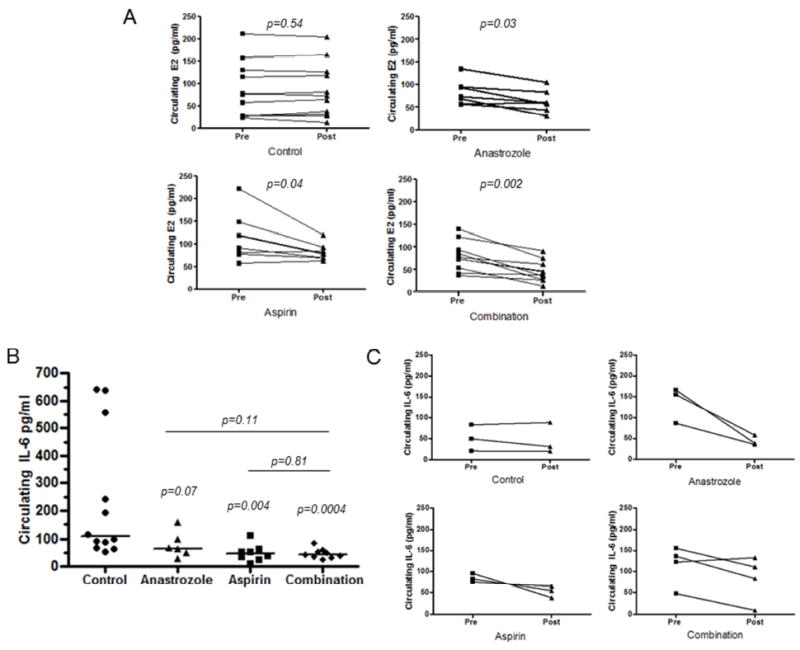
A, Serum β-estradiol was measured in duplicate by ELISA from pre-treatment and post-treatment blood samples from mice in the anastrozole/aspirin experiment. P-values from B, Serum IL-6 levels from anastrozole/aspirin experiment. P-values from Kruskal-Wallis and Mann-Whitney tests; each point represents single mouse; line represents median. C, Serum IL-6 before treatment (initiated 5 weeks after 4 weeks of NNK injections) and after 2 weeks of treatment (3-4 per group, separate animals from Figure 2A).
Pro-inflammatory cytokine IL-6 is decreased by AI and aspirin treatment
In order to evaluate effects of circulating inflammatory cytokines by experimental treatment, we analyzed a panel of markers including IL-1β, IL-6, Il-10, IL-12, IL-17A, IL-23, TNF-α, VEGF-A and IFNγ in mouse serum collected at the end of treatment. We found significant differences between the 4 treatment groups for IL-6 only (Fig. 2B) (Kruskal-Wallis test, p=0.001; results for the other markers not shown). The median circulating IL-6 level was 107.7 pg/ml for the control group, and 39% lower (median 65.2 pg/ml; p=0.07) and 56% lower (median 47.0 pg/ml; p=0.004) for the anastrozole and aspirin treatment groups, respectively (Fig. 2B). Median IL-6 level (42.6 pg/ml) was 60% lower for the combination treatment (p=0.0004) compared to vehicle control, but did not differ significantly from single agent treatments. Individual IL-6 values were highly variable, particularly in the control treatment group (range: 53.3-641.3 pg/ml). While the mouse with the largest tumor had the highest IL-6 value, overall total tumor burden did not correlate with the 3 mice with >500 pg/ml IL-6. However, to rule out the possibility that changes in IL-6 could be solely due to tumor burden rather than experimental treatment, we measured IL-6 in a separate short-term experiment before tumor formation. We measured pre-treatment and 2-week post-treatment IL-6 levels and observed decreases in IL-6 levels in individual mice (n=3-4) after only 2-weeks of anastrozole or aspirin treatment (Fig. 2C).
Cell proliferation, MAPK and STAT3 signaling is decreased in tumors arising after AI or aspirin treatment
To determine if tumors that occurred in the AI or aspirin treatment groups were less proliferative, we analyzed Ki67 positive tumor cells in FFPE lung tissue collected at the end of experiment (Fig. 3). Anastrozole and aspirin treatments alone both had significantly lower median Ki67 proliferation index than controls, each by approximately 30% (17.6 and 17.1 versus 27.1; p=0.03 and p=0.03, respectively). Tumors that arose in the combination treatment group had a 64% (p=0.03) lower median proliferative rate compared to control (Fig. 3B) which was reflective of their decreased tumor size in this experiment as well (Fig. 1B). Moreover, tumors that arose in mice that received the combination were significantly less proliferative than tumors arising from single agent treatments based on Ki67 labeling (p<0.05). The anti-proliferative effects of anastrozole and aspirin alone or in combination were accompanied by decreased activation of the MAPK and STAT3 signaling pathways. In the same FFPE tissues, we measured P-MAPK and P-STAT3 positive tumor cells by IHC (Fig. 3A) and observed 37% (p=0.03) and 27% (n.s.) lower median P-MAPK positivity with anastrozole or aspirin alone and a 61% decrease with the combination (p=0.03) compared to control (Fig. 3B). Similarly for P-STAT3, we found 68% (p=0.03) and 78% (p=0.03) decreases with anastrozole or aspirin alone, respectively, and almost a complete reduction (95%; p=0.03) with the combination compared to control (Fig. 3B).
Figure 3.
