

ABSTRACT
During forebrain development, a telencephalic organizer called the cortical hem is crucial for inducing hippocampal fate in adjacent cortical neuroepithelium. How the hem is restricted to its medial position is therefore a fundamental patterning issue. Here, we demonstrate that Foxg1-Lhx2 interactions are crucial for the formation of the hem. Loss of either gene causes a region of the cortical neuroepithelium to transform into hem. We show that FOXG1 regulates Lhx2 expression in the cortical primordium. In the absence of Foxg1, the presence of Lhx2 is sufficient to suppress hem fate, and hippocampal markers appear selectively in Lhx2-expressing regions. FOXG1 also restricts the temporal window in which loss of Lhx2 results in a transformation of cortical primordium into hem. Therefore, Foxg1 and Lhx2 form a genetic hierarchy in the spatiotemporal regulation of cortical hem specification and positioning, and together ensure the normal development of this hippocampal organizer.
KEY WORDS: LHX2, FOXG1, Telencephalon, Patterning, Hippocampus, Hem, Mouse
Summary: The cortical hem, a telencephalic organizer that induces the hippocampus, is positionally restricted by the transcription factor FOXG1 acting directly and also by positively regulating LHX2.
INTRODUCTION
The cortical primordium gives rise to the entire cerebral cortex, including both neocortex and hippocampus. The early patterning of the primordium is crucial for setting up a regulatory system that will control fundamental steps in neocortical and hippocampal development. Before cortical neurogenesis commences, a WNT- and BMP-expressing signalling centre called the cortical hem forms at the medial edge of the cortical primordium (Furuta et al., 1997; Grove et al., 1998). The hem is a secondary organizer in the embryo, necessary and sufficient for inducing the hippocampus in adjacent cortical neuroepithelium. Loss of the hem or of WNT3A, a hem-specific signal, results in loss of the hippocampus (Lee et al., 2000; Yoshida et al., 2006; Caronia-Brown et al., 2014), whereas ectopic hems are capable of inducing ectopic hippocampi (Mangale et al., 2008). This extrinsic signalling from the hem arises from the intrinsic identity of hem cells. Thus, the cell-intrinsic mechanisms that control the formation of the hem and restrict it to its medial location are of crucial importance because they determine the position of the hippocampus in the brain, and thus offer fundamental insights into the basic framework of early cortical patterning.
Based on studies of individual null mutant phenotypes, the transcription factors FOXG1 and LHX2 have been shown to regulate the formation of the hem. When either of these factors is constitutively lost, much of the dorsal telencephalic neuroepithelium transforms into hem instead of cortical primordium (Bulchand et al., 2001; Monuki et al., 2001; Vyas et al., 2003; Muzio and Mallamaci, 2005). There are, however, important differences in the two null mutant phenotypes. Although both display an expanded hem, the Lhx2 mutant displays an expanded hem juxtaposed to an expanded anti-hem, with no cortical primordium in between them (Mangale et al., 2008). In contrast, loss of Foxg1 spares only medial-dorsal fates, so the telencephalon contains an expanded hem and some hippocampal primordium, but no lateral cortical tissue or anti-hem (Vyas et al., 2003; Muzio and Mallamaci, 2005). Another important difference is that the specification of the ventral telencephalon appears normal in the Lhx2 mutant, whereas this structure is entirely lost in the Foxg1 mutant (Xuan et al., 1995; Huh et al., 1999; Martynoga et al., 2005).
Although FOXG1 and LHX2 have each been described as suppressors of hem fate, little attention has been paid to the interactions between them. Here, we uncovered new regulatory functions that position FOXG1 genetically upstream of LHX2 in the cascade responsible for the proper positioning of the hem. We also demonstrate, through analysis of Foxg1 conditional loss of function, that LHX2 is the proximal suppressor of hem fate. Finally, we show that hippocampal specification always occurs in Lhx2-expressing tissue and is seen only adjacent to patches of hem. Our results provide insight into how a major component of the dorsal telencephalon, the hippocampus, is positioned based on its proximity to the hem.
