

Abstract
Despite the importance and promise of cancer vaccines for broader prevention and treatment of cancer, limited clinical responses are observed, suggesting that key rational designs are required for inducing potent immune responses against cancer. Here we report a mesoporous silicon vector (MSV) as a multi-functional microparticle for formulating an efficient cancer vaccine composed of B16 melanoma derived-tyrosinase related protein 2 (TRP2) peptide and dual toll-like receptor (TLR) agonists. We demonstrated that MSV microparticles protected the peptide from rapid degradation for prolonged antigen presentation to immune cells. Moreover, MSV enabled co-delivery of two different TLR agonists [CpG oligonucleotide and monophosphoryl lipid A (MPLA)] along with TRP2 peptide into the same dendritic cell (DC), thus increasing the efficiency and capacity of DCs to induce potent TRP2-specifc CD8+ T cell responses against B16 melanoma. Furthermore, this MSV-based DC vaccine could significantly prolong the median survival of tumor-bearing mice by orchestrating effective host immune responses involving CD8+ T cells, CD4+ T cells and macrophages. Our study provides rational and potentially translational approach to develop durable and potent immunotherapy for patients with cancer by delivering various combinations of tumor antigens, neoantigens and innate immune agonists.
Keywords: cancer vaccine, mesoporous silicon microparticles, melanoma immunotherapy, dual TLR signaling, TRP2 peptide
Graphical abstract
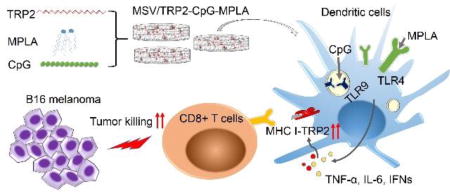
Mesoporous silicon microparticles enabled co-delivery of tumor antigen and dual toll-like receptor ligands provide rational and potentially translational approach to develop durable and potent immunotherapy against cancer.
1. Introduction
Cancer immunotherapy is considered as the most promising approach to cancer treatment, with a goal to potentiate patient’s own immune system to fight against cancer [1–9]. Despite great progresses in checkpoint blockade therapy and engineered T cell receptor (TCR) or chimeric antigen receptor (CAR) immunotherapy, not all cancer patients could benefit from these immunotherapies [2–4, 7, 10, 11]. For example, anti-PD1 or anti-PD-L1 checkpoint blockade therapy shows impressive and durable clinical response (generally 20–38% patients across different tumor types) [10, 11], but more than 60% cancer patients fail to respond to this checkpoint therapy. CD19-CAR immunotherapy shows impressive clinical response aganist blood cancers, including B cell lymphoma and leukemia, but CAR-T technology does not work well in solid tumors [2–4, 7]. Cancer vaccines utilize cancer-specific antigens to induce tumor-specific T cell responses against cancer [2]. The breadth of cancer vaccine application is projected to be largely broadened by the identification of cancer-shared antigens as well as mutation-derived neoantigens [7, 9, 10]. Therefore, cancer vaccines have great potential to benefit patients, but the efficiency of cancer vaccine is relatively low due to the weak and transient immune responses with current vaccine strategies [5, 12]. Synthetic tumor antigenic peptides are widely applied for tumor vaccine formulation because of their safety and ease of production, but poor immunogenicity and T cell stimulation potency are common barriers. In 2010, Sipuleucel-T, an autologous cellular immunotherapy, was the first cancer vaccine approved by FDA to treat metastatic prostate cancer [13]. Sipuleucel-T is composed of autologous peripheral blood mononuclear cells (PBMCs) and activated with a recombinant human protein constructed by linking GM-CSF to prostatic acid phosphatase (PAP-GM-CSF) [13]. Treatment of patients with the autologous tumor antigen loaded dendritic cells (DCs) did not show clinical response, yet the median survival of patients extended by 4.1 months as evident from a randomized double-blind placebo controlled multicenter trial [13].
In order to overcome these limitations, researchers have attempted to develop different strategies to improve the efficacy of vaccines by optimizing the tumor vaccine formulation with potent adjuvants. To this end, nano- and microparticles for vaccine delivery were designed to enhance antigen uptake by APCs, protect the antigen from proteolysis, and/or enable sustained release of antigen for pro-longed presentation by APCs [14–18]. Lipid nanoparticles mediated RNA delivery could enable efficient tumor antigen-coding RNA uptake and expression by DCs, and induced potent cytotoxic and memory T cell responses against cancer [19, 20]. In addition, nano- and microparticles delivery system could effectively co-deliver different immuno -stimulatory components and/or siRNAs along with antigens to elicit an improved antigen-specific immune response [21–25]. We recently reported that DCs loaded with mesoporous silicon vector (MSV) microparticles/peptide enhanced antitumor immunity [26]. It is known that stimulation of the innate arm of the immune system, such as induction of Toll like receptor (TLR) signaling in APCs, can promote antigen presentation to the adaptive immune system to enhance the antigen-specific immune responses [8, 16, 27, 28]. Therefore, in this study, we report that immunization of DC/MSV containing antigen (tyrosinase-related protein 2 peptide, TRP2 peptide, SYVDFFVWL) and dual TLR agonists (CpG and MPLA) can elicit synergistic antitumor T cell responses against B16 melanoma. We hypothesize that DCs loaded with TRP2 peptides and dual TLR agonists using MSV microparticles could markedly enhance antitumor immunity by prolonging antigen processing and presentation, thus triggering potent innate immune signaling and T cell responses.
2. Materials and Methods
2.1 Reagents and antibodies
MPLA and FITC-CpG1826 were obtained from Invivogen. CpG1826, PMA, ionomycin and t-butanol were purchased from Sigma. Fluorescent antibodies including anti-CD3/PerCp-Cy5.5 (145-2C11), anti-CD8/APC (53-6.7), anti-granzyme B/PE and anti-IFNγ/PE (XMG1.2) were all purchased from eBioscience. CD4 antibody for CD4+ T cell immunohistochemistry staining was obtained from eBioscience. CD8 antibody for CD8+ T cell immunohistochemistry staining was obtained from Bioss. Antibodies for IL-6, TNF-α and IFN-γ ELISA were purchased from eBioscience. GolgiSTOP is from BD Biosciences. DOPC liposome was obtained from Avanti. OVA257–264, TRP2 and β-Gal peptide were customized and obtained from Peptide 2.0. Hoechst 33342, LysoTracker Red DND-99 and LysoTracker Blue DND-22 were purchased from Thermo Fisher.
