

Abstract
The prefrontal cortex (PFC) is critical for memory formation, but the underlying molecular mechanisms are poorly understood. Clinical and animal model studies have shown that changes in PFC excitation and inhibition are important for cognitive functions as well as related disorders. Here, we discuss recent findings revealing the roles of the excitatory and inhibitory synaptic proteins neuroligin 1 (NLGN1) and NLGN2 in the PFC in memory formation and modulation of memory strength. We propose that shifts in NLGN1 and NLGN2 expression in specific excitatory and inhibitory neuronal subpopulations in response to experience regulate the dynamic processes of memory consolidation and strengthening. Because excitatory/inhibitory imbalances accompany neuropsychiatric disorders in which strength and flexibility of representations play important roles, understanding these mechanisms may suggest novel therapies.
Introduction
Strong and long-lasting memories are created by transforming fragile, newly learned information into stable and persistent biological representations, a process known as memory consolidation. In addition to post-translational modifications, consolidation requires a temporally limited phase of gene expression, which is accompanied by reorganization and strengthening of synaptic connections in specific neural circuits [1,2]. Consolidation is a highly dynamic process that allows for regulation of memory strength that can occur either through repetition of learning events or, in the case of single emotionally relevant experiences, via modulation [2-4]. Consolidated memories are not permanently stable; they can destabilize again and undergo re-consolidation if they are retrieved in certain conditions [5]. Reconsolidation is important because it provides flexibility and opportunities to strengthen or weaken the memory. Understanding the mechanisms and circuitry that underlie the strength and flexibility of memory through regulation of consolidation and reconsolidation is of clinical importance: several cognitive impairments are associated with either too little (e.g., aging and Alzheimer's disease) or too much memory strength (e.g. posttraumatic stress disorder [PTSD], addiction, obsessive compulsive disorder [OCD], autism spectrum disorder [ASD], and schizophrenia).
The consolidation process involves different neural circuits depending on the type of memory. For episodic memories, which process information about contexts, spaces, things, time, and conspecifics, consolidation involves interplay between the hippocampus and regions of the prefrontal cortex (PFC) [6,7]. With time (weeks in rodents, and up to years in humans), this interplay shifts the network supporting the memory representation, disengaging the hippocampus and redistributing the memory representation over cortical areas, a process known as system consolidation [2,8,9]. The biological, cellular and neural plasticity mechanisms recruited in the hippocampus for memory consolidation have been extensively investigated, but much less is known about the cortical mechanisms.
Typical experimental paradigms used to model episodic memories in rats and mice are based on emotionally arousing experiences, which elicit long-term memory after a single experience, e.g., contextual fear conditioning and inhibitory avoidance (IA). Using these paradigms, molecular, electrophysiological, optogenetic and pharmacogenetic investigations have revealed that biological changes induced by learning and required for consolidation progress differently in the hippocampus and cortical areas. Furthermore, these changes are more persistent in cortical regions [10-15]. The nature of these persistent molecular and cellular changes, and where in the PFC they occur is unclear.
The rodent PFC, which contains divisions that are anatomically and functionally similar to those of humans/primates, comprises the medial PFC (mPFC, further divided into prelimbic [PL] and infralimbic [IL] subregions), orbitofrontal cortex (OFC) and anterior cingulate cortex (ACC) [16]. As most biological characterizations on mPFC functions have been carried out in mice and rats, it is important to note that although evolutionarily more complex functional specializations are likely to exit in rats compared to mice, the cytoarchitectonic definitions of mouse and rat prefrontal cortical areas appear to be similar [17,18]. In this review, we will report studies done in both rats and mice and specify the species used.
Like all other areas of the cerebral cortex, the PFC circuitry is organized in layers and shaped by multiple subpopulations of excitatory and inhibitory GABAergic neurons, the latter representing 15-20% of the total neuronal population. Little is known about how these various cell types in the PFC respond to experience. One hypothesis proposes that experience changes the overall ratio of excitation to inhibition (E/I; e.g., [19]), and that E/I dysregulation makes a major contribution to many neuropsychiatric disorders, including PTSD, depression, addiction, anxiety, schizophrenia, and ASD [20-24]. Notably, in this regard, all these disorders are characterized by impaired behavioral flexibility.
However, invoking a change in the overall E/I ratio in the PFC to explain neuropsychiatric disorders is rather simplistic, and not commensurate with the specific organization of brain structures and the complexity of their associated cognitive functions. While the E/I shift model provides an important starting point, it begs for a deeper mechanistic understanding, and especially how experience changes E/I.
Here, we will discuss recent studies investigating PFC excitation and inhibition mechanisms in memory processes. We will focus on three questions: First, do both excitatory and inhibitory neurons in the PFC critically contribute to memory consolidation, and if so, how? Second, how do PFC excitatory and inhibitory synapses change upon learning or memory consolidation? And third, are these changes affected by memory retrievals that lead to strengthening or weakening of the memory? The answers to these questions would provide valuable insight into the mechanisms of memory strength and flexibility.
Do both excitatory and inhibitory neurons in the PFC critically contribute to memory consolidation? If so, how?
The combined use of cell type–specific transgenic rodent lines and optogenetic/chemogenetic techniques allows for cell type–specific control of neuronal activation. Using these approaches, research in the field has begun to identify how different neuronal populations in the PFC are involved in long-term memory formation, modulation, and flexibility. In addition to excitatory pyramidal neurons, the microcircuits of the PFC include several types of inhibitory neuron subpopulations that have distinct regulatory functions: fast-spiking parvalbumin positive (PV+) neurons, which provide strong perisomatic inhibition onto excitatory pyramidal neurons; somatostatin positive (SST+) neurons, which inhibit the dendritic branches of both excitatory and inhibitory neurons; and a heterogeneous subpopulation that expresses 5HT3aR, including vasoactive intestinal peptide positive (VIP+) neurons that specifically target PV+ and SST+ neurons, thereby disinhibiting excitatory neurons [25] (Figure 1).
