

Abstract
Background and Purpose
Aseptic prosthesis loosening, caused by wear particles, is one of the most common causes of arthroplasty failure. Extensive and over‐activated osteoclast formation and physiological functioning are regarded as the mechanism of prosthesis loosening. Therapeutic modalities based on inhibiting osteoclast formation and bone resorption have been confirmed to be an effective way of preventing aseptic prosthesis loosening. In this study, we have investigated the effects of sophocarpine (SPC, derived from Sophora flavescens) on preventing implant loosening and further explored the underlying mechanisms.
Experimental Approach
The effects of SPC in inhibiting osteoclastogenesis and bone resorption were evaluated in osteoclast formation, induced in vitro by the receptor activator of NF‐κB ligand (RANKL). A rat femoral particle‐induced peri‐implant osteolysis model was established. Subsequently, micro‐CT, histology, mechanical testing and bone turnover were used to assess the effects of SPC in preventing implant loosening.
Key Results
In vitro, we found that SPC suppressed osteoclast formation, bone resorption, F‐actin ring formation and osteoclast‐associated gene expression by inhibiting NF‐κB signalling, specifically by targeting IκB kinases. Our in vivo study showed that SPC prevented particle‐induced prosthesis loosening by inhibiting osteoclast formation, resulting in reduced periprosthetic bone loss, diminished pseudomembrane formation, improved bone‐implant contact, reduced bone resorption‐related turnover and enhanced stability of implants. Inhibition of NF‐κB signalling by SPC was confirmed in vivo.
Conclusion and Implications
SPC can prevent implant loosening through inhibiting osteoclast formation and bone resorption. Thus, SPC might be a novel therapeutic agent to prevent prosthesis loosening and for osteolytic diseases.
Abbreviations
- ALP
alkaline phosphatase
- B.Ar/T.Ar
bone area/total area
- BIC
bone‐implant contact
- BMD
bone mineral density
- BMM
bone marrow‐derived macrophage
- BV/TV
bone volume/total volume
- Conn.D
connective density
- CTX‐1
type I collagen cross‐linked C‐terminal telopeptide
- IKKβ
IκB kinase β
- M‐CSF
macrophage colony‐stimulating factor
- NFATc1
nuclear factor of activated T cells c1
- OCN
osteocalcin
- PFA
paraformaldehyde
- RANKL
receptor activator of NF‐κB ligand
- ROI
region of interest
- RUNX2
runt‐related transcription factor 2
- SEM
scanning electron microscope
- SMI
structural model index
- SPC
sophocarpine
- TAK1
transforming growth factors‐β‐activated kinase 1
- Tb.N
trabecular number
- Tb.Sp
trabecular separation
- Tb.Th
trabecular thickness
- TBST
tris‐buffered saline tween‐20
- TJA
total joint arthroplasty
- TRAP
tartrate‐resistant acid phosphatase
- UHMWPE
ultra‐high molecular weight polyethylene
Introduction
Aseptic implant loosening is one of the most common complications of long‐term total joint arthroplasty (TJA), which would eventually require a revision surgery. There were 38 310 hip and 16 711 knee revision surgeries performed in the UK due to aseptic loosening, between April 1, 2003 and December 31, 2015 (Porter, 2016). This is generally considered to be caused by an adverse response to implant‐derived wear particles emanating from material components, including ultra‐high molecular weight polyethylene (UHMWPE) and metal biomaterials (Jiang et al., 2013; Gallo et al., 2014; Pajarinen et al., 2014). These wear particles stimulate the secretion of chemokines and pro‐inflammatory cytokines, including TNF‐α, IL‐1β, IL‐6, IL‐11 and PGE2, which may provoke a local inflammatory reaction that increases osteoclastogenesis and bone resorption, resulting in periprosthetic osteolysis, dense pseudomembrane formation and reduced implant fixation properties (Ingham and Fisher, 2005; Holt et al., 2007; Purdue et al., 2007; Athanasou, 2016). Therefore, a potential therapeutic strategy to prevent implant loosening is to suppress osteoclast over‐formation and its functions.
Osteoclasts, originating from haematopoietic monocytes and macrophages, are fused multinucleated giant cells with the principal function of bone resorption. The differentiation of osteoclasts require stimulation by macrophage colony‐stimulating factor (M‐CSF) and receptor activator of NF‐κB ligand (RANKL) (Kong et al., 1999; Takahashi et al., 2003) and involve activation of downstream signalling pathways, such as the MAPK and NF‐κB signalling pathways (Pearson et al., 2001; Takaesu et al., 2001). This in turns further leads to the activation and up‐regulation of two crucial transcription factors for osteoclast formation, c‐Fos and nuclear factor of activated T cells c1 (NFATc1) (Takayanagi et al., 2002; Boyle et al., 2003). The osteoclasts play their role in bone resorption through membrane polarization and the formation of an actin‐rich sealing zone, which lead to the secretion of acids and proteolytic enzymes that dissolve and degrade the mineralized bone matrix (Boyle et al., 2003; Jurdic et al., 2006).
Due to the key role of osteoclasts in periprosthetic osteolysis and related implant loosening, drugs targeting the suppression of osteoclastic signalling pathways are found to be effective in preventing and treating the disorder. These include bisphosphonates, sclerostin antibody and strontium ranelate (Liu et al., 2012b, 2014; Qu et al., 2013). Sophocarpine (SPC) (Figure 1A) is one of the major bioactive compounds derived from the natural plant Sophora flavescens, which is widely distributed in Russia, Japan, India and China. Anti‐inflammatory, anti‐tumour, anti‐allergic, cardioprotective and neuroprotecive effects have been demonstrated in previous studies (Yang et al., 2011; Yifeng et al., 2011; Gao et al., 2012; Wang et al., 2012; Li et al., 2014; Zhang et al., 2016a). SPC can inhibit LPS‐induced inflammation in RAW 264.7 cells via the NF‐κB and MAPKs signalling pathways (Gao et al., 2009, 2012). Additionally, the cardioprotective effects SPC as reported in another study, involved suppression of the NF‐κB signalling pathway (Li et al., 2011). However, little is known about the effects of SPC on osteoclasts and osteolytic‐related diseases. As the NF‐κB and MAPKs signalling pathways are important in osteoclast formation and SPC is known to suppress these signalling pathways in macrophages, we hypothesized that SPC might prevent particle‐induced implant loosening by inhibiting osteoclast formation and functioning.
Figure 1.
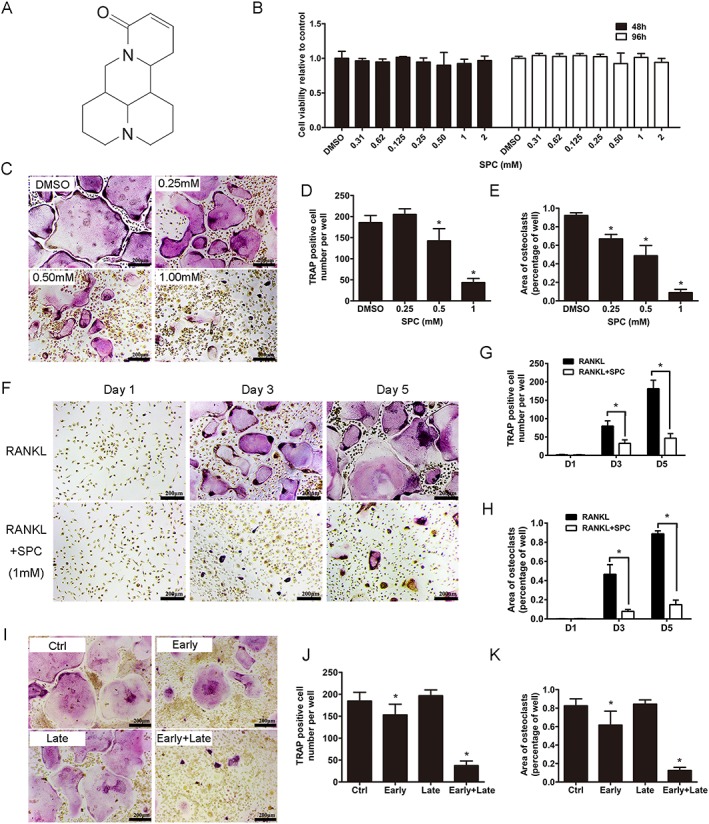
SPC attenuated RANKL‐induced osteoclast formation with negligible cytotoxicity in vitro. (A) Chemical structure of SPC. (B) BMMs were plated at a density of 8000 cells per well in a 96‐well plate and treated with different dilutions of SPC (0.31–2 mM) in the presence of 30 ng·mL−1 M‐CSF for 48 or 96 h. The viability of SPC‐exposed BMM cells were quantified by the CCK8 assay. (C) TRAP staining was performed on BMMs after treatment with different concentrations of SPC in the presence of M‐CSF (30 ng·mL−1) and RANKL (50 ng·mL−1) for 5 days. (D) The number and (E) the area of TRAP positive osteoclasts were quantified, as described. (F) BMMs were exposed to 1 mM SPC/DMSO with M‐CSF (30 ng·mL−1) and RANKL (50 ng·mL−1), and cells were fixed and TRAP staining was performed on days 1, 3 and 5. (G) The number and (H) the area of osteoclasts were quantified. (I) BMMs were cultured with treatment of 1 mM SPC for days 1–3 (early‐stage), days 3–5 (late‐stage) or days 1–5 (early + late stage) in the presence of M‐CSF (30 ng·mL−1) and RANKL (50 ng·mL−1). (J) The number and (K) the area of osteoclasts were measured. Values expressed are means ± SD; n = 5. *P < 0.05, significantly different from control group.
Our study showed that SPC attenuated osteoclast formation and function by suppressing the NF‐κB signalling pathway in vitro, and its likely target might be the IκB kinase (IKKs). We further confirmed that SPC prevented particle‐induced prosthesis loosening by inhibiting osteoclast formation via suppression of the NF‐κB signalling pathway in vivo.
Methods
Bone marrow‐derived macrophage (BMM) culture and cell viability assay
Primary rat bone monocyte/macrophage precursors were isolated from the long bones of male Sprague‐Dawley rats weighing about 100 g and cultured in α‐MEM supplemented with 10% (v/v) FBS, 2 mM L‐glutamine, 100 U·mL−1 penicillin/streptomycin (complete α‐MEM) and 30 ng·mL−1 M‐CSF for 2 days to induce differentiation into BMMs. Cell culture was carried out within a 37°C humidified incubator containing 5% (v/v) CO2, until cells were fully confluent. To explore the ratio of macrophage population in the whole cell population after 2 day culture, we collected the cells and analysed using flow cytometry.
Cells were seeded into 96‐well plates at a density of 2 × 104 cells per well for 24 h and subsequently treated with or without different dilutions of SPC (0.31–2 mM) for 48 or 96 h. Subsequently, the CCK‐8 assay was used to assess the cytotoxic effects of SPC by adding 10 μL of CCK‐8 buffer into each well, and the plate was incubated for another 4 h. The OD was measured by using an ELX800 absorbance microplate reader (Bio‐Tek Instr., Winooski, VT, USA) at a wavelength of 450 nm (650 nm reference).
Osteoclast differentiation and TRAP staining
A cell count of 6 × 103 cells per well of BMMs were seeded into a 96‐well plate with complete α‐MEM containing 30 ng·mL−1 M‐CSF, 50 ng·mL−1 RANKL and SPC (0, 0.25, 0.50 or 1.00 mM), followed by changes of culture media every 2 days for 5 days. Then, cells were fixed with 4% (w/v) paraformaldehyde (PFA) at 4°C for 30 min, washed twice with PBS and stained with the TRAP staining kit according to the manufacturer's protocol. TRAP‐positive cells with more than three nuclei were considered as mature osteoclasts, and their number and spread area were quantified under microscopy.