RESULTS AND DISCUSSION
We used mice carrying floxed alleles for Foxg1 (Miyoshi and Fishell, 2012) and Lhx2 (Mangale et al., 2008), and crossed them with a line expressing CreERT2 constitutively from the Rosa26 locus. Foxg1 and Lhx2 expression begins between E8.0 and E8.5 (Walther and Gruss, 1991; Hébert and McConnell, 2000; Tetreault et al., 2009), and by E12.5 both genes display robust expression in the dorsal telencephalon but are excluded from the hem (Bulchand et al., 2001; Muzio and Mallamaci, 2005) (Fig. S1). Hence, for experiments in which tamoxifen (Tam) was administered between E8.5 and E11.5, the age of examination was fixed at E12.5.
First, we examined the time window in which the hem and the anti-hem are sensitive to the loss of LHX2, as this transcription factor is known to suppress both fates. Administering tamoxifen to CreER;Lhx2lox/lox mice at E8.5 results in a phenotype that closely approximates that seen in Lhx2-null embryos in which the hem and anti-hem are both expanded and are seen juxtaposed to each other in the dorsal telencephalon, and no cortical primordium is detectable (Fig. 1F-J) (Mangale et al., 2008). Therefore, we chose E8.5 as the starting point for our analysis of loss of Lhx2, and E10.5 as the endpoint, as we and others had established in earlier work that loss of Lhx2 after E10.5 does not result in expansion of the hem (Mangale et al., 2008; Chou et al., 2009). Disruption of Lhx2 at E9.0 (Fig. 1K-O) reveals a disparity between the expansion of the hem and the anti-hem, and this disparity becomes more pronounced when tamoxifen is administered at E9.5 (Fig. 1P-T). Whereas the anti-hem continues to extend up to a characteristic morphological ‘kink’ in the mutant neuroepithelium (Fig. 1G,L,Q; black lines), the hem appears to be only minimally expanded, such that some medial cortical neuroepithelium is spared between the hem and the anti-hem. These data suggest that E9.5 represents a time point after which the medial neuroepithelium no longer requires LHX2 to prevent it from transforming into hem. By E10.5, the lateral neuroepithelium also appears to be insensitive to the loss of Lhx2. Administering tamoxifen at E10.5 does not appear to alter the normal extent of either the hem or the anti-hem (Fig. 1U-Y).
Fig. 1.

Temporal analysis of hem and anti-hem suppression by LHX2. (A-E) The expression of Wnt3a (hem), Dbx1 (anti-hem) and Lhx2 exon2/3 at E12.5 in control brains. (F-Y) Tamoxifen (Tam) was administered to CreERT2; Lhx2lox/lox embryos at E8.5 (F-J), E9.0 (K-O), E9.5 (P-T) or E10.5 (U-Y), and the embryos were harvested at E12.5. Both the anti-hem and hem expand up to a characteristic morphological ‘kink’ (black lines in F,G) when Lhx2 is disrupted by tamoxifen administration at E8.5 (F-J). If administered at E9.0 or E9.5, the anti-hem continues to display a striking expansion up to the morphological kink (black lines in L,Q), but the hem appears progressively less expanded (K,P). Lhx2 disruption at E10.5 does not cause either the anti-hem or hem to expand (U-Y). Lhx2 exon 2/3 expression identifies the non-recombined cells in serial sections. False-colour overlays of the hem and anti-hem marker expression in each condition (D,I,N,S,X) and a schematized representation (E,J,OT,Y) are shown in each row. Dbx1 expression above the hem in G and I is consistent with the presence of Cajal-Retzius cells from the expanded anti-hem, which line the surface of the entire dorsal telencephalon together with those derived from the expanded hem (Roy et al. 2014). Scale bar: 200 µm.
In summary, we demonstrate that the critical period for LHX2-mediated suppression of the hem ends earlier than that for suppression of the anti-hem (summarized in Table S1). This is surprising as the neurogenetic gradient in the dorsal telencephalon progresses in a lateromedial direction; therefore, one would expect lateral fates to be established prior to medial fates. One possible explanation for this is that the anti-hem, but not the hem, is subject to patterning interactions between PAX6 and GSX2 (Torresson et al., 2000; Yun et al., 2001), and these modulatory effects may affect the timing of its regulation by LHX2 through mechanisms that are not yet clear.
