

Abstract
Introduction
A major clinical challenge in treating allergic contact dermatitis (ACD) is that the first line of treatment is based on the use of corticosteroids. In this study, we aimed to develop a formulation that is devoid of steroids.
Methods
We used mouse ears treated with dinitrofluorobenzene (DNFB) to induce ACD. The efficacy of the test formulation to ameliorate and to prevent induced ACD was determined.
Results
To treat this experimentally induced ACD, we developed a formulation containing BIPxine (a mixture of Rosa moschata and Croton lechleri (antioxidants) and Aloe vera and d-panthenol (moisturizers), and hydroglycolic solutions of disodium cromoglycate. Our results show that clear inhibition of ACD took place. The target of this formulation was PAR-2, TRPV4, and other mediators of the inflammatory and pain responses. However, this formulation must be evaluated in other models besides the mouse to confirm its effectiveness.
Conclusion
The formulation presented here may provide new ACD therapies that do not involve the use of corticosteroids.
Electronic supplementary material
The online version of this article (10.1007/s13555-018-0223-8) contains supplementary material, which is available to authorized users.
Keywords: Cromoglycate, Ear thickness, Experimental allergic contact dermatitis, Inflammation, Nonsteroidal therapy
Introduction
Dermatitis, a general term for skin inflammation, is complex, with many subtypes, each with different etiologies, which may include dry skin, severe pruritus, pain, or lesions that, subsequently, may become infected [1, 2]. Atopic dermatitis (AD), resulting in inflamed, itchy, cracked skin, commences early in life, worsening as it progresses into adulthood [3], requiring ongoing, long-term, periodic treatments. The cause of AD is associated with environmental exposure, skin barrier dysfunction, immunological responses, and a complex interplay of susceptibility genes [4], affecting up to 20% of children and 3% of adults worldwide [5, 6]. Allergic contact dermatitis (ACD), on the other hand [7], represents 5–10% of consultations, including those related to irritation, and periodic treatment. Sensitizing allergens are diverse, can include metals such as nickel and gold, components of perfumes and soaps, or organic compounds, and once sensitized, the presence of the allergen will induce the characteristics of ACD [8–10]. Removal of the allergen results in loss of symptoms, but recovery can take weeks, during which treatment of symptoms is desirable [10]. Treatment of severe reactions includes corticosteroids. However, their mechanism of action is not well defined; it is complex and affects the immune system and skin barrier, and long-term use may result in dermal atrophy, telangiectasia, striae, hypopigmentation, or corticosteroid acne. Barrier disruption may allow percutaneous absorption that could initiate adrenal suppression, altered growth, hypertension, hyperglycemia, insulin resistance, and cataracts [11–13].
Controversy remains as to whether long-term corticosteroid use poses increased risk for lymphoma or skin cancer, or whether it activates herpes simplex. However, calcineurin inhibitors have been developed as corticosteroid alternatives [14, 15]. Also, immunosuppressants tacrolimus and pimecrolimus complexing with FK-binding proteins (FKBP-12/macrophilin-12) reduce calcineurin and T lymphocyte activity [14–16]. Thus, cytokines are inhibited and inflammatory signaling is halted or reduced.
Exogenous proteases or endogenous tryptases from mast cells activate protease-activated receptor-2 (PAR-2), localized in skin C-fiber terminals, mediating local inflammation [17–19]. Activated PAR-2 subsequently activates transient receptor potential vanilloids 1 and 4 (TRPV1, TRPV4) [20]. Ca2+ then enters these non-selective ion channels to promote inflammation and pain through calcitonin gene-related peptide (CGRP) and substance P [21]. PAR-2 sensitizes TRPV ion channels [22, 23], which are associated with itch [24].
Sodium cromoglycate is a potent free radical scavenger [25]; some naturally occurring chromones exhibit free radical inhibition [26]. Significantly, adverse effects from cromoglycate treatment have not been reported. We have evaluated treatments based on compounds with antioxidant properties, a physical barrier effect, and a protective capability of inhibiting inflammatory mediator release, as a possible alternative to steroid and immunosuppressant-related treatments in our mouse model by induction of ACD with repeat application of dinitrofluorobenzene (DNFB).
Methods
Animals
Male Balb/C mice (23–25 g) were obtained from Charles River Laboratories (Wilmington, MA) and acclimated to the Louisiana State University Health Sciences Center (LSUHSC) animal colony for 1 week. This facility is maintained on a 12:12 h light/dark cycle (06:00 h ON; 18:00 h OFF), with an average light intensity at cage level of 20 lx. Mice were supplied with normal mouse chow and water ad libitum. All animal procedures were approved by the Institutional Animal Care and Use Committee and Institutional Review Board, LSUHSC, New Orleans, all institutional and national guidelines for the care and use of laboratory animals were followed, and this study has conformed with the Helsinki Declaration of 1964, as revised in 2013, concerning human and animal rights.
General Reagents and Antibodies
Acetone was purchased from Fisher Scientific (Waltham, MA), 2,4-dinitrofluorobenzene (DNFB) was purchased form Sigma-Aldrich (St. Louis, MO), and olive oil was purchased locally. The following lists detail where the primary and secondary antibodies were obtained. Primary antibodies: PAR-2 (sc-8207) 1:1000, Santa Cruz; TRPV4 (GTX54764) 1:500, GeneTex; TNFα (LS-C104816) 1:1000, LS Biosciences; GAPDH (MAB374) 1:500, EMD Millipore. Secondary antibodies: Donkey anti-goat IgG-HRP (sc-2020) 1:5000; Donkey anti-rabbit IgG-HRP (sc-2313) 1:5000; Donkey anti-mouse IgG-HRP (sc-2314) 1:5000, Santa Cruz.
Ear Skin Sensitization and Induction of ACD
An established method for sensitization and ACD induction was utilized [27, 28]. Three protocols were utilized (Fig. 1). Mice were sensitized by application of 0.15% 2,4-dinitrofluorobenzene (DNFB) (Sigma-Aldrich, St Louis, MO) in acetone and olive oil (3:1) (vehicle) with cotton applicators to the outer surface of both ears every other day.
Fig. 1.

Outlines of experimental protocols. Protocol for the a curative, b preventative, and c experimental corticosteroid treatments. These protocols generally cover a 2-week period. Ear tissue was harvested at the times indicated by “killed”
Four test compounds were supplied as creams (Dermogen Farma, Madrid, Spain): (1) BIPxine (a mixture of Rosa moschata and Croton lechleri (antioxidants) and Aloe vera and d-panthenol (moisturizers), referred to hereafter as “mixture of antioxidants and moisturizers” (MAM); (2) 2% hydroglycolic solution of disodium cromoglycate (CGDS); (3) BIPxine and CGDS 2% (MAM + CGDS 2%, formulation A); (4) BIPxine and CGDS 5% (MAM + CGDS 5%, formulation B).