Figure 1.
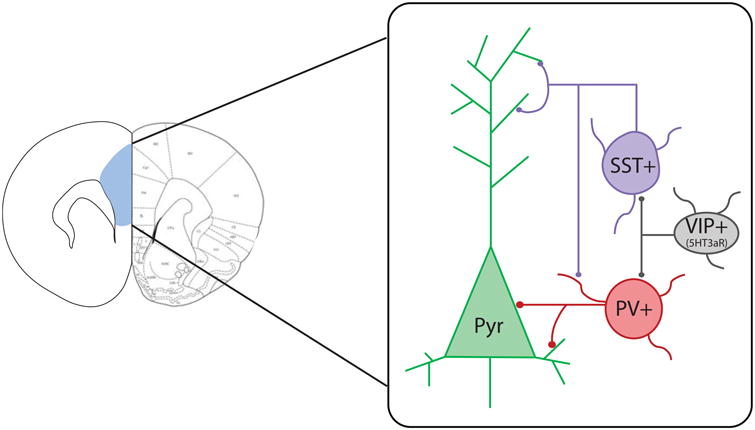
Representation of excitatory and inhibitory neuron types in the prefrontal cortex (highlighted in blue, left). Excitatory pyramidal neurons (Pyr) receive perisomatic inhibition from parvalbumin-positive interneurons (PV+) and dendritic inhibition from somatostatin-positive interneurons (SST+). SST+ neurons also provide dendritic inhibition onto PV+ neurons. Vasoactive intestinal peptide–positive neurons (VIP+), a subpopulation of 5-hydroxytryptamine-3a receptor (5HT3aR)-expressing interneurons, inhibit PV+ and SST+ inhibitory neurons, resulting in disinhibition of Pyr neurons.
Studies selectively targeting either excitatory or inhibitory neurons in the PFC, particularly PV+ interneurons, in a variety of behavioral paradigms led to the general conclusion that both neuronal populations play a critical role in memory formation and expression. Pioneering optogenetic experiments by Yizhar et al. (2011) [19] revealed that prolonged activation of mPFC excitatory neurons, resulting in an increased E/I ratio, impairs social interaction and Pavlovian fear conditioning, and that the compensatory elevation of inhibitory cell excitability partially rescues social deficits. These observations supported the hypothesis that elevated E/I contributes to neuropsychiatric diseases. More recently, Courtin et al. (2014) [26] showed that phasic inhibition of PV+ neurons in the PL cortex during fear memory retrieval enhances fear expression and synchronization of pyramidal neuron output upon presentation of a conditioned stimulus, whereas activation of PV+ neurons partially suppresses fear expression. These data suggested that PV+ neurons coordinate the activity of prefrontal projection neurons to the basolateral amygdala (BLA) to drive fear expression, again underscoring the conclusion that inhibitory neurons shape behavioral responses. Using the activating version of Designer Receptors Exclusively Activated by Designer Drugs (DREADD-hM3Gq), a chemogenetic tool that is used to increase the excitability of the target neurons in the presence of the synthetic ligand clozapine-N-oxide (CNO), Warthen et al. (2016) [27] found that increasing the excitability of the mPFC is sufficient to enhance memory for food reward. However, they observed no effects on social interaction, locomotion, or anxiety behavior. Similarly, Yau et al. (2015) [28], using a conditioned suppression of lever pressing for food as a behavioral output to assess Pavlovian conditioned fear, showed that enhanced excitability of mPFC excitatory neurons does not affect fear learning, consolidation or retrieval, but is important for fear prediction error. On the other hand, using the silencing version of DREADD (DREADD-hM4Di) to decrease the excitability of PV+ neurons, Perova et al. (2015) [29] reported that elevation of E/I in the mPFC promotes learned helplessness under stress. In sum, notwithstanding some discrepancies possibly due to differences in the techniques used for stimulation or silencing and/or the choice of behavioral paradigms, all of these studies converged onto the idea that E/I balance in the PFC makes an important contribution to memory formation and retention and/or behavioral flexibility.
These studies have only begun to reveal the contributions of specific mechanisms in each neuronal population. To achieve deeper insights into the mechanisms in the PFC, we must also take into consideration the fact that different PFC subregions have distinct, even opposing, functions in behavioral responses. One clear example is the mPFC in the context of fear memories: the PL cortex promotes fear expression, whereas the IL cortex promotes fear extinction, the decrease in the expression of fear-conditioned response upon repeated non-reinforced exposures [30,31].
How do changes in PFC excitatory and inhibitory synapses drive memory consolidation and storage? Are these changes affected by memory retrievals that lead to strengthening or weakening of the memory?
Given the fact that different inhibitory neuronal subpopulations can synapse onto excitatory and different types of inhibitory neurons, which can result in different E/I outcomes, it is important to understand the underlying precise network of synapses at each cell type. To better understand the mechanisms operating in the PFC in association with learning, memory consolidation, and memory strengthening, we must therefore identify the mechanisms that drive experience-dependent changes in the E/I balance of neurotransmission at glutamatergic and GABAergic synapses of specific neuronal subpopulations. Key synaptic proteins highly conserved in evolution between rodents and humans that control excitation and inhibition are the post-synaptic cell adhesion molecules neuroligin 1 (NLGN1) and NLGN2. NLGN1 is enriched at excitatory synapses while NLGN2 is enriched at inhibitory, dopaminergic and cholinergic synapses [32]. NLGNs form homodimers, and their extracellular domain binds to neurexins (NRXNs) present at presynaptic terminals, while their intracellular regions anchor to scaffolding proteins, such as the postsynaptic density protein-95 (PSD95) and gephyrin [32]. In addition to NLGN1 and NLGN2, two other NLGNs, NLGN3 and NLGN4, have been identified. NLGN3 is found at both excitatory and inhibitory synapses, and may form hetereodimers with both NLGN1 and NLGN2 [33]; NLGN4 is poorly conserved from rodents to humans, has low level of expression in the mouse and found to localize to glycinergic synapses [32]. These structural features enable NLGN1 and NLGN2 to coordinate the assembly of glutamate and GABA/glycine receptors at the postsynaptic site with the maturation and function of the presynaptic specialized structures (Figure 2). As their dysregulation alters the properties of synapses and disrupts neural networks without completely abolishing synaptic transmission, it appears that NLGN1 and 2 are required for synapse function, rather than synapse formation [34].