Immunofluorescence staining for F‐actin ring formation
To observe F‐actin ring formation, BMM were cultured on bovine bone slices with M‐CSF and RANKL for 5 days and were subsequently treated with serial dilutions of SPC (0, 0.25, 0.50 or 1.00 mM) for another 48 h. The cells were then fixed in 4% (w/v) PFA for 30 min and subsequently permeabilized for 5 min with 0.2% (v/v) Triton X‐100, followed by being incubation with rhodamine‐conjugated phalloidin diluted (1:100) in 0.2% (w/v) BSA‐PBS for 1 h at room temperature. Then the cells were stained with 4′, 6‐diamidino‐2‐phenylindole (KeyGen Biotech, Nanjing, China) for 5 min to visualize their nuclei and mounted with ProLong Gold anti‐fade mounting medium (Invitrogen, Life Technologies Corp, Carlsbad, CA, USA). Samples were analysed, and fluorescence images were captured using a NIKON A1Si spectral detector confocal system equipped with 40 × (dry) lenses. Five wells were randomly chosen in each group to calculate the number and size of the F‐actin ring cells by using the Image J software.
Bone resorption pit assay
BMM cells were treated with 30 ng·mL−1 M‐CSF and 50 ng·mL−1 RANKL for 4 days. Equal numbers of osteoclasts were seeded overnight on bovine bone slices in 96‐well plates. Cells were then cultured in complete α‐MEM containing 30 ng·mL−1 M‐CSF and 50 ng·mL−1 RANKL with different concentrations of SPC (0, 0.25, 0.50 or 1.00 mM) for another 48 h. Cells were gently removed by mechanical agitation, and resorption pits were observed under a scanning electron microscope (SEM) (S‐4800, Hitachi, Japan). Five random fields per bone slice were selected for quantitative analysis, and independent experiments were repeated at least five times. The area of bone resorption was quantified using Image J software.
Cell culture, RNA isolation and quantitative PCR
To detect the effects of SPC on osteoclast formation and functions, BMM cells were seeded at a density of 10 × 104 cells per well in 6‐well plates and cultured in osteoclastogenic medium (complete α‐MEM containing 30 ng·mL−1 M‐CSF and 50 ng·mL−1 RANKL) with different ways of SPC treatment. To detect its effects on osteoclast formation, cells were cultured in osteoclastogenic medium with varying doses of SPC (0, 0.25, 0.50 or 1.00 mM) for 5 days or 1.00 mM SPC for 0, 1, 3 or 5 days. To explore its effects of SPC on osteoclast function, cells were cultured for 5 days until mature osteoclasts formed and subsequently treated with indicated doses of SPC for another 48 h.
To evaluate the effects of SPC on osteogenic differentiation, osteoblasts were cultured in 6‐well plates in osteoinductive medium supplemented with varying concentrations of SPC (0, 0.25, 0.50 or 1.00 mM) for 7 or 14 days.
To investigate the effects of SPC on inflammation induced by titanium (Ti), BMMs were cultured in 6‐well plates with complete medium containing 30 ng·mL−1 M‐CSF, 0.1 mg·mL−1 Ti particles and varying doses of SPC (0, 0.25, 0.50 or 1.00 mM) for 4 h.
Total cellular RNA was extracted according to the manufacturer's instructions using the Qiagen RNeasy Mini kit (Qiagen, Valencia, CA, USA), and total RNA (≤1000 ng) was reverse‐transcripted into cDNA in a 20 μL‐reaction volume using a Double‐Strand cDNA Synthesis Kit (Takara, Dalian, China). The PCR reaction was performed with 1 μL of cDNA as template in triplicates using the Power SYBR® Green PCR Master Mix (Takara) on the ABI StepOnePlus System (Applied Biosystems, Warrington, UK). The cycling conditions were as follows: 95°C for 30 s and then 40 cycles of 95°C for 5 s and 60°C for 30 s. GAPDH was utilized as the housekeeping gene and independent experiments were repeated at least three times. The rat primer sequences were as follows: GAPDH: forward, 5′‐AGG GCT GCC TTC TCT TGT GAC‐3′and reverse, 5′‐TGG GTA GAA TCA TAC TGG AAC ATG TAG‐3′; Cathepsin K: forward, 5′‐CCC AGA CTC CAT CGA CTA TCG‐3′ and reverse, 5′‐CTG TAC CCT CTG CAC TTA GCT GCC‐3′; TRAP: forward, 5′‐GTG CAT GAC GCC AAT GAC AAG‐3′ and reverse, 5′‐TTT CCA GCC AGC ACG TAC CA‐3′; NFATc1: forward, 5′‐CAG CTG CCG TCG CAC TCT GGT C‐3′ and reverse, 5′‐CCC GGC TGC CTT CCG TCT CAT A‐3′; c‐Fos: forward, 5′‐CAG CCT TTC CTA CTA CCA TTC C‐3′ and reverse, 5′‐ACA GAT CTG CGC AAA AGT CC‐3′; dendritic cell‐specific transmembrane protein (DC‐STAMP): forward, 5′‐TTC ATC CAG CAT TTG GGA GT‐3′ and reverse, 5′‐CAT CCA GAC AGC AGA GAG CA‐3′; V‐ATPase d2: forward, 5′‐CAT TTG GCA CTG AAC TGA GC‐3′ and reverse, 5′‐GTC TTC GGC TTG GGC TAAC‐3′; iNOS: forward, 5′‐AGC GGC CCA TGA CTC TCA‐3′ and reverse, 5′‐CTG CAC CCA AAC ACC AAG GT‐3′; COX‐2: forward, 5′‐CCT TGA AGA CGG ACT TGC TCA C‐3′ and reverse, 5′‐TCT CTC TGC TCT GGT CAA TGG A‐3′; TNF‐α: forward, 5′‐TAC TCC CAG GTT CTC TTC AAG G‐3′ and reverse, 5′‐GGA GGC TGA CTT TCT CCT GGT A‐3′; IL‐6: forward, 5′‐GAG TTG TGC AAT GGC AAT TC‐3′ and reverse, 5′‐ACT CCA GAA GAC CAG AGC AG‐3′; IL‐1β: forward, 5′‐GGG CCT CAA GGG GAA GAA TC‐3′ and reverse, 5′‐ATG TCC CGA CCA TTG CTG TT‐3′; alkaline phosphatase (ALP): forward, 5′‐CAC GTT GAC TGT GGT TAC TGC TGA‐3′ and reverse, 5′‐CCT TGT AAC CAG GCC CGT TG‐3′; Runt‐related transcription factor 2 (RUNX2): forward, 5′‐GAC TGT GGT TAC CGT CAT GGC‐3′ and reverse, 5′‐ACT TGG TTT TTC ATA ACA GCG GA‐3′; osteocalcin (OCN): forward, 5′‐CCG GCC ACG CTA CTT TCT T‐3′ and reverse, 5′‐TGG ACT GGA AAC CGT TTC AGA‐3′; sp7: forward, 5′‐CAT CTA ACA GGA GGA TTT TGG TTT G‐3′ and reverse, 5′‐AAG CCT TTG CCC ACC TAC TTT T‐3′.
Western blot analysis
To investigate the exact signalling pathway which SPC suppressed, rat BMM cells were seeded into 6‐well plates with complete α‐MEM containing 30 ng·mL−1 M‐CSF for 24 h at a density of 80 × 104 per well and subsequently pretreated with or without 1.00 mM SPC for 4 h before being stimulated with 50 ng·mL−1 RANKL for 0, 5, 10, 20, 30 or 60 min. In addition, BMMs were seeded at a density of 10 × 104 cells per well were seeded in a 6‐well plate with complete α‐MEM in the presence of 30 ng·mL−1 M‐CSF and 50 ng·mL−1 RANKL for 0, 1, 3 or 5 days, under treatment with DMSO or 1.00 mM SPC, to examine the effects of SPC on NFATc1 and cathepsin K expression,. To investigate the effects of SPC on Ti‐induced expression of iNOS and COX‐2, rat BMMs were cultured in 6‐well plates at a density of 80 × 104 cells per well in complete α‐MEM containing 30 ng·mL−1 M‐CSF and different doses of SPC (0, 0.25, 0.50 or 1.00 mM) with or without 0.1 mg·mL−1 Ti particles for 24 h. Additionally, BMMs were seeded into 6‐well plates with complete α‐MEM containing 30 ng·mL−1 M‐CSF for 24 h at a density of 80 × 104 cells per well and were subsequently pretreated with or without 1.00 mM SPC for 4 h before being stimulated with 0.1 mg·mL−1 Ti particles for 0, 5, 10, 20, 30 or 60 min. Cells were collected and lysed using radioimmunoprecipitation assay (RIPA) buffer (Sangon Biotech Co. Ltd, Shanghai, China) on ice for 30 min, and the lysates were centrifuged at 9391 x g for 15 min, followed by the collection of supernatants. Total protein (30 μg per lane) was separated on 12% SDS polyacrylamide gels and transferred to PVDF membranes (Millipore, Merck KGaA, Germany). After non‐specific blocking with 5% (w/v) BSA‐TBS‐tween (TBST) for 1 h, membranes were incubated with primary antibodies diluted in TBST containing 5% (w/v) BSA at 4°C overnight. Subsequently, we incubated membranes with the appropriate secondary antibodies at 4°C for 2 h after rinsing three times in PBS and detected immunoreactive bands with a Bio‐Rad XRS chemiluminescence detection system (Bio‐Rad, Hercules, CA, USA).
Immunocytochemistry of NF‐κB nuclear translocation
BMMs were cultured on cell glass slides in a 24‐well plate at a density of 3 × 104 per well. BMMs were stimulated with 50 ng·mL−1 RANKL for 10 min, with or without pretreatment with 1.00 mM SPC for 4 h, followed by fixation in 4% (w/v) PFA for 30 min at 4°C. After being permeabilized and blocked for 30 min in 0.2% (w/v) Triton X‐100 and 5% (w/v) BSA at 4°C, cells were incubated with anti‐p65 antibody overnight. BMMs were then incubated with an appropriate fluorescence‐conjugated secondary antibody for 2 h and stained with DAPI for 10 min. The nuclear translocation of NF‐κB was subsequently examined under fluorescence microscopy (Leica, Wetzlar, Germany).
Animals
All animal care and experimental protocols complied with the Guide for the Care and Use of Laboratory Animals promulgated by the United States National Institutes of Health and was approved by the Animal Care and Use Committee of Zhejiang University. Male adult Sprague–Dawley rats weighing 350–400 g were obtained from the Experimental Animal Center of Zhejiang University. All animals were held in a room at 24 ± 2°C, 60% humidity and 12/12 h light/dark cycle with free access to food and water, with two animals per cage. Animals were evaluated daily for signs of pain, distress or morbidity visually, while their weights were recorded weekly. Animals with such signs or with 10% acute weight loss were killed humanely prior to the endpoint. In this study all efforts were made to minimize animal suffering and the number of animals used. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Establishment of implant loosening model in rats
Titanium (Ti) rods (1.5 mm in length and 15 mm in diameter) were supplied by the Experimental Research Center of Mechanics, Zhejiang University, and commercially available pure Ti particles were obtained from Johnson Matthey (Ward Hill, MA, USA). Ti rods and Ti particles were prepared by baking at 180°C and subsequent mixing with 70% (v/v) ethanol for 48 h to remove endotoxin and ensure sterility (Liu et al., 2009). A suspension of Ti particles was prepared as reported previously (Liu et al., 2012a; Bi et al., 2015).
