

Abstract
Asna1, also known as TRC40, is implicated in the delivery of tail-anchored (TA) proteins into the endoplasmic reticulum (ER), in vesicle-mediated transport, and in chaperoning unfolded proteins during oxidative stress/ATP depletion. Here, we show that Asna1 inactivation in pancreatic progenitor cells leads to redistribution of the Golgi TA SNARE proteins syntaxin 5 and syntaxin 6, Golgi fragmentation, and accumulation of cytosolic p62+ puncta. Asna1−/− multipotent progenitor cells (MPCs) selectively activate integrated stress response signaling and undergo apoptosis, thereby disrupting endocrine and acinar cell differentiation, resulting in pancreatic agenesis. Rescue experiments implicate the Asna1 ATPase activity and a CXXC di-cysteine motif in ensuring Golgi integrity, syntaxin 5 localization and MPC survival. Ex vivo inhibition of retrograde transport reproduces the perturbed Golgi morphology, and syntaxin 5 and syntaxin 6 expression, whereas modulation of p53 activity, using PFT-α and Nutlin-3, prevents or reproduces apoptosis in Asna1-deficient and wild-type MPCs, respectively. These findings support a role for the Asna1 ATPase activity in ensuring the survival of pancreatic MPCs, possibly by counteracting p53-mediated apoptosis.
KEY WORDS: Asna1/TRC40, Pancreatic hypoplasia, Pancreatic progenitor cell, Apoptosis, Impaired differentiation, Integrated stress response, Mouse
Summary: Conditional inactivation of Asna1/TRC40 in pancreatic progenitor cells results in pancreatic agenesis resulting from pancreatic progenitor cell apoptosis, thus revealing a crucial role for Asna1/TRC40 in pancreatic progenitor cell survival.
INTRODUCTION
Asna1, also known as TRC40, and its yeast homolog GET3 have been implicated in a variety of cellular processes, including proteasome assembly and degradation of ubiquitylated proteins, growth under oxidative or metal stress, membrane trafficking within the secretory pathway, and insulin secretion (Akahane et al., 2013; Auld et al., 2006; Costanzo et al., 2010; Jonikas et al., 2009; Norlin et al., 2016; Schuldiner et al., 2005; Shen et al., 2003). Many of these phenotypes have been attributed to the ability of Asna1/GET3 to mediate the ATP-dependent delivery of tail-anchored (TA) proteins for post-translational insertion into the ER via the CAML/WRB receptor complex, a pathway that has been delineated in cell-free systems (Favaloro et al., 2008; Johnson et al., 2013; Schuldiner et al., 2008; Stefanovic and Hegde, 2007). The physiological range of TA-protein clients that critically rely on Asna1/GET3 for their membrane insertion may, however, be limited (Norlin et al., 2016; Rivera-Monroy et al., 2016). In addition to its ATPase-dependent functions, studies in yeast have suggested an additional role for Asna1/GET3 as a holdase chaperone under conditions of oxidative stress and ATP depletion (Powis et al., 2013; Voth et al., 2014). The shift from an ATPase-dependent function to the holdase function is associated with a structural reorganization of Asna1/GET3 from a dimer to a tetramer, and involves the oxidation of two di-cysteine motifs (Voth et al., 2014). In Get3 yeast mutants, the holdase function can be selectively rescued by Asna1/Get3 constructs that carry mutations in the ATPase domain or hydrophobic groove, i.e. domains that mediate TA-protein insertion (Voth et al., 2014), suggesting that the part of Asna1 that ensures the holdase function is distinct from that required for the ATPase-dependent and TA-targeting activities. In asna-1-deficient C. elegans, re-expression of asna-1 mutated in the CXXC di-cysteine motif rescues the severe growth phenotype displayed by worms lacking asna-1 but not the sensitivity to cisplatin, an oxidative stress-inducing drug (Hemmingsson et al., 2010), suggesting that Asna1 also performs multiple functions in higher eukaryotes. The relative contribution of these functions in mammalian cells in vivo remains, however, unknown.
Through conditional inactivation of Asna1 in insulin-producing β-cells of mice, we recently demonstrated a role for Asna1 in ensuring retrograde transport and thereby ER homeostasis and insulin biosynthesis in β-cells (Norlin et al., 2016). Notably, the proposed Asna1 target TA proteins syntaxin 5 (Stx5) and syntaxin 6 (Stx6) were redistributed from their Golgi compartments both in Asna1 mutant β-cells and after pharmacological inhibition of retrograde transport using Retro-2 (Norlin et al., 2016; Stechmann et al., 2010). Together, these findings suggested a key role for Asna1 in ensuring retrograde transport and Golgi localization of Stx5 and Stx6 in adult β-cells. To gain further insight into the role(s) for Asna1 in mammalian cells in vivo, we have here conditionally inactivated Asna1 in pancreatic progenitor cells.
Pancreatic development is initiated as two evaginations from the primitive gut epithelia. Over time, the specified pancreatic epithelia grow into the surrounding mesenchyme and form a tubular epithelium that undergoes extensive branching morphogenesis. Mouse pancreatic progenitor cells undergo two major rounds of differentiation (Shih et al., 2013). During the early phase between E9-12 (i.e. 1st transition), multipotent progenitor cells (MPCs), which are capable of generating acinar, endocrine and ductal cell lineages, proliferate and generate a small number of endocrine cells primarily expressing glucagon. During the 2nd transition between E12-14, pancreatic progenitor cells undergo extensive growth and branching morphogenesis, and the initial Ptf1a+/Sox9+ MPC population segregates into two populations: a branch tip population containing Ptf1aHigh/Sox9Low proacinar cells; and a bipotential Ptf1a−/Sox9High branch trunk population containing Ngn3+ proendocrine cells and Ngn3− duct progenitor cells (Schaffer et al., 2010; Solar et al., 2009). After E14.5, Ptf1aHigh proacinar tip cells differentiate and initiate expression of mature acinar cell markers, e.g. amylase. In the branch trunks, duct progenitor cells form the pancreatic ducts that connect the acinar cells to the intestine, whereas the Ngn3+ proendocrine cells migrate into the surrounding mesenchyme and initiate expression of endocrine hormones as they differentiate into α-, β-, δ-, γ- and ε-cells that eventually form the endocrine islets. Thus, the different cell types in the developing pancreas serves as a model for evaluating the requirement for Asna1 in several cellular processes, including proliferation, differentiation, morphogenesis and hormone secretion.
Here, we show that inactivation of Asna1 in pancreatic progenitor cells results in severe pancreatic agenesis. Loss of Asna1 in pancreatic progenitor cells leads to rapid redistribution of the TA proteins Stx5 and Stx6, followed by perturbed Golgi morphology, apoptotic cell death, and impaired acinar and endocrine cell differentiation from E13 onwards. In contrast, Asna1-deficient Sox9+ duct/MPC-like cells, as well as differentiated α-cells and β-cells specified at earlier stages of pancreatic development, survive. Apoptotic cell death in embryonic Asna1-deficient pancreatic epithelium correlates with an increase in integrated stress response (ISR) signaling, whereas the adaptive UPR and oxidative stress responsive pathways appear unaltered. Pharmacological inhibition of retrograde transport reproduces the redistribution of Stx5 and Stx6, and the fragmentation of the Golgi, but is insufficient to provoke substantial apoptosis and impaired acinar and endocrine cell differentiation. Re-introduction of an ATPase-deficient mutant of Asna1 failed to restore Golgi integrity and differentiation of pancreatic progenitor cells lacking Asna1, suggesting that the ATPase-dependent and TA-targeting activities of Asna1 are required for pancreatic progenitor cell survival.
RESULTS
Deletion of Asna1 in pancreatic progenitor cells leads to severe pancreatic hypoplasia due to apoptosis
Asna1 was broadly expressed in the developing pancreatic epithelium from ∼E10.5 and by E13.5 the expression became prominent in the pro-acinar branch tip cells (Fig. S1A). To elucidate a potential functional role of Asna1 in mouse pancreatic progenitor cells in vivo, we crossed Asna1flox/flox mice (Norlin et al., 2016) with Ipf1-nlsCre mice (Svensson et al., 2009), yielding Asna1flox/flox;Ipf1-nlsCre+ mice (denoted Asna1Panc−/−) (Fig. S1B). Ipf1-nlsCre ensures pancreas- and duodenal-specific Cre-mediated recombination in Rosa26loxP-stop-loxPLacZ reporter mice (Soriano, 1999) as early as E10.5 (Fig. S1C) and, in agreement with this, qRT PCR analysis revealed 68% reduction of Asna1 expression in pancreatic epithelium of Asna1Panc−/− embryos at E11.5 (Fig. S1D). Asna1Panc WT and Asna1Panc+/− embryos did not show any apparent phenotype at any stage examined and were thus used as controls, collectively denoted Asna1Panc:Ctrl. The resulting Asna1Panc−/− mice were born alive but died soon after birth due to severe pancreatic and duodenal agenesis (Fig. 1A).
Fig. 1.