Two approaches were tested: a curative and a preventative approach. In the curative model, ACD was induced first, followed by application of the test compounds to determine their efficacy at restoring homeostasis (Fig. 1a).
In the curative model, as ACD developed and then as the test compounds were applied, ear thickness (millimeters) of 36 mice was measured daily with a digital caliper; ears gradually thickened as the ACD response developed. In this first experiment, DNFB treatment was halted at 19 days, when ear thicknesses surpassed 0.50 mm, and application of test formulations commenced.
In addition to the mice undergoing treatment with the test compounds, two additional sets of mice served as controls. One received no treatment throughout the experiment (control), while the second set was sensitized with DNFB only (ACD control), but received no test cream application. Test substances were applied by cotton applicators for 7 days and the ears collected for analysis. Six mice (12 ears) were employed for each set of conditions.
In a second repeat experiment, mice were similarly sensitized by application of DNFB to the outer surface of both ears. However, the sensitization period was shortened to 7 days. Test compounds were again applied for 7 days and the ears collected for analysis. During this experiment, ear thicknesses were collected only on days 0, 7, and 14 (Fig. 1a). Here, seven mice (14 ears) were employed for each set of conditions.
The second approach was designed to determine the ability of the test compounds to prevent ACD in the presence of the sensitizing agent. In this preventative experiment, mice were sensitized, as before, for 1 week. However, the test compounds were applied throughout the 2-week period. Ear thickness was measured daily and the results compared to untreated controls (Fig. 1b). Seven mice (14 ears) were employed for each set of conditions here as well.
A final experiment was designed to test the ACD model with intermediate- and low-strength steroids. Two groups of mice were sensitized with DNFB for 7 days, but one group received simultaneous treatment with desoximetasone or hydrocortisone. The other group was sensitized for 7 days and then treated with desoximetasone or hydrocortisone for an additional 7 days (Fig. 1c). Ear thicknesses were obtained at days 0, 7, and 14. Each group consisted of 5 mice each.
Immunoblot Analysis of Ear Tissue
Two proteins that are associated with the inflammatory and pain responses, PAR-2 and the transient receptor potential vanilloid receptor 4 (TRPV4), were quantitated by comparison to an unchanging house-keeping protein GAPDH. Because ACD is associated with the inflammatory response, the inflammatory marker TNFα was also quantitated. Ear tissue was minced and homogenized (glass-on-glass) at 4 °C using RIPA buffer (Thermo-Fisher Scientific) consisting of Complete Protease and Phosphatase Inhibitor Cocktail (Roche Diagnostics, Indianapolis, IN). Samples were sonicated and centrifuged for 10 min at 14,000g. The protein concentration was determined with a Bradford assay (Bio-Rad, Hercules, CA). The supernatant was used for SDS-PAGE and the pellet stored at − 20 °C. Equal amounts of boiled protein (20 μg per lane) were diluted with the SDS sample buffer, loaded onto BioRad Criterion Pre-cast Gels (4–12%) for SDS-PAGE (BioRad, Hercules, CA), and electrophoresed at 125 V for 1.5 h on ice. Proteins were electro-blotted to polyvinylidene difluoride (PVDF) membranes using the BioRad Criterion TransBlot system. Transference took place in a Turbo Transfer system (BioRad) following the manufacturer’s instructions. Gel retention was assessed by staining with Coomassie blue (Pierce, Rockford, IL). Nonspecific binding was blocked for 1 h at room temperature with SuperBlock blocking solution (Thermo-Fisher Scientific). Membranes were incubated overnight at 4 °C with primary antibodies to PAR-2, TRPV4, and TNFα in TBS. Bound primary antibody was detected by HRP-linked secondary antibody at 55, 98, and 17 kDa, respectively. Protein bands were visualized with a Fujifilm LAS-3000 digital scanner, following the manufacturer’s instructions, and quantitated using ImageQuant TL software (GE Healthcare). Membranes were then stripped for 30 min, re-probed with GAPDH (38 kDa, mouse monoclonal IgG) as a loading control (EMD Millipore), and detected with HRP anti-mouse IgG (Santa Cruz).
Labeling and Quantification of the Primary Afferent Arborizations
We found that neither PGP 9.5 nor beta III tubulin alone (both good markers for neuronal fibers) would label the fine neuronal processes within the ear. However, by combining both markers and incubating for prolonged periods in briefly fixed tissue (1 h), labeling was detectable with a 63× objective. While beta III tubulin will also label epidermal Langerhans cell dendrites, suggesting a mixed population of labeled fibers here, this method likely labeled all neuronal fibers. It is important to mention here that we were interested in learning whether any fibers near the surface were altered, not whether sensitization affected specific fiber subgroups. To determine whether inflammation had been induced following sensitization with DNFB, the protein PAR-2 was also probed. Briefly, vertical cryosections of mouse pinna (30 μm thickness) were prepared for immunolocalization of primary afferent fiber arborizations in fixed control and treated ears. Following Triton X-100 permeablization and blocking, samples were incubated with PGP 9.5 (1:500, Abcam, ab108986), beta III tubulin (1:200, Abcam, ab52901), and PAR-2 (1:200, Santa Cruz) in blocking buffer for 3 days, washed, and then incubated for 1 h in donkey anti-rabbit IgG Alexa Fluor 488 (Invitrogen) for simultaneous labeling of both PGP 9.5 and beta III tubulin, and donkey anti-goat IgG Alexa Fluor 633 (Invitrogen) for PAR-2. After labeling with DAPI for 10 min, slides were coverslipped and viewed with an Olympus Fluoview 1200 confocal microscope.
To determine the area of nerve fibers within sections of the sensitized ear, the green channel was isolated to reveal only the labeled fibers, and the contrast adjusted until the background was black. ImageJ was used to outline the arborized region within the image, and the number of green pixels, determined by the Color Pixel Counter plugin, was presented as the percentage of total specified area. Percentages were averaged, standard errors were calculated, and p values were determined by comparing control untreated ears with ears that underwent sensitization and subsequent treatment.
Lipidomic Analysis of Ear Tissue by LC–MS/MS
Mouse samples were homogenized in MeOH (3 ml) followed by the addition of CHCl3 (6 ml) containing an internal standard mixture of AA-d8, PGD2-d4, EPA-d5, 15HETE-d8, LTB4-d4, and PC(28:0), sonicated in an ice water bath, and then stored at − 80 °C overnight. After vortexing and centrifugation, the supernatant was removed and the pellet washed with CHCl3/MeOH (1 ml, 2:1), centrifuged, and the supernatants combined. Distilled water (2 ml, pH 3.5) was then added to the supernatant, centrifuged, and the pH of the upper phase adjusted to 3.5–4.0 with HCl. The lower phase was then dried down under N2, resuspended in methylformate (200 µl), transferred to a vial, dried under N2, resuspended in MeOH (10 µl), and then water (5 µl) was added.