Figure 2.

Neuroligin 1 (NLGN1) and NLGN2 cell adhesion molecules at excitatory and inhibitory synapses, respectively. NLGN1 and NLGN2 are enriched are postsynaptic membranes, and their extracellular domains adhere to distinct isoforms of neurexins (NRXN) at presynaptic membranes. Intracellularly, NLGN1 binds to the scaffolding protein postsynaptic density protein 95 (PSD95), and regulates the synaptic localization of glutamate receptors (GluR) α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) and N-methyl-D-aspartate receptors (NMDARs). NLGN2 forms a complex with collybistin (CB) and gephyrin (GPHN) and regulate the synaptic localization of GABA receptors (GABAR). The cell-surface molecules MAM domain-containing glycosylphosphatidylinositol anchor proteins (MDGAs) form post-synaptic cis-complexes with NLGN1 and NLGN2, and negatively regulate their trans-synaptic adhesion with NRXNs [60-62]. Additional protein abbreviations: GlyR, glycine receptor; P, PDZ binding domain; S, Src homology domain (SH3 domain); GK, guanylate kinase domain; D, Dbl homology domain; P, Pleckstrin homology domain (Adapted from Bemben et al., 2015 [48]).
Recent studies have provided important insights about the regulatory mechanisms that NLGNs can provide. Conditional genetic deletions of all three major NLGN isoforms, individually and in combination, in cultured mouse hippocampal and cortical neurons indicated that lack of NLGNs causes small or no change in synapse numbers, but significantly impairs synapse functions [35]. Conditional knockout of NLGN1 in newborn or juvenile mice results in a significant impairment in NMDAR- and L-type Ca2+ channel-dependent LTPs [36]. Finally, activity and learning leads to histone modifications regulating NRXN1 alternative splicing, hence controlling NRXN1 binding to NLGN1 in the mouse dentate gyrus [37]. Together, these results suggest that NLGN1 and NLGN2 have isoform-specific functions at excitatory and inhibitory synapses. Moreover, at least for NLGN1, these functions appear to contribute to a variety of activity-dependent responses [32].
The regulation and role of NLGN1 in learning and memory are still in the process of being understood. Clinical studies have revealed that NLGN1 genetic variants are associated with neuropsychiatric disorders such as ASD [38,39], memory loss and depression in Alzheimer's disease [40], and PTSD [41], indicating that NLGN1 plays a role in cognition. Notably, carriers of the NLGN1 variant strongly associated with PTSD exhibited greater neural activation in limbic and prefrontal regions, as well as increased functional connectivity between the amygdala and the dorsal–lateral PFC in response to fearful face stimuli [41]. In transgenic mouse models, loss or overexpression of NLGN1 impairs spatial memory, as determined by performance in the Morris water maze test, indicating the importance of optimal NLGN1 level in hippocampal-dependent memory tasks [42,43]. Notably in this regard, NLGN1 knockout mice also exhibit increased repetitive behavior, with small deficits in social interaction and pain sensation, accompanied by deficits in hippocampal LTP [42]. These findings paralleled studies in rats, in which local viral expression was used to target the amygdala in fear conditioning [44]. Therefore, NLGN1 in multiple brain regions plays a critical role in memory formation.
Regarding memory disorders, recent studies in rats showed that NLGN1 interacts with amyloid-β oligomers (AβO), forming a complex with GluN2B-containg N-methyl-D-aspartate (NMDA) receptors [45]. In mice, AβO interaction with NLGNs appears to mediate hippocampal synapse damage and memory loss [46], and, in rats, amyloid-induced neuroinflammation enhances epigenetic-mediated inhibition of NLGN1 expression, leading to glutamatergic dysfunction in the hippocampus and memory loss [47]. Given the fundamental roles of NLGN1 in memory and memory disorders, a next important question to be addressed is how experience regulates the expression of NLGN1 in specific cell populations, and how long the changes persist in order to promote memory consolidation and flexibility, or their dysregulation in memory disorders.
The role of NLGN2 in memory formation and storage also remains poorly characterized. As with NLGN1, genetic alterations in NLGN2 are linked to severe cognitive disorders, implying that it is also important for cognitive functions. Genome-wide analyses in humans have identified copy number variations (CNVs) and missense single nucleotide polymorphisms (SNPs) in NLGN2 that are associated with developmental disorders, including ASD and schizophrenia [48,49]. These human phenotypes have been in part recapitulated in transgenic mouse models. Nlgn2 knockout mice exhibit profound developmental delays in multiple metrics such as tail length, age of eye opening, and body length, as well as increased ultrasonic vocalization in pups [50]. NLGN2-overexpressing mice, on the other hand, have reduced lifespan and offspring viability, as well as limb clasping, repetitive behaviors, and impaired social interactions that resemble aspects of Rett syndrome and ASD [51]. Both knockout and overexpression of NLGN2 in mice increase anxiety-like behaviors and alter social behaviors, suggesting that synaptic inhibition plays a critical role in anxiety regulation [50-53]. Finally, viral-mediated overexpression of NLGN2 in the mouse hippocampus increases adult neurogenesis, while decreasing performance in the water maze task [54]. In sum, NLGN2 has emerged as a key molecule in brain development, anxiety as well as cognition.