2.2 Cells and mice
B16 melanoma cells (C57BL/6 origin) were grown and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% pen/strep in a humidified incubator at 37°C and 5% CO2 condition. BMDCs were prepared from bone marrow of C57BL/6 mice. Briefly, bone marrow cells were first flushed from femurs and tibias of mice. BMDC culture medium is based on RPMI-1640 medium with L-glutamine and then supplemented with 10% FBS, 1% Pen/Strep, 20 ng mL−1 GM-CSF, 10 ng/mL IL-4 and 0.5 µM β-ME. Bone marrow cells were plated in 6-well plates at a concentration of 2 × 106 cells/well in 5 mL above BMDC culture medium. Medium was replaced on day 1, 3 and 5 and non-adherent cells were discarded. On day 6, half-attached cell colonies were gently de-attached and suspended at 1 × 106 cells mL−1 in BMDC culture medium and plate in 10 cm2 dish. The half-attached cells and floating cells were collected on the second day of re-plating. BMDCs could achieve 85% purity (CD11c+ population) as determined by flow cytometry.
6 to 8 weeks old female C57BL/6 mice were purchased from Jackson Lab. Mice were housed at the animal facility at the Houston Methodist Research Institute (HMRI). 0.2 × 106 B16 tumor cells were suspended in 0.2 mL HBSS and intravenously (i.v.) injected into C57BL/6 mice to establish B16 lung metastases model, according to the protocol previously described with a minor modification [29]. All experiments were performed with compliance with procedures and protocols approved by the Animal Care and Use Committee of HMRI.
2.3 MSV/TRP2-CM vaccine preparation
MSV were kindly provided by the Nano-Core facility (directed by Dr. Xuewu Liu) at HMRI. To load TRP2, CpG and MPLA into DOPC liposome, TRP2 (10 µg µL−1) alone or together with CpG (4 µg µL−1), MPLA (1 µg µL−1) were mixed with 0.1% Tween-20 and DOPC (20 µg µL−1) in t-butanol (Figure 1a). For in vitro and in vivo studies, below volume ratio was used (peptide: CpG: MPLA: 0.1% Tween-20: DOPC: t-butanol = 5:1:2:4:5:336). Based on the mass concentration of stock solution for each component (TRP2 10 µg µL−1, CpG 4 µg µL−1, MPLA 1 µg µL−1, Tween 1.1 g mL−1, DOPC 20 µg µL−1, t-butanol 0.781 g mL−1), the weight ratio of TRP2:CpG:MPLA:Tween:DOPC is 125:10:5:11:250:655. Characterization of surface charge of various vaccine combinations are listed (Table S1). The criteria for loading optimization is as follow: we first optimized the ratio of TRP2 to CpG. A volume ratio of 5:1 (10 µL TRP2: 2 µL CpG) showed the best result in balancing TRP2 loading and encapsulation efficiency into liposome under different TRP2 to CpG ratios (Table S2). Next, we tried to maximize the loading of MPLA into liposome with the criteria of liposome size below 50 nm, since the diameter of MSV pore is 50 nm. As shown in Table S3, a volume ratio of 5:1:2 for TRP2:CpG:MPLA (10 µL TRP2: 2 µL CpG: 4 µL MPLA) was the optimized ratio for maximizing the loading of each component (Table S3). The mixtures were vortexed thoroughly for 1 min then lyophilized for reconstitution. After lyophilizing, sterile MilliQ H2O was added for reconstitution of TRP2-CpG-MPLA loaded DOPC liposomes, and the reconstituted liposomes were added into MSV (100 µg TRP2: 0.6 billion MSV) through gentle sonication for 3 times (3 × 10 s each time), 20 mins for each interval. After loading Lipo/TRP2-CM into MSV, MSV were washed using sterilized water and cent rifuged at 10000g for three times (5 min each time) to remove free unencapsulated Lipo/TRP2-CM. The size distribution and zeta potential were characterized (Figure 1b–d).
Figure 1.

Scheme and characterization of mesoporous silicon vector (MSV) loaded TRP2-CpG-MPLA (MSV/TRP2-CM) vaccine. (a) Schematic representation of TRP2-CM loading into DOPC liposome (Lipo/TRP2-CM), and then loading into MSV (MSV/TRP2-CM). (b) Transmission electron microscope (TEM) image of MSV particle and Lipo/TRP2-CM. (c) The sizes of liposome (left) and MSV (right) were measured by DLS before and after loading TRP2-CM. (d) Zeta potential of TRP2 peptide, CpG, MPLA, MSV, Lipo/TRP2-CM and MSV/TRP2-CM. (e) Cumulative TRP2 peptide release from Lipo/TRP2-CM and MSV/TRP2-CM in PBS (pH 7.4).
2.4 Vaccine administration in B16 melanoma-bearing mice
TRP2 peptide alone or along with CpG-MPLA, were encapsulated in liposome for loading into MSV particles. Different vaccines were directly i.v. injected into mice or incubated with BMDCs (1 × 106 cells) at 37°C in serum-free RPMI 1640 medium for 3 h prior to injection. BMDCs incubated with vaccines were washed and collected by centrifuge for i.v. injection. C57BL/6 mice were inoculated with B16 melanoma cells (0.2 × 106 cells) on day 0, followed by vaccine immunization at day 3. On day 18, mice were sacrificed and the lungs were collected, rinsed with PBS briefly, and fixed with Fekete’s buffer (70 mL of 75% alcohol, 10 mL of formalin, and 5 mL glacial acetic acid). After 48 h fixation, pulmonary tumor nodules were imaged and counted.
2.5 Intracellular IFN-γ and granzyme B staining for TRP2 specific CD8+ T cells
Splenocytes were prepared from immunized mice for intracellular IFN-γ staining. Briefly, splenocytes were stimulated with TRP2 or control peptide in the presence of GolgiSTOP for 5 h. Surface marker CD3 and CD8 were stained, then fixed and permeabilized for intracellular IFN-γ staining. Samples were analyzed with Flow cytometry (BD SRII) and the data were analyzed by FlowJo.