A Xevo TQ-S, equipped with Acquity I Class UPLC (Waters), was used for liquid chromatography–mass spectrometry (LC–MS/MS) analysis. An Acquity UPLC HSS T3 1.8 µm 2.1 × 50 mm column was used for fatty acids and their derivatives. Seventy-five percent of solvent A (H2O + 0.1% acetic acid) and 25% of solvent B (90% acetonitrile, 10% isopropanol) with a 0.6 ml/min flow rate were used for the first minute, then graduated to 100% of solvent B for 7.5 min, followed by 100% solvent B for 2.5 min. The column was then re-equilibrated to 75% A and 25% B for 2 min. The capillary voltage was − 2.5 kV, the desolvation temperature was set at 600 °C, the desolvation gas flow at 1100 l/h, the cone gas at 150 l/h, and the nebulizer pressure at 7.0 bar with the source temperature at 150 °C.
Eicosapentaenoic acid (EPA), arachidonic acid (AA), docosahexaenoic acid (DHA), 14HDHA, 17HDHA, neuroprotectin D1 (NPD1) and its isomers, 12HETE, 15HETE, PGE2, LTB4, and LTC4 were quantified for each of the treatments.
Statistics
Ear Measurements and Western Blotting
All results were expressed as mean ± SEM. Data from all experiments were evaluated using one-way ANOVA (analysis of variance) (n = 28; n = 6, respectively) followed by Sidak’s multiple comparisons post hoc test. Statistical analyses were performed using Graphpad Prism software Version 7.04. A value of p < 0.05 was considered to be statistically significant.
Mass spectrometric analysis
Results were expressed as means + SEM. p values were obtained with Student’s t test (two-tailed, two-sample equal variance conditions). Calculations were done with Microsoft Excel program.
Results
Ear Thickness
In our initial curative experimental design, as DNFB treatment proceeded, ear thicknesses increased from 0.20 mm to about 0.48 mm (a 0.28 mm increase, or 100%) by 22 days. When DNFB treatment was stopped and test substances applied, the increase in ear thickness ceased. Thicknesses remained relatively constant for 3 days, then decreased. However, the ears of the ACD control mice (DNFB alone; no test substances applied) continued to thicken for 5 days (up to 0.68 mm) and began to thin afterwards. Ear thickness in the MAM, CGDS 2%, and formulation A (MAM CGDS 2%) treatment mice declined similarly, reaching 0.30 mm (a decrease of 0.18 mm, or 64%) by 6 days. However, treatment with formulation B (MAM CGDS 5%) declined more rapidly to 0.27 mm (a decrease of 0.21 mm, or 75%). At this point, ear tissue was collected for further analysis (Fig. 2a, Suppl 1a–f). To corroborate these results, this experiment was repeated using four mice (eight ears). The same treatments produced similar results (Suppl 1g).
Fig. 2.
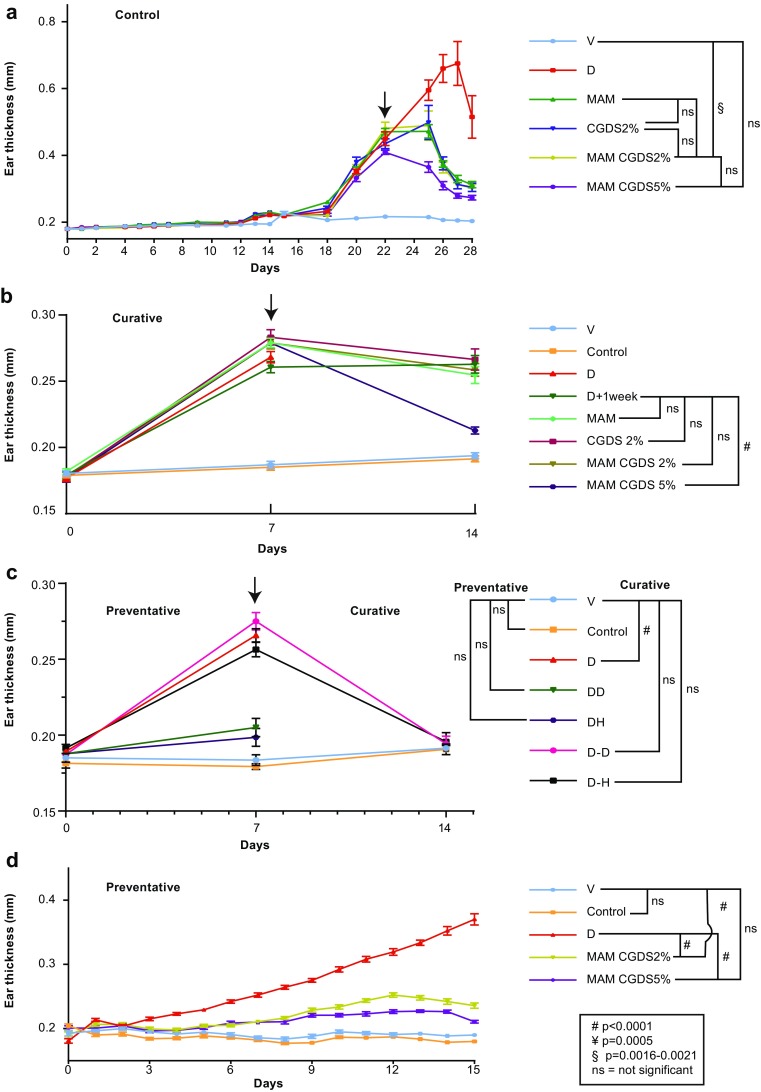
Ear thickness profiles for curative, preventative, and corticosteroid experimental treatments: novel formulations promote resolution of inflammation-induced ear thickening. a Ear thickness averages of individual responses (Suppl S1) for each curative experimental treatment showed increases induced by ACD. Following onset of secondary treatments, thickness rapidly declined with MAM, formulation A, and formulation B. Formulation B produced the best effect. No changes were observed in controls, while ear thickness in ACD-alone animals continued to increase. b Ear thickness averages for each curative experimental treatment based on the 14-day protocol (Fig. 1a; see Suppl S2 for additional graphs). Ear thickness averages for each experimental treatment showed increases of about 0.1 mm induced by ACD during the first week. Following onset of secondary treatments during week 2, thicknesses generally stabilized or were reduced. Treatment with a combination of MAM and cromoglycate 5% induced the most dramatic reduction, returning thicknesses to near starting levels. Onset of the secondary treatment occurred after 1 week. Measurements were collected at time zero and at 1 and 2 weeks from both ears of seven mice for each treatment. Graphs represent two independent experiments (total n = 28). The efficacy of formulation B in reducing the effects of ACD was similar to that shown for experiments in a. c Parallel curative and preventative experiments induced ACD for 1 week, with simultaneous treatment, or was followed by 1 week of treatment with two steroids, hydrocortisone (H) and desoximetasone (D) (Fig. 1c). Both compounds maintained or reduced ear thickness to naïve levels. Measurements were collected at time zero and at 1 and 2 weeks from both ears of five mice for each treatment (n = 10). d A preventative experiment was performed by simultaneously treating ears with the DNFB ACD inducer and the test compounds (Fig. 1b). ACD alone increased ear thickness as in all other experiments while co-treatment with MAM plus cromoglycate 2% or 5% maintained ear thicknesses very close to naïve control and vehicle-alone ears, demonstrating a preventative effect on the development of ACD. Arrows indicate onset of treatment. Control (no treatment), D (dermatitis), V (vehicle), MAM (mixture of antioxidants and moisturizer), CGDS 2% (cromoglycate 2%), formulation A (MAM + CGDS 2%), and formulation B (MAM + CGDS 5%). ± SEMs are indicated, #p < 0.0001, §p 0.0016–0.0021, ns not significant