A few recent studies explored the roles of NLGN1 and NLGN2 specifically in the PFC. Liang et al. (2015) [55] reported that virus-mediated conditional knockout of Nlgn2 in the mPFC of adult mice reduces anxiety or increases impulsivity-like behavior in the open arms of an elevated plus maze, and impairs fear conditioning. These behavioral changes are accompanied by an increased ratio of evoked E/I synaptic currents. Together, these data suggest that anxiety and fear learning may be highly sensitive to subtle changes in inhibitory synaptic function or plasticity in the mPFC [56]. Moreover, conditional Nlgn2 knockout, despite reducing inhibition, decreases experience-dependent induction of immediate-early genes, including c-Fos, Egr1 and Npas4 in the mPFC, suggesting that chronic reduction in local inhibition may impede experience-evoked mPFC activation. Tzanoulinou et al. (2016) [57] found that peripubertal stress in rats reduces NLGN2 expression in the mPFC, whereas virus-mediated overexpression of NLGN2 in mPFC rescues the attention deficits induced by peripubertal stress. In control rats that did not experience peripubertal stress, NLGN2 overexpression in the mPFC also impaired attention, indicating the importance of optimal inhibition in attention tasks.
In sum, although ablation or overexpression of NLGN1 and NLGN2 in the PFC indicate that these proteins are necessary for memory retention and responses to stress and attention [58], their roles in specific processes that regulate memory strength and flexibility, such as memory consolidation, reconsolidation, and modulation, remain to be understood.
To dissect the PFC mechanisms involved in memory consolidation and strengthening, our laboratory used inhibitory avoidance (IA) in rats [11]. IA is a contextual fear conditioning-based paradigm in which the animals learn to avoid a context previously paired with a footshock. This episodic type of memory undergoes hippocampal–cortical system consolidation, and lends itself well for molecular and behavioral investigations. We found that IA training led to a significant increase in both NLGN1 and NLGN2 levels in the PL cortex 6 days after training (Figure 3A). Blocking NLGN2 function with an excess of NLGN2 extracellular domain injected into the PL cortex once every 2 days after training significantly disrupted IA memory consolidation, but the same manipulation targeting NLGN1 had no effect. These data implied that a prolonged action of NLGN2, but not NLGN1, during the first week after training is required in the PL cortex for IA memory consolidation. This lingering effect of NLGN2 raises a few questions. First, is PL NLGN1 involved at all in memory consolidation? If so, what is its critical temporal window of action? Second, why is lasting synaptic inhibition necessary for consolidation of an IA memory? One possible explanation is that the increase in NLGN2-mediated inhibition disinhibits excitatory neurons, leading to an increase in excitation that mediates IA memory consolidation. This hypothesis is plausible, as it was supported by our experiments showing that IA training is accompanied by the induction of immediate early genes (such as activity-regulated cytoskeletal-associated protein, Arc/Arg3.1) in the PL cortex.
Figure 3.
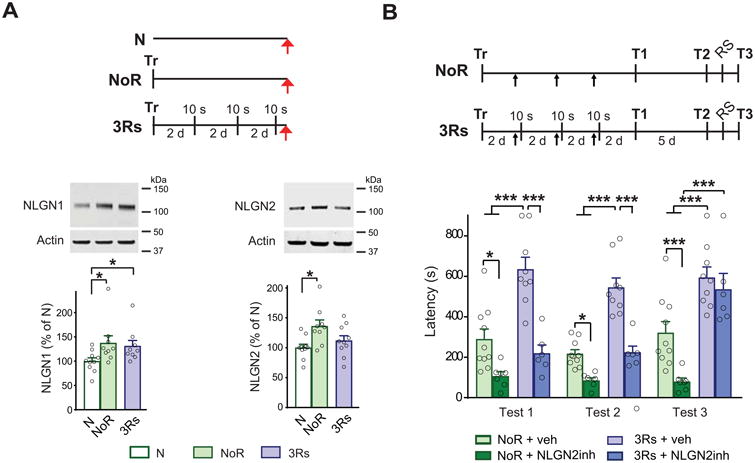
Neuroligin 1 (NLGN1) and NLGN2 play distinct roles in memory consolidation, strengthening, and extinction inhibition. (A) Schema of behavioral procedures, representative images and relative quantitative western blot analyses of PL cortex. Protein extracts were obtained from rats trained (Tr) in inhibitory avoidance (IA) and given 3 brief memory retrievals (3Rs), which consisted of 10 sec exposures to the training context without footshock, every two days. Rats were euthanized (red arrows) one hour after 3Rs or at the matched time point for the group that underwent training without retrievals and remained in the home cage after training (NoR). Naïve rats (N) served as reference controls. Data are presented as mean percentage ± s.e.m. of the mean values of the N group. One-way ANOVA followed by Newman-Keuls post hoc test; NLGN1 n = 9–10, NLGN2 n = 9–10; *p < 0.05 for both comparisons. (B) Schema of behavioral procedures is given above the graphs. Rats were trained (Tr) in IA and given 3Rs every two days or left in the home cage without retrieval (NoR) after training. Thirty minutes before each reactivation, or at matched timepoints in the NoR group, the animals received a bilateral PL cortex injection (black arrows) of NLGN2 recombinant extracellular domain (NLGN2inh) to inhibit NLGN2 function. Animals were tested for memory retention two days after the last retrieval (T1), and again five days later (T2), as shown in the schema. A reminder footshock (RS) was given in a different context with the same shock intensity one day after T2 and memory was tested one day later (T3). Data are expressed as mean latency ± s.e.m. Two-way ANOVA followed by Bonferroni post hoc test; *p<0.05, ***p<0.001 n = 7-10 (Adapted from Ye et al., 2017 [11]).
We previously showed that reconsolidation significantly strengthens IA memory if the memory is retrieved (or reactivated) with three brief context exposures given once every two days during the first week after IA training [59]. Using this paradigm, we found that, in comparison with memory consolidation (rats trained and remaining in the home cage following training), memory strengthening caused no additional changes in NLGN1 levels in the PL cortex.