2.6 Confocal microscope
TRP2 peptide, CpG and MPLA were labeled with fluorescence probe (FITC, Rhodamine 6G or quantum dot 633) for confocal imaging. BMDCs were incubated with fluorescence probe-labeled TRP2-CM, Lipo/TRP2-CM or MSV/TRP2-CM for 3 h, then washed extensively with PBS for further staining. For lysosome staining, cells were incubated with LysoTracker for 20 min according to manufacturer’s instruction. Cell nucleuses were labeled using Hoechst 33342 for 5 min according to manufacturer ’s instruction. Cells were then mounted and observed under confocal microscope (Olympus, FV1000).
2.7 ELISA for cytokine detection
BMDCs were incubated with control peptide (β-Gal), OVA257–264 or various vaccine formulations for different time points. Cell supernatants were collected for IL-6 and TNF-α measurement using ELISA. For OT-I T cell recognition assay, BMDCs were incubated with OVA257–264 peptide or various vaccine formulations in serum- free RPMI-1640 medium for 3 h, then washed with PBS for 5 times. OT-I specific CD8+ T cells were then co-cultured with BMDCs at 0, 24, 48, 72 and 96 h after washing for 18 h. Cell supernatants were collected for IFN-γ ELISA.
2.8 Depletion of cell subsets
To explore the functions of different cell populations in antitumor immunity, CD4+ T cells, CD8+ T cells, NK cells and macrophages were depleted using antibody or chemical agents. Briefly, CD4+ T cell depletion antibody (L3T4 antibody, TIB207, ATCC) or CD8+ T cell depletion antibody (Lyt-2.2 antibody, TIB210, ATCC) was i.p. injected every three days at 300 µg/mouse. For macrophage depletion, clodronate liposome (Liposoma B.V.) was applied at 150 µL per mouse. NK cells were depleted using NK1.1 antibody (600 µg per mouse, HB-191, ATCC). Flow cytometry showed that more than 90% cells were depleted (data not shown).
2.9 Immunohistochemistry staining
Serial tissue sections of 5 µm thickness were cut from the formalin fixed, paraffin embedded tissue blocks and mounted onto charged glass slides. All sections for immunohistochemistry were deparaffinized and hydrated using graded concentrations of ethanol to deionized water.
2.10 Statistical analysis and combination index calculation
Combination index (CI) for synergistic effect was calculated using the Bliss Independence model [30, 31]. CI >1, CI <1 and CI =1 denote antagonistic, synergistic, and additive combination effects, respectively. Statistical analysis was performed using GraphPad Prism v6.0. A one-way ANOVA or Student’s t test was applied for in vitro data comparisons. Kruskal-Wallis test applied for the tumor burden and Kaplan-Meier analysis applied for survival curve analysis. *p<0.05, **p<0.01, ***p<0.001
3. Results and Discussion
3.1 Characterization of MSV/TRP2-CpG-MPLA Vaccine
The fabrication of microparticles with specific shapes, sizes, and porosities was achieved by proper design and by adapting efficient silicon technologies. MSV microparticle delivery system containing multiple components was manipulated to execute the ultimate goal of efficient therapeutics. We developed MSV microparticles to load TRP2 peptide and dual TLR agonists to formulate the complete vaccine for systemic delivery (Figure 1a, b). MSV was a highly porous and hemispherical microparticle with a mean diameter of ~ 800 nm (Figure 1b, c) as previously described [26, 32]. MSV microparticles were fabricated with straight nanopores (nano-channels) crossing the particles perpendicular to the surface, and the mean diameter of the nanopore is around 50 nm [32]. Synthetic CpG oligonucleotide (CpG ODN) and MPLA were selected as dual TLR agonists for TLR9 and TLR4 signaling activation, respectively [33, 34]. To facilitate the components loading into MSV porous, TRP2 peptide, CpG ODN and MPLA were first loaded into dioleoylphosphatidylcholine (DOPC) liposome, followed by loading into MSV particles through electrostatic attractions and sonication to formulate the complete vaccine MSV/TRP2-CpG-MPLA (MSV/TRP2-CM for short) (Figure 1a). The average diameters of DOPC liposome before and after loading TRP2-CpG-MPLA (Lipo/TRP2-CM) were 21 nm and 28 nm, respectively. (Figure 1b, c). The average sizes of MSV before and after loading Lipo/TRP2-CM were 832 nm and 966 nm, respectively, as determined by transmission electron microscopy (TEM) and dynamic light scattering (DLS) analysis (Figure 1b, c). A volume ratio of TRP2:CpG:MPLA (5:1:2, v:v:v) was selected for vaccine preparation after optimization. After loading TRP2-CM into liposome, the anionic surface of Lipo/TRP2-CM could facilitate the loading into (3-Aminopropyl) triethoxysilane (APTES) modified cationic MSV microparticles (Figure 1d). The zeta potential of each component and the conversion of surface charge after loading of indicated components into liposome or MSV were characterized (Figure 1d). The cumulative TRP2 peptide release was measured. TRP2 release from MSV was markedly sustained compared with liposome (Figure 1e).