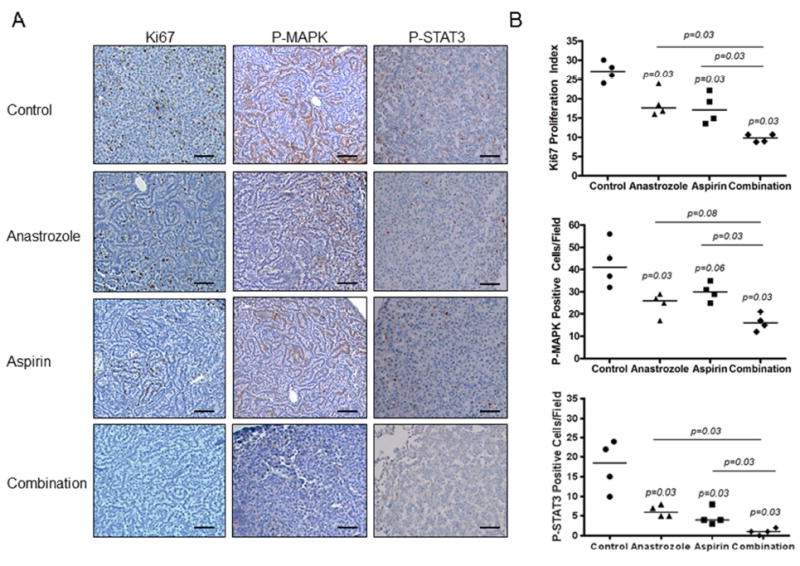
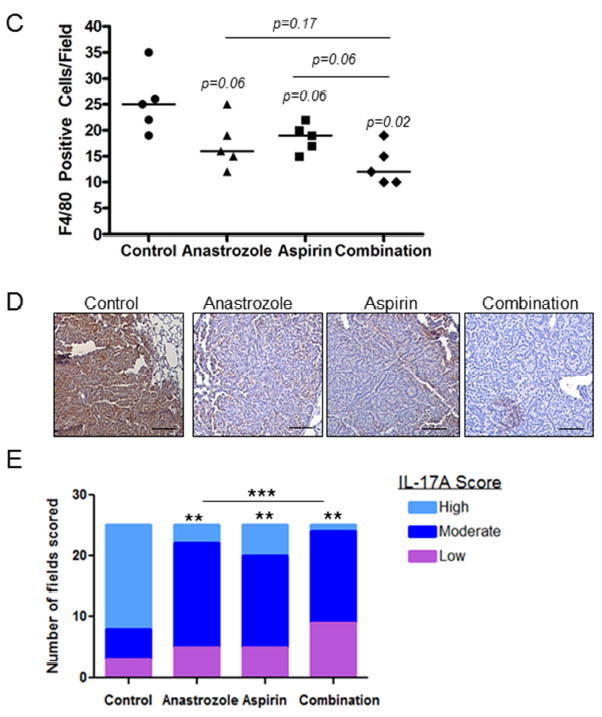
Representative mouse lung tumor sections stained A, and quantitated B, for Ki67, P-MAPK and P-STAT3 in mice exposed to NNK and treated with AI, aspirin or a combination of AI and aspirin. Scale bar= 50μm. P-values from Kruskal-Wallis and Mann-Whitney tests; each point represents the average quantitative value of 5 high-powered fields per tumor; horizontal line represents median. C, Tumor associated macrophages in lung tissue from each treatment group were measured by F4/80 immunostaining and quantitated by the number of F4/80 positive cells per field. P-values from Kruskal-Wallis and Mann-Whitney tests; each point represents the average quantitative value of 5 high-powered fields per tumor; horizontal line represents median. D, Representative IL-17A IHC by treatment group. Scale bar= 50μm. E, Quantitation of low (purple bars, <30% positive cells per field), moderate (dark blue bars, 30-60% positive cells per field) or high grade (light blue bars, >60% positive cells per field) fields by treatment group in lung tumors. P-values as analyzed by Chi-square and Fisher’s exact test. **p<0.01; ***p<0.001.
Tumor associated-macrophage infiltration is negatively correlated with AI and aspirin treatment
Macrophage infiltration is a hallmark of chronic inflammation. We therefore investigated the number of tumor infiltrating macrophages in FFPE lung tissues by F4/80 immunostaining (Fig. 3C). We observed lower median F4/80 positive cells for anastrozole (median=16; 32% lower; p=0.06) and aspirin (median=19; 26% lower; p=0.06) compared to control (median=25), while the combination significantly decreased macrophage number (median=12; 49% lower; p=0.02). These results correlated with lower tumor expression of IL-17A, a cytokine associated with M2 macrophages. Fig. 3D shows representative IL-17A immunostaining and quantitation of low, moderate and high grade staining is shown in Fig. 3E. A significant (p<0.001) shift towards increased low grade scoring and less high grade scoring was found with single agent treatments with the highest frequency of low grade scores in the combination treatment group compared to control. For example, anastrozole, aspirin and the combination had 12% (p<0.01), 20% (p<0.01) and 4% (p<0.01) of fields with high grade scores compared to 68% in the control group. Circulating IL-17A in this animal model was very low with <10pg/ml detected under all conditions (not shown).
Sox-2 and VEGF are decreased in airway preneoplasias by treatment with AI and aspirin
We previously reported that expression of the lung cancer stem-cell marker Sox-2 was significantly increased in the lungs of mice exposed to estrogen and down-regulated by anti-estrogens [22]. Next, we investigated the effect of AI and aspirin treatment on Sox-2 expression in normal bronchial epithelium and preneoplastic areas of mouse lungs from NNK-treated animals. This allowed for a determination of treatment effects on the cell compartments that give rise to tumors. FFPE lung tissues were used for IHC evaluation of Sox-2 after 8 weeks of control, anastrozole, aspirin or combination treatment (Fig. 4A). IHC staining from 25 microscopic fields per treatment group were scored as low, moderate or high and the distribution of scores was significantly different across all treatment groups compared to placebo (p<0.001) in both the bronchial epithelium as well as preneoplastic areas with a shift towards lower IHC scores (Fig. 4B). After treatment with aspirin, while Sox-2 scores were shifted towards more low and less high grade scores, this was not significantly modified in the normal bronchial epithelium or preneoplasias compared to vehicle control. However, after anastrozole and combination treatment, there was a greater proportion of low Sox-2 scores (p<0.05) in both normal bronchial epithelium and preneoplasias. In bronchial epithelium, the proportion of fields with a low score was 56% (14/25) for anastrozole treatment and 48% (12/25) for combination treatment, compared to only 12% (3/25) for control. Aspirin had a slightly greater proportion of low scores (16%, 4/25). Similar results were found in preneoplasias with 40% (9/25), 80% (20/25) and 76% (19/25) low scoring fields in aspirin, anastrozole and combination treatments, respectively compared to only 16% (4/25) in the control group. No fields with Sox-2 high scores were observed with anastrozole or combination treatment and only 2/25 (8%) had high scores in the aspirin group compared to 24% (6/25) in the control group. These results demonstrate lung stem cell marker reduction in murine airways by blocking the estrogen or inflammatory pathways.