A second, modified curative experiment was conducted, but measurements of ear thickness were obtained only at the start of the experiment at day 0, 7 days after the DNFB sensitization at the start of the test treatments, and after 1 week of treatments at the conclusion (day 14). Seven mice were included in each treatment. As before, one naïve control group received no treatments. A second group received only vehicle for the first week, then no treatment for the second week. All other groups were sensitized with DNFB for 1 week and then treated with test compounds for the second week. Treatments were DNFB for 1 week only; DNFB for 1 week with no treatment for the second week; and DNFB for 1 week then MAM + CGDS 2% (formulation A), or MAM + CGDS 5% (formulation B) (Figs. 1a, 2b, Suppl 2). This was repeated a second time. Figure 2b illustrates the average results of this experiment (two replicates of 7 mice per group, n = 28 ears). Formulation B was remarkable in reducing ear thickness to almost naïve control levels.
As an additional experiment, ACD was induced and hydrocortisone (H) or desoximetasone (D) was applied every other day either simultaneously with DNFB (D) sensitization (1 week only, DH, DD) or for 1 week following the sensitization period (D–H, D–D) (Fig. 1c). Both compounds reduced ear thickness to control levels by the end of the second week or prevented the onset of ear thickness by 1 week (Fig. 2c).
In a modification of the experimental protocol, DNFB sensitization was begun and treatment with the test compounds was applied simultaneously to determine whether these compounds would prevent the onset of ACD (Fig. 1b). Except with DNFB sensitization alone, ear thickening generally failed to take place. Only minimal thickening occurred in the DNFB-sensitized mice treated with either formulation A or formulation B. The best effect was obtained again with formulation B. No changes were observed in controls (Fig. 2d).
Protein Abundance by Immunoblotting
Mice were treated as in Fig. 1a. The inflammatory response indicators PAR-2 and TRPV4, while low in the naïve control animals, were upregulated in all ACD animals (Fig. 3). In the presence of formulation B (MAM + CGDS 5%), PAR-2 levels were about 50% below the ACD mouse values, whereas TRPV4 remained at control levels. TNFα, also an inflammatory marker, was upregulated in the ACD mice but was maintained at naïve mouse levels by formulation B.
Fig. 3.

Novel formulation treatment attenuated inflammatory markers. a Sensitization of skin promoted upregulation of PAR-2, while novel formulation attenuated PAR-2 production. Ears treated with vehicle alone did not develop ACD, while DNFB sensitization (D) triggered upregulation of the itch marker PAR-2. Test substances attenuated the PAR-2 response by 22% and 69%. Typical Western blots are shown. GAPDH, house-keeping protein. D, ACD; MAM + CGDS 2% (formulation A); MAM + CGDS 5% (formulation B). PAR-2 means + SEM, n = 7. *p = 0.002, #p = 0.016. b Skin sensitization promoted upregulation of TRPV4, while novel formulation prevented TRPV4 production. Similarly, ears treated with vehicle alone did not develop ACD; however, DNFB sensitization (D) triggered upregulation of the itch marker TRPV4. Test substance formulation B reduced TRPV4 formation to naïve control levels. Two typical Western blots are shown. GAPDH, house-keeping protein. D, ACD; MAM + CGDS 2% (formulation A); MAM + CGDS 5% (formulation B). TRPV4 means + SEM, n = 7. *p = 0.007, #p = 0.026. c Skin sensitization induced an inflammatory response, while novel formulation blocked inflammation onset. DNFB sensitization triggered upregulation of the inflammatory marker TNFα, but formulation B prevented inflammation onset. Two typical Western blots are shown. GAPDH, house-keeping protein. D, ACD; MAM + CGDS 2% (formulation A); MAM + CGDS 5% (formulation B). TNFα means + SEM, n = 7. *p = 0.249, #p = 0.210
Immunohistochemistry
Mice were treated as in Fig. 2a, and ears were collected and cryosectioned. Immunolabeling with antibodies containing a mixture of PGP 9.5 and beta III tubulin delimited fibers in the epidermis which exhibited extensive branching (Fig. 4a–d). The percentage of these arborizations within the dermatitic ears, including all ears that had undergone 1 week of treatment with the test compounds, was reduced by about 30% (data not shown). However, values from the naïve (untreated) ears matched those from the vehicle-alone treatment (p = 0.03), indicating that the acetone/olive oil mixture did not induce the dermatitic response.
Fig. 4.