3.2 In Vitro Distribution of MSV/TRP2-CM Vaccine and Sustained Release of TRP2 Peptide
To determine the cellular uptake of the MSV/TRP2-CM vaccine and the intracellular location, we used fluorescent probe-labeled TRP2 peptide, CpG, and MPLA to formulate the vaccine particles. After incubation with free TRP2-CM, Lipo/TRP2-CM or MSV/TRP2-CM, the localization of TRP2 peptide (Rhodamine 6G-labeled, yellow), CpG (FITC-labeled, green), MPLA (QD633- labeled, red) in bone marrow-derived DCs (BMDCs) were evident within endo-lysosomal compartments (LysoTracker, blue) (Figure 2a, left). Semi-quantitative co-localization analysis showed that the percentages of co- localization of TRP2, CpG and MPLA over lysotracker were 80.6%, 90.8% and 62.6% in MSV/TRP2-CM treated DCs, respectively (Figure 2a, right). These results suggest that TRP2 peptide, CpG and MPLA could be delivered simultaneously by MSV through DC phagocytosis. We hypothesized that after phagocytosis, the TRP2 peptides would be gradually released by MSV for subsequent processing and presentation through major histocompatibility complex (MHC) class I molecule. To compare the retention of TRP2 peptide within BMDCs, we incubated BMDCs with free TRP2-CM, Lipo/TRP2-CM, or MSV/TRP2-CM for 3 h. After extensive washes, we maintained the BMDCs in culture condition and monitored them at different time points (0, 24, 48, 72 and 96 h after initial incubation). We found that MSV microparticle could maintain the TRP2 peptide for antigen processing for over 96 h, whereas both free TRP2-CM and Lipo/TRP2-CM could be detected for 24–48 h probably due to rapid degradation (Figure 2b). Flow cytometry analysis consistently showed that MSV microparticles retained strongest fluorescent signals from FITC-labeled TRP2 peptide within BMDCs over a 96 h period (Figure 2c, d). Taken together, our data suggest that TRP2 peptide within the MSV particle can be protected from rapid degradation and sustained release through silicon hydrolysis, thus prolonging antigen presentation.
Figure 2.

In vitro distribution of MSV/TRP2-CM vaccine and sustained release of TRP2 peptide. (a) Confocal inages of co- localization of TRP2 (Rhodamine 6G-labeled, yellow), CpG (FITC-labeled, green) and MPLA (QD633-labeled, red) with lysosome (blue) in mouse BMDCs after 3 h uptake of free TRP2-CM, Lipo/TRP2-CM or MSV/TRP2-CM. Scale bar, 10 µm. Semi-quantitative analysis of TRP2, CpG and MPLA co-localization with lysosome are shown in right panel using Image J co-localization finder. Analyses were performed on three individual images of each. (b) Confocal images of TRP2 peptide retained in BMDCs and intracellular location at 0, 24, 48, 72 and 96 h after 3 h incubation with indicated formulations. Scale bar, 10 µm. Data shown are representative of three experiments. Lysosome, lysotracker (red); Nucleus, H33342 (blue), TRP2, FITC-labeled (green). (c, d) FACS analysis for the intensity of FITC- labeled TRP2 in BMDCs at 0, 24, 48, 72 and 96 h post-3 h incubation with indicated formulations. Cells were gated on CD11c+ population. BMDCs incubated with PBS served as control (gray). Mean fluorescence intensity (MFI) subtracting the background was quantified (d).
3.3 Enhanced Antigen-Specific T Cell Responses by Co-delivery of MSV Microparticles Containing Peptide with CpG and MPLA
Intrinsic specialized characteristics of DCs are to capture, process and present antigens through MHC I and II molecules. DC/peptide vaccine induces T cell response by presenting antigen peptide through MHC complex for T cell recognition. We tested our hypothesis that dual TLR signaling can improve the antigen-presentation for enhanced T cell activity. For this purpose, ovalbumin (OVA)257–264 was selected as a model peptide for in vitro T cell recognition assay. OVA257–264 is a class I (Kb)-restricted peptide epitope of OVA and is recognized by OVA-specific CD8+ T cells (OT-I cells). BMDCs were first incubated with different OVA257–264 vaccine formulations, followed by washing and subsequently maintained in culture condition for 0, 24, 48, 72 and 96 h prior to co-culture with OT-I cells. We found that the secretion of interferon (IFN)-γ from OT-I CD8+ T cells in all CpG-MPLA (CM) groups were much higher than groups without CM (Figure 3a), demonstrating that dual TLR signaling triggered by CM was important for antigen presentation for enhanced T cell activity. Our results are consistent with previous findings that antigen-selection and antigen presentation efficiency in DC could be enhanced in the presence of TLR ligands [35]. MPLA is a TLR4 ligand that leads to signaling through the TIR-domain-containing adapter-inducing interferon-β (TRIF) adaptor and activates NF-κB signaling to produce tumor necrosis factor (TNF)-α and interleukin-6 (IL-6) cytokine; CpG is a TLR9 ligand that leads to signaling through myeloid differentiation primary response gene 88 (MyD88) and activate both NF-κB and interferon regulatory factor 7 (IRF7) for TNF-α, IL-6 and interferon (IFN) cytokines. We found that TNF-α and IL-6 were only produced by DCs with CM treatment (Figure 3b). Furthermore, type I interferon signaling, but not inflammasome signaling, could also be activated by CM (Figure S1a, b). We also demonstrated that synergistic effect could be generated by dual ligands for OVA257–264-specific T cell response as well as the cytokine production from DCs (Figure S2a–e). The cytokine productions, including TNF-α and IFN-β, were markedly enhanced by dual ligands stimulation, compared with single ligand (Figure S2a, c). We next examined whether TRP2-CM delivered by MSV microparticles was superior in inducing T cell activity than other vaccine delivery methods. We found that MSV/TRP2-CM was the best for the persistence of T cell activity and sustained IL-6 production, compared with other groups (Figure 3a, b). These data suggest that dual TLR ligands and MSV delivery strategy collectively contribute to the enhanced capacity and efficiency of DC to present antigen for T cell response.
Figure 3.

Co-delivery of dual TLRs agonists with peptide enhances antigen-specific T cell responses in vitro. (a) OT-I T cell IFN-γ release after recognition of OVA257–264 presented by BMDCs. Multiple OVA257–264 containing formulations were incubated with BMDCs for 6 h, followed by washing for 3 times. OT-I T cells were added (0.1 million T cell: 0.02 million BMDCs) at 0, 24, 48, 72 and 96 h after washing, and incubated with BMDCs for 18 h. The supernatants were collected for IFN-γ measurement by ELISA. OVA stands for OVA257–264 peptide. (b) Cytokine production of BMDCs after incubation with indicated formulations for different time points. Error bars represent standard deviation. Data are plotted as means ±SD of three independent experiments. *p<0.05, **p<0.01, ***p<0.001 by comparison with control using Student’s t-test.