Figure 4.
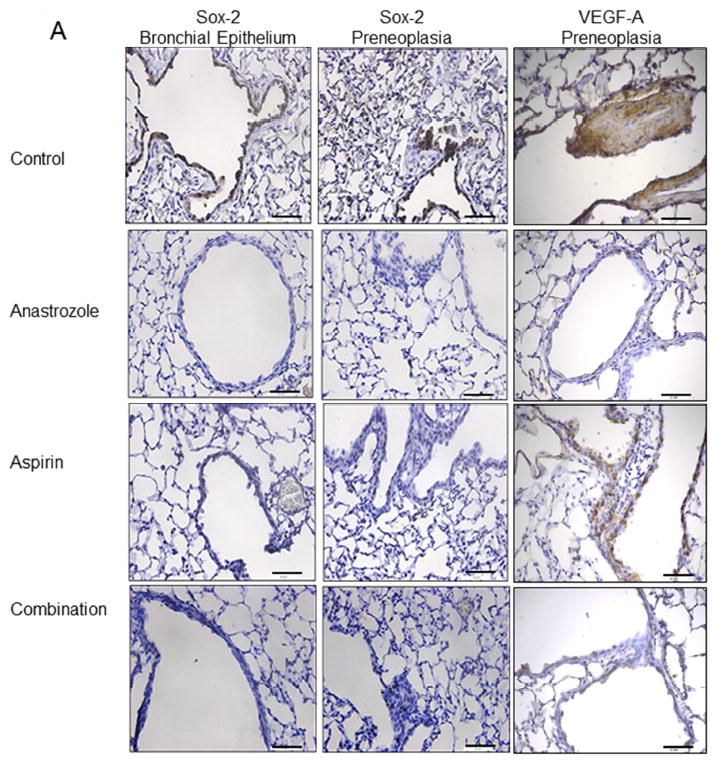
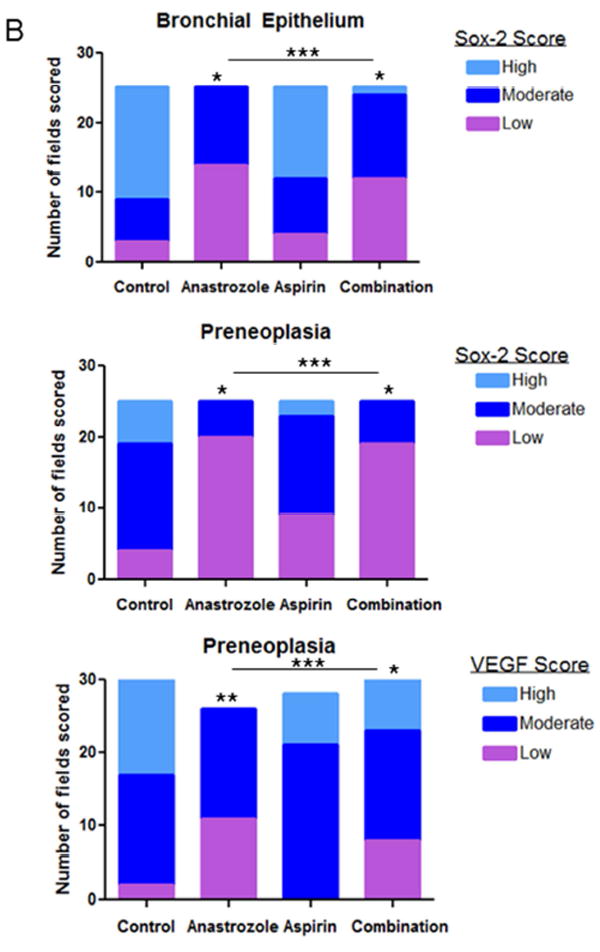
A, Representative Sox-2, VEGF immunostaining in bronchial epithelium or preneoplastic lung regions from control, anastrozole, aspirin or combination treatment. Scale bar= 71.4μm. B, Sox-2 and VEGF IHC quantitation of low (purple bars), moderate (dark blue bars) or high grade (light blue bars) scores per field by treatment group in bronchial epithelium and preneoplasias. P-values as analyzed by Chi-square and Fisher’s exact test. *p<0.05; **p<0.01; ***p<0.001.
We also measured protein expression of the E2-responsive gene, vascular endothelial growth factor-A (VEGF-A) [25]. VEGF-A is secreted by both tumors and macrophages in the tumor microenvironment. Using the same tissues and IHC low-high grade scoring system, we found no expression changes by experimental treatment in the bronchial epithelium (not shown). However in preneoplastic areas, VEGF expression was reduced by anastrozole (p<0.01) and combination treatment (p<0.05) but not aspirin treatment with a shift to more low-grade scoring and less high grade scoring (Fig. 4A-B). For example, 45% of the fields had high scores in the vehicle control group, compared to 0%, 25% and 23% for anastrozole, aspirin and combination treatment groups, respectively.
Ibuprofen elicits anti-tumorigenic effects similar to aspirin when combined with anastrozole
To determine if the association between aspirin and decreased tumor burden was specific to aspirin or if other NSAIDs could exert a similar anti-tumor effect, we evaluated the effect of the reversible COX inhibitor, ibuprofen. Using the same NNK animal model, we examined the effect on lung tumor burden and size with anastrozole, ibuprofen or the combination of the two agents compared to control. Similar to the aspirin and anastrozole experiment, we observed a 36-37% decrease in mean tumor burden per mouse with single-agent anastrozole or ibuprofen compared to control and 84% lower when both anastrozole and ibuprofen were administered together (p<0.001) (Supplemental Table 2 and Supplemental Fig. 1A). Furthermore, mice that received both agents developed 75% lower average number of lung tumors per mouse compared to mice treated with anastrozole or ibuprofen as single agent (p<0.001) (Supplemental Fig. 1A). Although we observed a strong decrease in tumor number with these treatments, there was no effect on tumor size (Supplemental Table 2 and Supplemental Fig. 1B).