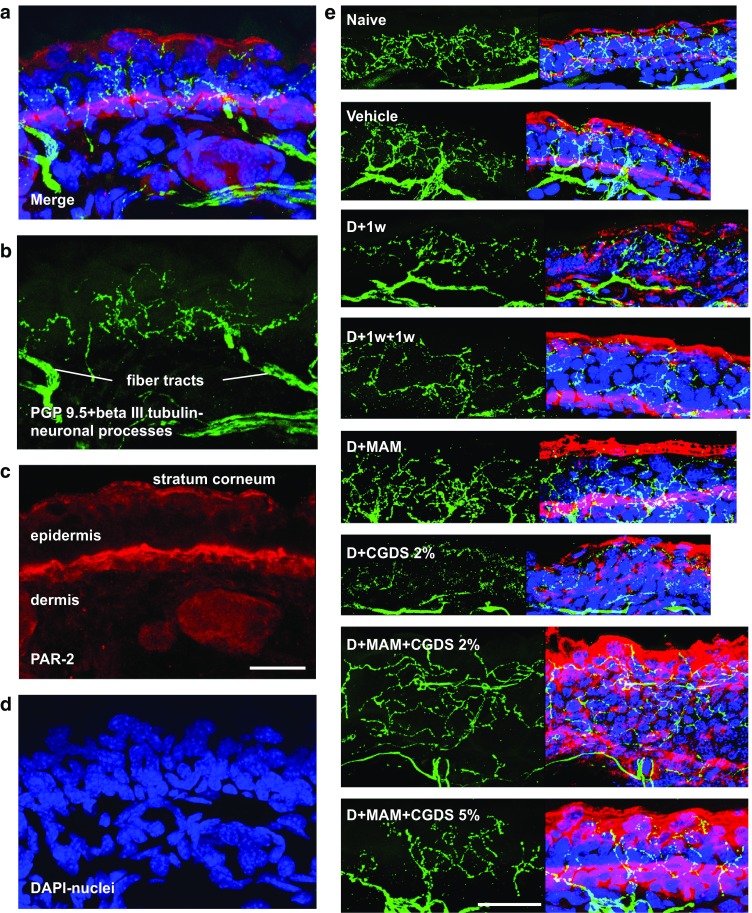
Naïve control ears exhibit dense sensory fibers and a distinctive PAR-2 band. a Merged immunohistochemistry image showing sensory fibers (green) within the epidermis and distinctive PAR-2 labeling (red). b Green fluorescence channel revealing PGP 9.5 plus beta III tubulin-labeled complex sensory fibers within the epidermis, arising from underlying fiber tracts within the dermis. c Red fluorescence channel demonstrating a narrow bright dense PAR-2-labeled band near the epidermis/dermis interface. Light background label also appears within the stratum corneum of all immunopreparations. d Blue fluorescence channel showing the DAPI-labeled nuclei within this section. Scale bar, 50 μm. e Dermatitic ears generally exhibit increases in PAR-2 deeper within the tissue, when compared to naïve control and vehicle-alone ears (red label), indicating that inflammation has been triggered. The green channel reveals the extent of the sensory fibers within the epidermis, also seen within the merged images at the right. Scale bar, 25 μm
PAR-2 is expressed in mast cells and increases in chronic skin inflammation; therefore, we looked for indications of PAR-2 in ear tissue that had undergone treatment with the test compounds. PAR-2 localized to two regions within the ear sections of all treated animals. The stratum corneum consistently demonstrated non-specific labeling, as did the upper-most layer of the epidermal cells. Moreover, intense labeling was often roughly correlated with the inner-most layer of epidermal cells near the basement membrane (Fig. 4c, e). This inner label appeared as a thin band in the naïve (untreated) and vehicle-alone-treated ears, but appeared to thicken and somewhat diffuse in the 2-week ACD ears.
Lipid Mediators
Neuroprotectin D1 (NPD1), a docosanoid, is derived from docosahexaenoic acid (DHA) under conditions of oxidative stress and inflammation, and it is highly neuroprotective in the brain and retina [29–31]. Analysis shows that treatment with formulation A (MAM + CGDS 2%) and formulation B (MAM + CGDS 5%) resulted in some production of NPD1, as well as its isomers (Fig. 5).
Fig. 5.
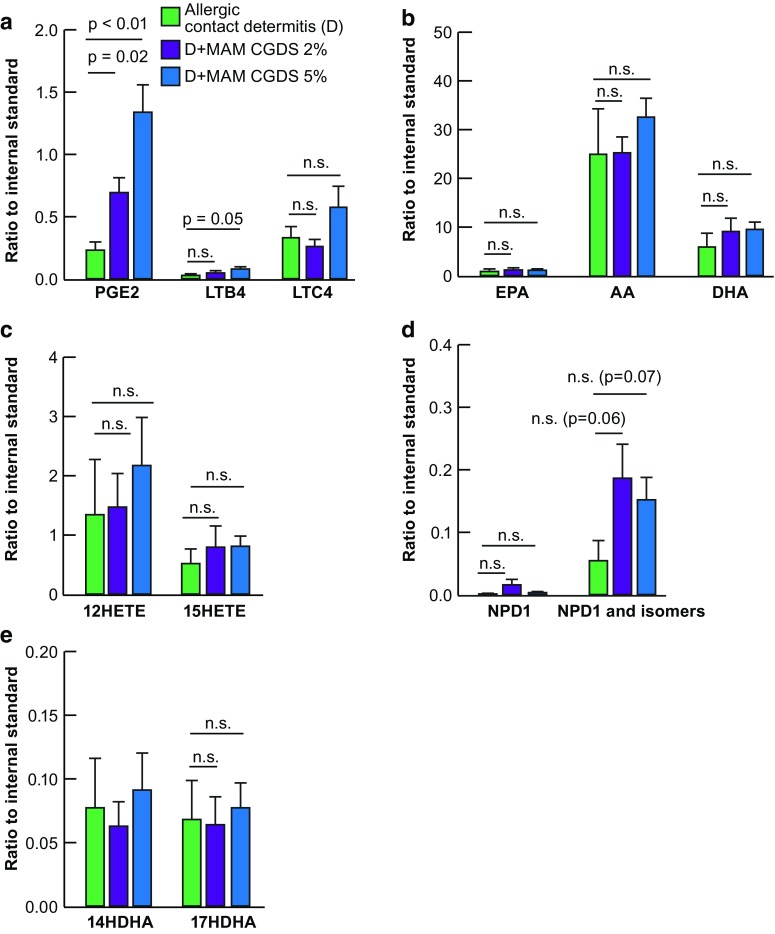
Mass spectrometric lipidomic analysis of ear tissue revealed synthesis of neuroprotective mediators. Mass spectrometric analysis of ear tissues permitted quantification of the inflammatory markers a PGE2, LTB4, and LTC4, b the precursor molecules initiating the stress-induced fatty acid signaling cascades, EPA, AA, and DHA, c products of the AA cascade 12HETE and 15HETE, d products of the DHA cascade 14HDHA, 17HDHA, and e the DHA-derived neuroprotective compounds NPD1 and its isomers. Following 1 week of treatment after ACD induction, tissues responded to test compounds in unexpected manners. Inflammatory markers increased, AA, DHA, and their associated metabolites remained unchanged. However, NPD1 and its isomers tended to increase as expected of protectant molecules. Means + SEM, n = 8
Following 1 week of treatment after ACD induction, tissues responded to test compounds in unexpected ways. Inflammatory markers increased (PGE2, LTB4, and LTC4) while arachidonic acid (AA), DHA, and their associated metabolites remained unchanged. NPD1 and its isomers tended to increase. When the NPD1 isomers were totaled, treatment with formulation A and formulation B resulted in borderline production of these docosanoids after only 1 week.