Sixty‐six male Sprague–Dawley rats weighing 350–400 g were randomly allocated into three groups (n = 22 per group): the Ctrl group, the Ti group and the SPC group. Ti particles were used in the Ti group and the SPC group in the surgery, while vehicle was applied in the Ctrl group. After surgery, rats were treated with 20 mg·kg−1·day−1 SPC in the SPC group, while PBS was administered to the Ctrl and Ti groups. The rat model was established as previously reported (Liu et al., 2012b; Bi et al., 2015). Briefly, rats were anaesthetized with intramuscular xylazine (2 mg·kg−1) and ketamine (50 mg·kg−1), and bilateral intramedullary Ti rod implantation was performed in distal femurs under aseptic conditions. An implant was inserted into the medullary canal created parallel to the long axis of the femur to achieve a press fit fixation. Before Ti rod insertion, the suspension with 30 mg Ti (about 5 × 107 particles) was injected into the canal of rats in the Ti and SPC groups to induce implant loosening, while PBS was injected in the Ctrl group. SPC/PBS was injected i.p. every day after surgery, and the body weight of the animals was measured weekly to adjust the drug dosage. Buprenorphine (0.3 mg·kg−1 s.c.) and carprofen (4 mg·kg−1 p.o.) were administered pre‐ and post‐operation for analgesia respectively. Penicillin G Procaine 6000U•kg−1) was injected i.m. daily to prevent infection after operation for 3 days; 30 mg Ti suspension/PBS was re‐injected into the knee joint cavities every other week from the second week postoperatively. Four and 12 weeks after surgery, rats (n = 11 per group) were killed with administration of an overdose of pentobarbital (90 mg·kg−1, Sigma Chemical Co., St. Louis, MO, USA), and specimens were collected for further examination. Six femurs were collected for biomechanical testing, and they were wrapped with gauze in normal saline and stored at −20°C at each respective analysis point. The remaining femurs were fixed in 4% (w/v) PFA for bone mineral density (BMD), micro‐CT, histological and immunohistochemical analyses. The blood of animals was also collected, and serum was stored at −80°C for biochemical analysis (Supporting Information Figure S2A, B).
Peri‐implant site BMD measurement
The BMD values of the peri‐implant site (n = 11 per group) (Figure 5A) were assessed by using dual‐energy X‐ray absorptionmetry (iDXA; GE Lunar Prodigy, Madison, WI, USA). Femurs were placed in the same position, and the area beside the implant was regarded as the region of interest (ROI) for determining the BMD values.
Figure 5.
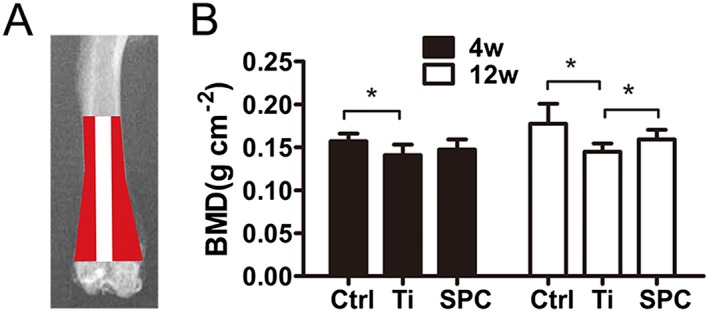
SPC prevented peri‐prosthesis bone loss in a rat model of aseptic implant loosening. (A) The sketch shows the peri‐prosthesis region measured for BMD values in the distal femurs. (B) All samples were quantified by iDXA at 4 or 12 weeks (4W or 12W) post‐surgery. Values expressed are means ± SD, n = 11. *P < 0.05, significantly different from the control group.
Micro‐CT scanning and analysis
Seven femurs from each group at each observation time point were subjected to micro‐CT scanning with an isometric resolution of 14.8 μm, using a Scanco μCT100 instrument (Scanco Medical, Bassersdorf, Switzerland). Three dimensional (3D) reconstructions of distal femurs were performed with the constrained Gaussian filter (σ = 1.2, support = 2) to partly suppress the noise in the volumes. A multilevel thresholds procedure (thresholds ranging from 220 to 520) was used to distinguish bone from other tissues and metal in vivo (Gabet et al., 2008). The area at a distance of 500 μm from the surface of implant was regarded as ROI. The parameters of bone trabecula surrounding the implant were quantified and analysed as previously reported (Bi et al., 2015), including the bone volume/total volume (BV/TV), connective density (Conn.D), structural model index (SMI), trabecular number (Tb.N), trabecular thickness (Tb.Th) and trabecular separation (Tb.Sp).
Biomechanical test of the implant pulling‐out strength
A total of 30 femurs (n = 6 per group at each observation time) being stored at −20°C were thawed completely at 4°C overnight before biomechanical testing. Pull‐out testing was performed as previously described (Liu et al., 2012a; Bi et al., 2015). Briefly, as shown in Figure 7A, the distal femur with implant inside was fixed vertically in dental cement and positioned on the platform with a canal in the middle. A 1.5‐mm‐diameter indenter was used to push the implant out of the bone cavity with a speed of 1 mm·min−1, and the maximum fixation strength was recorded by using a Zwick/Roell 2.5 material testing system (Zwick, Ulm, Germany).
Figure 7.
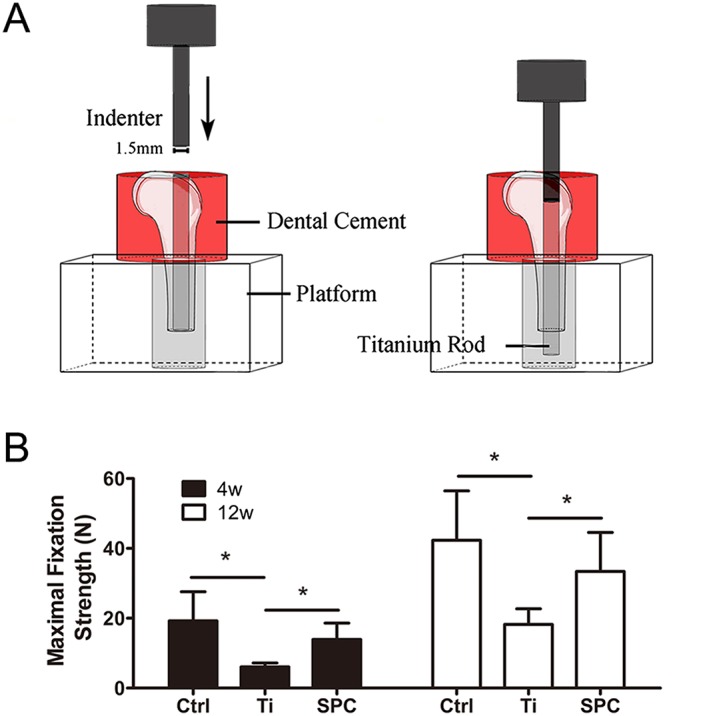
SPC inhibited the decrease in fixation strength of the implant, induced by wear particles, in the distal femurs. (A) The distal femurs were fixed vertically in dental cement and positioned on the platform with a canal, and a 1.5‐mm‐diameter indenter was subsequently used to push the implant out of the bone cavity. (B) Summary data. from samples taken at 4 weeks (4W) and 12 weeks (12W). of the maximal fixation strength. Values expressed are means ± SD; n = 6. *P < 0.05, significantly different as indicated.
Histological and immunohistochemical analysis
After complete decalcification of the bone samples in 10% (w/v) EDTA (pH = 7.4), the rods in the remaining specimens were removed and specimens were dehydrated and embedded in paraffin and then cut into five‐micron thick sections. Sections at a distance of 1 mm proximal to the distal femoral growth plate were selected for the histological analysis. Haematoxylin and eosin (H&E), Masson and TRAP staining were performed on the tissue sections, as previously described (Chen et al., 2015; Zhang et al., 2016b), and specimens were examined and measured under light microscopy (Olympus BX51, Tokyo, Japan). Parameters that were used for histological evaluation of osteolysis around the implant, including bone‐implant contact (BIC), ratio of bone area/total area (B.Ar/T.Ar) and mean thickness of the pseudomembrane. The area at a distance of 500 μm from the implant surface was regarded as ROI. Quantitative analysis of the mean thickness of pseudomembrane, BIC and B.Ar/T.Ar were performed in accordance with a previous study (Li et al., 2013). Briefly, B.Ar/T.Ar was measured through the trabecular area in the visual field/total area (Supporting Information Figure S3B); BIC was measured through the length of contact perimeter of the implant (green line), with the red line representing the uncontacted portion (Supporting Information Figure S3C), while mean thickness of the pseudomembrane was measured as the total area of the pseudomembrane surrounding the implant/perimeter of the implant (Supporting Information Figure S3D). The quantitative data of TRAP‐positive multinucleated osteoclasts formed on the bone surface were collected for each sample.
The levels of NLS‐p65 around the implants were analysed using an immunohistochemistry staining accessory kit (Boshide, Wuhan, China) according to the manufacturer's suggested protocol. At least five images (magnification 40×) from the fields at a distance of 300 μm from the implant in each section per femur were randomly selected and measured as a means of quantitatively analysing p65‐positive cells. The Image‐Pro Plus 6.0 software (Media Cybernetics Inc, Maryland, USA) was utilized for quantification.
Serum biomarkers
After death, rat blood (n = 10) was collected via the abdominal aorta and centrifuged to separate serum at 425× g for 5 min at 4°C as described previously (Liu et al., 2012a) and then stored at −80°C. Serum CTX‐1 level was analysed according to the manufacturer's protocol using specific ELISA kits. All samples, diluted appropriately, were assayed three times.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All experiments were measured by investigators blinded to the treatment. Data were expressed as mean ± SD. Each experiment was repeated at least five times independently and the results were analysed with SPSS 16.0 software (SPSS, Chicago, IL, USA). An unpaired t‐test was used for the comparisons between two groups. One‐way ANOVA with post hoc Newman–Keuls test was used to analyse differences in multiple comparisons. A probability level of P < 0.05 was considered statistically significant. A post hoc statistical power calculation was performed using G*Power 3.1 software (Dept. of Psychology, Univ Bonn, Germany), which showed that differences of 20% could be reliably detected with a power of more than 80%. Note that the values of the B.Ar/T.Ar ratio at 4 weeks were not covered by this calculation.
Materials
Modified Eagle's medium (α‐MEM), FBS and penicillin/streptomycin were purchased from Gibco‐BRL (Sydney, Australia). SPC, purchased from Selleck Chemicals (Houston, USA), was dissolved in DMSO and stored at −20°C in the dark, prior to being utilized in experiments. The Prime Script RT reagent kit and SYBR® Premix Ex Taq™ II were purchased from TaKaRa Biotechnology (Otsu, Shiga, Japan). The cell counting kit (CCK‐8) was purchased from Dojindo Molecular Technology (Kumamoto, Japan). Recombinant human M‐CSF and human RANKL were supplied by R&D systems (Minneapolis, MN, USA). Specific primary antibodies against ERK (#4695), JNK (#9252), p38 (#9212), IκBα (#4814), NF‐κB p65 (#8242), IκB kinase β (IKKβ) (#8943), phospho‐ERK (Thr202/Tyr204) (#4370), phospho‐JNK (Thr183/Tyr185) (#4668), phospho‐p38 (Thr180/Tyr182) (#4511), phospho‐IκBα (Ser32) (#2859), phospho‐NF‐κB p65 (Ser536) (#3033), phospho‐IKKα/β (Ser176/180) (#2697), phospho‐transforming growth factors‐β‐activated kinase 1 (TAK1) (#9339), TAK1 (#5206) and α‐tubulin (#2144) were obtained from Cell Signaling Technology (Cambridge, MA, USA); specific primary antibodies against GAPDH (sc‐20358), NFATc1 (sc‐7294), cathepsin K (sc‐48353), iNOS (sc‐7271) and COX‐2 (sc‐166475) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA); primary antibody against active NF‐κB‐p65(MAB3026) was obtained from Millipore (Merck KGaA, Germany). HRP‐conjugated secondary antibodies against rabbit IgG, mouse IgG and goat IgG were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Bone slices and type I collagen cross‐linked C‐terminal telopeptide (CTX‐1) were purchased from Immunodiagnostic Systems Limited (Boldon, UK). Commercial ELISA kits for rat PGE2 were purchased from R&D systems. Tris, glycine, SDS, rhodamine‐conjugated phalloidin for F‐actin staining, Diagnostic Acid Phosphatase kit for tartrate‐resistant acid phosphatase (TRAP), DMSO, Griess reagent and all other reagents were obtained from Sigma‐Aldrich (St. Louis, MO, USA)., unless stated otherwise.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18(Alexander et al., 2017a,b).