Asna1Panc−/− mice develop pancreatic and duodenal agenesis due to apoptosis. (A) Upper gastrointestinal tract dissected from neonatal Asna1Panc:Ctrl and Asna1Panc−/− littermates showing pancreatic and duodenal agenesis. (B) X-gal staining of E15.5 Asna1Panc:Ctrl and Asna1Panc:−/− embryos on a Rosa26loxP−stop-loxP-LacZ background. (C) Quantification of the dorsal pancreatic epithelia (E-cad+) area of Asna1Panc−/− and control littermates at E12.5 (n=5 and 7, respectively), E13.5 (n=6 and 7, respectively) and E15.5 (n=5). (D) Representative immunohistochemistry of dorsal pancreatic sections from E13.5 Asna1Panc:Ctrl and Asna1Panc−/− embryos using antibodies against E-cadherin (E-cad, green), cleaved caspase 3 (c.Casp.3, red) and phospho-Histone H3 (pH3, red). Inset shows magnification of Asna1Panc−/− epithelia with cleaved caspase 3+ cells at epithelial protrusions (arrows). Scale bar: 100 µm in D. (E) Quantification of cleaved caspase 3+ cells relative to E-cad+ epithelial area in the dorsal pancreatic epithelia of Asna1Panc−/− and control littermates at E13.5 (n=5 and 7, respectively) and E15.5 (n=5). (F) Quantification of phospho-histone H3+ (pH3+) cells relative to E-cad+ epithelial area in the dorsal pancreatic epithelia of Asna1Panc−/− and control littermates at E13.5 (n=5 and 7, respectively) and E15.5 (n=5). dp, dorsal pancreas; du, duodenum; vp, ventral pancreas; sp, spleen; st, stomach. Data are presented as mean±s.e.m.; *P<0.05, **P<0.01, ns, not significant (Student's t-test).
To identify the cause of the pancreatic agenesis in Asna1Panc−/− embryos, we next characterized the developing dorsal pancreatic epithelia (DPE) in more detail. X-gal staining of Asna1Panc−/−;Rosa26loxP-stop-loxPLacZ embryos revealed that severe dorsal and ventral pancreatic agenesis and reduced branching complexity were already evident by E15.5 (Fig. 1B). Analyses of earlier developmental stages showed that the size of Asna1Panc−/− DPE was normal at E12.5, but reduced by 28% at E13.5 (Fig. 1C). Moreover, the rapid expansion of the pancreatic epithelia area of control mice between E13.5 and E15.5 was severely impaired in the Asna1Panc−/− DPE (Fig. 1C). Notably, the growth defect of Asna1Panc−/− DPE coincided with the prominent appearance of apoptotic cells at E13.5, preferentially in branch tips along the epithelial/mesenchymal border of the Asna1Panc−/− DPE (cleaved caspase 3+ cells in Fig. 1D,E and TUNEL+ cells in Fig. S2A). In contrast, proliferation, as judged by the amount of phospho-H3 (pH3)-positive cells in the Asna1Panc−/− DPE, was unaffected at E13.5. (Fig. 1F). At E15.5, apoptosis was still drastically increased in Asna1Panc−/− DPE when compared with Asna1Panc:Ctrl DPE and, in addition, proliferation was reduced in Asna1Panc−/− DPE at this stage (Fig. 1E,F). Apoptosis was also evident in E13.5 Asna1Panc−/− ventral pancreatic and E14.5 Asna1Panc−/− duodenal epithelia (Fig. S2B). These data show that Asna1Panc−/− mice develop pancreatic and duodenal epithelial hypoplasia as a consequence of apoptosis.
Loss of Asna1 impairs pancreatic acinar, endocrine and ductal cell lineages
To identify the pancreatic cell types that undergo apoptosis in Asna1Panc−/− DPE, we next analyzed the expression of cell specification and differentiation markers in E12.5-E16.5 Asna1Panc−/− DPE. Cells in the branch trunk regions of E12.5 Asna1Panc−/− DPE showed normal Sox9 expression that is characteristic of endocrine and ductal progenitor cells (Fig. 2A). Moreover, like that of control littermates, branch tips of Asna1Panc−/− DPE contained a subset of cells that showed Ptf1aHigh expression characteristic of pro-acinar cells, and Ptf1aLow and carboxypeptidase A+ (CPA+) cells that may represent Ptf1aLow/Sox9+/CPA+ MPCs (Pan et al., 2013; Zhou et al., 2007) (Fig. 2A). Ngn3+ endocrine progenitor cells and differentiated glucagon+ α-cells were dispersed within and around the branch trunk region (Fig. 2A), and quantification revealed no significant difference in the numbers of these cells at E12.5, i.e. prior to the onset of apoptosis, when comparing Asna1Panc−/− and control littermates (Fig. 2B,C). Thus, the initial lineage specification of MPCs, endocrine and ductal progenitor cells, as well as differentiation of α-cells is unaffected in Asna1Panc−/− mice up to E12.5.
Fig. 2.

Impaired endocrine and acinar differentiation in Asna1Panc−/− DPE. (A) Immunohistochemistry of dorsal pancreatic sections from E12.5 Asna1Panc:Ctrl and Asna1Panc−/− embryos (n=3) using antibodies against Sox9, Ptf1a, CPA and Ngn3 (all red), and E-cadherin (E-cad) and glucagon (both green). Insets show Ptf1aHigh acinar progenitors (white arrowheads) and Ptf1aLow putative MPCs (black arrowheads). (B,C) Quantification of total pro-endocrine (Ngn3+) area (B) and total endocrine (glucagon+) area (C) in the DPE from Asna1Panc:Ctrl and Asna1Panc−/− embryos at E12.5 (n=3). (D-F) Immunohistochemistry of dorsal pancreatic sections from E14.5 (D), E15.5 (E) and E16.5 (F) Asna1Panc:Ctrl and Asna1Panc−/− embryos (n=3) using antibodies against Ptf1a, Sox9, Ipf1, Ngn3, insulin, amylase and phospho-histone H3 (pH3) (all red), and E-cadherin (E-cad) and glucagon or DBA lectin (DBA) (all green). Inset in E shows Ptf1aLow cells (black arrowhead). (G,H) Quantification of total pro-endocrine (Ngn3+) area (G) and total endocrine (glucagon++insulin+) area (H) in the DPE from Asna1Panc:Ctrl and Asna1Panc−/− embryos at E13.5 and E15.5 (n=5, respectively). Data are mean±s.e.m., *P<0.05, **P<0.01, ns, not significant (Student's t-test). Scale bars: 100 µm in A,D-F
Following the onset of apoptosis at E13.5, branching morphogenesis was clearly perturbed by E14.5 onwards (Fig. 2D-F). The expression of Sox9 and Ipf1/Pdx1 appeared unaltered in the bipotential branch trunk progenitors, arguing against a role for these transcription factors in mediating impaired morphogenesis and progenitor cell apoptosis in E14.5 Asna1Panc−/− DPE (Fig. 2D). Consistent with the localization of apoptotic cells in branch tips of the E13.5 DPE (Fig. 1D, Fig. S2A), Ptf1a+ tip cells appeared reduced in number in Asna1Panc−/− DPE at E14.5 (Fig. 2D). Moreover, although some Ptf1aLow cells remained at E15.5 (Fig. 2E insets), Ptf1aHigh pro-acinar cells were essentially absent (Fig. 2E) and the number of pro-endocrine Ngn3+ cells was severely reduced at this stage (Fig. 2E,G). Together, these results provide evidence that Asna1 is required for survival of MPCs and consequently their progression into Ptf1aHigh pro-acinar and Ngn3+ pro-endocrine cells. Coherent with this scenario, definitive amylase+ acinar cells were essentially absent, and numbers of differentiated glucagon+ α-cells and insulin+ β-cells were significantly reduced in E15.5-E16.5 Asna1Panc−/− DPE (Fig. 2F,H, Fig. S3). Notably, the number of α-cells and β-cells still increased in Asna1Panc−/− DPE between E13.5 and E15.5 (Fig. 2H), suggesting that they derive from Ngn3+ pro-endocrine cells already specified at E13.5. The remaining E16.5 Asna1Panc−/− epithelium consisted of proliferating Sox9Low/DBA-lectinLow cells (Fig. 2F), which survive and differentiate in the absence of Asna1. Taken together, these data suggest that MPCs and/or their immediate offspring undergo apoptosis from E13.5 onwards in Asna1Panc−/− embryos, which impairs the generation of pro-endocrine and pro-acinar cells, and, consequently, the subsequent differentiation of endocrine and acinar cell types.
Asna1Panc−/− pancreatic agenesis is associated with p53 activity and integrated stress response signaling
Mouse genetic models presenting with severe pancreatic agenesis and acinar hypoplasia appear to be preferentially associated with premature differentiation, impaired proliferation or altered specification (Apelqvist et al., 1999; De Vas et al., 2015; Fukuda et al., 2008; Jensen et al., 2000; Krapp et al., 1996; Murtaugh et al., 2005; Nakhai et al., 2008; Papadopoulou and Edlund, 2005; Seymour et al., 2007) and not primarily with apoptosis. However, pancreas-specific ablation of DNA methyltransferase 1 (Dnmt1) (Georgia et al., 2013) was shown to provoke pancreatic progenitor cell apoptosis, which involved the de-repression of p53, an oncogene that induces cell cycle block or apoptosis in response to cellular stress such as genotoxic and ER stress (Brooks and Gu, 2010; Vogelstein et al., 2000). To investigate a potential role for p53-mediated apoptosis in Asna1Panc−/− DPE, we modulated p53 activity in explants of wild-type and Asna1Panc−/− pancreas. Exposure of E10.5 Asna1Panc:Ctrl explants to the p53 inhibitor pifithrin-α (PFT-α) (Komarov et al., 1999) resulted in a reduced Ngn3+, insulin+ and amylase+ cell area, implying a requirement for p53 in β-cell and acinar cell differentiation (Fig. 3A,B). Similarly, β-cell differentiation was reduced in Asna1Panc−/− explants exposed to PFT-α (Fig. 3A,C). In addition, glucagon+ area was also reduced, suggesting that p53 may play a role in α-cell survival and/or differentiation in Asna1Panc−/− embryos. In contrast, however, amylase+ cell differentiation was, albeit partially, restored in Asna1Panc−/− explants exposed to PFT-α (Fig. 3A,C), suggesting that excess p53 activation may provoke the apoptosis of Asna1Panc−/− MPCs. To test this idea, we exposed wild-type E12.5 explants to the p53 agonist Nutlin-3 (Vassilev et al., 2004) (Fig. 3D), which resulted in increased epithelial apoptosis after 24 h (Fig. 3E) and a dramatic reduction of epithelial area and in amylase+ cell number after 3 days (i.e. E15.5) (Fig. 3D,F). Insulin+ cells appeared unaffected (Fig. 3F). Total p53 protein levels in E13.5 dorsal pancreas buds, however, were not increased (Fig. S4), suggesting that p53 activity, rather than expression levels, were increased in Asna1Panc−/− DPE. Taken together, these results leave open the possibility that apoptosis in Asna1Panc−/− DPE, at least in part, is mediated by p53.