Discussion
DNFB sensitization of ears induces thickening, which will continually progress, eventually causing ear tissue loss. Ear thickness measurements reveal the extent of sensitization, and the efficacy of treatment. We show that ear thickness changes from 0.2 to 0.9 mm. In the first sets of experiments in which sensitization preceded treatment, ear thickness of mice treated with test substances gradually decreased. Moreover, in the second group of experiments, with simultaneous sensitization and treatment, test substances prevented ear thickening. Both methods demonstrated an improving or preventive effect of the test compounds. These results were corroborated by Western blot analysis and mass spectrometry.
Overall, immunoblotting revealed that upregulation of inflammation-related molecules was greatly reduced by the MAM and CGDS compounds (formulation B, MAM + CGDS 5%, greatest; formulation A, MAM + CGDS 2%, little effect).
PAR-2, a mediator of neurogenic inflammation and pain, is activated by injury and inflammation. This activates TRPV4, resulting in mediation of the PAR-2 proinflammatory response. Consequently, both PAR-2 and TRPV4 are good indicators of inflammation and pain [21, 32]. These indicators were upregulated with ACD, but PAR-2 far exceeded TRPV4. The TRPV4 ion channel can enable Ca2+ entry, permitting inflammation- and pain-associated CGRP and substance P release [21]. While inflammation was indicated, we observed minimal upregulation of the inflammatory marker TNFα, perhaps due to reduced TRPV4 activation. These findings suggest that pain and inflammation may be separately triggered and progress independently in this ACD model.
PAR-2 is associated with inflammation and ichthyosis, but these are completely negated with a PAR-2 knockout genotype [33]. Trypsin and tryptase activate PAR-2, but, while topical application of trypsin or agonists of PAR-2 peptides trigger itch, the response is histamine-independent [34, 35]. The Mas-related GPCR itch receptor MrgprC11 [36] triggers scratching, but PAR-2 elicits thermal pain hyperalgesia. Combinations of several PARs can exist; PAR-2 can be activated by thrombin-cleaved PAR-1 [37], suggesting a link between MrgprC11 and PAR-2 [38]. PAR-2 is greatly increased in DNFB-sensitized skin and decreased by formulation B (MAM + CGDS 5%).
PAR-2 is expressed by mast cells [39] but is increased in chronic skin inflammation [40] and psoriatic lesions [41]. In humans, cell types localized throughout the dermis and epidermis also produce PAR-2 [39], and PAR-2 is also highly expressed in epidermal keratinocytes [41] within this stratum. PAR-2 is localized to the stratum corneum and basal cells within the mouse ear epidermis; its expression is increased by DNFB, and formulation B (MAM + CGDS 5%) reduces PAR-2 to control levels. Since PAR-2 is intimately associated with inflammation, reduced expression indicates amelioration of ACD.
Neuronal processes from the dermis extend into the epidermis, ramify, and terminate just below the stratum corneum. Noxious compounds on the skin activate PAR-2 on mast cells and initiate inflammation. These sensory C-fibers respond to PAR-2 signaling compromise [17–19]. The branching pattern of these fibers is complex, but changes in their density suggest that DNFB-induced ACD decreases their mass below naïve levels within all treatments. However, though treatment with formulation B (MAM + CGDS 5%) reduced PAR-2, TRPV4, and TNFα to near naïve values (indicating reduction of pain and resolution of inflammation), 1 week of treatment may not be enough for full tissue recovery. Importantly, it should be noted that proteins can be quantified by Western blot, but the same proteins can only be localized by immunohistochemistry without extreme care in quantifying fluorescence, so while Western blots indicated a reduction in signal (e.g., PAR-2), our immunohistochemistry only revealed that PAR-2 was present.
PAR-2, TRPV4, and TNFα upregulation indicates pain and inflammation induction by DNFB sensitization. Mass spectrometric analysis permitted quantification of inflammatory markers (PGE2, LTB4, and LTC4) and precursor molecules, which initiate stress-induced fatty acid signaling cascades from EPA, AA, and DHA (AA products: 12HETE and 15HETE; DHA products: 14HDHA, 17HDHA; and the DHA-derived neuroprotective compound NPD1 and its isomers). Following 1 week of treatment after ACD induction, tissues responded to test compounds in unexpected manners. Inflammatory markers increased while AA, DHA, and their metabolites remained unchanged. However, NPD1 and its isomers tended to increase, as expected of protectant molecules. NPD1, a highly protective docosanoid, is produced from DHA when cells undergo oxidative stress and homeostatic changes. When the NPD1 isomers were totaled, treatment with formulation A and formulation B resulted in borderline production of these docosanoids after only 1 week, suggesting that these experimental compounds add to the normal tissue protective response elicited by DNFB alone, stimulating additional upregulation of neuroprotective molecules. Importantly, increases in inflammatory markers PGE2, LTB4, and LTC4 indicate continual inflammation, suggesting that 1 week may be too short an interval to affect full ameliorative benefits from the test compounds, and that analysis of additional time points would resolve this issue. These lipid mediators reflect inflammatory cell influx plus activation of resident cells in this model. These results represent a snapshot of the ongoing changes from a steady state, and should progress as treatment continues; with additional time, indicators of inflammation should decrease and neuroprotective elements increase. These are issues that we are currently studying.
In summary, following DNFB treatment with no additional therapy, eventual decline in thickness occurred from normal tissue repair. However, subsequent treatment with MAM, disodium cromoglycate 2%, and formulation A (MAM + CGDS 2%) reduced ear thickness more rapidly by as much as 60%, whereas treatment with formulation B (MAM + CGDS 5%) caused a thickness reduction of up to 70%. Importantly, simultaneous sensitization and test compound treatment, especially with formulation B, prevented onset of the inflammation and pain response. Mass spectrometry suggests that both formulations are linked to the synthesis of NPD1 isomers, all of which are potent neuroprotective agents. Immunoblotting demonstrated that development of ACD is accompanied by upregulation of inflammatory and pain-related PAR-2 and TRPV4 and the inflammatory marker TNFα. Moreover, the presence of PAR-2, TRPV4, and TNFα was somewhat reduced in the presence of formulation A, but significantly prevented by formulation B. Finally, this supports studies which have shown that mediation of PAR-2 can regulate its proinflammatory actions [21]. Overall, this study suggests that the experimental compounds formulation A (MAM + CGDS 2%) and, particularly, formulation B (MAM + CGDS 5%) effectively decrease pain, reduce inflammation, ameliorate tissue swelling, and increase neuroprotective docosanoid levels.