Results
SPC suppresses RANKL‐induced osteoclastogenesis of rat BMMs with negligible cytotoxicity
As shown in Supporting Information Figure S1, flow cytometry results revealed that more than 90% of the whole cell population were CD11b positive cells, after 2 days culture in the complete medium with M‐CSF. To assess the cytotoxicity of the compound, BMMs were exposed to varying doses of SPC ranging from 0 to 2 mM for 48 and 96 h. No toxic effects of SPC were found up to the maximal concentration (Figure 1B). BMMs were then cultured with 30 ng·mL−1 M‐CSF and 50 ng·mL−1 RANKL with different doses of SPC (0.25, 0.5 and 1 mM) or DMSO of same volume. Dose‐dependent suppression of osteoclast formation was confirmed, as indicated by the decreasing number of and area occupied by TRAP‐positive multinucleated cells (Figure 1C–E). Moreover, TRAP‐positive osteoclasts began to fuse and form after two‐day RANKL treatment of the control, whereas significant inhibition of osteoclast differentiation was observed upon treatment with 1 mM SPC (Figure 1F–H).
To investigate which stage of osteoclastogenesis was impaired by SPC, cells were treated with 1 mM SPC at days 1–3 of RANKL stimulation (early stage), days 3–5 (late stage) or days 1–5 (early + late stage). As reported, BMMs proliferate in the early stage, and fuse with one another, forming a multinuclear cell in the late stage (Ikeda and Takeshita, 2016). As shown in Figure 1I–K, a small but marked reduction in the number and size of osteoclasts were observed with early‐stage SPC administration. However, no difference was found between the control group and late‐stage SPC administration. In the early + late stage SPC administration group, a significant decrease in the size and number of osteoclasts was observed. Collectively, these data confirmed the effects of SPC on inhibiting osteoclast differentiation with negligible cytotoxicity, specific to the early stage and the entire period of RANKL stimulation.
SPC attenuates the formation of F‐actin ring and bone resorption in vitro
To further investigate the effects of SPC on osteoclast function, we investigated whether SPC could suppress formation of F‐actins, which is generally regarded as a prerequisite for osteoclast bone resorption (Wilson et al., 2009). As shown in Figure 2A, a characteristic F‐actin ring formed in osteoclasts without SPC treatment, as visualized by phalloidin‐Alexa Fluor 647 staining. In contrast, the sizes of F‐actin rings were reduced, and more pleomorphic F‐actin rings appeared under SPC treatment in a dose‐dependent manner (Figure 2C).
Figure 2.
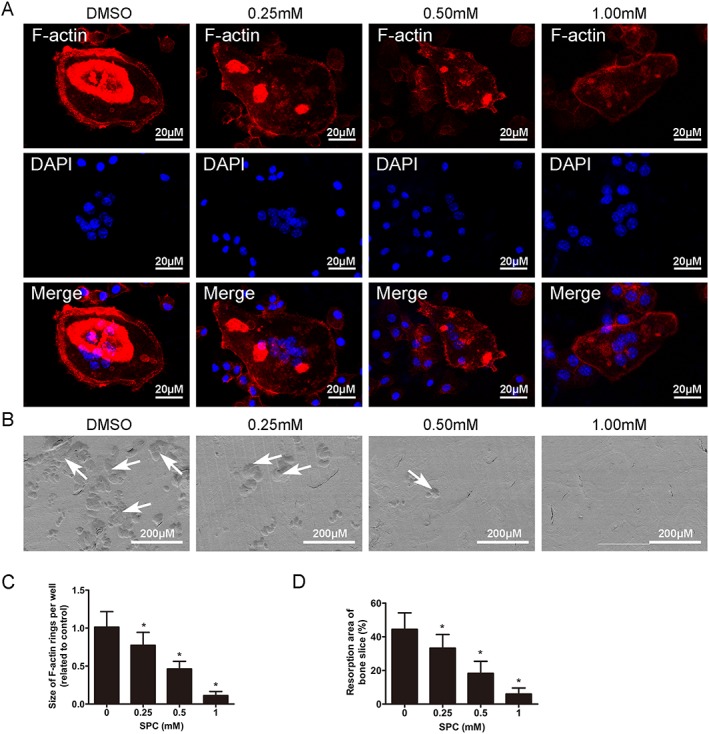
SPC inhibited osteoclast F‐actin ring formation and bone resorption in vitro. (A) BMMs were cultured in the presence of M‐CSF (30 ng·mL−1) and RANKL (50 ng·mL−1) until the mature osteoclasts were formed in the control group, followed by treatment with indicated dilutions of SPC for another 48 h. Cells were fixed and stained for F‐actin. The fluorescence images were detected by a NIKON A1Si spectral detector confocal system. (B) Equal number of osteoclasts were seeded on the bovine bone slices overnight, following by treatment with indicated doses of SPC for another 48 h. Cells were removed, and bone resorption pits were detected using SEM. (C) The area of F‐actin rings was measured. (D) The resorption areas were quantified. Values expressed are means ± SD, n = 5; *P < 0.05, significantly different from the control group.
With DMSO or different SPC concentrations, mature osteoclasts were next cultured on bone slices to perform an osteoclastic bone resorption assay. We detected large bone resorption pits on the bone surface by SEM analysis in the control group, that is, without SPC treatment (Figure 2B). This resorption area was reduced, in a dose‐dependent manner, after treatment with 0.25, 0.5 and 1 mM SPC, compared to the control group (Figure 2D). Therefore, these findings suggested that SPC reduced osteoclastic bone resorption in vitro.
SPC down‐regulates osteoclast‐specific gene expression levels in vitro
To further clarify the role of SPC on osteoclast differentiation and function, we utilized quantitative PCR (qPCR) to examine mRNA expression levels of osteoclast‐specific genes, which are generally up‐regulated during the process of osteoclastogenesis (Boyle et al., 2003). The data indicated that the mRNA expression levels of several markers of osteoclast differentiation, including cathepsin K, TRAP, NFATc1, c‐Fos, DC‐STAMP and V‐ATPase d2, were all significantly up‐regulated upon induction by RANKL during the osteoclast differentiation process. However, clear dose‐ and time‐dependent suppressions were observed after day 3, in the groups with SPC treatment during osteoclastogenesis (Figure 3A, B). Moreover, the mRNA expression levels of cathepsin K, TRAP and V‐ATPase d2, which are crucial for mature osteoclast functions, were also inhibited after 48 h of SPC treatment in mature osteoclasts (Figure 3C).
Figure 3.

SPC reduced osteoclast‐associated gene expression during RANKL‐induced osteoclast formation and bone resorption. (A) BMMs in the presence of M‐CSF (30 ng·mL−1) and RANKL (50 ng·mL−1) were treated with different concentrations of SPC for 5 days (B) or with or without 1.00 mM SPC for 0, 1, 3 or 5 days. Real‐time PCR was performed to analyse osteoclast‐specific gene expression (cathepsin K, TRAP, NFATc, c‐Fos, DC‐STAMP and V‐ATPase d2). (C) BMMs were cultured with indicated dilutions of SPC in the presence of M‐CSF (30 ng·mL−1) and RANKL (50 ng·mL−1) until mature osteoclasts were formed in the control group, followed by treatment with indicated dilutions of SPC for another 48 h. Osteoclast function‐related gene expression (TRAP, cathepsin K and V‐ATPase d2) were analysed, and all results were normalized to the expression of GAPDH. The data was obtained from five independent experiments. Values expressed are means ± SD. *P < 0.05, significantly different from the control group.
SPC inhibits rat osteoclast formation by suppressing activation of the NF‐κB signalling pathway without affecting MAPK signalling pathways in vitro
We next used Western blotting to explore the exact mechanism by which SPC inhibits osteoclastogenesis. As the NF‐κB and MAPK signalling pathways are recognized as the main signalling mechanisms in osteoclastogenesis (Boyle et al., 2003; Stevenson et al., 2011), we investigated further to confirm whether SPC suppressed osteoclast formation through these two signalling pathways. Upon RANKL stimulation, the phosphorylation of p65, ERK1/2, JNK1/2 and p38 in BMMs were elevated. However, in the SPC‐treated group (1 mM), there was an increase in p65 phosphorylation after 5 and 10 min stimulation with RANKL, but not to the same extent as RANKL alone, whereas no inhibition was observed in the phosphorylation of ERK1/2, JNK1/2 and p38 (Figure 4A, B and Supporting Information Figure S4A–C). The results thus showed that SPC impaired the NF‐κB signalling pathway rather than the MAPK signalling pathway.
Figure 4.