We also observed decreases in circulating IL-6 in the anastrozole/ibuprofen experiment. Either anastrozole or ibuprofen alone resulted in lower median IL-6 levels while the combination significantly decreased the median level by 83% (p=0.008) compared to control (Supplemental Fig. 2A). Pre- and post-treatment 2-week treatment IL-6 effects were also observed (Supplemental Fig. 2B). Ki67 proliferative effect by treatment was not as strong in the anastrozole/ibuprofen experiment (data not shown) compared to the anastrozole/aspirin experiment which corresponded to the lack of significant tumor size differences. These results suggest that NSAIDs show enhancement of the AI protective effect on the development of carcinogen-induced lung tumors and may reduce tumor size.
Estrogen triggers an immune inflammatory response in the NNK carcinogen-treated mouse model
To further evaluate the effect of estrogen signaling on inflammation, we used FFPE lung tissue as well as blood and bronchioalveolar lavage fluid from male mice treated with NNK that also received E2 in the drinking water for 4 weeks. We observed a 1.4-fold increase (p<0.05) in pulmonary macrophage content as measured by F4/80 IHC (Fig. 5A). Furthermore, we measured levels of the inflammatory markers, P-NFkB and IL-17A as well as the estrogen-response gene, VEGF. We observed a signficant increase in P-NFκB high-grade staining (40% 10/25 fields; p<0.001; Fig. 5B) and VEGF high-grade staining (44% 14/32 fields; p<0.001; Fig. 5C) in lung preneoplasias that developed after tobacco carcinogen and E2 exposure in mice compared to control (0% high-grade staining for each marker). Similar results were observed in normal bronchial epithelium in reponse to E2 (not shown). Furthermore, IL-17A expression was also increased in lung preneoplasias by E2 treatment in this animal model with 28% high scoring fields in the control group compared to 60% in the E2 group (p=0.06; Fig. 5D). These changes were accompanied by a 1.3-fold increase in blood vessel formation with estrogen treatment (data not shown). Finally, although very low levels, we observed a 3-fold increase in circulating levels of IL-17A in the mice treated with E2 (17.0 pg/ml; range 7.2 - 48.8 pg/ml) compared to control (5.7 pg/ml; range: 5.4 - 6.2 pg/ml). Ki67 proliferative index was also signficantly increased (p<0.001) in preneoplasias by E2 exposure (not shown).
Figure 5.
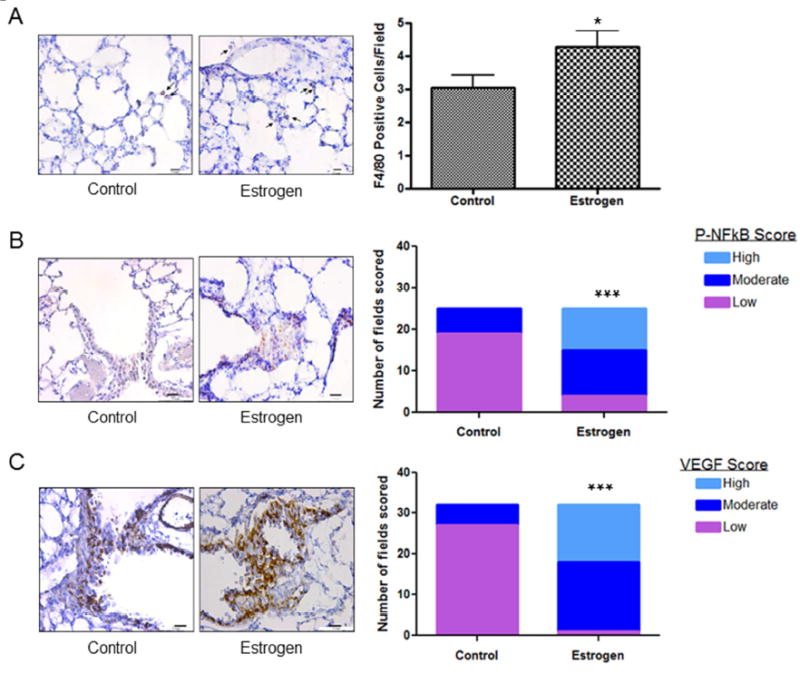
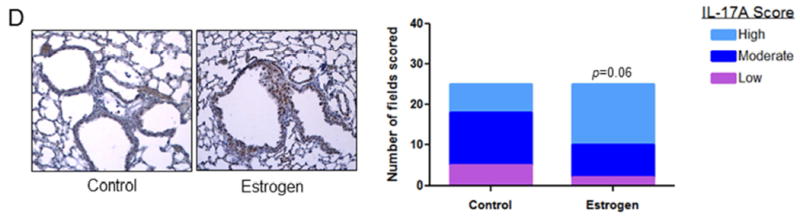
A, Pulmonary macrophages from either control or E2 treated mice were measured by F4/80 immunostaining and quantitated by the number of F4/80 positive cells per field. Results are the mean ± S.E. B, P-NFκB, C, VEGF and D, IL-17A and were measured in lung preneoplasias by immunostaining and each field quantitated for degree of staining. P-values as analyzed by Chi-square test. *P<0.05, ***p<0.001.
DISCUSSION
In human NSCLC lung tumors, more than 80% of lung tumors of all histological types are positive for ERs, and we and others have shown that estrogen is a driver of NSCLC proliferation in vitro and in vivo (reviewed in [26]). More than 60% of human lung tumors of different histologies are also are positive for aromatase expression, either in the carcinoma cells themselves, in tumor-associated macrophages, or in the tumor stroma [3, 5, 27]. Additionally, high expression of tissue aromatase is a poor prognostic marker in menopausal women with NSCLC [5]. There are limited studies evaluating the effect of AIs in lung cancer. Preclinically, exemestane has been shown to inhibit migration and invasion in NSCLC cell lines in vitro accompanied by changes in cell morphology [28] and dramatically reduced lung tumor xenograft growth, especially combined with standard chemotherapy [11]. Moreover, we have shown previously that anastrozole alone reduced the number of tobacco-induced lung tumors in mice by approximately 40% both when given during NNK exposure or after NNK exposure [13]. These preclinical AI studies along with evidence identifying estrogen as a driver of lung tumorigenesis have led to two ongoing clinical trials with AIs for lung cancer in the metastatic setting including a Phase I trial of exemestane in combination with chemotherapy (NCT01664754) as well as a Phase II single agent exemestane study (NCT02666105).