Fig. 3.

Apoptosis of pro-acinar progenitors in Asna1Panc−/− DPE is associated with p53 activity and activation of the IRS. (A) Representative immunohistochemistry of dorsal pancreas explants from E10.5 Asna1Panc:Ctrl and Asna1Panc−/− cultivated for 5 days exposed to vehicle (n=7 and 4, respectively) or PFT-α (30 µM) (n=11 and 4, respectively) using antibodies against amylase (red) and insulin (green). (B,C) Quantification of experiments described in A. Relative glucagon+ (Glu), Ngn3+, insulin+ (Ins) and amylase+ (Amy) areas over total E-cadherin+ area in explants from Asna1Panc:Ctrl (B) and Asna1Panc−/− (C) mice. (D) Representative immunohistochemistry of dorsal pancreas explants from E12.5 wild-type embryos cultivated for 3 days exposed to vehicle or Nutlin-3 (10 µM), using antibodies against E-cadherin (E-cad) and insulin (both green), and amylase (red). (E,F) Quantification of experiments described in D after 1 day (n=6 for each condition) (E) and 3 days (vehicle, n=4; Nutlin-3, n=5) (F) of cultivation; E-cadherin+ epithelial (Epi) and E-cadherin− mesenchymal (Mes) area; relative cleaved caspase 3+ (c.Casp.3) over E-cadherin+ area after 1 day; absolute insulin+ (Ins) and amylase+ (Amy) area after 3 days; arbitrary units (a.u). (G) qRT-PCR mRNA levels of the indicated ISR genes in dorsal pancreatic buds from E13.5 Asna1Panc:Ctrl (n=7) and Asna1Panc−/− (n=6) embryos. (H) Immunohistochemistry of dorsal pancreatic sections from E12.5 and E13.5 Asna1Panc:Ctrl and Asna1Panc−/− embryos (n=5) using antibodies against ATF4 (red) and E-cadherin (E-cad, green). Arrowheads indicate ATF4+ tip cells. (I) qRT-PCR mRNA levels of the indicated UPR genes in dorsal pancreatic buds from E13.5 Asna1Panc:Ctrl and Asna1Panc−/− (n=7) and Asna1Panc−/− (n=6) embryos. DAPI (blue) indicates nuclei in A,C. Scale bars: 50µm. Data are mean±s.e.m.; *P<0.05, **P<0.01, ***P<0.001; ns, not significant (Student's t-test).
As inactivation of Asna1 in mouse, C. elegans and yeast has been associated with both ER stress and increased sensitivity to oxidative stress, we next investigated stress signaling responses in Asna1Panc−/− embryonic pancreatic epithelium. Several cellular stress responses converge on the phosphorylation of eIF2α that, together with its downstream effector genes, are referred to collectively as the integrated stress response (ISR) pathway. qRT-PCR analysis of Asna1Panc−/− dorsal pancreatic buds, i.e. pancreatic epithelia and surrounding mesenchyme, revealed that the expression of genes associated with the ISR, such as Atf4 and its target genes Chop10 (Ddit3 – Mouse Genome Informatics), Atf3 and Trib3, was increased at E13.5 (Fig. 3G). Additionally, immunohistochemical analyses revealed strong nuclear expression of ATF4 in tip cells of Asna1Panc−/− DPE at E13.5, but not at E12.5 (Fig. 3H), thus correlating with the appearance of apoptotic progenitor cells (Fig. 1D). Taken together, these results suggest that ISR signaling and apoptosis occur in a spatially restricted subset of Asna1Panc−/− DPE cells.
The expression of oxidative stress genes, e.g. Nrf2 and its target genes Nqo1 and Hmox1 (previously HO-1), appeared unaltered at E13.5 (Fig. 3I). Although the expression of the UPR-regulated gene Herpud1, which encodes a component of the ER-associated degradation (ERAD) machinery, was upregulated in Asna1Panc−/− DPE, the expression of genes associated with the adaptive UPR, which encode chaperones [DnaJc3, Hsp90b1 (i.e. Grp94) and BiP (Hspa5) and oxidative folding enzymes (Pdia4)] rather showed a tendency to be reduced in E13.5 pancreatic buds (Fig. 3I), arguing against ER stress as the underlying cause for apoptosis observed in Asna1Panc−/− DPE. Together, these data suggest that in Asna1Panc−/− DPE tip cells, the ISR activates a program, possibly involving p53, that leads to MPC apoptosis and subsequent pancreatic agenesis.
Redistribution of Stx5 and Stx6 precedes Golgi fragmentation and apoptosis in Asna1Panc−/− pancreatic and duodenal epithelia
We next tried to establish a connection between the previously proposed functions of Asna1, ISR signaling and pancreatic progenitor cell apoptosis. As Asna1 is implicated in the targeting of TA proteins to the ER membrane, we first analyzed the structural integrity of endomembrane. Because apoptosis itself is associated with the breakdown of cellular structures, we analyzed the structural integrity of endomembrane compartments in Asna1Panc−/− DPE at E12.5, i.e. 1 day prior to the onset of apoptosis. Immunohistochemical analyses using markers for the ER (KDEL), endoplasmic reticulum Golgi-intermediate compartment (ERGIC53), endosome (EEA1) and lysosome (Lamp1) revealed that these compartments appeared structurally indistinguishable from that of controls (Fig. S5A). However, analyses using markers for the cis-Golgi (Gm130) and trans-Golgi network (TGN) (TGN46) showed that these markers were uncharacteristically dispersed in the cytoplasm of cells throughout the Asna1Panc−/− DPE (Fig. 4A). Transmission electron microscopy (TEM) revealed the presence of small distended membrane stacks in Asna1Panc−/− DPE, as opposed to the thin multilayered membrane stacks characteristic of the cis- and medial Golgi compartment observed in control littermates (Fig. 4B). However, no apparent difference was observed in ER or mitochondrial morphology at E12.5 (Fig. S5B). Together, these results show that cis-Golgi and TGN morphology is perturbed in cells throughout the Asna1Panc−/− DPE prior to apoptosis.
Fig. 4.

Altered morphology and Stx5 and Stx6 expression in the Golgi apparatus of Asna1Panc−/− DPE cells. (A) Immunohistochemistry of dorsal pancreatic sections from E12.5 Asna1Panc:Ctrl and Asna1Panc−/− embryos (n=3) using antibodies against Gm130 (green) and TGN46 (red). Boxed areas are shown at higher magnification on the right. (B) Transmission electron micrograph (TEM) of dorsal pancreatic sections from E12.5 Asna1Panc:Ctrl and Asna1Panc−/− embryos (n=3) showing compact Golgi stacks (red arrowheads) and distended Golgi stacks (white arrowheads). (C-E) Immunohistochemistry of dorsal pancreatic sections from E11.5 (C,E) or E12.5 (D) Asna1Panc:Ctrl and Asna1Panc−/− embryos (n=3) (C,D) or Asna1null/Panc+ and Asna1null/Panc− (E) embryos (n=3), using antibodies against Gm130 (green), Stx5 (red), Stx6 (green) and TGN46 (red). (F) Immunohistochemistry of dorsal pancreatic sections from E16.5 Asna1Panc:Ctrl and Asna1Panc−/− embryos (n=3) using antibodies against Stx5 (red) and Gm130 (green) together with either glucagon, insulin or DBA-lectin (all white). Merge panels show co-expression between Stx5 and Gm130 (yellow). (G) Immunohistochemistry of dorsal pancreas explants from E10.5 Asna1Panc−/− cultivated for 5 days (n=3) exposed to vehicle or PFT-α, using antibodies against Stx5 (red) and Gm130 (green) together with amylase (white). Merge panels show co-expression between Stx5 and Gm130 (yellow). DAPI (blue) indicates nuclei in A,C-G. Scale bars: 50 µm in A; 0.5 µm in B; 10 µm in C-E; 5 µm in F,G.
