

Abstract
Carcinomas contain tight junctions that can limit the penetration and therefore therapeutic efficacy of anticancer agents, especially those delivered by nano-carrier systems. The junction opener (JO) protein is a virus-derived protein that can transiently open intercellular junctions in epithelial tumors by cleaving the junction protein desmoglein-2 (DSG2). Co-administration of JO was previously shown to significantly increase the efficacy of various monoclonal antibodies and chemotherapy drugs in murine tumor models by allowing for increased intratumoral penetration of the drugs. To investigate the size-dependent effect of JO on nanocarriers, we used PEGylated gold nanoparticles (AuNPs) of two different sizes as model drugs and investigated their biodistribution following JO protein treatment. By inductively coupled plasma mass spectrometry (ICP-MS), JO was found to significantly increase bulk tumor accumulation of AuNPs of 35 nm but not 120 nm particles in both medium (200–300 mm3) and large (500–600 mm3) tumors. Image analysis of tumor sections corroborates this JO-mediated increase in tumor accumulation of AuNPs. Quantitative intratumoral distribution analyses show that most nanoparticles were found within 100 μm of the vasculature, and that the penetration profiles of AuNPs are not significantly affected by JO treatment at the 6 h timepoint.
Keywords: gold nanoparticles, tumor penetration, junction opening, image analysis
Graphical abstract
1. Introduction
Nanoparticles have the potential to impact cancer drug delivery by facilitating delivery to solid tumor sites while reducing drug distribution to other tissues, thus improving safety profiles.[1] Several recent studies have demonstrated the importance of small nanoparticle size in both tumor distribution and retention.[2–4] However, ultrasmall nanoparticles also have smaller drug loading capacities. Additionally, tumor accumulation and penetration remain significant challenges, especially because permeation into tumors is highly dependent on nanoparticle size and surface chemistry, and the incomplete distribution of therapeutics throughout the tumor tissue can lead to drug resistance.[5–7] In particular, carcinomas are characterized by the presence of intercellular tight junctions which restrict the paracellular transport of molecules, a primary interstitial transport mechanism for nanoparticles in solid tumors.[8,9] Thus, even after extravasation through the tumor vascular endothelium, nano-sized drug carriers encounter the additional penetration barrier of tight junctions. One such epithelial junction protein is desmoglein-2 (DSG2), which has been found to be upregulated in a number of malignant cell types including gastric cancer, [10] ovarian cancer, [11] and breast cancer.[12]
Recently, we identified DSG2 as the receptor used by several human adenoviruses (Ad), including Ad serotype 3, for cellular infection.[11] In subsequent studies, a recombinant protein derived from the Ad3 fiber knob was produced.[13] This protein, named junction opener-1 (JO-1), triggers the transient opening of intercellular junctions through binding and cleavage of the DSG2 dimers between epithelial cells.[13,14] This effect was observed in mice with epithelial tumors within one hour of intravenous (i.v.) injection of JO-1.[14] Co-administration of JO-1 facilitates intratumoral penetration and therapeutic efficacy of monoclonal antibodies (mAbs) such as the anti-Her2/neu mAb trastuzumab (Herceptin) and the EGFR inhibitor cetuximab (Erbitux).[14] Furthermore, JO-1 was tested in combination with several chemotherapeutic drugs including paclitaxel (Taxol), irinotecan (Camptosar), nanoparticle albumin-bound paclitaxel (Abraxane), and liposomal doxorubicin (Doxil). JO-1 co-therapy enhanced the efficacy of these drugs and overcame drug resistance in several models while reducing the drug doses necessary for therapeutic effect.[12] JO-1 and variants of this protein (such as the affinity-enhanced version, JO-4[15]) are therefore interesting for clinical application.
To evaluate the potential for JO protein-facilitated delivery of nano-sized carriers, we sought to investigate the effect of JO pretreatment on the in vivo biodistribution of gold nanoparticles of different sizes (Figure 1). Gold nanoparticles were selected as a surrogate for solid nano-sized drug carriers because they can be synthesized with defined sizes over a wide size range and surface-modified through reactions with thiol groups.[16] Additionally, gold nanoparticles can be quantified by inductively coupled plasma mass spectrometry (ICP-MS) with excellent sensitivity, as well as visualized by light microscopy after silver enhancement to determine tissue distribution.[5,17,18] Herein, we evaluate the impact of concurrent JO treatment on both the biodistribution and tumor distribution of polyethylene glycol (PEG)-modified gold nanoparticles in tumor-bearing mice. For the latter, we report a technique to investigate the intratumoral distribution of nanoparticles using microscopy and quantitative image analysis.