In breast cancer, a causal link between inflammation and E2 has been established. In this regard, triggers of inflammation, such as obesity, increase E2 production in the mammary microenvironment through the combined action of macrophages and adipocytes, driven by the cytokines and growth factors released at the site of inflammation that increase tissue aromatase expression [29]. This local E2 production through the action of tissue aromatase increases the risk of breast cancer development. Thus, AIs are now the standard of care for treatment of hormone receptor-positive breast cancer, both in the adjuvant setting and in advanced disease [30, 31], and are also considered as a chemoprevention option for women at high-risk to develop breast cancer [32, 33]. Similarly in lung cancer, tobacco carcinogens produce an inflammatory response that causes an influx of macrophages into the lungs, with stimulation of cytokine production. Our previous studies in both animal models and human tumor tissues provide evidence that the lung microenvironment is a site of local E2 synthesis through the action of tissue aromatase. Thus a similar link between inflammation and E2 production in the tumor microenvironment also plays a role in development of lung cancer.
Confirming the association between E2 and inflammation, use of anti-inflammatory drugs has been shown to reduce E2 levels in postmenopausal women [23, 24] and have been associated with decreased risk of hormone-driven cancers such as breast and ovarian cancer [34]. Use of an NSAID such as aspirin was associated with lower circulating E2 and mechanistic studies suggest that aspirin has a direct effect to reduce aromatase expression by inhibiting COX2 expression and prostaglandin synthesis [35]. The combination of the anti-inflammatory agent celecoxib with the AI exemestane reduced E2 levels and pro-tumor biomarkers such as Ki67 in post-menopausal women with ductal carcinoma-in-situ compared to single treatment [36]. Thus reducing aromatase expression via an NSAID and inhibiting tissue aromatase activity with an AI may be an effective strategy to reduce E2 levels thereby inhibiting the proliferative effects of E2. COX2 inhibition itself also results in a decrease in VEGF, IL-6, and IL-8, and is associated with decreased angiogenesis, decreased resistance to apoptosis, and increased anti-tumor cellular immunity [37-39]. These effects should also contribute independently to cancer prevention, and add to effects on the estrogen pathway. In this report, we sought evidence that such a strategy would be effective for lung cancer prevention using a carcinogen-induced murine lung tumor model.
Using our well-characterized NNK lung tumor model where tissue aromatase expression and E2 production is increased at inflammatory sites with NNK exposure [13], our results show that the AI anastrozole maximally prevents lung tumor development after carcinogen exposure when combined with an NSAID. Effects were observed with both aspirin and ibuprofen. Mean lung tumor number per mouse and tumor size were lower with combined anastrozole and NSAID treatment after tobacco-carcinogen exposure. These combination effects were accompanied by lower circulating E2 as well as a change in the inflammatory environment and proliferative and signaling capacity of the tumors with all endpoints examined, compared to controls. Examining all inflammatory and proliferative endpoints together as a group, individual treatments modulated some but not all endpoints compared to vehicle control, whereas with combination treatment all endpoints were significantly different from vehicle. This finding strongly suggests that combining an AI with an NSAID will produce a greater preventive effect against lung tumor formation. Decreased estrogen-mediated crosstalk with growth factor signaling pathways that leads to decreased MAPK and STAT3 activation may contribute to the lower proliferative rate, in addition to downstream signaling from the estrogen receptor itself.
Out of the panel of cytokines that we examined in the circulating blood, only IL-6 levels differed significantly by treatment group. Increased systemic and pulmonary production of IL-6 is commonly observed in lung adenocarcinoma patients and correlates with poor patient survival [40-43]. IL-6 is a pleotropic cytokine, considered a biomarker of ongoing inflammation with potential to modulate the immune response. This effect was observed not only after 8 weeks of AI and/or NSAID treatment, but also after only 2 weeks of treatment in our short-term exposure experiment. Furthermore, we observed that tumoral IL-17A expression and macrophage content was downregulated by both anastrozole and aspirin treatments. IL-17A is a pro-inflammatory cytokine produced by both Th-17 cells and macrophages. High levels of IL-17A expression is also associated with poor survival in lung cancer patients [44] IL-17A has also been linked to lung tumor progression [45, 46] and initiates an inflammatory response (including recruitment of macrophages and neutrophils) by inducing production of other cytokines such as IL-6 [47]. IL-17A is also thought to promote EMT in lung cancer and can also promote tumor growth through the IL-6/STAT3 signaling pathway. A recent study found that lung cancer patients with both elevated IL-6 and IL-17A had significantly worse survival compared to patients with low levels of these cytokines or high levels of only one of these cytokines [48]. Both IL-6 and IL-17A can be tumor-derived or released from tumor associated macrophages. Together the decreases observed in IL-17A, IL-6, and macrophage recruitment suggest a role for E2-driven regulation of the inflammatory environment in the lung. Furthermore, downregulating the lung stem cell phenotype may be an important part of this combined effect as we observed significant down-regulation of Sox-2 expression in the lungs (both in the bronchial epithelium and preneoplasias) of mice treated with anastrozole or aspirin. This is consistent with our previous observation that both Sox-2 and Nanog expression were decreased with anastrozole treatment and were positively correlated [22]. A novel role of ERβ and estrogen sensitivity in breast cancer stem cells has recently been described which may contribute to the importance of estrogens for tumor growth [49]. Our results are in agreement with other studies demonstrating a link between Akt signaling through the IL-6/STAT3 axis to maintaining stemness and this signaling axis is highly active in cancer stem cells [50, 51].
Preneoplasias also had reduced VEGF-A expression after anastrozole and combination treatment, in accordance with inhibition of E2-mediated pathways. Since IL-6 is also known to promote the release of VEGF to regulate tumor angiogenesis, the overall effect of reduction in IL-6 by anastrozole, aspirin or the combination could be directly driving reduction in the VEGF/angiogenesis pathway, contributing to the overall chemoprevention effect [52-54].