Loss of Asna1/GET3 function in both yeast and mouse is associated with altered subcellular localization of the TA-SNAREs Stx5/Sed5 and Stx6, which undergo a striking re-localization from the cis-Golgi and TGN, respectively (Jonikas et al., 2009; Norlin et al., 2016; Schuldiner et al., 2008). In Asna1Panc−/− DPE at E11.5, both Golgi integrity, as well as Stx5 and Stx6 expression, was normal (Fig. 4C). However, at E12.5, Stx5 and Stx6 immunoreactivity was severely reduced and virtually absent in the fragmented cis-Golgi and TGN compartments of Asna1Panc−/− DPE (Fig. 4D). However, total Stx5 protein levels were unaltered in E12.5 pancreas buds (Fig. S5C), suggesting that, like Asna1-deficient β-cells (Norlin et al., 2016), Stx5 protein becomes redistributed rather than downregulated in Asna1Panc−/− DPE. An intermediate phenotype was observed in embryos carrying a germline Asna1null allele in combination with a conditionally inactivated Asna1flox allele, denoted Asna1null/Panc−, in which Stx5 and Stx6 immunoreactivity was already perturbed at E11.5, whereas Golgi compartments appeared intact (Fig. 4E). Nonetheless, this earlier depletion of Asna1 in Asna1null/Panc− embryos was insufficient to provoke an earlier onset of increased apoptosis at E12.5 (Fig. S5D). Taken together, these results provide evidence that redistribution of Stx5 and Stx6 precedes Golgi fragmentation, which in turn is followed by overt apoptosis at E13.5. Moreover, similar changes in Golgi morphology and syntaxin distribution also preceded apoptosis in the ventral pancreas and the duodenum epithelia (Fig. S5E,F), suggesting a general Asna1-dependent function(s) in endodermal progenitor cells.
We next investigated whether Stx5 redistribution and Golgi fragmentation was retained in surviving cell types at E16.5. The Sox9Low/DBA-lectinLow duct-like cells, β-cells and most α-cells all showed redistribution of Stx5 and fragmented Golgi compartments (Fig. 4F), suggesting that Stx5 redistribution and Golgi fragmentation does not affect survival of these cells. Notably, as the Ipf1-nlsCre transgene is inactive in α-cells, these results further suggest that most α-cells are derived from Asna1-deficient Ngn3+ pro-endocrine cells. However, a minor fraction of the α-cells showed intact Golgi structure and Stx5 localization (data not shown), suggesting that they derive from the early pool of α-cells that are generated prior to E11.5, and may thus have escaped Ipf1-nlsCre-mediated inactivation of Asna1. Finally, treatment of Asna1Panc−/− explants with the p53 inhibitor PFT-α did not restore Stx5 distribution in epithelial cells, including the rescued amylase+ cells (Fig. 4G), suggesting that p53 inhibition did not prevent apoptosis by restoring Stx5 localization or by interfering with CRE-mediated inactivation of Asna1. However, the integrity of the Golgi compartment in the rescued amylase+ cells appeared partially restored (Fig. 4G). These results demonstrate that Asna1 inactivation, Stx5 redistribution and Golgi fragmentation is tolerated by most cell types in the developing pancreas, thus highlighting the selective sensitivity of MPCs.
p62+ protein aggregates accumulate in Asna1Panc−/− DPE
Apart from a role in TA-protein targeting, endomembrane transport and ER homeostasis, Asna1/GET3 is also implicated as a redox-regulated holdase chaperone during oxidative stress and ATP depletion (Powis et al., 2013; Voth et al., 2014). Thus, one consequence of Asna1 inactivation might be the aggregation of misfolded TA, as has been suggested for Stx5 (Rivera-Monroy et al., 2016), and/or non-TA Asna1 target proteins, which may be potentially cytotoxic. In keeping with this idea, the E13.5 pancreatic epithelium showed increased numbers of distinct puncta positive for p62 (Fig. 5A), an ubiquitin-like protein conjugate that targets proteins for autophagic degradation. These results suggest that protein inclusions destined for proteasomal and/or autophagic degradation accumulates in Asna1Panc−/− DPE at the time of apoptotic onset. An alternative explanation could be that autophagic degradation per se is impaired in Asna1Panc−/− DPE as autophagy is dependent on vesicular transport. To separate these possibilities, we first investigated whether autophagic degradation was decreased by analyzing the accumulation of LC3+ autophagosomes in GFP-LC3 reporter mice on an Asna1Panc−/− background. Although occasional cells exhibited increased GFP-LC3 levels, a general accumulation in LC3-GFP levels was not observed in Asna1Panc−/− DPE (Fig. 5B). Next, we specifically inactivated autophagy in the developing pancreas by generating Atg5flox/flox, Ipf1-nlsCre mice (denoted Atg5Panc−/−). At E14.5, Atg5Panc−/− DPE exhibited an increase in cytosolic p62 protein in the pancreatic trunk epithelia but not in pro-acinar tip cells (Fig. 5C, insets). Notably, no corresponding increase in p62+ puncta or apoptosis was observed in any part of the Atg5Panc−/− DPE (Fig. 5C). Taken together, these results strongly argue against decreased autophagic degradation as the primary cause for the accumulation of p62+ puncta in Asna1Panc−/− cells. Instead, it appears likely that these p62+ puncta consist of aggregating Asna1 client TA- or non-TA-proteins, which in turn may contribute to apoptotic cell death.
Fig. 5.
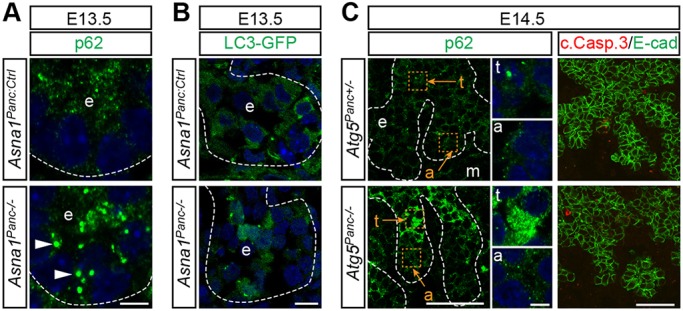
Accumulation of autophagy markers in Asna1Panc−/− DPE. (A) Immunohistochemistry of dorsal pancreatic sections from E13.5 Asna1Panc:Ctrl and Asna1Panc−/− embryos (n=5) using antibodies against p62 (green). Arrowheads indicate p62+ puncta. (B) Dorsal pancreatic sections from E13.5 Asna1Panc:Ctrl and Asna1Panc−/− embryos on a LC3-GFP transgenic background, showing LC3-GFP fluorescence in green (n=2). (C) Immunohistochemistry of dorsal pancreatic sections from E14.5 Atg5Panc+/− and Atg5Panc−/− embryos (n=3), using antibodies against p62 (green), E-cadherin (E-cad; green) and cleaved caspase3 (c.Casp.3; red). Insets show magnification of selected trunk (t) and acinar (a) epithelia areas (orange squares, letters and arrows). DAPI (blue) indicates nuclei in A-C. Dashed lines delineate pancreatic epithelium (e) and mesenchyme (m). Scale bars: 5 µm in A,B and insets in C; 50 µm in C. All image pairs were captured using the same settings.
Progenitor cell survival and exocrine cell differentiation depend on the ATPase activity of Asna1
Rescue experiments in yeast have demonstrated that both the ATPase activity and the TA-binding ability are required for the TA-targeting function of GET3/Asna1, as defined by the Golgi localization of Sed5/Stx5, but not for growth under oxidative stress (Voth et al., 2014). The GET3/Asna1 holdase activity is associated with a tetramer configuration and involves the formation of di-sulfide bonds within two di-cysteine motifs, CXC(246-248) and CXXC(285-288) (Voth et al., 2014). Accordingly, the CXXC motif is required for growth under oxidative stress in yeast (Metz et al., 2006). Moreover, in C. elegans, the CXXC motif of Asna1 is required for resistance to the oxidative stress-inducing drug cisplatin, but not for rescuing the growth defect of Asna1 mutant worms (Hemmingsson et al., 2010). In vitro, mutations in the CXXC(285-288) motif affect GET3/Asna1 homodimerization, TA-protein binding and ATPase activity (Mateja et al., 2009; Metz et al., 2006), suggesting a possible dual role for the CXXC(285-288) motif in both configurations of Asna1. The ability of the Asna1 CXXC(285-288) mutant to restore Stx5 localization in vivo has, however, not been directly tested. The more N-terminal CXC(246-248) motif is not required for dimer formation in vitro or for growth under oxidative stress conditions (Metz et al., 2006). To explore which of the activities of Asna1, i.e. the ATPase-dependent chaperone (TA-targeting) or holdase function, is required for pancreatic progenitor survival and differentiation, we next used a lentivirus delivery system to reintroduce Asna1 versions carrying mutations in different functional domains into Asna1Panc−/− DPE. E11.5 Asna1Panc−/− pancreatic explants infected with empty lentivirus control vector and cultivated for 5 days did not show any evidence of exocrine differentiation, the Golgi compartments were fragmented and Stx5 expression was faint (Fig. 6A), thus reproducing the phenotypes of E16.5 Asna1Panc−/− DPE.
Fig. 6.