Consistently, increased TRP2-specific CD8+ T cell response observed in vitro was also augmented in B16 melanoma inoculated C57BL/6 mice after administration of MSV/TRP2-CM treated DCs (DC/MSV/TRP2-CM). We checked TRP2-specific T cell activity on day 8 post-vaccination by intracellular IFN-γ staining, and found that co-delivery of TRP2 peptide with single TLR agonist CpG (DC/MSV/TRP2-C) could significantly enhance TRP2-specific CD8+IFNγ+ cell population compared with control peptide (β-Gal) immunization group (Figure 4a, b). Degranulation of TRP2-specific CD8+ T cells was also observed for their cytotoxicity to B16 melanoma (Figure 4c, d). In addition, delivery of dual TLRs agonists (CpG and MPLA) along with TRP2 peptide could generate synergistic effects for TRP2-specific CD8+ T cell response, as CD8+IFN-γ+ cell population of DC/MSV/TRP2-CM immunized group was further increased compared to groups with single TLR agonist (Figure 4a, b). The combination index for T cell response induced by DC/MSV/TRP2-CM is 0.51 (<1 indicates synergism). These data demonstrate the essential role of dual TLR agonists for the effective antigen presentation and T cell response.
Figure 4.

Co-delivery of dual TLRs agonists with TRP2 peptide using MSV enhances antigen-specific T cell responses in vivo. (a) FACS analysis for the percentage of CD8+ IFN-γ+ T cells in splenocytes of B16 tumor-bearing mice at day 8 after immunization with indicated TRP2 vaccine formulations. All vaccine formulations were pre-incubated with BMDCs for intravenous (i.v.) immunization. Statistical analysis of TRP2-specific CD8+ IFN-γ+ T cells are shown in (b). (c, d) FACS analysis for the percentage of CD8+Granzyme B+ T cells in splenocytes of B16 tumor-bearing mice at day 8 after immunization with indicated TRP2 vaccine formulations. Statistical analysis of TRP2-specific CD8+ Granzyme B+ T cells are shown in (d). Error bars represent standard deviation. *p<0.05, **p<0.01, ***p<0.001
3.4 Potent Antitumor Immunity against B16 Melanoma Lung Metastasis Generated by DC/MSV/TRP2-CM Vaccine and Safety Assessment
In order to validate the vaccine efficacy for tumor immunotherapy in vivo, we used an established B16 pulmonary metastases model. On day 3 post-inoculation of B16 melanoma cells, different vaccine formulations were either pre-incubated with DCs for 3 h before injection or directly injected into mice for systemic immunization. Tumor burdens of lung metastasis were checked on day 18 (Figure 5a). We found that the number of B16 lung metastasis nodule was significantly decreased after single immunization of MSV/TRP2-CM vaccine. Of note, immunization with MSV/TRP2-CM pre-incubated with DC prior to injection resulted in further reduction of pulmonary melanoma metastases (Figure 5b, c). Consistently, tumor specific CD8+ T cell response was also enhanced for the inhibition of tumor burden (Figure 5d). Importantly, DC/MSV/TRP2-CM vaccine generated a more effective antitumor immunity than DC/Lipo/TRP2-CM vaccine (Figure 5e, f). Moreover, DC/MSV/TRP2-CM showed a much stronger antitumor immunity than DC/MSV/TRP2 plus soluble CpG and MPLA (CM) (separately) (Figure 5e, f). These data suggest that MSV enabled co-delivery of TRP2 with dual ligands to DCs is critical for inducing potent antitumor immunity. We next examined biodistribution of DC/MSV/TRP2-CM in mice at 0,1, 6, 24, 48 and 72 h post i.v. injection. We found that DC/MSV/TRP2-CM were mainly distributed in the liver and lung, and cleared after 72 h (Figure 5g). Consistent with increased T cell response, we found that the median survival of DC/MSV/TRP2-CM immunized mice were significantly increased, compared with other groups (Figure 5h). To determine the therapeutic time window for a successful vaccination, we performed experiments with the DC/MSV/TRP2-CM vaccination on day 3, 5 and 7 after tumor inoculation (Figure S3a). Vaccination on day 5 still showed significant antitumor effect, but vaccination on day 7 failed to inhibit the tumor growth (Figure S3b, c). This could be attributed to the fact that B16 melanoma was a highly metastatic and aggressive tumor model with all tumor-bearing mice dying around day 18 to 21 (Figure 5h). Thus, vaccination before day 5 after tumor injection is the best therapeutic window for a successful vaccination in B16 melanoma lung metastasis model.
Figure 5.

DC/MSV/TRP2-CM vaccine generates potent antitumor immunity against B16 melanoma lung metastasis. (a) Scheme for the establishment of B16 melanoma lung metastasis model in C57BL/6 mice and vaccination timeline. C57BL/6 mice were inoculated with B16 melanoma cells (0.2 million/mouse, i.v.) and immunized with the following vaccines: DC/MSV, DC/TRP2, DC/MSV/TRP2, DC/MSV/TRP2-CM, MSV/TPR2, and MSV/TRP2-CM on day 3. Lung metastases were examined on day 18 (b). (c) Numbers of the lung tumor nodules were counted. (d) FACS analysis of TRP2 specific CD8+ IFN-γ+ T cells in splenocytes of B16 tumor-bearing mouse at day 18. (e, f) C57BL/6 mice were inoculated with B16 melanoma cells (0.2 million/mouse, i.v.) and immunized with the following vaccines: DC/MSV, DC/MSV/TRP2-CM, DC/Lipo/TPR2-CM, and DC/soluble CM+MSV/TRP2 (DC incubated with soluble CpG, MPLA and MSV/TRP2) on day 3. Lung metastases were examined on day 18 (e). Numbers of the lung tumor nodules were counted (f). (g) Biodistribution of DC/MSV/TRP2-CM (MSV labeled with Rhodamine 6G) in mice. Ex vivo fluorescent images of mouse tissues were taken post 0, 1, 6, 24, 48 and 72 h of i.v. injection of DCs. (h) Survival of B16 tumor-bearing mouse vaccinated with indicated groups. Error bars represent standard deviation. *p<0.05, **p<0.01, ***p<0.001
To further evaluate whether delivery of TRP2 peptide and CM to the same DC is required for the improved antitumor immunity, we used two groups of DCs loaded with either MSV/TRP2 or MSV/CM, then mixed together, and injected into the tumor-bearing mice for antitumor immunotherapy. Even though all the components were delivered into mice via DCs, TRP2 peptide and CM were separated for their function. We found that no significant antitumor response was generated when TRP2 peptide and CM were delivered into different DCs (Figure 6a). This finding suggests that delivery of both TRP2 peptides and TLR agonists to the same DC is critically required for a highly efficient immunotherapy. The likelihood of using dual TLR agonists could achieve a better adaptive immunity than single agonist was shown for yellow fever vaccine YF-17D [36]. Immunization of mice with polymeric nanoparticles containing antigens for YF -17D plus ligands for TLR4 and TLR7 synergistically magnify the antigen-specific antibody compared to single TLR ligand, and could enhance persistence of germinal centers of plasma -cell responses for more than 1.5 years [37, 38]. In this case, the YF-17D antigen and TLR agonists were delivered by two separate nanoparticle vehicles to increase the antibody responses for YF-17D in targeting different cell types [37, 38]. YF-17D antigen aims to target specific B cell receptor, and the TLR agonists aim to target DCs for augmenting CD4+ T cells assisted antibody response. In contrast, for a DC-targeting tumor vaccine in our study, both antigen and TLR agonists must be delivered into the same DC through MSV for inducing robust T cell antitumor immunity.