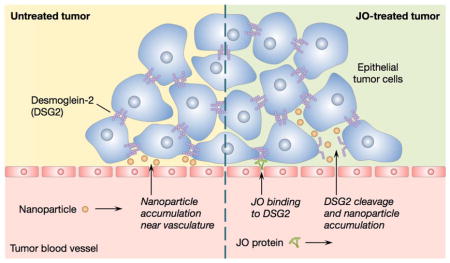
Figure 1.

Schematic illustrations of nanoparticle accumulation in untreated and JO-treated tumors. (a) Perivascular nanoparticle accumulation in an untreated tumor due to the presence of tight junctions. (b) Proposed mechanism of JO binding and cleaving DSG2 and subsequent nanoparticle accumulation beyond the vasculature.
2. Materials and Methods
2.1. AuNP Surface Modification
AuNPs with diameters of 5 and 100 nm were purchased from Ted Pella (Redding, CA) and Nanopartz (Loveland, CO), respectively (actual diameters: 5.5 and 103 nm as determined by the manufacturers). AuNPs were surface-modified with PEG5000-thiol (Laysan Bio, Arab, AL) following the known reaction of thiols with gold; PEG-SH was added in excess (4 PEG molecules per nm2 gold surface area following our previously reported protocol[17], assuming spherical particles) and allowed to react for 30 min at room temperature prior to characterization or use.
2.2. Nanoparticle Characterization
The effective diameters of AuNPs were measured by dynamic light scattering (DLS) with a ZetaPlus analyzer (Brookhaven Instruments, Holtsville, NY) at a detection angle of 90°. Unmodified or PEGylated AuNPs with initial diameters of 5 or 100 nm were first measured in nanopure water. Particle size measurements were acquired for 6 independent samples, using 5 1-minute runs for each sample.
To confirm particle stability in the presence of physiological salt concentrations, unmodified or PEGylated AuNPs were diluted with an equal volume of 2× PBS (or nanopure water as a negative control) immediately before sizing. Size was measured again after 2 h and 24 h; measurements were completed using 3 independent samples. Similarly, particle stability in serum was assessed by diluting AuNPs with an equal volume of 2× PBS containing 20% fetal bovine serum (FBS), for a final serum concentration of 10%. Serum stability was monitored by DLS or by measuring absorbance spectra for the samples from 400–800 nm using a Tecan Safire2 plate reader (Männedorf, Switzerland).
Doxil (0.2 mg/mL) and 120 nm PEGylated AuNPs were injected through a 20 nm filter membrane using a syringe pump with a flow rate of 0.1 mL/min. The filtered solution was collected and absorbance was measured at wavelengths of 498 nm and 560 nm for Doxil and PEGylated AuNPs, respectively. The recovery efficiency was calculated by normalizing the absorbance of the filtered solution to that of the initial solution.
2.3. Animals
Scid-beige (CB17) mice (8–10 weeks) were obtained from The Jackson Laboratory (strain name: NOD.CB17-Prkdcscid298/J). All experimental procedures were performed in accordance with protocols approved by the University of Washington Institutional Animal Care and Use Committee.
2.4. Cell Culture
A549 human lung epithelial cells (ATCC CCL-185) were maintained in F-12K medium (Corning cellgro) supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were cultured as a monolayer in a 37°C, 5% CO2 environment. Medium was replaced every 2–3 days. Cells were passaged at ~70–80% confluence by incubation with Trypsin-EDTA, followed by resuspension in complete growth medium.
2.5. Protein Production
JO-4 was produced in E. coli with an N-terminal 6-His tag using the pQE30 expression vector (Qiagen, Valencia, CA) and purified by Ni-NTA agarose chromatography. JO-4 preparations used in animals were depleted of bacterial endotoxin using EndoTrap blue 1/1 columns (Hyglos GmbH, Bernried, Germany).
2.6. Biodistribution of AuNPs
To develop xenograft tumors, mice were inoculated subcutaneously in the right flank with 5×106 A549 cells in 100 μL of F-12K medium without serum. Biodistribution studies were initiated when the tumors reached the specified volumes (200–300 mm3 or 500–600 mm3). For each tumor volume, mice were randomly distributed into four groups of 5–6 mice each (n = 5 per group, total of 20 mice for smaller tumors; n = 6 per group, total of 24 mice for larger tumors). Mice received JO-4 pretreatment or no pretreatment, followed by administration of either 35 or 120 nm PEGylated AuNPs.
Mice receiving JO-4 pretreatment were first injected with 2 mg JO-4 protein/kg mouse in PBS via tail vein injection. One hour later, mice were injected with 35 or 120 nm PEGylated AuNPs in PBS at a dose of 100 μg gold/kg mouse via tail vein injection. After 6 hours, mice were anesthesized by intraperitoneal injection with 2.5% Avertin solution (300 μL/20 g mouse). Mice were then perfused with PBS, and tumors and organs were harvested.