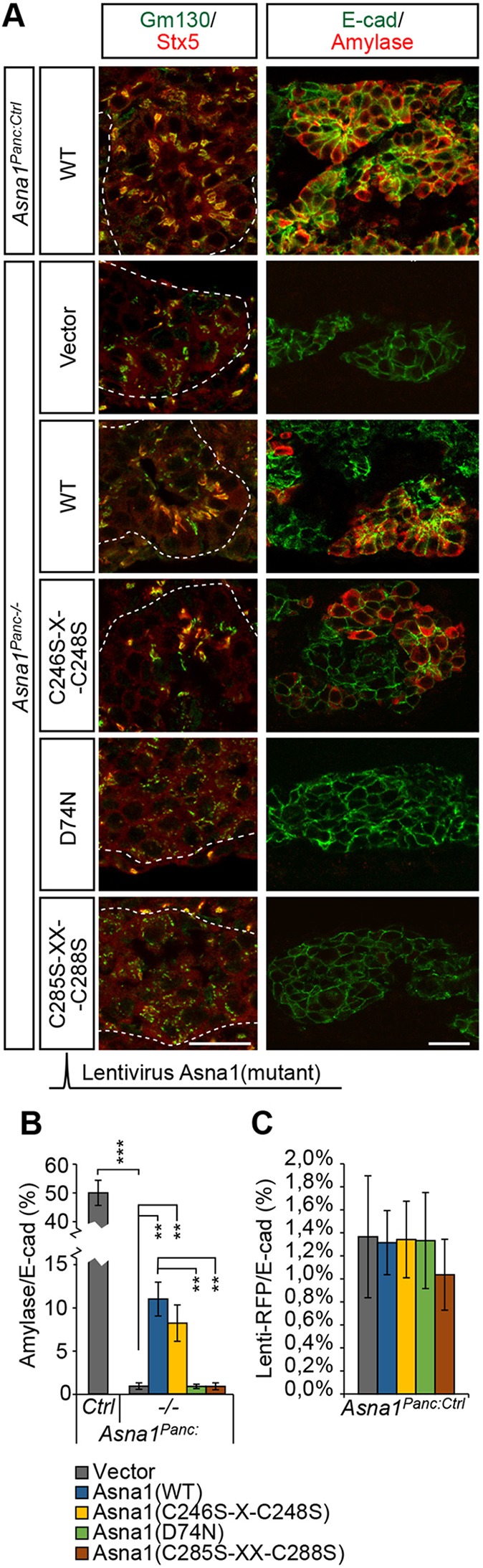
Normal Golgi morphology, Stx5 distribution and acinar differentiation requires the ATPase domain and CXXC(285-288) di-cysteine motif of Asna1. (A) Representative immunohistochemistry of lentivirus-infected dorsal pancreas explants using antibodies against Gm130 (green), Stx5 (red), E-cadherin (E-cad; green) and amylase (red). Dorsal pancreas explants from E11.5 Asna1Panc:Ctrl or Asna1Panc−/− embryos were transduced with lentivirus particles carrying either empty expression vector (Vector) (n=6 and 5, respectively) or constructs encoding wild-type Asna1 (WT) (n=5), Asna1 (D74N) (n=4), Asna1 (C246S-X-C248S) (n=13) or Asna1 (C285S-XX-C288S) (n=4), as indicated, and grown for 5 days. Dashed lines delineate pancreatic epithelium. Scale bar: 25 µm. (B) Quantification of relative amylase+ epithelial area (amylase+/total E-cad+) from experiments described in A. (C) Quantification of relative RFP+ epithelial area (RFP+/Ecad+) in control explants infected with the different lentivirus expression constructs from experiments described in A. Data are presented as mean±s.e.m., **P<0.01, ***P<0.001 (Student's t-test).
Infection of Asna1Panc−/− pancreatic explants with Asna1 wild-type construct partly restored acinar cell differentiation as well as Stx5 expression in the Golgi (Fig. 6A,B). Similar results were obtained when re-expressing the Asna1(C246S-X-C248S) mutant (Fig. 6A,B). In contrast, an Asna1(D74N) construct, which carries a mutation in the ATPase domain, failed to restore Golgi morphology, Stx5 expression and acinar cell differentiation (Fig. 6A,B). Similar to the Asna1(D74N) construct, re-expression of Asna1(C285S-XX-C288S) failed to rescue the Asna1 deficiency in E11.5 Asna1Panc−/− pancreatic explants (Fig. 6A,B). These results suggest that, in agreement with its proposed role in the dimerization of Asna1 (Mateja et al., 2009; Metz et al., 2006), the CXXC(285-288) motif also affects the ATPase-dependent activities of the Asna1 dimer, and thus cannot be used to assess the contribution of a putative ATPase-independent holdase function of Asna1 in Asna1Panc−/− mice. Thus, these data show that the ATPase-dependent chaperone functions of Asna1 are required for ensuring both Stx5 expression/localization, as well as the survival of pancreatic progenitor cells from E13.5 onwards.
Inhibition of retrograde transport in pancreatic progenitor cells mimics Stx5 redistribution and Golgi fragmentation but does not provoke apoptosis
The loss of ATPase/CXXC-dependent activities in Asna1Panc−/− mice is likely to affect an array of cellular processes, including vesicle-mediated transport of various protein cargo. In yeast, the GET complex, including GET3/Asna1, genetically interacts with retrograde transport pathways (Jonikas et al., 2009; Schuldiner et al., 2005) and β-cell-specific inactivation of Asna1 result in impaired endosome (EE)-to-TGN as well as COPI-independent Golgi-to-ER transport (Norlin et al., 2016). Thus, we next investigated whether inhibition of retrograde transport with the small molecule inhibitor Retro-2 (Norlin et al., 2016; Stechmann et al., 2010) could reproduce Asna1Panc−/− phenotypes. Exposure of E11.5 wild-type (wt) dorsal pancreatic bud explants to Retro-2 for 48 h resulted in reduced Stx5 and Stx6 expression in the cis-Golgi and TGN compartments (Fig. 7A). In analogy with Asna1Panc−/− DPE, the cis-Golgi of Retro-2-treated wild-type DPE appeared partly fragmented and the TGN more diffuse (Fig. 7A, insets). However, Retro-2 exposure did not induce prominent ATF4 expression in DPE explants (Fig. 7B) and extended exposure of E10.5 wild-type DPE explants to Retro-2 for 5 days (i.e. equivalent to E15.5) did not significantly affect epithelial size or the differentiation of endocrine or acinar cell lineages (Fig. 7C,D), although it did provoke apoptosis in both epithelial and mesenchymal cells (Fig. 7E). Taken together, these data show that inhibition of retrograde transport by Retro-2 is sufficient to reproduce the redistribution of Stx5 and Stx6 and the fragmentation of the Golgi compartments but not the loss of acinar and endocrine cell types observed in Asna1Panc−/− DPE.
Fig. 7.

Inhibition of retrograde transport by Retro-2 disrupts Golgi morphology and Stx5 distribution but allows for acinar differentiation. (A,B) Immunohistochemistry of E11.5 dorsal pancreas explants from wild-type embryos grown for 48 h and exposed to vehicle (n=4) or Retro-2 (50 µM) (n=5), using antibodies against Gm130, Stx6 and E-cad (all green), and Stx5, TGN46 and ATF4 (all red). Insets in A show magnification of green and red channels for selected areas (dashed squares). Dashed lines delineate pancreatic epithelium. Scale bars: 10 µm in A; 50 µm in B. (C-E) Quantification of total epithelial (Epi) and mesenchymal (Mes) area (C), relative endocrine (Ngn3+, glucagon+ and insulin+) and exocrine (amylase+) area (% of total E-cad+ area) (D), and relative apoptotic cell area [% cleaved caspase 3+ (c.Casp.3+) of E-cad+ epithelium (Epi) and E-cad− mesenchyme (Mes)] (E) in E10.5 dorsal pancreas explants grown for 5 days exposed to vehicle (n=8) or Retro-2 (50 µM) (n=7). Data are presented as mean± s.e.m.; *P<0.05; ns, not significant (Student's t-test).
DISCUSSION
Here, we show that inactivation of Asna1 in pancreatic progenitor cells leads to selective apoptosis of multipotent progenitor cells, thereby depleting the pancreatic progenitor cell pool and perturbing subsequent growth and differentiation of the developing pancreatic epithelium, which ultimately results in severe pancreatic agenesis. Embryonic pancreatic epithelial cells lacking Asna1 exhibited perturbed localization of Stx5 and Stx6 in the cis-Golgi and TGN compartments, respectively, aberrant Golgi morphology, activation of the ISR and accumulation of cytosolic p62+ protein aggregates. Finally, we demonstrate that the ATPase activity of Asna1 is required to restore Golgi morphology and Stx5 expression as well as progenitor cell survival.
The most striking effect of Asna1 deficiency in pancreatic progenitor cells is the severe pancreatic agenesis that appears to be caused by selective apoptosis of multipotent progenitor cells at E13.5. However, the reduced pancreatic epithelial cell proliferation observed at E15.5, which likely is secondary to the apoptosis of MPC at E13.5, might also contribute to the pancreatic hypoplasia of Asna1Panc−/− mice. Whereas Asna1 seems to be functionally depleted throughout the E12.5 Asna1Panc−/− DPE, as indicated by perturbed Golgi morphology and Stx5 expression, the first signs of apoptosis and increased ISR signaling, i.e. ATF4 expression, is observed 1 day later and is primarily evident in the branch tips of the growing pancreatic epithelium that contain MPCs for both acinar and endocrine cell linages, thus providing an explanation for the perturbed endocrine and acinar cell differentiation in Asna1Panc−/− pancreatic epithelium beyond E13.5 (Zhou et al., 2007). Moreover, the few remaining endocrine cells observed in Asna1Panc−/− embryos do not appear to escape recombination as Stx5 and Stx6 are mis-localized also in these cells, providing evidence that loss of Asna1 per se does not affect survival of already specified lineage-specific progenitor cells nor their subsequent differentiation. Thus, the severe pancreatic hypoplasia observed in Asna1Panc−/− embryos appears to be the consequence of massive cell death within the domain of multipotent progenitor cells or their immediate offspring from E13.5 onwards.