We also showed the opposite effect on the inflammatory microenvironment when we used male mice treated with NNK and E2. In this model, a 4-week exposure was used to simulate the inflammatory environment that is present in smokers before tumors develop. We found increased macrophage content and increased inflammatory markers such as VEGF and p-NFκB in lung tissue. These results confirm our previous studies in NSCLC cell lines whereby E2 induced VEGF-A mRNA and protein secretion, where combining an anti-estrogen with a VEGFR inhibitor showed enhanced anti-tumorigenic effects in a NSCLC xenograft model [55]. VEGF has also been shown to induce macrophage recruitment and regulate the M1 to M2 macrophage phenotypic switch [56]. Previous studies have also shown that the tumor promoting immunosuppressive M2 phenotype is also promoted by E2 [14]. The exact mechanism of macrophage infiltration from E2 exposure is still unclear but may be partly responsible for estrogen-driven tumorigenesis of the lung. These findings provide a new perspective on the effects of estrogen in the lung tumor microenvironment by increasing macrophage production and pro-inflammatory markers to drive proliferation, in addition to direct stimulation of ER-positive pulmonary epithelial cells. The role that estrogens play in pro- vs. anti-inflammatory activities appears to be disease-state dependent with estrogen-induced anti-inflammatory effects identified in bone remodeling, CNS conditions and auto-immune disorders [57-59]. This may be due to selective actions of E2 on specific immune cell types in different organs. It is clear that the inflammatory milieu, while complex in nature, plays a critical role in the lung carcinogenic process. Further studies examining the effects of E2 inhibition on different immune cell types in the lung microenvironment are needed to fully understand the overall effect of targeting estrogen on lung tumorigenesis.
In conclusion, our results suggest that the combination of low-dose NSAID with an AI may reduce cancer risk in post-menopausal women who are at elevated risk for lung cancer. Women at high-risk for breast cancer may also benefit from this combination treatment approach as low-dose aspirin alone has been shown to reduce risk of hormone receptor positive breast cancer development by 20% [60]. This strategy for lung cancer chemoprevention would be particularly effective in post-menopausal women who have inflammatory lung diseases such as COPD. Furthermore, this combination would be an inexpensive and well-tolerated preventive measure that could be easily implemented into clinical practice. Future studies to delineate the mechanism of estrogen-mediated immune and inflammatory modulation in the lung tumor microenvironment are necessary.
Supplementary Material
Acknowledgments
This work was supported by NIH grant P50 CA090440 and a grant to LPS from the Lung Cancer Research Foundation. This project used the UPCI Animal Facility and UPCI Biostatistics Facility and was supported in part by award P30CA047904. This project also used the Masonic Cancer Center Animal Facility and was supported in part by award P30CA077598.
Abbreviations
- ER
estrogen receptor
- E2
β-estradiol
- NSCLC
non-small cell lung cancer
- HRT
hormone replacement therapy
- AI
aromatase inhibitor
- NNK
4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone
Footnotes
Conflicts of Interest: None
Disclosures: Dr. Diergaarde reports grants from NIH, during the conduct of this study. Dr. Kurland reports grants from NIH, during the conduct of the study; personal fees from Celidex, outside the submitted work. Dr. Siegfried reports grants from NIH, during the conduct of this study. The other authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Laura P. Stabile, Department of Pharmacology & Chemical Biology, University of Pittsburgh, and UPMC Hillman Cancer Center, Pittsburgh, PA
Mariya Farooqui, Department of Pharmacology, University of Minnesota, Minneapolis, MN.
Beatriz Kanterewicz, UMPC Hillman Cancer Center, Pittsburgh PA.
Shira Abberbock, UPMC Hillman Cancer Center, Pittsburgh PA.
Brenda F. Kurland, Department of Biostatistics, University of Pittsburgh School of Public Health, Pittsburgh, PA
Brenda Diergaarde, Department of Human Genetics, Graduate School of Public Health, University of Pittsburgh, and UPMC Hillman Cancer Center, Pittsburgh PA.
Jill M. Siegfried, Department of Pharmacology, University of Minnesota Minneapolis, MN and Department of Pharmacology & Chemical Biology, University of Pittsburgh
REFERENCES CITED
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Stabile LP, Davis AL, Gubish CT, et al. Human non-small cell lung tumors and cells derived from normal lung express both estrogen receptor alpha and beta and show biological responses to estrogen. Cancer Res. 2002;62:2141–2150. [PubMed] [Google Scholar]
- 3.Stabile LP, Dacic S, Land SR, et al. Combined analysis of estrogen receptor beta-1 and progesterone receptor expression identifies lung cancer patients with poor outcome. Clin Cancer Res. 2011;17:154–164. doi: 10.1158/1078-0432.CCR-10-0992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu CT, Chang YL, Shih JY, Lee YC. The significance of estrogen receptor beta in 301 surgically treated non-small cell lung cancers. J Thorac Cardiovasc Surg. 2005;130:979–986. doi: 10.1016/j.jtcvs.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 5.Mah V, Seligson DB, Li A, et al. Aromatase expression predicts survival in women with early-stage non small cell lung cancer. Cancer Res. 2007;67:10484–10490. doi: 10.1158/0008-5472.CAN-07-2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stabile LP, Lyker JS, Gubish CT, et al. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced antiproliferative effects. Cancer Res. 2005;65:1459–1470. doi: 10.1158/0008-5472.CAN-04-1872. [DOI] [PubMed] [Google Scholar]
- 7.Marquez-Garban DC, Chen HW, Fishbein MC, et al. Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids. 2007;72:135–143. doi: 10.1016/j.steroids.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hershberger PA, Vasquez AC, Kanterewicz B, et al. Regulation of endogenous gene expression in human non-small cell lung cancer cells by estrogen receptor ligands. Cancer Res. 2005;65:1598–1605. doi: 10.1158/0008-5472.CAN-04-2694. [DOI] [PubMed] [Google Scholar]