By contrast, PL NLGN2 levels decreased to control levels, suggesting that memory strengthening is accompanied by a sustained increase in excitation and a reduction of inhibition in the PL cortex (Figure 3A). These results were confirmed by a survey of Arc induction. The decrease in NLGN2 expression after retrievals suggested that changes in NLGN2 levels dynamically accompany memory strengthening. Furthermore, it suggested that the reduction in NLGN2 after retrieval occurs in cell subpopulations and synapses distinct from those in which NLGN2 was upregulated during consolidation: if NLGN2 during consolidation serves to disinhibit inhibitory neurons, a decrease in NLGN2 expression in the same synapses would reverse that effect, leading to reduced excitation, which is the opposite of what the data showed. Indeed, memory strengthening evoked by retrievals was accompanied by increased Arc induction in the PL cortex. One model that may explain these data is that training and retrieval-induced memory strengthening are paralleled by dynamic regulation of NLGN1 and NLGN2 in distinct cell subpopulations (Figure 4). Consistent with this idea, we found that blocking NLGN2 at each memory reactivation did not disrupt memory consolidation (unlike when given without the retrievals), but only blocked memory strengthening (Figure 3B). Notably, we also observed that the blocking effect of NLGN2 on memory strengthening was reversed by a reminder shock (RS), and the memory fully reinstated. Because reinstatement after RS is a typical behavioral response associated with memory extinction, we reasoned that the mechanisms mediated by NLGN2 allow for memory strengthening actually by inhibiting extinction. This explanation was confirmed experimentally using an extinction paradigm [11]. Therefore, we concluded that the experience-dependent regulations of NLGN1 and NLGN2 in distinct cell populations is key for a flexible response to fear and aversive experiences [11]. Cell population-specific changes may therefore accompany the lack of extinction flexibility, as is found in PTSD.
Figure 4.

Schematic representation of our working model for cell type–specific changes in NLGN1 and NLGN2 after IA training and retrieval-induced memory strengthening in the PL cortex. IA training may increase NLGN2 expression in parvalbumin-positive interneurons (PV+), which would lead to disinhibition of pyramidal (Pyr) cells, as well as NLGN1 in Pyr cells. These changes would result in increased E/I. Memory retrieval-induced strengthening would decrease NLGN2 levels in Pyr cells, while maintaining NLGN2 increased levels in PV+ neurons. These changes would further increase excitation. Together, these NLGN1 and NLGN2 changes on different neuronal populations would differentially regulate E/I balance in memory consolidation and following retrievals leading to memory strengthening.
In sum, our results demonstrate that it is important to identify the subregions, cortical layers and cell subpopulations of the PFC in which NLGN1 and NLGN2 expression shifts in response to experience. These synaptic maps reflect the stability, strength, and flexibility of specific behaviors; hence, their dysregulation may underlie neuropsychiatric disorders.
Conclusions
Our understanding of the local PFC mechanisms underlying cognitive function remains limited. An important goal for future work will be the mapping of NLGN1 and NLGN2 regulation in relation to memory encoding, consolidation, and strength modulation. Furthermore, manipulations of neuron subpopulation-specific NLGN1 and NLGN2 could be used to drive memory strengthening or weakening. Because the PFC does not function in isolation, local changes will influence and be influenced by connected brain regions.
Here, we discussed the role of NLGN1 and NLGN2 in the modulation of fear memory strength, which is critical for adaptive behavior: while it is important to form long-lasting adaptive memories for the fittest behavior, overconsolidated or overstrengthened memories are maladaptive and associated with psychopathologies. Therefore, the identification of the specific circuitry and molecular mechanisms of excitation and inhibition underlying long-term memory consolidation and strengthening is important for developing specific therapeutic strategies to address the inflexibility of neuropsychiatric disorders.
Highlights.
Learning-induced molecular changes in the PFC are critical for long-term memory.
Neuronal excitation and inhibition in the PFC are important for memory formation.
NLGN1 and NLGN2 cooperate to regulate memory strength.
Acknowledgments
We thank Xiaojing Ye and Dana Kapeller-Libermann for discussions on an earlier draft of this manuscript. This work was supported by MH065635 and MH074736 to CMA and T32-MH019524 to AK.