The Asna1/GET3 pathway has been implicated in multiple functions in yeast and mammalian cells, including that of (1) an ATPase-dependent chaperone function required for Sed5/Stx5 localization to the Golgi (Schuldiner et al., 2008; Voth et al., 2014); (2) an ATP independent holdase chaperone function that supports growth under conditions of oxidative/metal stress (Powis et al., 2013; Voth et al., 2014); and (3) a proteostatic quality control network ensuring cellular proteostasis (Akahane et al., 2013; Auld et al., 2006). Accordingly, Asna1Panc−/− pancreatic progenitor cells display reduced Stx5 expression in the Golgi, which may be attributed to the loss of ATPase-dependent chaperone activity of Asna1, and increased prevalence of cytosolic p62+ puncta, which may be interpreted as an accumulation of protein aggregates in the absence of the holdase activity of Asna1 and/or due to the proposed role for Asna1 in ensuring cellular proteostasis. Notably, our results clearly demonstrate that the ATPase activity of Asna1 is required for restoring Golgi morphology and Stx5 and Stx6 expression as well as acinar cell differentiation, whereas a putative holdase function, which should be retained in the Asna1(D74N) mutant (Voth et al., 2014), is unable to rescue these phenotypes. We further find that the CXXC(285-288) motif, which is predicted to affect the holdase function (Hemmingsson et al., 2010; Metz et al., 2006), also interferes with the ATPase-dependent Stx5 localization, presumably by affecting dimerization of Asna1/GET3 (Metz et al., 2006), thus precluding a direct test of the putative role for the holdase function of Asna1.
The redistribution of Stx5/Sed5 is a common hallmark phenotype observed in yeast GET3 mutants, Asna1-deficient pancreatic progenitor and β-cells, as well as in cardiomyocytes and hepatocytes in which the Asna1/TRC40 receptor WRB was disrupted, and thus indicates the functional loss of the ATPase-dependent GET3/Asna1/TRC40 pathway activity (Norlin et al., 2016; Powis et al., 2013; Rivera-Monroy et al., 2016; Schuldiner et al., 2008; Voth et al., 2014). The notion that membrane integration of Stx5, as well as that of other TA proteins, is directly mediated by the ATPase-dependent function of Asna1/GET3 relies heavily on studies in cell-free systems, whereas in vivo analysis show that many TA proteins are not dependent on Asna1/GET3 for their membrane insertion (Norlin et al., 2016; Rivera-Monroy et al., 2016; Schuldiner et al., 2008). However, the ability of Retro-2 to phenocopy the redistribution of Stx5 and Stx6 raises the possibility that these Golgi syntaxins are appropriately inserted into the ER membrane in Asna1-deficient cells but subsequently redistributed from the Golgi due to impaired vesicle transport. These two explanations are not mutually exclusive and additional experiments are required to fully resolve the role of Asna1 and retrograde transport for the localization of Stx5 and Stx6.
Regardless of the exact mechanism, several phenotypes of Asna1-deficient pancreatic progenitors and β-cells can be attributed to perturbed Retro-2-sensitive retrograde transport. First, as mentioned above, the redistribution of Stx5 and Stx6 can be reproduced by exposure of both islets (Norlin et al., 2016) and pancreatic explants to Retro-2. Second, impaired COPI-independent Golgi-to-ER retrograde transport and increased UPR signaling in Asna1-deficient β-cells is recapitulated by treatment with Retro-2 (Norlin et al., 2016). Finally, Golgi fragmentation, which is observed in progenitor cells but not in mature β-cells, is selectively reproduced by Retro-2 in progenitor cells but not in β-cells, further emphasizing the overlap between Retro-2-sensitive and Asna1-dependent processes, albeit in an apparent cell context-specific manner. In contrast, the induction of ISR signaling and the apoptosis observed in Asna1Panc−/− MPCs cannot easily be attributed exclusively to impaired Retro-2-sensitive retrograde transport. Like mature β-cells of Asna1ß−/− mice (Norlin et al., 2016), Sox9+ duct-like cells, Ngn3+ pro-endocrine cells and differentiated α-cells and β-cells present in the Asna1Panc−/− DPE remain refractory to apoptosis, despite apparent Stx5 and Stx6 redistribution as well as Golgi fragmentation. In addition, inhibition of retrograde transport by Retro-2 mimics the redistribution of Stx5 and Stx6, and the Golgi fragmentation in wild-type MPCs, but does not trigger extensive apoptosis or impair acinar cell differentiation. Thus, it appears that the requirement for Asna1 ATPase function for progenitor cell survival is mediated by pathways that are independent of Stx5- and Retro-2-sensitive retrograde transport, but might mediate membrane insertion of other TA protein(s) that still remain to be defined. We cannot, however, exclude the possibility that poor solubility of Retro-2 precludes achieving a concentration that is sufficient complete inhibition of Asna1-dependent retrograde transport in pancreatic progenitor cells.
Several possibilities may account for the cell-specific differences in Asna1−/− phenotypes. For example, the absence of Golgi fragmentation as observed in β-cells of Asna1ß−/− mice may be explained by the activation of UPR signaling that may help to maintain Golgi integrity (Norlin et al., 2016). Another possibility is that, dependent on the proliferative status of cells (i.e. pancreatic progenitor cells as compared with differentiated β-cells), the redistribution of Stx5 may prevent the re-assembly of the Golgi compartments after cell division (Rabouille et al., 1998). The proliferative status may also influence the apoptotic response. Cell types with limited proliferative potential, such as specified pro-endocrine cells and differentiated endocrine cells, appear refractory to apoptosis. In contrast, Asna1Panc−/− progenitor cell apoptosis, which may be partly p53 dependent, is observed among the proliferating cells in the MPC domain (tip cells) in both the dorsal and ventral pancreatic, as well as the proliferating duodenal, epithelia in Asna1Panc−/− mice. The link between p53, an oncogene that can induce cell cycle arrest and/or apoptosis in response to a variety of cellular stresses, and pancreatic progenitor cell apoptosis in Asna1Panc−/− mice, suggests that Asna1 inactivation somehow interferes with cell cycle progression. Interestingly, cells deficient for one of the Asna1 ER-receptor subunits, CAML, exhibit mitotic defects that include chromosome mis-segregation (Liu et al., 2009). The causal link between Asna1 deficiency and p53 activation, however, remains unknown and will require further analyses. To this end, it is worth noting that inactivation of RINT1, a component of the DSL1/NRZ and the RZZ complexes (Tagaya et al., 2014), interferes with retrograde transport and chromosome segregation, resulting in Golgi fragmentation, genomic instability and apoptosis (Grigaravicius et al., 2016), indicating a possible connection between the vesicle transport machinery and cell division control.
In summary, we show that Asna1 in pancreatic progenitors has two functions: (1) ensuring retrograde transport pathway(s), thereby maintaining Golgi integrity; and (2) ensuring progenitor cell survival, possibly by preventing p53 activation. Both these functions rely, directly or indirectly, on the ATPase activity of Asna1, presumably by promoting insertion of a TA protein. We cannot, however, rule out putative ATPase-dependent functions of Asna1 that are unrelated to the actual membrane insertion of TA proteins, such as influencing protein degradation, or direct effects of Asna1 in regulating retrograde transport. Elucidation of the molecular mechanism(s) underlying the redistribution of Stx5 and Stx6 as well as the progenitor cell apoptosis observed in Asna1Panc−/− pancreatic and duodenal epithelium will require further analyses.
MATERIALS AND METHODS
Data reporting
For animal experiments, no sample-size estimate was calculated before the study was executed. The experiments were not randomized unless otherwise stated. Investigators were not blinded to allocation during experiments and outcome assessment. For in vivo data, each n value corresponds to a single mouse or mouse embryo. For ex vivo cultures, each n value corresponds to independent explants. If technical replicates were performed, then their mean was considered as n=1.
Mouse strains and generation of Asna1Panc mice
Ipf1-nlsCre transgenic mice (Svensson et al., 2009) were bred with Asna1+/flox mice (Norlin et al., 2016), yielding Asna1+/flox, Ipf1-nlsCre+ mice (denoted Asna1Panc+/−), which thus exhibit pancreas- and duodenum-specific Cre-mediated recombination from at least E10.5. Unless otherwise stated, male Asna1Panc+/− males were crossed with Asna1flox/flox females to yield Asna1Panc−/− mice. To achieve earlier inactivation of Asna1, we took advantage of the leakiness of the Ipf1-nlsCre transgene (Svensson et al., 2009) in the germ cell lineage of female Asna1Panc+/− mice, which thus transmit wild-type Asna1 (Asna1+) or recombined Asna1null alleles. Hence, crossing male Asna1flox/flox mice with female Asna1+/flox;Ipf1-nlsCre+ mice yields Asna1null/flox;Ipf1-nlsCre+ mice (denoted Asna1null/Panc−). Cre-mediated recombination was visualized in situ by breeding a Rosa26loxP-stop-loxP-LacZ transgene (Soriano, 1999) onto an Ipf1-nlsCre+ or Asna1Panc+/− or Asna1Panc−/− background. For evaluation of autophagic degradation, Asna1Panc−/− mice were bred onto a background of GFP-LC3 transgenic mice (Mizushima et al., 2004) [provided by RIKEN BRC (RBRC00806)] and pancreas progenitor-specific deletion of Atg5 was performed by breeding Atg5flox mice (Kuma et al., 2004) [provided by RIKEN BRC (RBRC02231)] with Ipf1-nlsCre mice. The genotypes of mice were determined by PCR analyses of genomic DNA samples extracted from tail biopsy specimens. For PCR primer sequences, see Table S3. PCR primers Asna-exon2-196F, ASNA_G_WT-A and Asna-G4R were used to detect the Asna1+ (407 bp), Asna1flox (525 bp) and Asna1− (267 bp) alleles. PCR primers IPF1-5′3, IPF1-AR and CRE1 were used to detect the Ipf1-nlsCre transgene (950 bp) and endogenous Ipf1 allele (700 bp). The GFP-LC3 transgenic construct and the endogenous LC3 allele were detected using PCR primers GFP(LC3) and LC3*rc3 (400 bp), and mLC3ex3GT and mLC3ex4AG (550 bp), respectively. The PCR primers ATG5exon3-1, ATG5check and ATG5short were used together to amplify the Atg5flox (700 bp) and Atg5WT (350 bp) alleles. Genetically modified mice were kept on a mixed background. Embryonic wild-type tissue for in situ hybridization and explants were obtained from crossing CBA males with B6 females. The animal studies were approved by the Institutional Animal Care and Use Committee of Umeå University and were conducted in accordance with the guidelines for the care and use of laboratory animals.