Figure 6.

DC/MSV/TRP2-CM vaccine orchestrates effective host antitumor immunity. (a) Number of the B16 lung tumor nodules by vaccination of MSV/TRP2, DC/MSV/TRP2-CM, or DC/MSV/TRP2 and DC/MSV/CM separately. (b) Number of the B16 lung tumor nodules by vaccination of DC/MSV/TRP2-CM in mice depleted CD4+ T cells, CD8+ T cells, CD4+ and CD8+ T cells, NK1.1 cells and macrophages, respectively. (c) Immunohistochemistry of CD4+ and CD8+ T cells infiltration in B16 tumor lung metastasis site. Scale bar, 10 µm. (d) Number of the B16 lung tumor nodules after vaccination using DC/MSV/TRP2-CM vaccines from different DC-donors. B16 melanoma inoculated mice were administered with MSV/TRP2-CM by pre- incubation of C57BL/6 mouse-derived DCs, Balb/c mouse-derived DCs, or irradiated Balb/c mouse-derived DCs. C57BL/6 mouse-derived DCs incubated with MSV particles alone served as a control. Error bars represent standard deviation. *p<0.05, **p<0.01, ***p<0.001.
To determine the cell types that are essential in augmenting DC/MSV/TRP2 -CM vaccine induced antitumor immunity, we depleted CD4+ T cells, CD8+ T cells, macrophages, or natural killer (NK) cells, respectively. We showed that both CD4+ and CD8+ T cell were required for the antitumor immunity, since depletion of either CD4+ or CD8+ T cell could completely abolish the antitumor immune response (Figure 6b). Consistently, immunohistochemistry staining showed that the infiltration of CD8+ and CD4+ T cells within melanoma lung metastases were significantly increased after DC/MSV/TRP2-CM vaccination (Figure 6c), suggesting that the DC/MSV/TRP2-CM vaccine enhanced the tumor-specific CD8+ T cell response and orchestrated host immune network involving both CD4+ and CD8+ T cells. Our results were consistent with previous findings that CD4+ T cells provide help for priming CD8+ T cells response and maintaining CD8+ T cell memory [39–41], even though CD4+ T cells were not activated since TRP2 peptide is a MHC class I restricted epitope (Figure S4). On the contrary, the depletion of macrophages by clodronate-containing liposomes could partially compromise the vaccine efficacy. However, NK1.1 cell depletion did not affect the antitumor immune response (Figure 6b). Although clodronate treatment was intended for depletion of macrophages, dendritic cells may also affected [42]. Thus, further studies are needed to address the importance of macrophages in mice by using CD11b-DTR mice treated with diphtheria toxin [43]. To determine whether both exogenous DC and endogenous DC are essential for the antigen presentation and synergistically activate T cells, we found that exogenous DCs were required for potent antitumor immunity, since antitumor effect was improved when MSV/TRP2-CM was delivered into DCs before injection (DC/MSVTRP2-CM) (Figure 5b–d). To determine whether endogenous DCs and macrophages are required for antigen presentation, we used MHC I-mismatched (Balb/c mice) BMDCs for MSV/TRP2-CM loading and immunization, B16 melanoma metastases could partially be inhibited (Figure 6d), suggesting that endogenous APCs were involved in antigen uptake and presentation to T cells since Balb/c-derived DCs (Kd) are unable to present antigen to activate T cells in C57BL/6J (Kb) mice. We reasoned that endogenous DCs might take up the MSV/TRP2-CM released from dying exogenous Balb/c DCs and present antigens for T cell activation. To further test this possibility, we applied irradiated Balb/c-derived DC for MSV/TRP2-CM immunization, and found that tumor inhibition effect was almost identical to non- irradiated Balb/c-derived DC group (Figure 6d). These results suggest that endogenous DCs and macrophages could take up MSV/TRP2-CM vaccine released from exogenous DCs for subsequent activation of T cells for antitumor responses.
Finally, we performed the toxicity assessment of MSV/TRP2-CM vaccine. MTT assay was used for the cytotoxicity assessment of MSV and Tween 20. We found a dose-dependent mild cytotoxicity to DCs (Figure 7a, b). Red arrows indicated that the dosage we used for preparation of MSV/TRP2-CM vaccine formulation did not generate obvious cytotoxicity. In vivo toxicity was assessed by hematoxylin and eosin (HE) staining of different tissues, measurement of body weight, and in vivo clearance of MSV (Figure 7c–e). Histopathological examination showed no significant changes in tissues of DC/MSV/TRP2-CM treated mice in comparison to both untreated healthy and tumor-bearing mice (Figure 7c). We also measured the body weight of tumor-bearing mice, and found that DC/MSV/TRP2-CM administration could prevent the mice from body weight loss. Tumor-bearing mice receiving DC/MSV/TRP2-CM could gain as much body weight as the healthy mice (Figure 7d). Biodistribution of MSV (labeled with Rhodamine 6G) particles in mice tissues were also examined. Fluorescent images were taken 0, 1, 6, 24, 48 h post i.v. injection of MSV particles. MSV were predominately distributed in the liver and lung, and typically cleared after 48 h (Figure 7e), suggesting the biosafety of DC/MSV/TRP2-CM and potential application for clinical uses.