Gold content in tissue samples was measured by ICP-MS at the Environmental Health Laboratory & Trace Organics Analysis Center at the University of Washington. Briefly, tissue samples were pre-digested in concentrated nitric acid and concentrated hydrochloric acid (Fisher Trace Metal Grade, in a 1:0.88 volume ratio) overnight at room temperature. After overnight pre-digestion, samples were vortexed, centrifuged at 2000 rpm for 5 min, and microwaved using a CEM MARS microwave-assisted digestion oven (Matthews, NC) until fully digested. Samples were diluted with nanopure water to a final concentration of 12.5% HNO3 and 11% HCl and centrifuged to remove cell debris and fatty deposits. The supernatants were analyzed for gold using an Agilent 7500CE ICP-MS (Santa Clara, CA).
Gold content in the tumor and liver was analyzed for all mice. Gold content in the brain, colon, heart, intestine, kidney, lung, and spleen was analyzed for a subset of 3 mice per group, selecting for mice with tumor weights closest to the average overall tumor weight (~240 mg). The gold content of each sample was normalized to sample mass. Statistical significance was assessed using a Student’s two-tailed t-test to compare the mean gold content of each organ with and without JO-4 pretreatment.
2.7. Light and Fluorescence Microscopy
For imaging studies, JO-4 and AuNP injections were completed as described above, with the tumors harvested following perfusion. Tumors were embedded in optimal cutting temperature (OCT) compound (Tissue-Tek, Sakura Finetek USA, Torrance, CA) in cryomolds, flash frozen, and cryosectioned into 8 μm-thick sections. Tumor sections were post-fixed in 4% paraformaldehyde (PFA) in PBS for 15 min at room temperature and stained for blood vessels with rat anti-mouse CD31 antibody (clone MEC 13.3, BD Pharmingen) and Alexa Fluor 488-conjugated donkey anti-rat IgG secondary antibody (Jackson ImmunoResearch). After immunofluorescence staining, sections were stained for AuNPs by incubating with silver enhancement solution (Ted Pella) for 20 min at room temperature. Finally, sections were washed, coverslipped using Fluoromount-G (eBioscience), and imaged on a Nikon E800 upright microscope with a 60x objective.
2.8. Image Analysis
A total of 11 image pairs (brightfield for AuNPs, fluorescence for CD31) were obtained of each tumor section. Imaging fields were selected such that at least one CD31-stained blood vessel was visible in order to facilitate subsequent analysis. Images were thresholded using Fiji image processing software. MATLAB was then used to overlay the thresholded brightfield and fluorescence images, perform particle detection, and calculate the penetration distance for each AuNP, defined as the distance from the particle to the nearest CD31-stained pixel. Distance data were aggregated from all images of a tumor section and binned in MATLAB. Sample MATLAB code can be found in the supplementary information.
3. Results and Discussion
3.1. Gold Nanoparticle Sizing and Stability
Gold nanoparticles (AuNPs) of 2 different diameters, 5 and 100 nm, were surface-modified by reaction with PEG5000-thiol as reported previously;[17] PEGylation of nanoparticles is commonly employed to reduce aggregation and protein adsorption, thereby increasing circulation half-life in vivo. Particle sizing by dynamic light scattering (DLS) was determined for AuNPs before and after PEGylation (Table 1). The final diameters of the PEGylated AuNPs were measured to be 33.2 and 121.1 nm, demonstrating a moderate size increase after PEG modification consistent with previous reports.[19,20] These particle sizes were selected based on the sizes of other nanocarriers that have been clinically approved or are in late-stage clinical trials, such as Genexol-PM (~24 nm)[21] and similar polymeric micelles (20–40 nm)[22] at the lower size range and Doxil (80–100 nm)[23] and BIND-014 (~100 nm)[24] at the higher range.
Table 1.
Particle sizing of unmodified and PEGylated AuNPs and Doxil by DLS.
| Formulation | Effective diameter (nm)a | |
|---|---|---|
|
| ||
| Before PEGylation | After PEGylation | |
| 5 nm AuNP core + PEG-SH | 10.8b ± 1.2 | 33.2 ± 1.4 |
| 100 nm AuNP core + PEG-SH | 103.9 ± 1.7 | 121.1 ± 1.2 |
| Doxil | N/A | 82.1 ± 0.31 |
Data are reported as the mean ± S.D., n = 6.
Particle size as determined by DLS; reported as 5.5 nm by manufacturer using a combination of DLS, TEM, and UV-Vis spectroscopy.