Footnotes
The authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alberini CM, Kandel ER. The regulation of transcription in memory consolidation. Cold Spring Harbor perspectives in biology. 2014;7:a021741. doi: 10.1101/cshperspect.a021741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dudai Y, Karni A, Born J. The Consolidation and Transformation of Memory. Neuron. 2015;88:20–32. doi: 10.1016/j.neuron.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Kandel ER, Dudai Y, Mayford MR. The Molecular and Systems Biology of Memory. Cell. 2014;157:163–186. doi: 10.1016/j.cell.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiological reviews. 2009;89:121–145. doi: 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alberini CM. The role of reconsolidation and the dynamic process of long-term memory formation and storage. Frontiers in behavioral neuroscience. 2011;5:12. doi: 10.3389/fnbeh.2011.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Euston DR, Gruber AJ, McNaughton BL. The Role of Medial Prefrontal Cortex in Memory and Decision Making. Neuron. 2012;76:1057–1070. doi: 10.1016/j.neuron.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eichenbaum H. Prefrontal-hippocampal interactions in episodic memory. Nature reviews Neuroscience. 2017;18:547–558. doi: 10.1038/nrn.2017.74. [DOI] [PubMed] [Google Scholar]
- 8.Squire LR, Genzel L, Wixted JT, Morris RG. Memory consolidation. Cold Spring Harbor perspectives in biology. 2015;7:a021766. doi: 10.1101/cshperspect.a021766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Z, Wilson MA. Deciphering Neural Codes of Memory during Sleep. Trends in Neurosciences. 2017;40:260–275. doi: 10.1016/j.tins.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bambah-Mukku D, Travaglia A, Chen DY, Pollonini G, Alberini CM. A positive autoregulatory BDNF feedback loop via C/EBPβ mediates hippocampal memory consolidation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:12547–12559. doi: 10.1523/JNEUROSCI.0324-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11**.Ye X, Kapeller-Libermann D, Travaglia A, Inda MC, Alberini CM. Direct dorsal hippocampal-prelimbic cortex connections strengthen fear memories. Nature Neuroscience. 2017 doi: 10.1038/nn.4443. Using phamacologic and phamacogenetic tools with an inhibitory avoidance task in rats, the authors demonstrate that memory retrieval engages dorsal hippocampal-prelimbic cortex (PL) projections to regulate memory strength. In the PL cortex, NLGN1 and NLGN2 cooperate to promote memory strengthening: while NLGN1 supports memory strengthening, NLGN2 suppresses extinction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12*.Bero AW, Meng J, Cho S, Shen AH, Canter RG, Ericsson M, Tsai LH. Early remodeling of the neocortex upon episodic memory encoding. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:11852–11857. doi: 10.1073/pnas.1408378111. Using RNA-sequencing analyses the authors reveal a learning-induced transcriptomic program in the prefrontal cortex of mice 1 hour after contextual fear conditioning training. This research was among the first to suggest that long-term episodic memory is associated with rapid molecular and structural changes in the neocortex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13**.Kitamura T, Ogawa SK, Roy DS, Okuyama T, Morrissey MD, Smith LM, Redondo RL, Tonegawa S. Engrams and circuits crucial for systems consolidation of a memory. Science. 2017;356:73–78. doi: 10.1126/science.aam6808. Using optogenetic manipulation of activated neuronal networks, the authors demonstrate that the prefrontal cortex rapidly activates an ensemble of cells driven by inputs from the hippocampal-entorhinal cortex network and the basolateral amygdala. They show that this network of activated cells change over time, and become necessary for memory retreival only at remote time points, at which point the hippocampal network is no longer necessary. This study supports the idea that the prefrontal cortex is rapidly engaged with training and forms an immature memory trace, which functionally matures over time through hippocampal and amygdala inputs. This maturation results in a switch of recall circuitry with time, engaging the prefrontal cortex with memory retrieval at later time points. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winocur G, Moscovitch M, Bontempi B. Memory formation and long-term retention in humans and animals: convergence towards a transformation account of hippocampal-neocortical interactions. Neuropsychologia. 2010;48:2339–2356. doi: 10.1016/j.neuropsychologia.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 15.Holloway CM, McIntyre CK. Post-training disruption of Arc protein expression in the anterior cingulate cortex impairs long-term memory for inhibitory avoidance training. Neurobiology of Learning and Memory. 2011;95:425–432. doi: 10.1016/j.nlm.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 16.Jin J, Maren S. Prefrontal-Hippocampal Interactions in Memory and Emotion. Frontiers in systems neuroscience. 2015;9:170. doi: 10.3389/fnsys.2015.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van De Werd HJJM, Rajkowska G, Evers P, Uylings HBM. Cytoarchitectonic and chemoarchitectonic characterization of the prefrontal cortical areas in the mouse. Brain Structure and Function. 2010;214:339–353. doi: 10.1007/s00429-010-0247-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uylings HBM, Groenewegen HJ, Kolb B. Do rats have a prefrontal cortex? Behavioural Brain Research. 2003;146:3–17. doi: 10.1016/j.bbr.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 19.Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson SM, Kallarackal AJ, Kvarta MD, Van Dyke AM, LeGates TA, Cai X. An excitatory synapse hypothesis of depression. Trends in Neurosciences. 2015;38:279–294. doi: 10.1016/j.tins.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosal S, Hare B, Duman RS. Prefrontal Cortex GABAergic Deficits and Circuit Dysfunction in the Pathophysiology and Treatment of Chronic Stress and Depression. Current opinion in behavioral sciences. 2017;14:1–8. doi: 10.1016/j.cobeha.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson SB, Valakh V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron. 2015;87:684–698. doi: 10.1016/j.neuron.2015.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volkow ND, Morales M. The Brain on Drugs: From Reward to Addiction. Cell. 2015;162:712–725. doi: 10.1016/j.cell.2015.07.046. [DOI] [PubMed] [Google Scholar]