- 9.Chlebowski RT, Schwartz AG, Wakelee H, et al. Oestrogen plus progestin and lung cancer in postmenopausal women (Women’s Health Initiative trial): a post-hoc analysis of a randomized controlled trial. Lancet. 2009;374:1243–1251. doi: 10.1016/S0140-6736(09)61526-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu SC, Hsieh CJ, Wang TF, et al. Antiestrogen use in breast cancer patients reduces the risk of subsequent lung cancer: A population-based study. Cancer Epidemiol. 2017;48:22–28. doi: 10.1016/j.canep.2017.02.010. [DOI] [PubMed] [Google Scholar]
- 11.Marquez-Garban DC, Chen HW, Goodglick L, et al. Targeting aromatase and estrogen signaling in human non-small cell lung cancer. Ann N Y Acad Sci. 2009;1155:194–205. doi: 10.1111/j.1749-6632.2009.04116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garon EB, Pietras RJ, Finn RS, et al. Antiestrogen fulvestrant enhances the antiproliferative effects of epidermal growth factor receptor inhibitors in human non-small-cell lung cancer. J Thorac Oncol. 2013;8:270–278. doi: 10.1097/JTO.0b013e31827d525c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stabile LP, Rothstein ME, Cunningham DE, et al. Prevention of tobacco carcinogen-induced lung cancer in female mice using antiestrogens. Carcinogenesis. 2012;33:2181–2189. doi: 10.1093/carcin/bgs260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toniolo A, Fadini GP, Tedesco S, et al. Alternative activation of human macrophages is rescued by estrogen treatment in vitro and impaired by menopausal status. J Clin Endocrinol Metab. 2015;100:E50–58. doi: 10.1210/jc.2014-2751. [DOI] [PubMed] [Google Scholar]
- 15.Bolego C, Cignarella A, Staels B, Chinetti-Gbaguidi G. Macrophage function and polarization in cardiovascular disease: a role of estrogen signaling? Arterioscler Thromb Vasc Biol. 2013;33:1127–1134. doi: 10.1161/ATVBAHA.113.301328. [DOI] [PubMed] [Google Scholar]
- 16.Svoronos N, Perales-Puchalt A, Allegrezza MJ, et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017;7:72–85. doi: 10.1158/2159-8290.CD-16-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris RE, Beebe-Donk J, Alshafie GA. Reduced risk of human lung cancer by selective cyclooxygenase 2 (COX-2) blockade: results of a case control study. Int J Biol Sci. 2007;3:328–334. doi: 10.7150/ijbs.3.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muscat JE, Chen SQ, Richie JP, Jr, et al. Risk of lung carcinoma among users of nonsteroidal antiinflammatory drugs. Cancer. 2003;97:1732–1736. doi: 10.1002/cncr.11242. [DOI] [PubMed] [Google Scholar]
- 19.Harris RE, Beebe-Donk J, Schuller HM. Chemoprevention of lung cancer by non-steroidal anti-inflammatory drugs among cigarette smokers. Oncol Rep. 2002;9:693–695. [PubMed] [Google Scholar]
- 20.Hochmuth F, Jochem M, Schlattmann P. Meta-analysis of aspirin use and risk of lung cancer shows notable results. Eur J Cancer Prev. 2016;25:259–268. doi: 10.1097/CEJ.0000000000000176. [DOI] [PubMed] [Google Scholar]
- 21.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. Faseb j. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 22.Siegfried JM, Farooqui M, Rothenberger NJ, et al. Interaction between the estrogen receptor and fibroblast growth factor receptor pathways in non-small cell lung cancer. Oncotarget. 2017;8:24063–24076. doi: 10.18632/oncotarget.16030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gates MA, Tworoger SS, Eliassen AH, et al. Analgesic use and sex steroid hormone concentrations in postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2010;19:1033–1041. doi: 10.1158/1055-9965.EPI-09-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hudson AG, Gierach GL, Modugno F, et al. Nonsteroidal anti-inflammatory drug use and serum total estradiol in postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2008;17:680–687. doi: 10.1158/1055-9965.EPI-07-2739. [DOI] [PubMed] [Google Scholar]
- 25.Applanat MP, Buteau-Lozano H, Herve MA, Corpet A. Vascular endothelial growth factor is a target gene for estrogen receptor and contributes to breast cancer progression. Adv Exp Med Biol. 2008;617:437–444. doi: 10.1007/978-0-387-69080-3_42. [DOI] [PubMed] [Google Scholar]
- 26.Burns TF, Stabile LP. Targeting the estrogen pathway for the treatment and prevention of lung cancer. Lung Cancer Manag. 2014;3:43–52. doi: 10.2217/lmt.13.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mah V, Marquez D, Alavi M, et al. Expression levels of estrogen receptor beta in conjunction with aromatase predict survival in non-small cell lung cancer. Lung Cancer. 2011;74:318–325. doi: 10.1016/j.lungcan.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giannopoulou E, Siatis KE, Metsiou D, et al. The inhibition of aromatase alters the mechanical and rheological properties of non-small-cell lung cancer cell lines affecting cell migration. Biochim Biophys Acta. 2015;1853:328–337. doi: 10.1016/j.bbamcr.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 29.Subbaramaiah K, Morris PG, Zhou XK, et al. Increased levels of COX-2 and prostaglandin E2 contribute to elevated aromatase expression in inflamed breast tissue of obese women. Cancer Discov. 2012;2:356–365. doi: 10.1158/2159-8290.CD-11-0241. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Burstein HJ, Prestrud AA, Seidenfeld J, et al. American Society of Clinical Oncology clinical practice guideline: update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Clin Oncol. 2010;28:3784–3796. doi: 10.1200/JCO.2009.26.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sainsbury R. The development of endocrine therapy for women with breast cancer. Cancer Treat Rev. 2013;39:507–517. doi: 10.1016/j.ctrv.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 32.Goss PE, Ingle JN, Ales-Martinez JE, et al. Exemestane for breast-cancer prevention in postmenopausal women. N Engl J Med. 2011;364:2381–2391. doi: 10.1056/NEJMoa1103507. [DOI] [PubMed] [Google Scholar]
- 33.Cuzick J, Sestak I, Forbes JF, et al. Anastrozole for prevention of breast cancer in high-risk postmenopausal women (IBIS-II): an international, double-blind, randomised placebo-controlled trial. Lancet. 2014;383:1041–1048. doi: 10.1016/S0140-6736(13)62292-8. [DOI] [PubMed] [Google Scholar]