There were some limitations in this study. It was initially determined that induction of dermatitis increased ear thickness from about 0.2 mm to more than 0.6 mm, with eventual tissue destruction and loss of ear tips. To avoid tissue loss, we chose an intermediate thickness of about 0.50 mm, at which to apply the test compounds, and then applied treatment for 7 days, during which ear thicknesses were decreased. However, there were indications that some inflammatory markers (e.g., PGE2 and arachidonic acid) remained elevated. This suggests that treatment had not completely ameliorated the dermatitis; by prolonging the treatment phase in some mice, more information could have been obtained about the action of these compounds and their effective time course. Also, there was variability between ears of the same animal and among the mice within each treatment, as evidenced by the size of error bars in some data groups. Obviously, more than 12 mice (24 ears) per group would likely have produced less variability within groups. Finally, to achieve visible neurite labeling we had to combined two markers, PGP 9.5 and beta III tubulin. However, tubulin also labels the dendritic components of Langerhans cells, which likely contribute to the green fluorescently labeled processes of Fig. 4. Also, with all neuronal processes labeled, subpopulations could not be distinguished.
Conclusions
The significance of this study is twofold: BIPxine (a mixture of Rosa moschata and Croton lechleri (antioxidants) and Aloe vera and d-panthenol (moisturizers), i.e., the mixture of antioxidants and moisturizers (MAM)), when combined with the hydroglycolic solution of disodium cromoglycate (CGDS) to produce the treatment formulations applied to the ACD, ameliorated inflammation, reduced swelling, and initiated docosanoid protection. Moreover, this treatment offers an alternative to steroid therapy and the possible side effects. However, additional studies are needed to determine the ideal duration of treatment and the most efficacious concentrations of the active components in the test formulations which will best alleviate ACD.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Funding
This work was supported by the National Institute of General Medical Sciences P30GM103340 (NGB). Article processing charges were funded by the LSU Neuroscience Center of Excellence.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Ricardo Palacios Peláez, Nicolas G. Bazan, Hélène Varoqui, and William C. Gordon designed the experiments. Concha Tiana Ferrer, Fernando Pineda de la Losa, and Gabriela Silvina Bacchini supplied the test compounds. William C. Gordon and Surjyadipta Bhattacharjee performed the experiments. Bokkyoo Jun performed the mass spectrometry analyses. William C. Gordon, Ricardo Palacios Peláez, Nicolas G. Bazan, Virginia García López, David Rodríguez Gil, Javier Alcover Díaz, and Concha Tiana Ferrer wrote the paper.
Disclosures
William C. Gordon, Virginia García López, Surjyadipta Bhattacharjee, David Rodríguez Gil, Javier Alcover Díaz, Fernando Pineda de la Losa, Ricardo Palacios Peláez, Concha Tiana Ferrer, Gabriela Silvina Bacchini, Bokkyoo Jun, Hélène Varoqui, and Nicolas G. Bazan have nothing to disclose.
Compliance with Ethics Guideline
All animal procedures were approved by the Institutional Animal Care and Use Committee and Institutional Review Board, LSUHSC, New Orleans, all institutional and national guidelines for the care and use of laboratory animals were followed, and this study has conformed with the Helsinki Declaration of 1964, as revised in 2013, concerning human and animal rights.
Data Availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Footnotes
Enhanced content
To view enhanced content for this article go to 10.6084/m9.figshare.5798148.
William C. Gordon, Virginia García López, and Surjyadipta Bhattacharjee contributed equally.
References
- 1.Furue M, Kadono T. New therapies for controlling atopic itch. J Dermatol. 2015;42:847–850. doi: 10.1111/1346-8138.13060. [DOI] [PubMed] [Google Scholar]
- 2.Wahlgren CF. Pathophysiology of itching in urticaria and atopic dermatitis. Allergy. 1992;47:65–75. doi: 10.1111/j.1398-9995.1992.tb05091.x. [DOI] [PubMed] [Google Scholar]
- 3.Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387:1109–1122. doi: 10.1016/S0140-6736(15)00149-X. [DOI] [PubMed] [Google Scholar]
- 4.Tamari M, Tanaka S, Hirota T. Genome-wide association studies of allergic diseases. Allergol Int. 2013;62:21–28. doi: 10.2332/allergolint.13-RAI-0539. [DOI] [PubMed] [Google Scholar]
- 5.Asher MI, Montefort S, Björkstén B, et al. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC phases one and three repeat multicountry cross-sectional surveys. Lancet. 2006;368:733–743. doi: 10.1016/S0140-6736(06)69283-0. [DOI] [PubMed] [Google Scholar]
- 6.Nutten S. Atopic dermatitis: global epidemiology and risk factors. Ann Nutr Metab. 2015;66:8–16. doi: 10.1159/000370220. [DOI] [PubMed] [Google Scholar]
- 7.Aquino M, Fonacier L. The role of contact dermatitis in patients with atopic dermatitis. J Allergy Clin Immunol Pract. 2014;2:382–387. doi: 10.1016/j.jaip.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Linauskienė K, Malinauskienė L, Blažienė A. Metals are important contact sensitizers: an experience from Lithuania. Biomed Res Int. 2017;2017:3964045. doi: 10.1155/2017/3964045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lidén C, Yazar K, Johansen JD, Karlberg AT, Uter W, White IR. Comparative sensitizing potencies of fragrances, preservatives, and hair dyes. Contact Dermat. 2016;75:265–275. doi: 10.1111/cod.12683. [DOI] [PubMed] [Google Scholar]
- 10.Kimber I, Basketter DA, Gerberick GF, Dearman RJ. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201–211. doi: 10.1016/S1567-5769(01)00173-4. [DOI] [PubMed] [Google Scholar]
- 11.Keipert JA, Kelly R. Temporary Cushing’s syndrome from percutaneous absorption of betamethasone 17-valerate. Med J Aust. 1971;1:542–544. doi: 10.5694/j.1326-5377.1971.tb87696.x. [DOI] [PubMed] [Google Scholar]
- 12.May P, Stein EJ, Ryter RJ, Hirsh FS, Michel B, Levy RP. Cushing syndrome from percutaneous absorption of triamcinolone cream. Arch Intern Med. 1976;136:612–613. doi: 10.1001/archinte.1976.03630050086014. [DOI] [PubMed] [Google Scholar]
- 13.Munro DD. Topical corticosteroid therapy and its effect on the hypothalamic–pituitary–adrenal axis. Dermatologica. 1976;152:173–180. doi: 10.1159/000257888. [DOI] [PubMed] [Google Scholar]