To confirm that the nanoparticles can avoid particle-particle aggregation under physiological conditions, nanoparticle size in salt and serum was determined. In the presence of physiological salt concentrations, unmodified AuNPs were shown to aggregate rapidly (Figure 2a and Figure 2b), whereas PEGylated AuNPs remained stable in PBS even after 24 h. Serum stability was also assessed by suspending particles in PBS containing 10% FBS. The addition of serum conferred increased stability on unmodified AuNPs, consistent with published reports.[17,25] Particle sizing indicated that unmodified 100 nm AuNPs did not form large aggregates over 24 h in serum conditions, although a slight increase in effective diameter suggests that some aggregation behavior or protein adsorption to the nanoparticles may occur (Figure 2a). No change in particle size was observed for PEGylated 100 nm AuNPs incubated in serum.
Figure 2.

Stability of unmodified and PEGylated 100 nm (a) and 5 nm (b, c) AuNPs incubated in PBS or PBS containing 10% serum as monitored by DLS (a, b) and red shift in absorbance (c). Data are reported as the mean ± S.D., n = 3.
Due to the low count rate of the smaller AuNPs on DLS and the presence of large protein aggregates in serum solutions, the serum stability of 5 nm AuNPs was confirmed by an alternate method. Because aggregation of AuNPs results in a red shift in their absorbance spectra, serum stability was assessed by monitoring absorbance over the 400–800 nm range.[17] Similar to the larger particles, PEGylated 5 nm AuNPs in serum displayed stable absorbance spectra over 24 h (Figure 2c). Significant aggregation behavior was observed only for unmodified 5 nm AuNPs incubated in PBS, as indicated by a noticeable shift in this spectrum toward longer wavelengths, corroborating the results obtained by DLS (Figure 2b). Overall, these results confirm that PEG coating yields AuNPs that are sterically stabilized in the presence of salt and serum.
3.2. Biodistribution of AuNPs with JO-4 Pretreatment
PEGylated AuNPs are referred to by their hydrodynamic sizes after PEG modification, 35 nm and 120 nm. The A549 lung cancer model was used to assess the in vivo biodistribution of nanoparticles administered alone or in conjunction with JO-4 protein pretreatment; A549 cells are known to overexpress DSG2, [26] and previous work indicates that the JO protein can potentiate chemotherapy in this model.[12,14] JO-4 pretreatment was used since nanoparticles are known to be cleared rapidly from the circulation and since JO pretreatment has been shown to increase tumor accumulation of antibody administered one hour later.[14] Studies were conducted in mice bearing two different sizes of tumors (tumor volumes of 200–300 mm3 or 500–600 mm3) to represent changes in extracellular matrix and tight junction development with tumor progression.[27,28]
AuNP biodistribution was initially assessed in the tumor and the liver (the primary site of nanoparticle clearance).[29,30] Biodistribution results indicated that JO-4 pretreatment significantly increased tumor accumulation of the smaller 35 nm AuNPs, from 2.2% to 4.7% of the injected dose (ID) of Au per g tissue for the 200–300 mm3 tumors and from 2.9% to 3.7% ID per g tissue for the 500–600 mm3 tumors (Figure 3). This result confirms prior reports of increased efficacy of irinotecan (586.7 Da), [12] the antibody cetuximab, [14] and Doxil (~90 nm)[12] with JO-1 co-therapy in the A549 tumor model. Interestingly, although the increase in tumor accumulation of 35 nm AuNPs was observed with both tumor volumes, the magnitude of the JO-mediated increase was less pronounced for the larger 500–600 mm3 tumors. This may be attributed in part to the fact that the larger tumors demonstrated greater AuNP accumulation even without pretreatment (gold content of 2.2% vs. 2.9% ID/g for untreated smaller and larger tumors, respectively; p < 0.05). This is in good agreement with recent work by Chan and coworkers demonstrating that AuNP accumulation generally increases with tumor volume due to the higher porosity and decreased rigidity of the extracellular matrix in larger tumors, and therefore diffusional barriers may be more compromised in the 500–600 mm3 tumors compared to the 200–300 mm3 tumors.[28]
Figure 3.