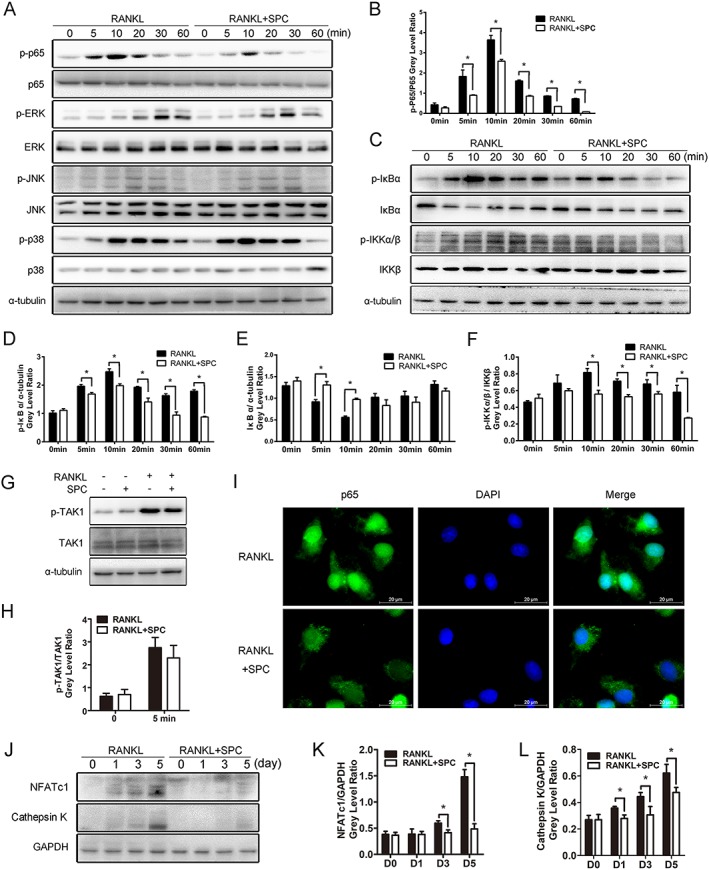
SPC inhibited osteoclastogenesis by suppressing RANKL‐induced activation of NF‐κB signalling without affecting MAPK pathways in vitro. (A, C) Rat BMMs were pretreated with 1.00 mM SPC or DMSO for 4 h and subsequently treated with 50 ng·mL−1 RANKL for 0, 5, 10, 20, 30 and 60 min. Cells were collected, and cell lysates were analysed by Western blotting using primary antibodies specific to phospho‐p65, p65, phospho‐ERK1/2, ERK, phospho‐JNK1/2, JNK, phospho‐p38, p38, phospho‐IκBα, IκBα, phospho‐IKKα/β, IKKα/β and α‐tubulin. (B, D, E, F) (n = 5). The grey levels of phospho‐p65 and phospho‐IKKα/β were analysed by being normalized to total p65 and IKKα/β. The grey levels of phospho‐IκBα and IκBα were normalized to α‐tubulin. (G) Rat BMMs were pretreated with 1.00 mM SPC or DMSO for 4 h and subsequently treated with 50 ng·mL−1 RANKL for 0 or 5 min. Cell lysates were analysed by Western blotting using primary antibodies specific to phospho‐TAK1 and TAK1 (n = 5). (H) The grey level of phospho‐TAK1 was normalized to TAK1. (I) Rat BMMs were pretreated with 1.00 mM SPC or DMSO for 4 h and then stimulated with 50 ng·mL−1 RANKL for 10 min. Cells were fixed and incubated with DAPI and a specific primary antibody against NF‐κB/p65. NF‐κB/p65 (green) and nuclei (blue) were detected under fluorescence microscopy (Leica) (n = 5). (J) Rat BMMs were cultured with or without 1.00 mM SPC in culture media containing M‐CSF (30 ng·mL−1) and RANKL (50 ng·mL−1) for 0, 1, 3 and 5 days. Cell lysates were analysed by Western blotting using specific antibodies against NFATc1 and cathepsin K (n = 5). (K, L) The grey levels of NFATc1 and cathepsin K were normalized relative to β‐actin using Image J Software. Values expressed are means ± SD; *P < 0.05, significantly different from the control group.
To elucidate the mechanism by which SPC inhibited osteoclastolgenesis through NF‐κB signalling, we explored the cascade of signalling proteins related to the pathway. In canonical NF‐κB signalling associated with osteoclast formation, activation of RANK results in the phosphorylation of the IκB kinase (IKK) complex, which could promote the phosphorylation and degradation of IκBα downstream, leading to phosphorylation and nuclear translocation of p65, which then initiates activation of the downstream pathway (Yu et al., 2014). As shown in Figure 4C–F, we found that the phosphorylation of IκBα and IKK was down‐regulated by SPC treatment, along with inhibition of degradation of IκBα. To confirm the exact molecular target of SPC, we explored its effect on the upstream regulatory factor TAK1. Interestingly, no change was observed when BMMs were incubated with RANKL or RANKL combined with SPC (Figure 4G, H), showing that IKKs might be the targets of SPC. Moreover, immunofluorescence staining revealed that the RANKL‐induced nuclear translocation of p65 was blocked upon pretreatment with SPC (Figure 4I).
NFATc1, the downstream transcription factor of NF‐κB signalling, serves as the master regulator of osteoclastogenesis (Yamashita et al., 2007). It induces the marker gene expression of mature osteoclasts, such as cathepsin K and TRAP. In our study, we found that protein expression levels of NFATc1 and cathepsin K increased in a time‐dependent manner at 1, 3 and 5 days in the presence of RANKL. However, SPC treatment strongly inhibited the elevation of protein expression levels of NFATc1 and cathepsin K (Figure 4J–L). Consequently, SPC inhibited rat osteoclastogenesis through the NF‐κB signalling pathway.
SPC has no effects on the osteogenic differentiation of rat primary osteoblasts
As suppression of the NF‐κB signalling pathway was previously reported to up‐regulate osteoblast differentiation (Novack, 2011), we next investigated the effects of SPC on the osteogenic differentiation of rat primary osteoblasts. No cytotoxicity of SPC was detected in osteoblasts up to 1 mM (Supporting Information Figure S5A). As shown in Supporting Information Figure S2B, C, no differences were observed between osteoblasts treated with osteoinductive medium in the presence of 0, 0.25, 0.50 or 1 mM SPC, as shown by ALP staining at 7 days and Alizarin Red S staining at 14 days (Supporting Information Figure S5B, C). The unchanged mRNA expression levels of ALP, sp7, RUNX2 and OCN in each group on day 14 further confirmed the previous observation (Supporting Information Figure S5D).
Administration of SPC inhibits implant loosening in a rat femoral model
To investigate the effects of SPC on wear‐particle implant loosening, we utilized a rat femoral model. The administration of 20 mg·kg−1 SPC daily was found to have the beneficial effect of preventing inflammation in rats (Yifeng et al., 2011). Therefore, this dose was utilized for subsequent experiments. As shown in Supporting Information Figure S6, significant reduction in body weight was found in the Ti group from week 5 to the end, as compared with the control group. Comparable increase in body weights were observed in the SPC group, relative to the Ti group in the last 3 weeks.
Bilateral femurs were collected and analysed by iDXA to determine the BMD of the peri‐implant bone. The results (Figure 5B) indicated that the BMD of the Ti group was markedly decreased in comparison with the control at 4 and 12 weeks post‐surgery. The BMD of the SPC group was higher than that of the Ti group at 12 weeks after administration, but no differences were found at 4 weeks post‐surgery.
In order to further confirm the protective effects of SPC on particle‐induced bone loss, specimens were analysed by micro‐CT imaging and histological staining. According to 3D reconstruction of micro‐CT images, peri‐implant bone loss was detected in the Ti group at 4 and 12 weeks post‐surgery, compared with the control group. However, bone resorption in the SPC group was considerably lower than that in the Ti group (Figure 6A, D) at both time points. Quantification of bone parameters presented as BV/TV, Conn.D, SMI, Tb.N, Tb.Sp and Tb.Th confirmed the beneficial effects of SPC in suppressing bone loss (Figure 6G). Notably, peri‐implant bone mass of the group with long‐term SPC administration (12 weeks) demonstrated the high efficacy of SPC in preventing bone loss. Histological assessment (Figure 6B, C, E, F) showed that the presence of Ti particles led to abnormal structure at the bone‐implant interface, with increased pseudomembrane and reduction of bone‐implant contact at 4 and 12 weeks post‐surgery. However, rats in the SPC group, sampled at the same times, exhibited attenuated formation of pseudomembrane and improved bone‐implant contact. The quantitative histomorphometry analysis supported these findings (Figure 6H). At 4 weeks post‐surgery, the Ti group exhibited less BIC and thicker pseudomembrane when compared to the control group, whereas SPC treatment significantly enhanced BIC and inhibited pseudomembrane formation. However, there were no significant differences in B.Ar/T.Ar among the three groups. At the late stage (12 weeks), lower B.Ar/T.Ar and BIC, as well as thicker pseudomembrane, were observed in the Ti group. Nevertheless, supplementation with SPC significantly inhibited peri‐implant trabecular loss and formation of interface membrane, as well as improved bone‐implant contact, consistent with the iDXA and micro‐CT results.
Figure 6.
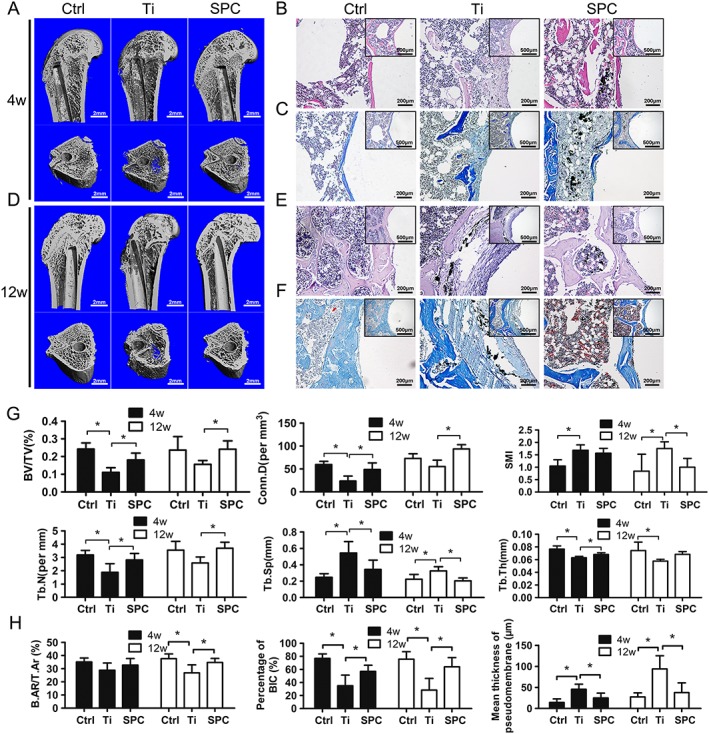
SPC prevented aseptic implant loosening in vivo, as demonstrated by micro‐CT and histological analysis at 4 weeks (4W) and 12 weeks (12W) post‐surgery . (A, D) Representative three‐dimensional micro‐CT images of bone trabecula around the implants were reconstructed from each group at each time point. (B, E) H&E staining and (C, F) Masson staining were performed for the histological analysis of at least three sections per group. (G) Using samples taken at4 weeks (4W) and 12 weeks (12W) post‐surgery, the bone volume/total volume (BV/TV), connective density (Conn.D), structural model index (SMI), trabecular number (Tb.N), trabecular thickness (Tb.Th) and trabecular separation (Tb.Sp) values were obtained and analysed. (H) Bone‐implant contact (BIC), bone area/total area (B.Ar/T.Ar) and mean thickness of the pseudomembrane were measured and quantified. Values expressed are means ± SD; n = 7. *P < 0.05, significantly different from the control group.
Biomechanical testing was performed to investigate the effects of SPC on maintaining implant stability within the medullary cavity. The outcomes are presented as the maximal fixation strength (Figure 7B). Particle‐induced implant loosening resulted in lower maximal fixation strength in the Ti group, as compared to the control group at 4 and 12 weeks post‐surgery respectively. However, in the SPC group, the maximal fixation strength improved significantly, compared with that in the Ti group at both 4 and 12 weeks post‐surgery.
Administration of SPC suppresses osteoclastogenesis and serum marker of bone resorption in vivo
TRAP staining and serum analysis were used at 12 weeks to investigate whether SPC prevented implant loosening by inhibiting osteoclast formation and activity at the bone‐implant surface. A significantly increased number of multinucleated osteoclasts being formed at the lining of the eroded bone surface in the presence of particles was observed, whereas the number and the size of osteoclasts were significantly reduced after the treatment with SPC (Figure 8A, B). Biochemical analysis of the serum bone resorption marker (CTX‐1) further confirmed that SPC suppressed osteoclast activity in vivo (Figure 8C).
Figure 8.

SPC prevented wear particle‐induced peri‐prosthesis loosening by suppressing osteoclast activity via inhibition of NF‐κB signalling pathway. (A) TRAP staining was performed on specimens (n = 5) after 12 weeks treatment with SPC, and three sections per sample were used for analysis. TRAP positive osteoclasts (black arrows) are indicated by arrows. (B) The number of TRAP positive osteoclasts normalized with respect to the bone surface and the percentages of osteoclast surface per bone surface were measured. (C) Serum levels of CTX‐1 were quantified by ELISA. Values expressed are means ± SD; n = 10. *P < 0.05, significantly different from the control group. (D) Sections of specimens around the implants from each group (at 4weeks and 12 weeks) were stained with a specific antibody against active p65, and images were captured under light microscopy. (E) Active p65 positive cells (red arrows) in the trabecular bone and surrounding bone marrow were quantified. At least five sections per group were analysed. Values expressed are means ± SD; n = 5. *P < 0.05, significantly different as indicated.