Figure 7.

Safety assessment and biodistribution of MSV/TRP2-CM vaccine. (a, b) MTT assay for cytotoxicity assessment of MSV (a) and Tween 20 (b) to DCs. Red arrows indicated the dosage currently used for MSV/TRP2-CM vaccine formulation and administration. (c) Haematoxylin and eosin (HE) staining of different tissues of DC/MSV/TRP2-CM treated mice. (d) Body weight of healthy mice and tumor bearing mice with or without DC/MSV/TRP2-CM administration. (e) Biodistribution of MSV (labeled with Rhodamine 6G) particles in mice. Ex vivo fluorescent images of mouse tissues were taken post-0, 1, 6, 24, 48 h of i.v. injection of MSV particles.
4. Conclusion
We have developed a highly efficient cancer vaccine approach by using MSV for the co-delivery of TRP2 peptide, CpG, and MPLA into the same DC. The MSV we used is a multi-stage delivery system which has more flexibility in loading molecules of interest compared to other delivery systems: the porous of MSV could be loaded with various small molecules as well as nanoparticles such as quantum dots and liposome. We show that MSV-based vaccine generated more effective and durable immune response as evidenced in our in vitro and in vivo study. Several previous studies have demonstrated silicon materials to be biodegradable and biocompatible [17, 44], our study provides further evidence on the biocompatibility and safety. Furthermore, innate immune signaling activated by dual TLR agonists (CpG and MPLA) for TRP2 vaccine formulation exerted superior effects on improving antigen-specific CD8+ T cell responses against B16 tumor cells, compared with TRP2 peptide alone or with a single TLR agonist. Importantly, we show that both antigen and TLR agonists must function within the same DC for enhanced antigen presentation and robust antitumor immunity. It is well known that TLR ligands are a class of innate immune signaling agonists and required for potent immune response in preclinical and clinical studies [28, 45, 46]. Activation of TLR signaling has been reported to regulate adaptive immune responses, including antibody production and antigen-presentation for specific T cell responses [27, 37]. We further show that both endogenous DCs and macrophages play critical role s in the uptake and presentation of antigen-TLR ligands complexes in MSV for inducing potent antitumor immunity. Using MSV microparticles as a high-loading capacity vehicle with the ability for co-delivery of multiple components (antigenic peptides and TLR ligands) may be developed for clinical application for augmenting strong antitumor immunity. This vaccine strategy may be further developed for cancer patients who do not respond to checkpoint blockade therapy to generate tumor-specific T cells, and then combined with checkpoint blockade therapy.
Supplementary Material
Acknowledgments
This work was supported by grants from the NCI and NIDA, NIH (R01CA101795, R01DA030338 and U54CA210181), Cancer Prevention and Research Institute of Texas (CPRIT; DP150099, RP150611 and RP170537), Department of Defense (DoD) CDMRP BCRP (BC151081), and Golfer against cancer foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: None.
Supplementary data is available online including Figure S1–S4 and Tables S1–S3.
References
- 1.Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, Mellstedt H. Therapeutic vaccines for cancer: an overview of clinical trials. Nat. Rev. Clin. Oncol. 2014;11:509–524. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- 2.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, Linette GP. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delamarre L, Mellman I, Yadav M. Neo approaches to cancer vaccines. Science. 2015;348:760–761. doi: 10.1126/science.aab3465. [DOI] [PubMed] [Google Scholar]
- 4.Gubin MM, Schreiber RD. The odds of immunotherapy success. Science. 2015;350:158–159. doi: 10.1126/science.aad4140. [DOI] [PubMed] [Google Scholar]
- 5.Ledford H. Therapeutic cancer vaccine survives biotech bust. Nature. 2015;519:17–18. doi: 10.1038/nature.2015.16990. [DOI] [PubMed] [Google Scholar]
- 6.Weber JS, Mule JJ. Cancer immunotherapy meets biomaterials. Nat. Biotechnol. 2015;33:44–45. doi: 10.1038/nbt.3119. [DOI] [PubMed] [Google Scholar]
- 7.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 8.De Vries J, Figdor C. Immunotherapy: Cancer vaccine triggers antiviral-type defences. Nature. 2016;534:329. doi: 10.1038/nature18443. [DOI] [PubMed] [Google Scholar]
- 9.Wang R-F, Wang HY. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res. 2017;27:11–37. doi: 10.1038/cr.2016.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Errico A. Immunotherapy: PD-1-PD-L1 axis: efficient checkpoint blockade against cancer. Nat. Rev. Clin. Oncol. 2015;12:63. doi: 10.1038/nrclinonc.2014.221. [DOI] [PubMed] [Google Scholar]
- 11.Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, Kertser A, David E, Amit I, Schwartz M. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat. Med. 2016;22:135–137. doi: 10.1038/nm.4022. [DOI] [PubMed] [Google Scholar]
- 12.Johnston SA. Cancer goal: Vaccine is cause for optimism. Nature. 2013;493:304. doi: 10.1038/493304c. [DOI] [PubMed] [Google Scholar]
- 13.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 14.Smith DM, Simon JK, Baker JR., Jr Applications of nanotechnology for immunology. Nat. Rev. Immunol. 2013;13:592–605. doi: 10.1038/nri3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan H, Jiang W, von Roemeling CA, Qie Y, Liu X, Chen Y, Wang Y, Wharen RE, Yun K, Bu G. Multivalent bi-specific nanobioconjugate engager for targeted cancer immunotherapy. Nat. Nanotechnol. 2017;12:763–769. doi: 10.1038/nnano.2017.69. [DOI] [PubMed] [Google Scholar]
- 16.Kim SY, Phuengkham H, Noh YW, Lee HG, Um SH, Lim YT. Immune Complexes Mimicking Synthetic Vaccine Nanoparticles for Enhanced Migration and Cross - Presentation of Dendritic Cells. Adv. Funct. Mater. 2016;26:8072–8082. [Google Scholar]