Quantitative AuNP biodistribution by ICP-MS. (a, b) Tumor and liver accumulation of two different sizes of AuNPs 6 h post-NP injection in control or JO-4 pretreated mice bearing 200–300 mm3 (a) or 500–600 mm3 (b) tumors. Data are reported as the mean ± S.D. 200–300 mm3: n = 5; 500–600 mm3: n = 6. (c) Biodistribution of two different sizes of AuNPs 6 h post-NP injection in control or JO-4 pretreated mice bearing 500–600 mm3 tumors. Data are reported as the mean ± S.D., n = 3. *p < 0.05
Meanwhile, JO-4 pretreatment did not significantly affect tumor accumulation of 120 nm AuNPs regardless of tumor size (Figure 3 and Figure S1). One possible explanation is that the primary barrier for the larger 120 nm particles is the extracellular matrix (ECM). While the tumor ECM has been reported to be poorly organized compared to normal ECM, the spacing has been observed to be around 75–130 nm.[31]
Furthermore, given the previous success of this strategy for enhancing delivery of Doxil, differences in particle rigidity are also likely to influence JO-facilitated transport through epithelial junctions, as Doxil, unlike rigid AuNPs, consists of a deformable lipid bilayer. To test this hypothesis, we applied Doxil and 120 nm AuNPs to a 20 nm filter (to mimic transport through a porous environment) under a 0.1 mL/min flow rate and monitored recovery of the two types of nanoparticles by absorbance measurements. Nearly half (47%) of Doxil particles were recovered while only 6.6% of 120 nm AuNPs were recovered after filtration. These results further support the possibility that Doxil can transit more easily through epithelial junctions and tumor ECM compared to the rigid 120 nm AuNPs.
Surprisingly, a significant increase in liver accumulation of 35 nm AuNPs was also observed for the JO-4 pretreated group with smaller tumors (Figure 3a), although DSG2 is not broadly accessible in the liver (DSG2 is not found on hepatocytes but is detectable in the intrahepatic bile duct epithelium).[32] This trend was not present in the mice bearing larger tumors, which demonstrate higher liver accumulation of 35 nm AuNPs even in the absence of JO-4 (Figure 3b).
To establish a complete picture of AuNP biodistribution, gold content in the colon, heart, intestine, kidney, lung, brain, and spleen was analyzed for mice bearing 500–600 mm3 tumors. JO-4 pretreatment had no significant effect on AuNP accumulation in any of the organs examined (Figure 3c), suggesting that the protein acts specifically on the DSG2-expressing tumor cells to facilitate delivery to the tumor. Overall, 120 nm AuNPs accumulated more in the liver and spleen compared to the 35 nm AuNPs. This phenomenon, attributed to rapid phagocytosis of larger sized nanoparticles by resident macrophages, has been previously reported in the literature.[17,33,34]
3.3. Intratumoral Penetration of AuNPs
Because ICP-MS provides only a bulk measurement of gold content in tissue and not spatial information, we developed an imaging technique to enable quantification of AuNP penetration into tumors. Tumors from mice injected with 35 nm AuNPs were selected for this detailed image analysis because biodistribution results from ICP-MS showed that they were significantly affected by JO-4 pretreatment. AuNPs and CD31 (a marker for blood vessels) were imaged by brightfield microscopy and fluorescence microscopy, respectively, and images were analyzed to determine the penetration distance for each AuNP, defined as the distance from the particle’s centroid to the nearest CD31-stained pixel (Figure 4). AuNP counts and penetration distances were measured in this way for multiple fields of view of each tumor section (Figure 5). A total of 6 or 7 distinct sections pooled from 3 mice was analyzed for each treatment group (1–3 sections from each tumor).
Figure 4.

Representative images of a tumor section and image analysis strategy. (a) 35 nm AuNPs (black) were visualized by silver enhancement and brightfield microscopy. (b) Blood vessels were stained with anti-CD31 antibody and fluorescent secondary antibody (green) and imaged by fluorescence microscopy for the same field of view. Images are shown before (a, b) and after (c, d) thresholding. (e) Composite image showing relative locations of AuNPs (cyan) and blood vessels (green) in a tumor section, with red lines indicating the shortest distance from each AuNP to a blood vessel.
Figure 5.

Quantitative image analysis of 35 nm AuNPs in the tumors of control or JO-4 pretreated mice. (a) Number of AuNPs detected per image. (b, c) Boxplots of the fraction (b) and total number (c) of AuNPs penetrating beyond given threshold distances from a blood vessel. ● indicates the mean of the data. n = 6 or 7 tumor sections pooled from 3 mice, data aggregated from 11 images per section. Statistical significance was assessed using a nonparametric (assuming nonnormal distributions) two-tailed Mann-Whitney test for (a) and two-tailed t-tests for (b) and (c). **p < 0.01, ***p < 0.001, ****p < 0.0001
Images of JO-4 treated tumors indicate that more than 5 times as many AuNPs were detected in the areas around blood vessels as compared to untreated tumors (Figure 5a). An analysis of AuNP penetration depth was performed by determining the fraction of particles found at varying distances from a blood vessel. Our previous work demonstrates that JO protein penetrates murine orthotopic breast tumors effectively within one hour after delivery.[14] JO-4 pretreatment was associated with a slight increase in penetration distance (higher fractions of AuNPs penetrating beyond the threshold for all threshold distances), although these differences were not statistically significant (Figure 5b). However, because the JO-4 treated tumors contained substantially more AuNPs, there were significant differences in the total numbers of AuNPs penetrating into the tumor (Figure 5c). Overall, nearly all AuNPs were within 100 μm of a blood vessel, a distance of approximately 6–7 cell layers based on a diameter of ~15 μm for an A549 cancer cell.[35] Of note, the increase in accumulation of AuNPs due to JO-4 appears more pronounced in imaging than in ICP-MS. The image analysis technique relies on the presence of at least one blood vessel per image in order to calculate a particle’s penetration distance; therefore, this data is inherently localized to vascularized regions of the tumor that likely have a higher density of nanoparticles. In comparison, ICP-MS provides a summary measurement of a heterogeneously perfused tumor.