SPC attenuates implant loosening through NF‐κB signalling in vivo
Our in vitro results indicate that SPC down‐regulates osteoclast formation and bone resorption through suppressing the NF‐κB signalling pathway. To confirm our findings in vivo, we used immunohistochemical staining for activated p65 to investigate the effects of SPC on NF‐κB. Our data demonstrated that NF‐κB activity in osteoclasts and bone marrow cells surrounding the implant was enhanced by wear particles at 4 and 12 weeks post‐surgery. In contrast, the expression of activated p65 was significantly attenuated upon treatment with SPC at both 4 and 12 weeks post‐surgery (Figure 8D, E). Hence, the inhibition of implant loosening by SPC was confirmed in vivo.
SPC suppresses Ti‐induced inflammation by inhibition of the NF‐κB signalling pathway in rat BMMs
The activation of NF‐κB signalling by the toll‐like receptors (TLRs)/MyD88‐dependent pathways in macrophage also plays a role in particle‐induced osteolysis through inflammatory activities (Landgraeber et al., 2014). Based on our observation, we next explored the effects of SPC on Ti‐induced activation of the NF‐κB signalling pathway in BMMs. The Western blot results demonstrated that SPC partly suppressed Ti‐induced activation of phosphorylated p65 (Supporting Information Figure S7A, B). Upon further investigating the effects of SPC on IκBα and IKK, we found that SPC also inhibited the activation of phosphorylated IκBα and IKK, as well as the degradation of IκBα (Supporting Information Figure S7C–F).
To elucidate the effects of SPC on Ti‐induced inflammation, we investigated the expression of inflammatory mediators (NO and PGE2) and pro‐inflammatory cytokines (Blaine et al., 1997; Hukkanen et al., 1997). As shown in Supporting Information Figure S8A, the production of NO and PGE2 were markedly increased after administration of Ti, while SPC successfully reduced their production in a dose‐dependent manner. COX‐2 and iNOS have been reported to be the key enzymes responsible for the production of NO and PGE2. Hence, we analysed the expression of COX‐2 and iNOS. The results showed that the Ti‐induced elevated mRNA and protein expression levels of COX‐2 and iNOS were inhibited by SPC in a concentration‐dependent manner (Supporting Information Figure S8B–D). Meanwhile, the effects of SPC on the Ti‐induced production of three pro‐inflammatory cytokines, TNF‐α, IL‐1β and IL‐6, were assayed using qPCR. Supporting Information Figure S9A–C showed that the increased mRNA expressions of TNF‐α, IL‐6 and IL‐1β induced by Ti were suppressed the administration of SPC in a concentration‐dependent manner. In summary, SPC might mediate anti‐inflammatory activity in Ti‐induced rat BMMs via suppression of the NF‐κB signalling pathway.
Discussion
Aseptic implant loosening is one of the most common complications of TJA, and over‐activated and increased osteoclast activity play a crucial role in this disease. In our study, we have shown that SPC exhibits anti‐osteoclastogenic and anti‐bone resorptive effects by suppressing RANKL‐induced activation of NF‐κB signalling in vitro. We have also demonstrated the preventive effects of SPC on particle‐induced implant loosening in a rat model by inhibiting osteoclast formation, resulting in reduced periprosthetic bone loss, diminished pseudomembrane formation, improved bone‐implant contact and reduced bone resorption‐related turnover, as well as enhancement in the stability of the implants. Moreover, we confirmed that, in vivo, SPC attenuated wear‐induced osteolysis by modulating NF‐κB signalling. Therefore, our study provides evidence that SPC may be a novel treatment for the prevention and treatment of particle‐induced implant loosening.
RANKL plays a critical role in driving osteoclast differentiation and function upon binding to RANK on osteoclast precursors, which subsequently triggers a series of cascade activation of downstream signalling pathways, including the ERK, p38, JNK and NF‐κB pathways. NF‐κB signalling is vital for osteoclast differentiation, as it has been reported that severe osteopetrosis was observed in the p50/p52 double‐knockout mouse by blocking osteoclastogenesis (Iotsova et al., 1997). After activation by RANKL, the RANK receptor associates its cytoplasmic domain with TNF receptor‐associated factor‐6 (TRAF6), which further forms a complex with Tak1 and Tak1 binding protein‐2 (Tab2) (Lomaga et al., 1999; Bai et al., 2008). This complex then phosphorylates and activates Tak1, which in turn leads to the phosphorylation of IKK (Mizukami et al., 2002). However, in its inactive state, a complex consisting of NF‐κB with IκB protein is retained within the cytoplasm (Lee and Kim, 2003). Upon RANKL stimulation, the two catalytic components of IKK, IKKα and IKKβ, work together, resulting in proteasomal degradation of IκB, which subsequently leads to nuclear translocation of NF‐κB p65/RelA to modulate the transcription of genes further downstream (Soysa and Alles, 2009). In our study, we observed that SPC could suppress the phosphorylation of p65. Further investigation showed that SPC inhibited the phosphorylation of IKK and IκBα, as well as the proteasomal degradation of IκBα. However, there were no significant effects of SPC on RANKL‐induced activation of TAK1. These data thus indicate that IKKs might be the molecular targets of SPC. Moreover, the results from immunofluorescence staining confirmed the inhibitory effects of SPC on the nuclear translocation of p65 in vitro. Immunohistochemistry of activated p65 around the implants revealed the inhibition of NF‐κB by SPC in vivo. However, SPC did not affect the phosphorylation of the MAPK signalling pathway.
NFATc1 is identified as the key molecular target of the NF‐κB signalling pathway for modulating terminal osteoclastogenic differentiation (Yamashita et al., 2007). The NF‐κB proteins are bind to the NFATc1 promoter after nuclear translocation, resulting in auto‐amplification of NFATc1 during osteoclastogenesis. This factor is a major regulator of osteoclast‐specific genes and protein expression, including TRAP, cathepsin K, DC‐STAMP and V‐ATPase d2. We demonstrated that SPC attenuated the expression levels of these genes at the mRNA and protein levels, as well as osteoclast differentiation and bone resorption.
To our knowledge, the osteoclast is regarded as the primary cell type causing implant loosening. We then established a rat model of titanium particle‐induced implant loosening in distal femurs, which serves as one of the classical experimental models of prosthesis loosening (Liu et al., 2012a,b; Bi et al., 2015). Local distribution of particles around the implants has been reported to diminish weight gain, due to inflammation (Liu et al., 2012a). Thick fibrous pseudomembranes, containing wear particles, are usually observed between loose implants and cortical or cancellous bone, which is the result of a variable lymphocyte reaction (Bauer and Schils, 1999a,b; Athanasou, 2007), resulting in the increase of osteoclast formation and activation of bone resorption, thus eventually leading to the reduction of implant fixation. Many studies have previously reported that SPC exerts anti‐inflammatory activity in vivo with different disease models (Gao et al., 2009; Song et al., 2015; Wang et al., 2016), but no one has yet investigated its pharmacological effects on aseptic loosening. Our results demonstrated inhibition by SPC of the reduction in weight gain, induced by Ti particles, an effect which may be due to its anti‐inflammatory action. The assays using iDXA and Micro‐CT revealed the protective effects of SPC on wear particles‐induced periprosthetic bone loss in vivo. Histological data of H&E and Masson staining further confirmed its effects on suppressing bone loss and pseudomembrane formation, as well as improving bone‐implant contact. As previously observed in similar models (Smith et al., 2010; Liu et al., 2012b), fixation strength of femoral implants were significantly reduced due to aseptic loosening induced by intraarticular administration of wear debris, whereas administration of SPC successfully inhibited the fixation reduction. Moreover, TRAP staining confirmed the inhibitory effect of SPC on osteoclastogenesis in vivo. As wear‐induced peri‐implant osteolysis is a long‐term process of net bone resorption that precedes aseptic loosening in the clinic, we chose to sample at 12 weeks of SPC administration in order to observe osteoclasts. Elevated levels of serum CTX‐1, a biomarker of bone resorption, have been reported in patients with loosened implants, as compared to those with stable implants (Wilkinson et al., 2003; Streich et al., 2009), which corroborates our results. Moreover, we found that elevated levels of serum CTX‐1 were reduced by SPC. Our results thus provide evidence that SPC could prevent peri‐implant bone loss and aseptic implant loosening through inhibiting the activation of osteoclasts and bone resorption in vivo.
The NF‐κF signalling pathway is crucial in inflammation, and wear debris could induce local inflammation by stimulating the release of inflammatory cytokines in macrophages (Ingham and Fisher, 2005; Landgraeber et al., 2014). When metallic wear particles activate TLR4, the adaptor molecules MyD88, IL‐1 receptor‐associated kinase (IRAK)1, IRAK2, IRAK4 and TRAF6, all take part in the subsequent pathway , resulting in the activation of NF‐κB signalling and the production of proinflammatory cytokines and inflammatory mediators(Hirayama et al., 2011). Our results showed that SPC inhibited the Ti‐induced activation of the NF‐κB signalling pathway and further suppressed the expression of NO, PGE2, iNOS, COX‐2, TNF‐α, IL‐1β and IL‐6.
Nevertheless, there are some limitations to this study. UHMWPE wear particles were a more common cause of TJA failure than metal particles (Hirakawa et al., 1996). However, metal particles are still a causative factor of implant loosening, and both metal and UHMWPE particles could induce osteoclastogenesis and osteolysis in vivo. Therefore, it is reasonable to utilize Ti particles in an animal model, even though UHMWPE particles would be more closely related to the clinical findings. As the statistical power value for the B.Ar/T.Ar ratio in the 4‐week samples is lower than 80%, this statistical defect might explain why no differences were detected between different groups in terms of B.Ar/T.Ar in the 4 weeks samples. However, as reliable differences on the BV/TV results of Micro‐CT in 4 week samples and B.Ar/T.Ar results of histology in the 12 week samples were detected, we did not increase the number of animals for histology.
Collectively, our study shows SPC exerts an inhibitory effect on osteoclast formation and function by suppressing NF‐κB signalling in vitro. We also show its efficacy, in vivo, in preventing particle‐induced prosthesis loosening and in improving prosthesis stability in a rat model. Moreover, the effect of SPC on NF‐κB signalling was confirmed in vivo. Therefore, we can conclude that SPC may provide a novel therapeutic approach for preventing prosthesis loosening and osteoclastic diseases.
Author contributions
S.‐g.Y., W.‐l.S. and C.‐h.Z. designed this study. C.‐h.Z. and J.‐h.M., Z.‐l.S. and B.H. performed most of the experiments. Y.‐t.Y., S.J. and H.‐x.Z. performed some of the molecular cell experiments. C.‐h.Z., Z.‐x.C., C.‐c.Z., H.‐b.W. and W.Y. performed the statistics analysis. V.‐J.A.P. and B.C.H. gave helpful suggestions on the manuscript. Experiments were performed under the supervision of S.‐g.Y. and W.‐l.S. C.‐h.Z. wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Flow cytometry analysis of CD11b positive expression was measured on BMMs. 91.3 ± 3.1% BMMs are CD11b positive cells. Data on graph are the mean ± SD of five independent experiments.
Figure S2 The schematic diagrams showed the design of the study and animal size for each time point in each group. (A) A schematic diagram for the timeline of the study is shown. (B) The table shows the detailed tests for the study and the animal size for each tests at each time point.