- 34.Takkouche B, Regueira-Mendez C, Etminan M. Breast cancer and use of nonsteroidal anti-inflammatory drugs: a meta-analysis. J Natl Cancer Inst. 2008;100:1439–1447. doi: 10.1093/jnci/djn324. [DOI] [PubMed] [Google Scholar]
- 35.Diaz-Cruz ES, Brueggemeier RW. Interrelationships between cyclooxygenases and aromatase: unraveling the relevance of cyclooxygenase inhibitors in breast cancer. Anticancer Agents Med Chem. 2006;6:221–232. doi: 10.2174/187152006776930873. [DOI] [PubMed] [Google Scholar]
- 36.Generali D, Buffa FM, Deb S, et al. COX-2 expression is predictive for early relapse and aromatase inhibitor resistance in patients with ductal carcinoma in situ of the breast, and is a target for treatment. Br J Cancer. 2014;111:46–54. doi: 10.1038/bjc.2014.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernard MP, Bancos S, Sime PJ, Phipps RP. Targeting cyclooxygenase-2 in hematological malignancies: rationale and promise. Curr Pharm Des. 2008;14:2051–2060. doi: 10.2174/138161208785294654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams CS, Tsujii M, Reese J, et al. Host cyclooxygenase-2 modulates carcinoma growth. J Clin Invest. 2000;105:1589–1594. doi: 10.1172/JCI9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Liu A, Li H, et al. Celecoxib inhibits interleukin-6/interleukin-6 receptor-induced JAK2/STAT3 phosphorylation in human hepatocellular carcinoma cells. Cancer Prev Res (Phila) 2011;4:1296–1305. doi: 10.1158/1940-6207.CAPR-10-0317. [DOI] [PubMed] [Google Scholar]
- 40.Yanagawa H, Sone S, Takahashi Y, et al. Serum levels of interleukin 6 in patients with lung cancer. Br J Cancer. 1995;71:1095–1098. doi: 10.1038/bjc.1995.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haura EB, Livingston S, Coppola D. Autocrine interleukin-6/interleukin-6 receptor stimulation in non-small-cell lung cancer. Clin Lung Cancer. 2006;7:273–275. doi: 10.3816/CLC.2006.n.006. [DOI] [PubMed] [Google Scholar]
- 42.Zhou B, Liu J, Wang ZM, Xi T. C-reactive protein, interleukin 6 and lung cancer risk: a meta-analysis. PLoS One. 2012;7:e43075. doi: 10.1371/journal.pone.0043075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pine SR, Mechanic LE, Enewold L, et al. Increased levels of circulating interleukin 6, interleukin 8, C-reactive protein, and risk of lung cancer. J Natl Cancer Inst. 2011;103:1112–1122. doi: 10.1093/jnci/djr216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu C, Hao K, Yu L, Zhang X. Serum interleukin-17 as a diagnostic and prognostic marker for nonsmall cell lung cancer. Biomarkers. 2014;19:287–290. doi: 10.3109/1354750X.2014.908954. [DOI] [PubMed] [Google Scholar]
- 45.Bao Z, Lu G, Cui D, et al. IL-17A-producing T cells are associated with the progression of lung adenocarcinoma. Oncol Rep. 2016;36:641–650. doi: 10.3892/or.2016.4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akbay EA, Koyama S, Liu Y, et al. Interleukin-17A Promotes Lung Tumor Progression Through Neutrophil Attraction to Tumor Sites and Mediating Resistance to PD-1 Blockade. J Thorac Oncol. 2017 doi: 10.1016/j.jtho.2017.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. 2012;11:763–776. doi: 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- 48.Meaney CL, Zingone A, Brown D, et al. Identification of serum inflammatory markers as classifiers of lung cancer mortality for stage I adenocarcinoma. Oncotarget. 2017 doi: 10.18632/oncotarget.16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma R, Karthik GM, Lovrot J, et al. Estrogen Receptor beta as a Therapeutic Target in Breast Cancer Stem Cells. J Natl Cancer Inst. 2017;109:1–14. doi: 10.1093/jnci/djw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malanga D, De Marco C, Guerriero I, et al. The Akt1/IL-6/STAT3 pathway regulates growth of lung tumor initiating cells. Oncotarget. 2015;6:42667–42686. doi: 10.18632/oncotarget.5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marotta LL, Almendro V, Marusyk A, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121:2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang SP, Wu MS, Shun CT, et al. Interleukin-6 increases vascular endothelial growth factor and angiogenesis in gastric carcinoma. J Biomed Sci. 2004;11:517–527. doi: 10.1007/BF02256101. [DOI] [PubMed] [Google Scholar]
- 53.Nilsson MB, Langley RR, Fidler IJ. Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine. Cancer Res. 2005;65:10794–10800. doi: 10.1158/0008-5472.CAN-05-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yeh HH, Lai WW, Chen HH, et al. Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene. 2006;25:4300–4309. doi: 10.1038/sj.onc.1209464. [DOI] [PubMed] [Google Scholar]
- 55.Siegfried JM, Gubish CT, Rothstein ME, et al. Combining the multitargeted tyrosine kinase inhibitor vandetanib with the antiestrogen fulvestrant enhances its antitumor effect in non-small cell lung cancer. J Thorac Oncol. 2012;7:485–495. doi: 10.1097/JTO.0b013e31824177ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Okizaki S, Ito Y, Hosono K, et al. Vascular Endothelial Growth Factor Receptor Type 1 Signaling Prevents Delayed Wound Healing in Diabetes by Attenuating the Production of IL-1beta by Recruited Macrophages. Am J Pathol. 2016;186:1481–1498. doi: 10.1016/j.ajpath.2016.02.014. [DOI] [PubMed] [Google Scholar]
- 57.Liu HB, Loo KK, Palaszynski K, et al. Estrogen receptor alpha mediates estrogen’s immune protection in autoimmune disease. J Immunol. 2003;171:6936–6940. doi: 10.4049/jimmunol.171.12.6936. [DOI] [PubMed] [Google Scholar]
- 58.Vegeto E, Benedusi V, Maggi A. Estrogen anti-inflammatory activity in brain: a therapeutic opportunity for menopause and neurodegenerative diseases. Front Neuroendocrinol. 2008;29:507–519. doi: 10.1016/j.yfrne.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weitzmann MN, Pacifici R. Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest. 2006;116:1186–1194. doi: 10.1172/JCI28550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clarke CA, Canchola AJ, Moy LM, et al. Regular and low-dose aspirin, other non-steroidal anti-inflammatory medications and prospective risk of HER2-defined breast cancer: the California Teachers Study. Breast Cancer Res. 2017;19:52. doi: 10.1186/s13058-017-0840-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.