Taken together, quantitative biodistribution data from ICP-MS and microscopy indicate that JO-4 pretreatment increases tumor accumulation of 35 nm AuNPs. At this 6 hour timepoint of analysis, intratumoral penetration is not significantly impacted by JO-4 treatment. One possible explanation for this finding is that the high-affinity interaction between JO-4 and its binding partner DSG-2 may restrict distribution of the JO-4 protein into the tumor. Models suggest that targeting ligands with high binding affinity can actually bind so strongly as to prevent effective intratumoral penetration, a phenomenon known as the “binding site barrier.”[36] Indeed, one study of transferrin (Tf)-containing AuNPs targeted to the brain indicated that nanoparticles with intermediate avidity were able to transcytose across the blood-brain barrier into the brain parenchyma, while high-avidity nanoparticles bound to brain endothelial cells but were unable to enter the brain parenchyma.[18] However, JO protein affinity has previously been shown to correlate with therapeutic effect in vivo, with JO-4 (KD, 11.4 nM) significantly enhancing irinotecan therapy as compared to either JO-2 (24.9 nM) or JO-1 (10100 nM).[37] Thus, it is likely not the case that the high binding affinity of JO-4 prevents nanoparticle penetration to greater depths within the tumor.
These findings have important implications for the clinical application of JO-4 and other epithelial junction-opening proteins. Firstly, because both junction opening and macromolecule penetration are dynamic processes, evaluation at later timepoints may reveal greater differences in nanoparticle penetration. In addition, intratumoral (i.t.) injection of the JO protein may increase protein distribution and consequently AuNP penetration distance. A previous study found that i.v. injection of JO-1 improved cetuximab therapy, but combining this with an additional i.t. injection of JO-1 did not further increase therapeutic efficacy, [14] although intratumoral drug distribution was not investigated. Ultimately, the JO-4-mediated increase in overall tumor localization may be sufficient for some chemotherapeutics such as high-affinity antibodies or liposomal formulations (for which free drug is able to diffuse out of the liposome); additional studies are needed to evaluate penetration effects at later timepoints.
4. Conclusions
The limited penetration of small molecule drugs or nanoparticulate drug carriers into solid tumors poses a critical barrier for chemotherapy efficacy, as exposure to subtoxic drug concentrations can give rise to resistance in distal tumor cells. In this work, JO-4 was found to significantly increase tumor delivery of 35 nm AuNPs in two different sizes of xenograft tumors. A trend toward increased tumor accumulation of 120 nm AuNPs after JO-4 pretreatment was also observed in mice with smaller 200–300 mm3 tumors, although this difference was not statistically significant. Importantly, junction opening appeared to be specific to tumor tissue without off-target effects in other organs. In addition, image analysis methods were developed to quantify nanoparticle penetration in tumor sections; this analysis indicated that JO treatment does not significantly impact the penetration distance of AuNPs from tumor blood vessels. The methods described here can also be applied to study a variety of tumor and animal models or evaluate other strategies for altering drug penetration into solid tumors. Overall, a mechanistic understanding of the size limitations of junction opening in vivo could elucidate design criteria for novel drug carriers that best exploit this delivery mechanism.
Supplementary Material
Acknowledgments
This work was supported by NIH 1R01CA177272, NSF DMR 1206426, a solid tumor translational research grant from the Fred Hutchinson Cancer Research Center, and a National Science Foundation Graduate Research Fellowship to C.E.W. ICP-MS analysis was conducted at the Environmental Health Laboratory & Trace Organics Analysis Center at the University of Washington with the assistance of Dr. Russell Dills, Dr. Susan Tao, and Rebecca Christ. The authors acknowledge the University of Washington Engineered Biomaterials (UWEB) Research Center and Dr. Mei Speer for assistance with microscopy.