Figure S3 The details for the quantification of B.Ar/T/Ar, BIC and the mean thickness of pseudomembrane. (A) An image of a slice for H&E staining. (B) The image shows the ROI and bone area (blue) for the histological analysis of B.Ar/T.Ar. (C) The bone‐implant contact part (red line) and out of contact part (green line). (D) The area of the pseudomembrane around the implant (yellow).
Figure S4 The quantification for SPC on the activation of MAPK signalling pathway. (A) The levels of phospho‐ERK were quantified by being normalized to total ERK. (B) The levels of phospho‐JNK were quantified by being normalized to total JNK. (C) The levels of phospho‐p38 were quantified by being normalized to total p38. Experiments were performed at least 3 times and values are expressed as mean ± SD.
Figure S5 SPC has no effects on osteogenic differentiation in vitro. (A) Rat primary osteoblasts were cultured in a 96‐well plated and treated with a range of concentrations of SPC (0.31‐2 mM) for 48 h or 96 h. The viability of cells was measured by the CCK8 kit. (B) Osteoblasts were cultured in osteoinductive medium in the presence of indicated dilutions of SPC for 7 days. ALP staining was performed and ALP activity was measured. (C) Osteoblasts were cultured in osteoinductive medium with different dilutions of SPC for 14 days. ARS staining and quantification were performed. (D) The mRNA expression of ALP, RUNX2, sp7 and OCN in the primary osteoblasts were quantified after osteogenic differentiation for 14 days in the presence of different concentrations of SPC. Experiments were performed at least 3 times and values are expressed as mean ± SD.
Figure S6 SPC prevented the decrease of weight gain in a rat model of aseptic implant loosening. Body weight of all rats was measured every week after SPC administration. n = 11 animals per group, Values are expressed as mean ± SD; *P < 0.05 versus the control group; #P < 0.05 versus the Ti group.
Figure S7 SPC impaired Ti‐induced activation of NF‐κB signalling pathway in vitro. (A, C) After 4 h pretreatment with 1 mM SPC or DMSO, BMMs were treated with 0.1 mg·mL−1 Ti for 0, 5, 10, 20, 30 and 60 min. Cells were collected and cell lysates were analysed by Western blotting using primary antibodies specific to phospho‐p65, p65, IκBα, phospho‐IKKα/β, IKKα/β,and α‐tubulin. (B, D, E) The levels of phospho‐p65 and phospho‐IKKα/β were normalized to total p65 and IKKα/β. IκBα was normalized to α‐tubulin. Three independent experiments were performed for the data analysis and values are expressed as mean ± SD; *P < 0.05 versus the control group.
Figure S8 SPC inhibited Ti‐induced NO and PGE2 production, as well as the expression of iNOS and COX‐2. (A) Rat BMMs were pretreated with different concentrations of SPC (0, 0.25, 0.50 or 1.00 mM) for 4 h and subsequently treated with 0.1 mg·mL−1 Ti or vehicle for 24 h. Supernatants were collected and the production of NO and PGE2 were measured. (B) After being pretreated with indicated concentrations of SPC for 4 h, BMMs were treated with or without 0.1 mg·mL−1 Ti for another 4 h. The mRNA expression of iNOS and COX‐2 were measured by RT‐PCR. (C) BMMs were pretreated with various dilutions of SPC for 4 h, following treated with 0.1 mg·mL−1 Ti particle or vehicle for another 24 h. The protein expression of iNOS and COX‐2 were explored using Western blotting. (D) The levels of iNOS and COX‐2 were normalized relative to α‐tubulin using Image J Software. Data are from three independent experiments and values are expressed as mean ± SD; *P < 0.05 versus Ti particles alone.
Figure S9 SPC suppressed Ti‐induced proinflammatory cytokine expression in rat primary macrophages. (A, B and C) After being pretreated with 30 ng·mL−1 M‐CSF and indicated dilutions of SPC for 4 h, rat primary BMMs were treated with or without 0.1 mg·mL−1 Ti for another 4 h. Real‐time PCR was performed to analyse the mRNA expression of TNF‐α, IL‐1β and IL‐6 and all results were normalized to the expression of GAPDH. All experiments were performed 5 times and values are expressed as mean ± SD; *P < 0.05 versus Ti particles alone.
Acknowledgements
This study was supported by research grants from the National Natural Science Foundation of China (81371954, 81472113, 81401785 and 81572157). We thank Dr Mengrui Wu and Dr Pei Ying Ng from the Harvard School of Dental Medicine and Dr An Qin from the Department of Orthopaedics, Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine for technical help and advice.
Zhou, C. , Shi, Z. , Meng, J. , Hu, B. , Zhao, C. , Yang, Y. , Yu, W. , Chen, Z. , Heng, B. C. , Parkman, V.‐J. A. , Jiang, S. , Zhu, H. , Wu, H. , Shen, W. , and Yan, S. (2018) Sophocarpine attenuates wear particle‐induced implant loosening by inhibiting osteoclastogenesis and bone resorption via suppression of the NF‐κB signalling pathway in a rat model. British Journal of Pharmacology, 175: 859–876. doi: 10.1111/bph.14092.
Contributor Information
Wei‐liang Shen, Email: wlshen@zju.edu.cn.
Shi‐gui Yan, Email: zrjwsj@zju.edu.cn.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The concise guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanasou NA (2007). Peri‐implant pathology – relation to implant failure and tumor formation. J Long Term Eff Med Implants 17: 193–206. [DOI] [PubMed] [Google Scholar]
- Athanasou NA (2016). The pathobiology and pathology of aseptic implant failure. Bone Joint Res 5: 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai S, Zha JK, Zhao HB, Ross FP, Teitelbaum SL (2008). Tumor necrosis factor receptor‐associated factor 6 is an intranuclear transcriptional coactivator in osteoclasts. J Biol Chem 283: 30861–30867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer TW, Schils J (1999a). The pathology of total joint arthroplasty. I. Mechanisms of implant fixation. Skeletal Radiol 28: 423–432. [DOI] [PubMed] [Google Scholar]
- Bauer TW, Schils J (1999b). The pathology of total joint arthroplasty.II. Mechanisms of implant failure. Skeletal Radiol 28: 483–497. [DOI] [PubMed] [Google Scholar]
- Bi F, Shi Z, Zhou C, Liu A, Shen Y, Yan S (2015). Intermittent administration of parathyroid hormone [1‐34] prevents particle‐induced periprosthetic osteolysis in a rat model. PLoS One 10: e0139793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaine TA, Pollice PF, Rosier RN, Reynolds PR, Puzas JE, O'Keefe RJ (1997). Modulation of the production of cytokines in titanium‐stimulated human peripheral blood monocytes by pharmacological agents. The role of cAMP‐mediated signaling mechanisms. J Bone Joint Surg Am 79: 1519–1528. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Simonet WS, Lacey DL (2003). Osteoclast differentiation and activation. Nature 423: 337–342. [DOI] [PubMed] [Google Scholar]
- Chen S, Jin G, Huang KM, Ma JJ, Wang Q, Ma Y et al (2015). Lycorine suppresses RANKL‐induced osteoclastogenesis in vitro and prevents ovariectomy‐induced osteoporosis and titanium particle‐induced osteolysis in vivo . Sci Rep 5: 12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabet Y, Kohavi D, Kohler T, Baras M, Muller R, Bab I (2008). Trabecular bone gradient in rat long bone metaphyses: mathematical modeling and application to morphometric measurements and correction of implant positioning. J Bone Miner Res 23: 48–57. [DOI] [PubMed] [Google Scholar]
- Gallo J, Vaculova J, Goodman SB, Konttinen YT, Thyssen JP (2014). Contributions of human tissue analysis to understanding the mechanisms of loosening and osteolysis in total hip replacement. Acta Biomater 10: 2354–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Jiang W, Dong C, Li C, Fu X, Min L et al (2012). Anti‐inflammatory effects of sophocarpine in LPS‐induced RAW 264.7 cells via NF‐kappaB and MAPKs signaling pathways. Toxicol In Vitro 26: 1–6. [DOI] [PubMed] [Google Scholar]
- Gao Y, Li G, Li C, Zhu X, Li M, Fu C et al (2009). Anti‐nociceptive and anti‐inflammatory activity of sophocarpine. J Ethnopharmacol 125: 324–329. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al, (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa K, Bauer TW, Stulberg BN, Wilde AH (1996). Comparison and quantitation of wear debris of failed total hip and total knee arthroplasty. J Biomed Mater Res 31: 257–263. [DOI] [PubMed] [Google Scholar]
- Hirayama T, Tamaki Y, Takakubo Y, Iwazaki K, Sasaki K, Ogino T et al (2011). Toll‐like receptors and their adaptors are regulated in macrophages after phagocytosis of lipopolysaccharide‐coated titanium particles. J Orthop Res 29: 984–992. [DOI] [PubMed] [Google Scholar]
- Holt G, Murnaghan C, Reilly J, Meek RM (2007). The biology of aseptic osteolysis. Clin Orthop Relat Res 460: 240–252. [DOI] [PubMed] [Google Scholar]
- Hukkanen M, Corbett SA, Batten J, Konttinen YT, McCarthy ID, Maclouf J et al (1997). Aseptic loosening of total hip replacement. Macrophage expression of inducible nitric oxide synthase and cyclo‐oxygenase‐2, together with peroxynitrite formation, as a possible mechanism for early prosthesis failure. J Bone Joint Surg 79: 467–474. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Takeshita S (2016). The role of osteoclast differentiation and function in skeletal homeostasis. J Biochem 159: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham E, Fisher J (2005). The role of macrophages in osteolysis of total joint replacement. Biomaterials 26: 1271–1286. [DOI] [PubMed] [Google Scholar]
- Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R (1997). Osteopetrosis in mice lacking NF‐kappaB1 and NF‐kappaB2. Nat Med 3: 1285–1289. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Jia T, Wooley PH, Yang SY (2013). Current research in the pathogenesis of aseptic implant loosening associated with particulate wear debris. Acta Orthop Belg 79: 1–9. [PubMed] [Google Scholar]
- Jurdic P, Saltel F, Chabadel A, Destaing O (2006). Podosome and sealing zone: specificity of the osteoclast model. Eur J Cell Biol 85: 195–202. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S et al (1999). Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 402: 304–309. [DOI] [PubMed] [Google Scholar]
- Landgraeber S, Jager M, Jacobs JJ, Hallab NJ (2014). The pathology of orthopedic implant failure is mediated by innate immune system cytokines. Mediators Inflamm 2014: 185150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ZH, Kim HH (2003). Signal transduction by receptor activator of nuclear factor kappa B in osteoclasts. Biochem Biophys Res Commun 305: 211–214. [DOI] [PubMed] [Google Scholar]
- Li C, Gao Y, Tian J, Shen J, Xing Y, Liu Z (2011). Sophocarpine administration preserves myocardial function from ischemia‐reperfusion in rats via NF‐kappaB inactivation. J Ethnopharmacol 135: 620–625. [DOI] [PubMed] [Google Scholar]
- Li J, Li L, Chu H, Sun X, Ge Z (2014). Oral sophocarpine protects rat heart against pressure overload‐induced cardiac fibrosis. Pharm Biol 52: 1045–1051. [DOI] [PubMed] [Google Scholar]
- Li YF, Li XD, Bao CY, Chen QM, Zhang H, Hu J (2013). Promotion of peri‐implant bone healing by systemically administered parathyroid hormone (1‐34) and zoledronic acid adsorbed onto the implant surface. Osteoporos Int 24: 1063–1071. [DOI] [PubMed] [Google Scholar]
- Liu F, Zhu Z, Mao Y, Liu M, Tang T, Qiu S (2009). Inhibition of titanium particle‐induced osteoclastogenesis through inactivation of NFATc1 by VIVIT peptide. Biomaterials 30: 1756–1762. [DOI] [PubMed] [Google Scholar]
- Liu S, Virdi AS, Sena K, Hughes WF, Sumner DR (2012a). Bone turnover markers correlate with implant fixation in a rat model using LPS‐doped particles to induced implant loosening. J Biomed Mater Res A 100: 918–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Virdi AS, Sena K, Sumner DR (2012b). Sclerostin antibody prevents particle‐induced implant loosening by stimulating bone formation and inhibiting bone resorption in a rat model. Arthritis Rheum 64: 4012–4020. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhu S, Cui J, Shao H, Zhang W, Yang H et al (2014). Strontium ranelate inhibits titanium‐particle‐induced osteolysis by restraining inflammatory osteoclastogenesis in vivo . Acta Biomater 10: 4912–4918. [DOI] [PubMed] [Google Scholar]
- Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A et al (1999). TRAF6 deficiency results in osteopetrosis and defective interleukin‐1, CD40, and LPS signaling. Genes Dev 13: 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizukami J, Takaesu G, Akatsuka H, Sakurai H, Ninomiya‐Tsuji J, Matsumoto K et al (2002). Receptor activator of NF‐kappaB ligand (RANKL) activates TAK1 mitogen‐activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Mol Cell Biol 22: 992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novack DV (2011). Role of NF‐κB in the skeleton. Cell Res 21: 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajarinen J, Lin TH, Sato T, Yao Z, Goodman S (2014). Interaction of materials and biology in total joint replacement – successes, challenges and future directions. J Mater Chem B Mater Biol Med 2: 7094–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K et al (2001). Mitogen‐activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22: 153–183. [DOI] [PubMed] [Google Scholar]
- Porter M (2016). National Joint Registry for England, Wales, Northern Ireland and the Isle of Man: 13th Annual Report.