- 17.Wang Y, Zhao Q, Han N, Bai L, Li J, Liu J, Che E, Hu L, Zhang Q, Jiang T, Wang S. Mesoporous silica nanoparticles in drug delivery and biomedical applications. Nanomedicine. 2015;11:313–327. doi: 10.1016/j.nano.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 18.Noh YW, Kim SY, Kim JE, Kim S, Ryu J, Kim I, Lee E, Um SH, Lim YT. Multifaceted Immunomodulatory Nanoliposomes: Reshaping Tumors into Vaccines for Enhanced Cancer Immunotherapy. Adv. Funct. Mater. 2017;27 [Google Scholar]
- 19.Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A, Anderson DG, Langer R, Blankschtein D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017;17:1326–1335. doi: 10.1021/acs.nanolett.6b03329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, Grunwitz C, Vormehr M, Husemann Y, Selmi A, Kuhn AN, Buck J, Derhovanessian E, Rae R, Attig S, Diekmann J, Jabulowsky RA, Heesch S, Hassel J, Langguth P, Grabbe S, Huber C, Tureci O, Sahin U. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396–401. doi: 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- 21.Gregory AE, Titball R, Williamson D. Vaccine delivery using nanoparticles. Front. Cell. Infect. Microbiol. 2013;3:13. doi: 10.3389/fcimb.2013.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silva JM, Videira M, Gaspar R, Preat V, Florindo HF. Immune system targeting by biodegradable nanoparticles for cancer vaccines. J. Control. Release. 2013;168:179–199. doi: 10.1016/j.jconrel.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 23.Li N, Peng LH, Chen X, Zhang TY, Shao GF, Liang WQ, Gao JQ. Antigen-loaded nanocarriers enhance the migration of stimulated Langerhans cells to draining lymph nodes and induce effective transcutaneous immunization. Nanomedicine. 2014;10:215–223. doi: 10.1016/j.nano.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 24.Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, Fiering S. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat. Nanotechnol. 2015;11:295–303. doi: 10.1038/nnano.2015.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson DS, Dalmasso G, Wang L, Sitaraman SV, Merlin D, Murthy N. Orally delivered thioketal nanoparticles loaded with TNF-alpha-siRNA target inflammation and inhibit gene expression in the intestines. Nat. Mater. 2010;9:923–928. doi: 10.1038/nmat2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia X, Mai J, Xu R, Perez JE, Guevara ML, Shen Q, Mu C, Tung HY, Corry DB, Evans SE, Liu X, Ferrari M, Zhang Z, Li XC, Wang RF, Shen H. Porous silicon microparticle potentiates anti-tumor immunity by enhancing cross-presentation and inducing type I interferon response. Cell Rep. 2015;11:957–966. doi: 10.1016/j.celrep.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watts C, West MA, Zaru R. TLR signalling regulated antigen presentation in dendritic cells. Curr. Opin. Immunol. 2010;22:124–130. doi: 10.1016/j.coi.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat. Med. 2013;19:1597–1608. doi: 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- 29.Overwijk WW, Restifo NP. B16 as a mouse model for human melanoma. Curr. Protoc. Immunol. 2001;Chapter 20(Unit 20):21. doi: 10.1002/0471142735.im2001s39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foucquier J, Guedj M. Analysis of drug combinations: current methodological landscape. Pharmacol. Res. Perspect. 2015;3:e00149. doi: 10.1002/prp2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang J, Wennerberg K, Aittokallio T. What is synergy? The Saariselka agreement revisited. Front. Pharmacol. 2015;6:181. doi: 10.3389/fphar.2015.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen J, Xu R, Mai J, Kim HC, Guo X, Qin G, Yang Y, Wolfram J, Mu C, Xia X, Gu J, Liu X, Mao ZW, Ferrari M, Shen H. High capacity nanoporous silicon carrier for systemic delivery of gene silencing therapeutics. ACS Nano. 2013;7:9867–9880. doi: 10.1021/nn4035316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316:1628–1632. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 34.Haas T, Metzger J, Schmitz F, Heit A, Muller T, Latz E, Wagner H. The DNA sugar backbone 2' deoxyribose determines toll-like receptor 9 activation. Immunity. 2008;28:315–323. doi: 10.1016/j.immuni.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 35.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 36.Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, Akira S, Ahmed R, Pulendran B. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J. Exp. Med. 2006;203:413–424. doi: 10.1084/jem.20051720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, Garcia-Sastre A, Compans R, Pulendran B. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470:543–547. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flemming A. Vaccines: nano-adjuvant: double TLR stimulation is the key. Nat. Rev. Drug Discov. 2011;10:258. doi: 10.1038/nrd3426. [DOI] [PubMed] [Google Scholar]
- 39.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J. Exp. Med. 1998;188:2357–2368. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang RF. The role of MHC class II-restricted tumor antigens and CD4+ T cells in antitumor immunity. Trends Immunol. 2001;22:269–276. doi: 10.1016/s1471-4906(01)01896-8. [DOI] [PubMed] [Google Scholar]
- 41.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 42.Weisser SB, van Rooijen N, Sly LM. Depletion and reconstitution of macrophages in mice. JoVE. 2012:4105. doi: 10.3791/4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu X, Cai B, Wang M, Tan P, Ding X, Wu J, Li J, Li Q, Liu P, Xing C, Wang HY, Su XZ, Wang RF. Cross-Regulation of Two Type I Interferon Signaling Pathways in Plasmacytoid Dendritic Cells Controls Anti-malaria Immunity and Host Mortality. Immunity. 2016;45:1093–1107. doi: 10.1016/j.immuni.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang F, Li L, Chen D. Mesoporous silica nanoparticles: synthesis, biocompatibility and drug delivery. Adv. Mater. 2012;24:1504–1534. doi: 10.1002/adma.201104763. [DOI] [PubMed] [Google Scholar]
- 45.Olive C. Pattern recognition receptors: sentinels in innate immunity and targets of new vaccine adjuvants. Expert Rev. Vaccines. 2012;11:237–256. doi: 10.1586/erv.11.189. [DOI] [PubMed] [Google Scholar]
- 46.Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33:492–503. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.