Footnotes
AuNP biodistribution data, MATLAB code for image analysis (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflict of Interest Statement
AL is a co-founder of Compliment Corp, a start-up company that has licensed the JO technology.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 2.Huang K, Ma H, Liu J, Huo S, Kumar A, Wei T, et al. Size-dependent localization and penetration of ultrasmall gold nanoparticles in cancer cells, multicellular spheroids, and tumors in vivo. ACS Nano. 2012;6:4483–4493. doi: 10.1021/nn301282m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang L, Gabrielson NP, Uckun FM, Fan TM, Cheng J. Size-dependent tumor penetration and in vivo efficacy of monodisperse drug-silica nanoconjugates. Mol Pharm. 2013;10:883–892. doi: 10.1021/mp300684a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang L, Yang X, Yin Q, Cai K, Wang H, Chaudhury I, et al. Investigating the optimal size of anticancer nanomedicine. Proc Natl Acad Sci USa. 2014;111:15344–15349. doi: 10.1073/pnas.1411499111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WCW. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009;9:1909–1915. doi: 10.1021/nl900031y. [DOI] [PubMed] [Google Scholar]
- 6.Tannock IF, Lee CM, Tunggal JK, Cowan DSM, Egorin MJ. Limited penetration of anticancer drugs through tumor tissue: a potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res. 2002;8:878–884. [PubMed] [Google Scholar]
- 7.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 8.Christiansen J, Rajasekaran AK. Biological impediments to monoclonal antibody-based cancer immunotherapy. Mol Cancer Ther. 2004;3:1493–1501. [PubMed] [Google Scholar]
- 9.Green SK, Karlsson MCI, Ravetch JV, Kerbel RS. Disruption of cell-cell adhesion enhances antibody-dependent cellular cytotoxicity: implications for antibody-based therapeutics of cancer. Cancer Res. 2002;62:6891–6900. [PubMed] [Google Scholar]
- 10.Biedermann K, Vogelsang H, Becker I, Plaschke S, Siewert JR, Höfler H, et al. Desmoglein 2 is expressed abnormally rather than mutated in familial and sporadic gastric cancer. J Pathol. 2005;207:199–206. doi: 10.1002/path.1821. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Li Z-Y, Liu Y, Persson J, Beyer I, Möller T, et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat Med. 2011;17:96–104. doi: 10.1038/nm.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beyer I, Cao H, Persson J, Song H, Richter M, Feng Q, et al. Coadministration of epithelial junction opener JO-1 improves the efficacy and safety of chemotherapeutic drugs. Clin Cancer Res. 2012;18:3340–3351. doi: 10.1158/1078-0432.CCR-11-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Li Z, Yumul R, Lara S, Hemminki A, Fender P, et al. Multimerization of adenovirus serotype 3 fiber knob domains is required for efficient binding of virus to desmoglein 2 and subsequent opening of epithelial junctions. J Virol. 2011;85:6390–6402. doi: 10.1128/JVI.00514-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beyer I, van Rensburg R, Strauss R, Li Z, Wang H, Persson J, et al. Epithelial junction opener JO-1 improves monoclonal antibody therapy of cancer. Cancer Res. 2011;71:7080–7090. doi: 10.1158/0008-5472.CAN-11-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richter M, Yumul R, Wang H, Saydaminova K, Ho M, May D, et al. Preclinical safety and efficacy studies with an affinity-enhanced epithelial junction opener and PEGylated liposomal doxorubicin. Mol Ther Methods Clin Dev. 2015;2:15005. doi: 10.1038/mtm.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daniel M-C, Astruc D. Gold nanoparticles: assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem Rev. 2004;104:293–346. doi: 10.1021/cr030698. [DOI] [PubMed] [Google Scholar]
- 17.Bergen JM, von Recum HA, Goodman TT, Massey AP, Pun SH. Gold nanoparticles as a versatile platform for optimizing physicochemical parameters for targeted drug delivery. Macromol Biosci. 2006;6:506–516. doi: 10.1002/mabi.200600075. [DOI] [PubMed] [Google Scholar]
- 18.Wiley DT, Webster P, Gale A, Davis ME. Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc Natl Acad Sci USa. 2013;110:8662–8667. doi: 10.1073/pnas.1307152110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosn B, van de Ven AL, Tam J, Gillenwater A, Sokolov KV, Richards-Kortum R, et al. Efficient mucosal delivery of optical contrast agents using imidazole-modified chitosan. J Biomed Opt. 2010;15:015003. doi: 10.1117/1.3309739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qian X, Peng XH, Ansari DO, Yin-Goen Q, Chen GZ, Shin DM, et al. In vivo tumor targeting and spectroscopic detection with surface-enhanced Raman nanoparticle tags. Nat Biotechnol. 2008;26:83–90. doi: 10.1038/nbt1377. [DOI] [PubMed] [Google Scholar]