- 14.Berger TG, Duvic M, Van Voorhees AS, VanBeek MJ, Frieden IJ, American Academy of Dermatology Association Task Force The use of topical calcineurin inhibitors in dermatology: safety concerns. Report of the American Academy of Dermatology Association Task Force. J Am Acad Dermatol. 2006;54:818–823. doi: 10.1016/j.jaad.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 15.Liu J, Farmer JD, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin–cyclosporin A and FKBP–FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-H. [DOI] [PubMed] [Google Scholar]
- 16.Meingassner JG, Kowalsky E, Schwendinger H, Elbe-Bürger A, Stütz A. Pimecrolimus does not affect Langerhans cells in murine epidermis. Br J Dermatol. 2003;149:853–857. doi: 10.1046/j.1365-2133.2003.05559.x. [DOI] [PubMed] [Google Scholar]
- 17.Carvalho RF, Nilsson G, Harvima IT. Increased mast cell expression of PAR-2 in skin inflammatory diseases and release of IL-8 upon PAR-2 activation. Exp Dermatol. 2010;19:117–122. doi: 10.1111/j.1600-0625.2009.00998.x. [DOI] [PubMed] [Google Scholar]
- 18.Zhu Y, Wang XR, Peng C, et al. Induction of leukotriene B(4) and prostaglandin E(2) release from keratinocytes by protease-activated receptor-2-activating peptide in ICR mice. Int Immunopharmacol. 2009;9:1332–1336. doi: 10.1016/j.intimp.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Zhu Y, Pan WH, Wang XR, et al. Tryptase and protease-activated receptor-2 stimulate scratching behavior in a murine model of ovalbumin-induced atopic-like dermatitis. Int Immunopharmacol. 2015;28:507–512. doi: 10.1016/j.intimp.2015.04.047. [DOI] [PubMed] [Google Scholar]
- 20.White JP, Cibelli M, Urban L, Nilius B, McGeown JG, Nagy I. TRPV4: molecular conductor of a diverse orchestra. Physiol Rev. 2016;96:911–973. doi: 10.1152/physrev.00016.2015. [DOI] [PubMed] [Google Scholar]
- 21.Poole DP, Amadesi S, Veldhuis NA, et al. Protease-activated receptor 2 (PAR2) protein and transient receptor potential vanilloid 4 (TRPV4) protein coupling is required for sustained inflammatory signaling. J Biol Chem. 2013;288:5790–5802. doi: 10.1074/jbc.M112.438184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai Y, Wang S, Tominaga M, et al. Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J Clin Investig. 2007;117:1979–1987. doi: 10.1172/JCI30951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grant AD, Cottrell GS, Amadesi S, et al. Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol. 2007;578:715–733. doi: 10.1113/jphysiol.2006.121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akiyama T, Ivanov M, Nagamine M, et al. Involvement of TRPV4 in serotonin-evoked scratching. J Invest Dermatol. 2016;136:154–160. doi: 10.1038/JID.2015.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Debbasch C, Pisella PJ, Rat P, Warnet JM, Baudouin C. Antioxidant effects of mast cell inhibitors in a human conjunctival cell line. J Fr Ophthalmol. 2001;24:121–128. [PubMed] [Google Scholar]
- 26.Kładna A, Berczyński P, Piechowska T, et al. Studies on the antioxidant activities of some new chromone compounds. Luminescence. 2014;29:846–853. doi: 10.1002/bio.2631. [DOI] [PubMed] [Google Scholar]
- 27.Jeon YD, Kee JY, Kim DS, et al. Effects of lxeris dentata water extract and caffeic acid on allergic inflammation in vivo and in vitro. BMC Complement Altern Med. 2015;15:196. doi: 10.1186/s12906-015-0700-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan XY, Ma HM, Li RZ, Wang RY, Liu W, Guo JY. Topical application of aloperine improves 2,4-dinitrofluorobenzene-induced atopic dermatitis-like skin lesions in NC/Nga mice. Eur J Pharmacol. 2011;658:263–269. doi: 10.1016/j.ejphar.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 29.Bazan NG. Homeostatic regulation of photoreceptor cell integrity: significance of the potent mediator neuroprotectin D1 biosynthesized from docosahexaenoic acid: the Proctor Lecture. Invest Ophthalmol Vis Sci. 2007;48:4866–4881. doi: 10.1167/iovs.07-0918. [DOI] [PubMed] [Google Scholar]
- 30.Bazan NG. Cellular and molecular events mediated by docosahexaenoic acid-derived neuroprotectin D1 signaling in photoreceptor cell survival and brain protection. Prostaglandin Leukot Essent Fatty Acids. 2009;81:205–211. doi: 10.1016/j.plefa.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bazan NG. Neuroprotectin D1-mediated anti-inflammatory and survival signaling in stroke, retinal degenerations, and Alzheimer’s disease. J Lipid Res. 2009;50:S400–S405. doi: 10.1194/jlr.R800068-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao P, Lieu T, Barlow N, et al. Neutrophil elastase activates protease-activated receptor-2 (PAR2) and transient receptor potential vanilloid 4 (TRPV4) to cause inflammation and pain. J Biol Chem. 2015;290:13875–13887. doi: 10.1074/jbc.M115.642736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frateschi S, Camerer E, Crisante G, et al. PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin. Nat Commun. 2001;2:161. doi: 10.1038/ncomms1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Costa R, Marotta DM, Manjavachi MN, et al. Evidence for the role of neurogenic inflammation components in trypsin-elicited scratching behaviour in mice. Br J Pharmacol. 2008;154:1094–1103. doi: 10.1038/bjp.2008.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimada SG, Shimada KA, Collins JG. Scratching behavior in mice induced by the proteinase-activated receptor-2 agonist, SLIGRL-NH2. Eur J Pharmacol. 2006;530:281–283. doi: 10.1016/j.ejphar.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 36.Dong X, Han S, Zylka MJ, Simon MI, Anderson DJ. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell. 2001;106:619–632. doi: 10.1016/S0092-8674(01)00483-4. [DOI] [PubMed] [Google Scholar]
- 37.O’Brien PJ, Prevost N, Molino M, et al. Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J Biol Chem. 2000;275:13502–13509. doi: 10.1074/jbc.275.18.13502. [DOI] [PubMed] [Google Scholar]
- 38.Liu Q, Weng HJ, Patel KN, et al. The distinct roles of two GPCRs, MrgprC11 and PAR2, in itch and hyperalgesia. Sci Signal. 2011;4:ra45. doi: 10.1126/scisignal.2001925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moormann C, Artuc M, Pohl E, et al. Functional characterization and expression analysis of the protease-activated receptor-2 in human cutaneous mast cells. J Invest Dermatol. 2006;126:746–755. doi: 10.1038/sj.jid.5700169. [DOI] [PubMed] [Google Scholar]
- 40.Harvima IT, Naukkarinen A, Paukkonen K, et al. Mast cell tryptase and chymase in developing and mature psoriatic lesions. Arch Dermatol Res. 1993;285:184–192. doi: 10.1007/BF00372007. [DOI] [PubMed] [Google Scholar]
- 41.Hou L, Kapas S, Cruchley AT, et al. Immunolocalization of protease-activated receptor-2 in skin: receptor activation stimulates interleukin-8 secretion by keratinocytes in vitro. Immunology. 1998;94:356–362. doi: 10.1046/j.1365-2567.1998.00528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.