Tissue isolation and preparation
Embryos were collected at selected stages. The day of the vaginal plug was considered embryonic day (E) 0.5. Embryonic tissue for immunohistochemical or in situ analysis were fixed in 4% PFA in PBS for 1 h, equilibrated in 30% sucrose in PBS, frozen and sectioned. For qPCR analysis or ex vivo culture experiments, the dorsal pancreatic buds were isolated using tungsten needles to remove as much of the surrounding mesenchyme as possible. Alternatively, as indicated in text, the mesenchyme was removed after proteolytic degradation with Dispase II (Roche).
Immunohistochemistry and in situ hybridization
Tissue sections or intact cells were incubated for 20 min with blocking buffer [10% fetal bovine serum diluted in Tris-HCl (pH7.4), 0.15 M NaCl, 0.1%Triton-X100 (TBST)]. For mouse monoclonal primary antibodies, sections were additionally blocked with MOM reagent (30-60 min, diluted 1:30 in TBST) (Vector labs, MKB-2213). Tissues were incubated with primary antibodies (Table S1) overnight at 4°C followed by washes and incubation with fluorochrome-labeled secondary antibodies for 1 h. All antibodies were diluted in blocking buffer. TUNEL assay to detect apoptosis was performed according to manufacturer's recommendations (in situ cell death detection kit, Fluorescein, Roche, 11684795). In situ hybridization using digoxigenin-labeled probes was performed as described previously (Schaeren-Wiemers and Gerfin-Moser, 1993).
Transmission electron microscopy (TEM)
Tissue samples were processed for TEM by Umeå Core Facility Electron Microscopy (UCEM). Briefly, tissue samples were fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH7.4), post-fixed in 1% osmiumtetroxide, dehydrated and finally embedded in Spurr resin according to standard procedures. Sections were contrasted with uranyl acetate and lead citrate, and examined using a Jeol 1230 TEM. Micrographs were acquired using a Gatan MSC 600CW. The presence of electron-dense tight junctions between epithelial cells was used to distinguish them from mesenchymal cells.
Ex vivo assays
Explants were cultivated on Millicell CM culture plate inserts (Merk, PICM01250) in explant medium: DMEM (Gibco, 21885) supplemented with 10% BCS, 10 mM HEPES (pH7.4) and 10 U/ml PEST (Gibco, 15140-122). Explant medium was further supplemented with vehicle (DMSO), Retro-2 (Calbiochem, 554715), pifithrin-α hydrobromide (PFT-α) (Tocris, 1267), Nutlin-3 (Tocris, #3984). To facilitate lentivirus infection, explants were split in half to expose the pancreatic epithelium and pre-incubated with concentrated virus particles for 2 h at 4°C.
Lentivirus expression
Asna1 mutant cDNA was cloned after the EF1-promoter of the lentivirus expression vector EF.CMV.RFP (Addgene plasmid 17619, deposited by Linzhao Cheng), which also contains a separate CMV-driven RFP reporter to monitor transduction efficiency (Yu et al., 2003). The various expression vectors were co-transfected with envelope and packaging plasmids, pMD2.G and psPax2 (Addgene plasmids 12259 and 12260, deposited by Didier Trono), to produce virus particles as previously described (Szulc et al., 2006). Infection efficiency of pancreatic epithelia was similar for all constructs (∼1% of Ecad+ area) as monitored by the CMV-RFP expression. However, owing to imperfect correlation between expression from the EF1 and CMV promoters (Yu et al., 2003), the true efficiency of Asna1 re-expression is probably underestimated using this method.
qRT PCR analyses
Individual dorsal pancreatic buds were used to prepare total RNA (RNeasy Micro Kit, Qiagen 74004) and cDNA (SuperScript III First-Strand Synthesis System, Life Technologies, 18080-051). Expression of the TBP was used to normalize expression levels. To ensure comparable Cq values, equal amounts (5 ng) of cDNA were used as template and Cq of the reference gene TBP was kept within ±1 cycle. Quantitative PCR (qPCR) analysis was performed using an ABI Prism 7000 sequence detection system and SYBR Green PCR Master Mix (ABI). Oligonucleotide sequences used for qPCR are listed in Table S2.
Western blot analysis
Western blot expression data were normalized using α-tubulin expression. See supplementary Materials and Methods for further details.
Image analysis and quantification
All comparisons were performed pairwise between Asna1Panc−/− and control tissue. For intensity comparisons and area quantification, images were captured using the same settings or modified to achieve similar background signal intensities. For quantification of immunopositive areas, the entire embryonic pancreas or pancreatic explant was sectioned (8 µm sections). Every 5th-15th section was collected for immunohistochemistry (5-12 sections/reaction) and all IHC sections were photographed and quantified using scripted procedures in ImageJ software, including software-based threshold algorithms to define immunopositive areas. Data from damaged sections were replaced by interpolation from adjacent sections.
Statistical analyses
All numerical data are presented as mean±s.e.m. All statistical analyses were performed by heteroscedastic two-tailed Student's t-test. P<0.05 was considered statistically significant and denoted with: *P<0.05, **P<0.01, ***P<0.001.
Supplementary Material
Acknowledgements
We thank Elisabet Kjellkvist, Fredrik Backlund and Jurate Straseviciene for technical help. We further acknowledge the facilities and technical assistance of the Umeå Core Facility Electron Microscopy (UCEM) and Umeå Center for Comparative Biology (UCCB) at Umeå University.
Footnotes
Competing interests
H.E. is a co-founder, shareholder and consultant of the unlisted biotech company Betagenon. S.N. and V.P. declare no competing financial interests.
Author contributions
Conceptualization: H.E., S.N.; Methodology: H.E., S.N., V.P.; Software: S.N.; Validation: H.E., S.N.; Formal analysis: H.E., S.N., V.P.; Investigation: H.E., S.N., V.P.; Resources: H.E.; Data curation: H.E., S.N.; Writing - original draft: H.E., S.N., V.P.; Writing - review & editing: H.E., S.N.; Visualization: H.E., S.N.; Supervision: H.E.; Project administration: H.E.; Funding acquisition: H.E.
Funding
The research leading to these results has received funding from the Vetenskapsrådet (K2014-55X-10367-19-3) and the Knut och Alice Wallenbergs Stiftelse (KAW 2010.0033) to H.E. We also acknowledge support from the Strategic Research Program in Diabetes at Umea University. Deposited in PMC for immediate release.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.154468.supplemental
References
- Akahane T., Sahara K., Yashiroda H., Tanaka K. and Murata S. (2013). Involvement of Bag6 and the TRC pathway in proteasome assembly. Nat. Commun. 4, 2234 10.1038/ncomms3234 [DOI] [PubMed] [Google Scholar]
- Apelqvist A., Li H., Sommer L., Beatus P., Anderson D. J., Honjo T., Hrabe de Angelis M., Lendahl U. and Edlund H. (1999). Notch signalling controls pancreatic cell differentiation. Nature 400, 877-881. 10.1038/23716 [DOI] [PubMed] [Google Scholar]
- Auld K. L., Hitchcock A. L., Doherty H. K., Frietze S., Huang L. S. and Silver P. A. (2006). The conserved ATPase Get3/Arr4 modulates the activity of membrane-associated proteins in Saccharomyces cerevisiae. Genetics 174, 215-227. 10.1534/genetics.106.058362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C. L. and Gu W. (2010). New insights into p53 activation. Cell Res. 20, 614-621. 10.1038/cr.2010.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M., Baryshnikova A., Bellay J., Kim Y., Spear E. D., Sevier C. S., Ding H., Koh J. L. Y., Toufighi K., Mostafavi S. et al. (2010). The genetic landscape of a cell. Science 327, 425-431. 10.1126/science.1180823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vas M. G., Kopp J. L., Heliot C., Sander M., Cereghini S. and Haumaitre C. (2015). Hnf1b controls pancreas morphogenesis and the generation of Ngn3+ endocrine progenitors. Development 142, 871-882. 10.1242/dev.110759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaloro V., Spasic M., Schwappach B. and Dobberstein B. (2008). Distinct targeting pathways for the membrane insertion of tail-anchored (TA) proteins. J. Cell Sci. 121, 1832 10.1242/jcs.020321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda A., Kawaguchi Y., Furuyama K., Kodama S., Horiguchi M., Kuhara T., Kawaguchi M., Terao M., Doi R., Wright C. V. E. et al. (2008). Reduction of Ptf1a gene dosage causes pancreatic hypoplasia and diabetes in mice. Diabetes 57, 2421-2431. 10.2337/db07-1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgia S., Kanji M. and Bhushan A. (2013). DNMT1 represses p53 to maintain progenitor cell survival during pancreatic organogenesis. Genes Dev. 27, 372-377. 10.1101/gad.207001.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigaravicius P., Kaminska E., Hübner C. A., McKinnon P. J., von Deimling A. and Frappart P.-O. (2016). Rint1 inactivation triggers genomic instability, ER stress and autophagy inhibition in the brain. Cell Death Differ. 23, 454-468. 10.1038/cdd.2015.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmingsson O., Kao G., Still M. and Naredi P. (2010). ASNA-1 activity modulates sensitivity to cisplatin. Cancer Res. 70, 10321-10328. 10.1158/0008-5472.CAN-10-1548 [DOI] [PubMed] [Google Scholar]