- 24.Foss-Feig JH, Adkinson BD, Ji JL, Yang G, Srihari VH, McPartland JC, Krystal JH, Murray JD, Anticevic A. Searching for Cross-Diagnostic Convergence: Neural Mechanisms Governing Excitation and Inhibition Balance in Schizophrenia and Autism Spectrum Disorders. Biological psychiatry. 2017;81:848–861. doi: 10.1016/j.biopsych.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Developmental neurobiology. 2011;71:45–61. doi: 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26**.Courtin J, Chaudun F, Rozeske RR, Karalis N, Gonzalez-Campo C, Wurtz H, Abdi A, Baufreton J, Bienvenu TCM, Herry C. Prefrontal parvalbumin interneurons shape neuronal activity to drive fear expression. Nature. 2014;505:92–96. doi: 10.1038/nature12755. Using optogenetic manipulations and in vivo single unit recordings, the authors identify parvalbumin positive interneurons in the prefrontal cortex as critical for fear expression. These cells, by disinhibiting projection neurons at the onset of the conditioned stimulus, reset local theta oscillations, which lead to coordinated communication with the basolateral amygdala and fear expression. [DOI] [PubMed] [Google Scholar]
- 27.Warthen DM, Lambeth PS, Ottolini M, Shi Y, Barker BS, Gaykema RP, Newmyer BA, Joy-Gaba J, Ohmura Y, Perez-Reyes E, et al. Activation of Pyramidal Neurons in Mouse Medial Prefrontal Cortex Enhances Food-Seeking Behavior While Reducing Impulsivity in the Absence of an Effect on Food Intake. Frontiers in behavioral neuroscience. 2016;10:63. doi: 10.3389/fnbeh.2016.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yau JOY, McNally GP. Pharmacogenetic excitation of dorsomedial prefrontal cortex restores fear prediction error. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:74–83. doi: 10.1523/JNEUROSCI.3777-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perova Z, Delevich K, Li B. Depression of excitatory synapses onto parvalbumin interneurons in the medial prefrontal cortex in susceptibility to stress. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:3201–3206. doi: 10.1523/JNEUROSCI.2670-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giustino TF, Maren S. The Role of the Medial Prefrontal Cortex in the Conditioning and Extinction of Fear. Frontiers in behavioral neuroscience. 2015;9:298. doi: 10.3389/fnbeh.2015.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sotres-Bayon F, Quirk GJ. Prefrontal control of fear: more than just extinction. Current opinion in neurobiology. 2010;20:231–235. doi: 10.1016/j.conb.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Südhof TC. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell. 2017;171:745–769. doi: 10.1016/j.cell.2017.10.024. This comprehensive review on neurexin (NRXN) complexes summarizes the current knowledge on structure and functions of the NRXN-NLGN complex involved in the trans-synaptic signaling network that controls synapse properties. It also describes mutations found in the related genes linked to neuropsychiatric diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poulopoulos A, Soykan T, Tuffy LP, Hammer M, Varoqueaux F, Brose N. Homodimerization and isoform-specific heterodimerization of neuroligins. The Biochemical journal. 2012;446:321–330. doi: 10.1042/BJ20120808. [DOI] [PubMed] [Google Scholar]
- 34.Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35**.Chanda S, Hale WD, Zhang B, Wernig M, Südhof TC. Unique versus Redundant Functions of Neuroligin Genes in Shaping Excitatory and Inhibitory Synapse Properties. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2017;37:6816–6836. doi: 10.1523/JNEUROSCI.0125-17.2017. This is the first systematic functional analysis of neuroligin isoforms (NLGN1, NLNG2 and NLGN3), based on imaging and electrophysiology approaches in cultured mouse hippocampal and cortical neurons. Using conditional genetic deletion of the NLGNs, individually or in combination, the authors show that genetic deletion caused small or no change in synapse number, but resulted in significant deficits in synaptic function. As these impairments were isoform-specific, these data suggest that NLGNs are not functionally redundant. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang M, Polepalli J, Chen LY, Zhang B, Südhof TC, Malenka RC. Conditional ablation of neuroligin-1 in CA1 pyramidal neurons blocks LTP by a cell-autonomous NMDA receptor-independent mechanism. Molecular psychiatry. 2017;22:375–383. doi: 10.1038/mp.2016.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding X, Liu S, Tian M, Zhang W, Zhu T, Li D, Wu J, Deng H, Jia Y, Xie W, et al. Activity-induced histone modifications govern Neurexin-1 mRNA splicing and memory preservation. Nature Neuroscience. 2017;20:690–699. doi: 10.1038/nn.4536. [DOI] [PubMed] [Google Scholar]
- 38.Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakanishi M, Nomura J, Ji X, Tamada K, Arai T, Takahashi E, Bucan M, Takumi T. Functional significance of rare neuroligin 1 variants found in autism. PLoS genetics. 2017;13:e1006940. doi: 10.1371/journal.pgen.1006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tristán-Clavijo E, Camacho-Garcia RJ, Robles-Lanuza E, Ruiz A, van der Zee J, Van Broeckhoven C, Hernandez I, Martinez-Mir A, Scholl FG. A truncating mutation in Alzheimer's disease inactivates neuroligin-1 synaptic function. Neurobiology of aging. 2015;36:3171–3175. doi: 10.1016/j.neurobiolaging.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 41**.Kilaru V, Iyer SV, Almli LM, Stevens JS, Lori A, Jovanovic T, Ely TD, Bradley B, Binder EB, Koen N, et al. Genome-wide gene-based analysis suggests an association between Neuroligin 1 (NLGN1) and post-traumatic stress disorder. Translational psychiatry. 2016;6:e820. doi: 10.1038/tp.2016.69. Applying gene based association testing to data from the Grady Trauma Project, the authors identified single nucleotide polymorphisms in NLGN1 as a risk factor for PTSD. The authors correlate risk alleles in NLGN1 with increased startle response as well as with increased neural activity in limbic and prefrontal brain regions in response to threat (fearful vs. neutral face stimuli). This study supports a contribution of NLGN1 to the neurobiological underpinning of PTSD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, Bolliger MF, Südhof TC, Powell CM. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:2115–2129. doi: 10.1523/JNEUROSCI.4517-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dahlhaus R, Hines RM, Eadie BD, Kannangara TS, Hines DJ, Brown CE, Christie BR, El-Husseini A. Overexpression of the cell adhesion protein neuroligin-1 induces learning deficits and impairs synaptic plasticity by altering the ratio of excitation to inhibition in the hippocampus. Hippocampus. 2010;20:305–322. doi: 10.1002/hipo.20630. [DOI] [PubMed] [Google Scholar]