- Purdue PE, Koulouvaris P, Potter HG, Nestor BJ, Sculco TP (2007). The cellular and molecular biology of periprosthetic osteolysis. Clin Orthop Relat Res 454: 251–261. [DOI] [PubMed] [Google Scholar]
- Qu SX, Bai YL, Liu XM, Fu R, Duan K, Weng J (2013). Study on in vitro release and cell response to alendronate sodium‐loaded ultrahigh molecular weight polyethylene loaded with alendronate sodium wear particles to treat the particles‐induced osteolysis. J Biomed Mater Res A 101: 394–403. [DOI] [PubMed] [Google Scholar]
- Smith RA, Maghsoodpour A, Hallab NJ (2010). In vivo response to cross‐linked polyethylene and polycarbonate‐urethane particles. J Biomed Mater Res A 93: 227–234. [DOI] [PubMed] [Google Scholar]
- Song CY, Zeng X, Wang Y, Shi J, Qian H, Zhang Y et al (2015). Sophocarpine attenuates toll‐like receptor 4 in steatotic hepatocytes to suppress pro‐inflammatory cytokines synthesis. J Gastroenterol Hepatol 30: 405–412. [DOI] [PubMed] [Google Scholar]
- Soysa NS, Alles N (2009). NF‐kappaB functions in osteoclasts. Biochem Biophys Res Commun 378: 1–5. [DOI] [PubMed] [Google Scholar]
- Stevenson DA, Schwarz EL, Carey JC, Viskochil DH, Hanson H, Bauer S et al (2011). Bone resorption in syndromes of the Ras/MAPK pathway. Clin Genet 80: 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streich NA, Gotterbarm T, Jung M, Schneider U, Heisel C (2009). Biochemical markers of bone turnover in aseptic loosening in hip arthroplasty. Int Orthop 33: 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaesu G, Ninomiya‐Tsuji J, Kishida S, Li X, Stark GR, Matsumoto K (2001). Interleukin‐1 (IL‐1) receptor‐associated kinase leads to activation of TAK1 by inducing TAB2 translocation in the IL‐1 signaling pathway. Mol Cell Biol 21: 2475–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Udagawa N, Tanaka S, Suda T (2003). Generating murine osteoclasts from bone marrow. Methods Mol Med 80: 129–144. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H et al (2002). Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 3: 889–901. [DOI] [PubMed] [Google Scholar]
- Wang D, Xu N, Zhang Z, Yang S, Qiu C, Li C et al (2016). Sophocarpine displays anti‐inflammatory effect via inhibiting TLR4 and TLR4 downstream pathways on LPS‐induced mastitis in the mammary gland of mice. Int Immunopharmacol 35: 111–118. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Deng HZ, Jiang B, Yao H (2012). The natural plant product sophocarpine ameliorates dextran sodium sulfate‐induced colitis in mice by regulating cytokine balance. Int J Colorectal Dis 27: 575–581. [DOI] [PubMed] [Google Scholar]
- Wilkinson JM, Hamer AJ, Rogers A, Stockley I, Eastell R (2003). Bone mineral density and biochemical markers of bone turnover in aseptic loosening after total hip arthroplasty. J Orthop Res 21: 691–696. [DOI] [PubMed] [Google Scholar]
- Wilson SR, Peters C, Saftig P, Bromme D (2009). Cathepsin K activity‐dependent regulation of osteoclast actin ring formation and bone resorption. J Biol Chem 284: 2584–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Yao Z, Li F, Zhang Q, Badell IR, Schwarz EM et al (2007). NF‐kappaB p50 and p52 regulate receptor activator of NF‐kappaB ligand (RANKL) and tumor necrosis factor‐induced osteoclast precursor differentiation by activating c‐Fos and NFATc1. J Biol Chem 282: 18245–18253. [DOI] [PubMed] [Google Scholar]
- Yang ZF, Li CZ, Wang W, Chen YM, Zhang Y, Liu YM et al (2011). Electrophysiological mechanisms of sophocarpine as a potential antiarrhythmic agent. Acta Pharmacol Sin 32: 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yifeng M, Bin W, Weiqiao Z, Yongming Q, Bing L, Xiaojie L (2011). Neuroprotective effect of sophocarpine against transient focal cerebral ischemia via down‐regulation of the acid‐sensing ion channel 1 in rats. Brain Res 1382: 245–251. [DOI] [PubMed] [Google Scholar]
- Yu B, Chang J, Liu Y, Li J, Kevork K, Al‐Hezaimi K et al (2014). Wnt4 signaling prevents skeletal aging and inflammation by inhibiting nuclear factor‐kappaB. Nat Med 20: 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang PP, Wang PQ, Qiao CP, Zhang Q, Zhang JP, Chen F et al (2016a). Differentiation therapy of hepatocellular carcinoma by inhibiting the activity of AKT/GSK‐3beta/beta‐catenin axis and TGF‐beta induced EMT with sophocarpine. Cancer Lett 376: 95–103. [DOI] [PubMed] [Google Scholar]
- Zhang W, Xue D, Yin H, Wang S, Li C, Chen E et al (2016b). Overexpression of HSPA1A enhances the osteogenic differentiation of bone marrow mesenchymal stem cells via activation of the Wnt/beta‐catenin signaling pathway. Sci Rep 6: 27622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Flow cytometry analysis of CD11b positive expression was measured on BMMs. 91.3 ± 3.1% BMMs are CD11b positive cells. Data on graph are the mean ± SD of five independent experiments.
Figure S2 The schematic diagrams showed the design of the study and animal size for each time point in each group. (A) A schematic diagram for the timeline of the study is shown. (B) The table shows the detailed tests for the study and the animal size for each tests at each time point.
Figure S3 The details for the quantification of B.Ar/T/Ar, BIC and the mean thickness of pseudomembrane. (A) An image of a slice for H&E staining. (B) The image shows the ROI and bone area (blue) for the histological analysis of B.Ar/T.Ar. (C) The bone‐implant contact part (red line) and out of contact part (green line). (D) The area of the pseudomembrane around the implant (yellow).
Figure S4 The quantification for SPC on the activation of MAPK signalling pathway. (A) The levels of phospho‐ERK were quantified by being normalized to total ERK. (B) The levels of phospho‐JNK were quantified by being normalized to total JNK. (C) The levels of phospho‐p38 were quantified by being normalized to total p38. Experiments were performed at least 3 times and values are expressed as mean ± SD.
Figure S5 SPC has no effects on osteogenic differentiation in vitro. (A) Rat primary osteoblasts were cultured in a 96‐well plated and treated with a range of concentrations of SPC (0.31‐2 mM) for 48 h or 96 h. The viability of cells was measured by the CCK8 kit. (B) Osteoblasts were cultured in osteoinductive medium in the presence of indicated dilutions of SPC for 7 days. ALP staining was performed and ALP activity was measured. (C) Osteoblasts were cultured in osteoinductive medium with different dilutions of SPC for 14 days. ARS staining and quantification were performed. (D) The mRNA expression of ALP, RUNX2, sp7 and OCN in the primary osteoblasts were quantified after osteogenic differentiation for 14 days in the presence of different concentrations of SPC. Experiments were performed at least 3 times and values are expressed as mean ± SD.
Figure S6 SPC prevented the decrease of weight gain in a rat model of aseptic implant loosening. Body weight of all rats was measured every week after SPC administration. n = 11 animals per group, Values are expressed as mean ± SD; *P < 0.05 versus the control group; #P < 0.05 versus the Ti group.
Figure S7 SPC impaired Ti‐induced activation of NF‐κB signalling pathway in vitro. (A, C) After 4 h pretreatment with 1 mM SPC or DMSO, BMMs were treated with 0.1 mg·mL−1 Ti for 0, 5, 10, 20, 30 and 60 min. Cells were collected and cell lysates were analysed by Western blotting using primary antibodies specific to phospho‐p65, p65, IκBα, phospho‐IKKα/β, IKKα/β,and α‐tubulin. (B, D, E) The levels of phospho‐p65 and phospho‐IKKα/β were normalized to total p65 and IKKα/β. IκBα was normalized to α‐tubulin. Three independent experiments were performed for the data analysis and values are expressed as mean ± SD; *P < 0.05 versus the control group.
Figure S8 SPC inhibited Ti‐induced NO and PGE2 production, as well as the expression of iNOS and COX‐2. (A) Rat BMMs were pretreated with different concentrations of SPC (0, 0.25, 0.50 or 1.00 mM) for 4 h and subsequently treated with 0.1 mg·mL−1 Ti or vehicle for 24 h. Supernatants were collected and the production of NO and PGE2 were measured. (B) After being pretreated with indicated concentrations of SPC for 4 h, BMMs were treated with or without 0.1 mg·mL−1 Ti for another 4 h. The mRNA expression of iNOS and COX‐2 were measured by RT‐PCR. (C) BMMs were pretreated with various dilutions of SPC for 4 h, following treated with 0.1 mg·mL−1 Ti particle or vehicle for another 24 h. The protein expression of iNOS and COX‐2 were explored using Western blotting. (D) The levels of iNOS and COX‐2 were normalized relative to α‐tubulin using Image J Software. Data are from three independent experiments and values are expressed as mean ± SD; *P < 0.05 versus Ti particles alone.
Figure S9 SPC suppressed Ti‐induced proinflammatory cytokine expression in rat primary macrophages. (A, B and C) After being pretreated with 30 ng·mL−1 M‐CSF and indicated dilutions of SPC for 4 h, rat primary BMMs were treated with or without 0.1 mg·mL−1 Ti for another 4 h. Real‐time PCR was performed to analyse the mRNA expression of TNF‐α, IL‐1β and IL‐6 and all results were normalized to the expression of GAPDH. All experiments were performed 5 times and values are expressed as mean ± SD; *P < 0.05 versus Ti particles alone.