- 21.Werner ME, Cummings ND, Sethi M, Wang EC, Sukumar R, Moore DT, et al. Preclinical Evaluation of Genexol-PM, a Nanoparticle Formulation of Paclitaxel, as a Novel Radiosensitizer for the Treatment of Non-Small Cell Lung Cancer. Int J Radiat Oncol Biol Phys. 2013;86:463–468. doi: 10.1016/j.ijrobp.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cabral H, Kataoka K. Progress of drug-loaded polymeric micelles into clinical studies. J Control Release. 2014;190:465–476. doi: 10.1016/j.jconrel.2014.06.042. [DOI] [PubMed] [Google Scholar]
- 23.Soundararajan A, Bao A, Phillips WT, Perez R. [186 Re] Liposomal doxorubicin (Doxil): in vitro stability, pharmacokinetics, imaging and biodistribution in a head and neck squamous cell carcinoma xenograft model. Nuclear Medicine and …. 2009 doi: 10.1016/j.nucmedbio.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Von Hoff DD, Mita MM, Ramanathan RK, Weiss GJ, Mita AC, LoRusso PM, et al. Phase I Study of PSMA-Targeted Docetaxel-Containing Nanoparticle BIND-014 in Patients with Advanced Solid Tumors. Clin Cancer Res. 2016;22:3157–3163. doi: 10.1158/1078-0432.CCR-15-2548. [DOI] [PubMed] [Google Scholar]
- 25.Brewer SH, Glomm WR, Johnson MC, Knag MK, Franzen S. Probing BSA binding to citrate-coated gold nanoparticles and surfaces. Langmuir. 2005;21:9303–9307. doi: 10.1021/la050588t. [DOI] [PubMed] [Google Scholar]
- 26.Cai F, Zhu Q, Miao Y, Shen S, Su X, Shi Y. Desmoglein-2 is overexpressed in non-small cell lung cancer tissues and its knockdown suppresses NSCLC growth by regulation of p27 and CDK2. J Cancer Res Clin Oncol. 2017;143:59–69. doi: 10.1007/s00432-016-2250-0. [DOI] [PubMed] [Google Scholar]
- 27.Pitteri SJ, Kelly-Spratt KS, Gurley KE, Kennedy J, Buson TB, Chin A, et al. Tumor microenvironment-derived proteins dominate the plasma proteome response during breast cancer induction and progression. Cancer Res. 2011;71:5090–5100. doi: 10.1158/0008-5472.CAN-11-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sykes EA, Dai Q, Sarsons CD, Chen J, Rocheleau JV, Hwang DM, et al. Tailoring nanoparticle designs to target cancer based on tumor pathophysiology. Proc Natl Acad Sci USa. 2016;113:E1142–51. doi: 10.1073/pnas.1521265113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsoi KM, MacParland SA, Ma XZ, Spetzler VN, Echeverri J, Ouyang B, et al. Mechanism of hard-nanomaterial clearance by the liver. Nature Materials. 2016;15:1212–1221. doi: 10.1038/nmat4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, di Tomaso E, et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proc Natl Acad Sci USa. 2001;98:4628–4633. doi: 10.1073/pnas.081626898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, Beyer I, Persson J, Song H, Li Z, Richter M, et al. A new human DSG2-transgenic mouse model for studying the tropism and pathology of human adenoviruses. J Virol. 2012;86:6286–6302. doi: 10.1128/JVI.00205-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho WS, Cho M, Jeong J, Choi M, Han BS, Shin HS, et al. Size-dependent tissue kinetics of PEG-coated gold nanoparticles. Toxicol Appl Pharmacol. 2010;245:116–123. doi: 10.1016/j.taap.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 34.Hirn S, Semmler-Behnke M, Schleh C, Wenk A, Lipka J, Schäffler M, et al. Particle size-dependent and surface charge-dependent biodistribution of gold nanoparticles after intravenous administration. Eur J Pharm Biopharm. 2011;77:407–416. doi: 10.1016/j.ejpb.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang R-D, Shen H, Piao Y-J. The morphometrical analysis on the ultrastructure of A549 cells. Rom J Morphol Embryol. 2010;51:663–667. [PubMed] [Google Scholar]
- 36.Fujimori K, Covell DG, Fletcher JE, Weinstein JN. Modeling analysis of the global and microscopic distribution of immunoglobulin G, F(ab′)2, and Fab in tumors. Cancer Res. 1989;49:5656–5663. [PubMed] [Google Scholar]
- 37.Wang H, Yumul R, Cao H, Ran L, Fan X, Richter M, et al. Structural and functional studies on the interaction of adenovirus fiber knobs and desmoglein 2. J Virol. 2013;87:11346–11362. doi: 10.1128/JVI.01825-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.