- 44.Kim J, Jung SY, Lee YK, Park S, Choi JS, Lee CJ, Kim HS, Choi YB, Scheiffele P, Bailey CH, et al. Neuroligin-1 is required for normal expression of LTP and associative fear memory in the amygdala of adult animals. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:9087–9092. doi: 10.1073/pnas.0803448105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dinamarca MC, Di Luca M, Godoy JA, Inestrosa NC. The soluble extracellular fragment of neuroligin-1 targets Aβ oligomers to the postsynaptic region of excitatory synapses. Biochemical and biophysical research communications. 2015;466:66–71. doi: 10.1016/j.bbrc.2015.08.107. [DOI] [PubMed] [Google Scholar]
- 46.Brito-Moreira J, Lourenco MV, Oliveira MM, Ribeiro FC, Ledo JH, Diniz LP, Vital JFS, Magdesian MH, Melo HM, Barros-Aragão F, et al. Interaction of amyloid-β (Aβ) oligomers with neurexin 2α and neuroligin 1 mediates synapse damage and memory loss in mice. The Journal of biological chemistry. 2017;292:7327–7337. doi: 10.1074/jbc.M116.761189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bie B, Wu J, Yang H, Xu JJ, Brown DL, Naguib M. Epigenetic suppression of neuroligin 1 underlies amyloid-induced memory deficiency. Nature Neuroscience. 2014;17:223–231. doi: 10.1038/nn.3618. [DOI] [PubMed] [Google Scholar]
- 48.Parente DJ, Garriga C, Baskin B, Douglas G, Cho MT, Araujo GC, Shinawi M. Neuroligin 2 nonsense variant associated with anxiety, autism, intellectual disability, hyperphagia, and obesity. American journal of medical genetics Part A. 2017;173:213–216. doi: 10.1002/ajmg.a.37977. [DOI] [PubMed] [Google Scholar]
- 49.Sun C, Cheng MC, Qin R, Liao DL, Chen TT, Koong FJ, Chen G, Chen CH. Identification and functional characterization of rare mutations of the neuroligin-2 gene (NLGN2) associated with schizophrenia. Human Molecular Genetics. 2011;20:3042–3051. doi: 10.1093/hmg/ddr208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wöhr M, Silverman JL, Scattoni ML, Turner SM, Harris MJ, Saxena R, Crawley JN. Developmental delays and reduced pup ultrasonic vocalizations but normal sociability in mice lacking the postsynaptic cell adhesion protein neuroligin2. Behavioural Brain Research. 2013;251:50–64. doi: 10.1016/j.bbr.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hines RM, Wu L, Hines DJ, Steenland H, Mansour S, Dahlhaus R, Singaraja RR, Cao X, Sammler E, Hormuzdi SG, et al. Synaptic imbalance, stereotypies, and impaired social interactions in mice with altered neuroligin 2 expression. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:6055–6067. doi: 10.1523/JNEUROSCI.0032-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blundell J, Tabuchi K, Bolliger MF, Blaiss CA, Brose N, Liu X, Südhof TC, Powell CM. Increased anxiety-like behavior in mice lacking the inhibitory synapse cell adhesion molecule neuroligin 2. Genes, brain, and behavior. 2009;8:114–126. doi: 10.1111/j.1601-183X.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van der Kooij MA, Fantin M, Kraev I, Korshunova I, Grosse J, Zanoletti O, Guirado R, García-Mompó C, Nacher J, Stewart MG, et al. Impaired hippocampal neuroligin-2 function by chronic stress or synthetic peptide treatment is linked to social deficits and increased aggression. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2014;39:1148–1158. doi: 10.1038/npp.2013.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krzisch M, Fülling C, Jabinet L, Armida J, Gebara E, Cassé F, Habbas S, Volterra A, Hornung JP, Toni N. Synaptic Adhesion Molecules Regulate the Integration of New Granule Neurons in the Postnatal Mouse Hippocampus and their Impact on Spatial Memory. Cerebral Cortex. 2016 doi: 10.1093/cercor/bhw217. [DOI] [PubMed] [Google Scholar]
- 55**.Liang J, Xu W, Hsu YT, Yee AX, Chen L, Südhof TC. Conditional neuroligin-2 knockout in adult medial prefrontal cortex links chronic changes in synaptic inhibition to cognitive impairments. Molecular psychiatry. 2015;20:850–859. doi: 10.1038/mp.2015.31. Using viral-mediated conditional knockout of NLGN2 in the medial prefrontal cortex of adult mice, the authors show that NLGN2 is important for social interaction, fear learning, and expression of anxiety-like phenotypes. These data highlight the important role of inhibitory synapses in the medial prefrontal cortex for cognitive functions and anxiety. [DOI] [PubMed] [Google Scholar]
- 56.Durieux AMS, Horder J, Petrinovic MM. Neuroligin-2 and the tightrope of excitation/inhibition balance in the prefrontal cortex. Journal of neurophysiology. 2016;115:5–7. doi: 10.1152/jn.00703.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tzanoulinou S, García-Mompó C, Riccio O, Grosse J, Zanoletti O, Dedousis P, Nacher J, Sandi C. Neuroligin-2 Expression in the Prefrontal Cortex is Involved in Attention Deficits Induced by Peripubertal Stress. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2016;41:751–761. doi: 10.1038/npp.2015.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maćkowiak M, Mordalska P, Wędzony K. Neuroligins, synapse balance and neuropsychiatric disorders. Pharmacological reports : PR. 2014;66:830–835. doi: 10.1016/j.pharep.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 59.Inda MC, Muravieva EV, Alberini CM. Memory retrieval and the passage of time: from reconsolidation and strengthening to extinction. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:1635–1643. doi: 10.1523/JNEUROSCI.4736-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Connor SA, Ammendrup-Johnsen I, Chan AW, Kishimoto Y, Murayama C, Kurihara N, Tada A, Ge Y, Lu H, Yan R, et al. Altered Cortical Dynamics and Cognitive Function upon Haploinsufficiency of the Autism-Linked Excitatory Synaptic Suppressor MDGA2. Neuron. 2016;91:1052–1068. doi: 10.1016/j.neuron.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 61.Elegheert J, Cvetkovska V, Clayton AJ, Heroven C, Vennekens KM, Smukowski SN, Regan MC, Jia W, Smith AC, Furukawa H, et al. Structural Mechanism for Modulation of Synaptic Neuroligin-Neurexin Signaling by MDGA Proteins. Neuron. 2017;95:896–913.e810. doi: 10.1016/j.neuron.2017.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thoumine O, Marchot P. A Triad of Crystals Sheds Light on MDGA Interference with Neuroligation. Neuron. 2017;95:729–732. doi: 10.1016/j.neuron.2017.08.001. [DOI] [PubMed] [Google Scholar]