- Jensen J., Pedersen E. E., Galante P., Hald J., Heller R. S., Ishibashi M., Kageyama R., Guillemot F., Serup P. and Madsen O. D. (2000). Control of endodermal endocrine development by Hes-1. Nat. Genet. 24, 36-44. 10.1038/71657 [DOI] [PubMed] [Google Scholar]
- Johnson N., Powis K. and High S. (2013). Post-translational translocation into the endoplasmic reticulum. Biochim. Biophys. Acta 1833, 2403-2409. 10.1016/j.bbamcr.2012.12.008 [DOI] [PubMed] [Google Scholar]
- Jonikas M. C., Collins S. R., Denic V., Oh E., Quan E. M., Schmid V., Weibezahn J., Schwappach B., Walter P., Weissman J. S. et al. (2009). Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 323, 1693-1697. 10.1126/science.1167983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarov P. G., Komarova E. A., Kondratov R. V., Christov-Tselkov K., Coon J. S., Chernov M. V. and Gudkov A. V. (1999). A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285, 1733-1737. 10.1126/science.285.5434.1733 [DOI] [PubMed] [Google Scholar]
- Krapp A., Knofler M., Frutiger S., Hughes G. J., Hagenbuchle O. and Wellauer P. K. (1996). The p48 DNA-binding subunit of transcription factor PTF1 is a new exocrine pancreas-specific basic helix-loop-helix protein. EMBO J. 15, 4317-4329. [PMC free article] [PubMed] [Google Scholar]
- Kuma A., Hatano M., Matsui M., Yamamoto A., Nakaya H., Yoshimori T., Ohsumi Y., Tokuhisa T. and Mizushima N. (2004). The role of autophagy during the early neonatal starvation period. Nature 432, 1032-1036. 10.1038/nature03029 [DOI] [PubMed] [Google Scholar]
- Liu Y., Malureanu L., Jeganathan K. B., Tran D. D., Lindquist L. D., van Deursen J. M. and Bram R. J. (2009). CAML loss causes anaphase failure and chromosome missegregation. Cell Cycle 8, 940-949. 10.4161/cc.8.6.7948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateja A., Szlachcic A., Downing M. E., Dobosz M., Mariappan M., Hegde R. S. and Keenan R. J. (2009). The structural basis of tail-anchored membrane protein recognition by Get3. Nature 461, 361-366. 10.1038/nature08319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz J., Wächter A., Schmidt B., Bujnicki J. M. and Schwappach B. (2006). The yeast Arr4p ATPase binds the chloride transporter Gef1p when copper is available in the cytosol. J. Biol. Chem. 281, 410-417. 10.1074/jbc.M507481200 [DOI] [PubMed] [Google Scholar]
- Mizushima N., Yamamoto A., Matsui M., Yoshimori T. and Ohsumi Y. (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101-1111. 10.1091/mbc.E03-09-0704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh L. C., Law A. C., Dor Y. and Melton D. A. (2005). Beta-catenin is essential for pancreatic acinar but not islet development. Development 132, 4663-4674. 10.1242/dev.02063 [DOI] [PubMed] [Google Scholar]
- Nakhai H., Siveke J. T., Mendoza-Torres L. and Schmid R. M. (2008). Conditional inactivation of Myc impairs development of the exocrine pancreas. Development 135, 3191-3196. 10.1242/dev.017137 [DOI] [PubMed] [Google Scholar]
- Norlin S., Parekh V. S., Naredi P. and Edlund H. (2016). Asna1/TRC40 controls beta-cell function and endoplasmic reticulum homeostasis by ensuring retrograde transport. Diabetes 65, 110-119. 10.2337/db15-0699 [DOI] [PubMed] [Google Scholar]
- Pan F. C., Bankaitis E. D., Boyer D., Xu X., Van de Casteele M., Magnuson M. A., Heimberg H. and Wright C. V. E. (2013). Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 140, 751-764. 10.1242/dev.090159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulou S. and Edlund H. (2005). Attenuated Wnt signaling perturbs pancreatic growth but not pancreatic function. Diabetes 54, 2844-2851. 10.2337/diabetes.54.10.2844 [DOI] [PubMed] [Google Scholar]
- Powis K., Schrul B., Tienson H., Gostimskaya I., Breker M., High S., Schuldiner M., Jakob U. and Schwappach B. (2013). Get3 is a holdase chaperone and moves to deposition sites for aggregated proteins when membrane targeting is blocked. J. Cell Sci. 126, 473-483. 10.1242/jcs.112151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouille C., Kondo H., Newman R., Hui N., Freemont P. and Warren G. (1998). Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell 92, 603-610. 10.1016/S0092-8674(00)81128-9 [DOI] [PubMed] [Google Scholar]
- Rivera-Monroy J., Musiol L., Unthan-Fechner K., Farkas A., Clancy A., Coy-Vergara J., Weill U., Gockel S., Lin S.-Y., Corey D. P. et al. (2016). Mice lacking WRB reveal differential biogenesis requirements of tail-anchored proteins in vivo. Sci. Rep. 6, 39464 10.1038/srep39464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N. and Gerfin-Moser A. (1993). A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry 100, 431-440. 10.1007/BF00267823 [DOI] [PubMed] [Google Scholar]
- Schaffer A. E., Freude K. K., Nelson S. B. and Sander M. (2010). Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev. Cell 18, 1022-1029. 10.1016/j.devcel.2010.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M., Collins S. R., Thompson N. J., Denic V., Bhamidipati A., Punna T., Ihmels J., Andrews B., Boone C., Greenblatt J. F. et al. (2005). Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 123, 507-519. 10.1016/j.cell.2005.08.031 [DOI] [PubMed] [Google Scholar]
- Schuldiner M., Metz J., Schmid V., Denic V., Rakwalska M., Schmitt H. D., Schwappach B. and Weissman J. S. (2008). The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell 134, 634-645. 10.1016/j.cell.2008.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour P. A., Freude K. K., Tran M. N., Mayes E. E., Jensen J., Kist R., Scherer G. and Sander M. (2007). SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc. Natl. Acad. Sci. USA 104, 1865-1870. 10.1073/pnas.0609217104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J., Hsu C.-M., Kang B.-K., Rosen B. P. and Bhattacharjee H. (2003). The Saccharomyces cerevisiae Arr4p is involved in metal and heat tolerance. Biometals 16, 369-378. 10.1023/A:1022504311669 [DOI] [PubMed] [Google Scholar]
- Shih H. P., Wang A. and Sander M. (2013). Pancreas organogenesis: from lineage determination to morphogenesis. Annu. Rev. Cell Dev. Biol. 29, 81-105. 10.1146/annurev-cellbio-101512-122405 [DOI] [PubMed] [Google Scholar]
- Solar M., Cardalda C., Houbracken I., Martín M., Maestro M. A., De Medts N., Xu X., Grau V., Heimberg H., Bouwens L. et al. (2009). Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev. Cell 17, 849-860. 10.1016/j.devcel.2009.11.003 [DOI] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70-71. 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- Stechmann B., Bai S.-K., Gobbo E., Lopez R., Merer G., Pinchard S., Panigai L., Tenza D., Raposo G., Beaumelle B. et al. (2010). Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 141, 231-242. 10.1016/j.cell.2010.01.043 [DOI] [PubMed] [Google Scholar]
- Stefanovic S. and Hegde R. S. (2007). Identification of a targeting factor for posttranslational membrane protein insertion into the ER. Cell 128, 1147-1159. 10.1016/j.cell.2007.01.036 [DOI] [PubMed] [Google Scholar]
- Svensson P., Bergqvist I., Norlin S. and Edlund H. (2009). MFng is dispensable for mouse pancreas development and function. Mol. Cell. Biol. 29, 2129-2138. 10.1128/MCB.01644-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulc J., Wiznerowicz M., Sauvain M.-O., Trono D. and Aebischer P. (2006). A versatile tool for conditional gene expression and knockdown. Nat. Methods 3, 109-116. 10.1038/nmeth846 [DOI] [PubMed] [Google Scholar]
- Tagaya M., Arasaki K., Inoue H. and Kimura H. (2014). Moonlighting functions of the NRZ (mammalian Dsl1) complex. Front. Cell Dev. Biol. 2, 25 10.3389/fcell.2014.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev L. T., Vu B. T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C. et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844-848. 10.1126/science.1092472 [DOI] [PubMed] [Google Scholar]
- Vogelstein B., Lane D. and Levine A. J. (2000). Surfing the p53 network. Nature 408, 307-310. 10.1038/35042675 [DOI] [PubMed] [Google Scholar]
- Voth W., Schick M., Gates S., Li S., Vilardi F., Gostimskaya I., Southworth D. R., Schwappach B. and Jakob U. (2014). The protein targeting factor Get3 functions as ATP-independent chaperone under oxidative stress conditions. Mol. Cell 56, 116-127. 10.1016/j.molcel.2014.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Zhan X., D'Costa J., Tanavde V. M., Ye Z., Peng T., Malehorn M. T., Yang X., Civin C. I. and Cheng L. (2003). Lentiviral vectors with two independent internal promoters transfer high-level expression of multiple transgenes to human hematopoietic stem-progenitor cells. Mol. Ther. 7, 827-838. 10.1016/S1525-0016(03)00104-7 [DOI] [PubMed] [Google Scholar]
- Zhou Q., Law A. C., Rajagopal J., Anderson W. J., Gray P. A. and Melton D. A. (2007). A multipotent progenitor domain guides pancreatic organogenesis. Dev. Cell 13, 103-114. 10.1016/j.devcel.2007.06.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.