The loss of Foxg1 also causes an expansion of the hem, but this is the only similarity it shares with the Lhx2 mutant phenotype. Upon loss of Foxg1, the entire dorsal telencephalon is respecified as medial pallium, so that only hem, hippocampus, and Cajal-Retzius cells that are derived from the hem are present (Muzio and Mallamaci, 2005). There is also a loss of the entire ventral telencephalon in the absence of FOXG1, owing to a profound deficit of cell proliferation in this tissue after E9.5, accompanied by a lack of ventral telencephalic SHH. Together, these deficits account for the severely abnormal morphology of the E12.5 brain (Xuan et al., 1995; Huh et al., 1999; Martynoga et al., 2005). Nonetheless, the presence of an expanded hem motivated the hypothesis that Lhx2 and Foxg1 may interact to restrict this domain. First, we ascertained that administration of tamoxifen at E8.5 to CreER; Foxg1lox/lox animals recapitulates the well-established Foxg1-null phenotype of loss of ventral and lateral telencephalic tissue. Indeed, we find that Wnt8b expression, which is normally restricted to the hem and hippocampal primordium, encompasses the entire telencephalic neuroepithelium in both Foxg1-null and Foxg1 lox/lox embryos (Fig. S2).
To test whether FOXG1 and LHX2 regulate expression of each other in the cortical primordium, we selected E9.5 for tamoxifen administration, as loss of Lhx2 at this stage spares the medial cortical primordium and does not cause it to become converted to hem (Fig. 1). When Lhx2 is lost from E9.5, Foxg1 expression is similar to that seen in controls. Therefore, loss of Lhx2 does not appear to affect Foxg1 expression in the cortical primordium (Fig. 2A-D; additional embryos in Fig. S3). This result is qualitatively distinct from our previous study (Mangale et al., 2008), in which Lhx2null patches among Lhx2+/+ tissue formed ectopic hems that did not express Foxg1, in the medial telencephalon. In the ectopic hems of Mangale et al. (2008), downregulation of Foxg1 may occur because it depends critically on LHX2 to maintain its expression in medial tissue prior to E8.5. After E9.5, other factors may regulate Foxg1 and compensate for loss of LHX2, resulting in no detectable change in Foxg1 expression.
Fig. 2.

FOXG1 regulates the expression of Lhx2 in the dorsal telencephalic neuroepithelium. (A-F) Tamoxifen (Tam) was administered at E9.5 to control, CreERT2;Foxg1lox/lox and CreERT2;Lhx2lox/lox embryos, which were examined at E12.5. (A,B) Foxg1 and Lhx2 are normally highly expressed in the dorsal telencephalon in control embryos. (C,D) The expression of Foxg1 is unaltered in the absence of Lhx2, and the extent of Lhx2 recombination is revealed by a probe against the floxed exon. (E,F) Foxg1 conditional mutants display no detectable expression of Foxg1, and a profound reduction in Lhx2 expression. (G) FOXG1 ChIP-seq revealed three MACS peaks (model-based analysis of ChIP-seq) denoted by a, b and c that are associated with the Lhx2 gene locus. (H) A luciferase assay was performed using constructs that contained either regions corresponding to the a, b or c peaks, upstream of a vector containing a minimal promoter and a luciferase reporter. Each of the constructs produces a significant upregulation of the reporter in response to the addition of FOXG1 (n=4, **P<0.01, ***P<0.001; Student's t-test). (I) Histone ChIP was performed on chromatin isolated from E12.5 CreERT2; Foxg1lox/lox tissue (Tamoxifen at E10.5), using anti-PanH3, anti- H3K27Ac and anti-H3K27me3 antibodies. A significant increase in the repressive H3K27me3 mark and an apparent decrease in the activatory H3K27Ac mark were seen at the LHX2 TSS region (n=3, *P<0.05, Student's t-test). PanH3 levels were used to normalize the data. Scale bar: 200 µm.
In contrast, loss of Foxg1 results in a marked reduction of Lhx2 expression (Fig. 2E,F; additional embryos in Fig. S4). These findings indicate FOXG1 may regulate Lhx2 directly or indirectly. We performed ChIP-seq using anti-FOXG1 antibody on chromatin isolated from E14.5 cortical tissue and identified three sites of FOXG1 occupancy in the region of the Lhx2 locus (Fig. 2G). Two of these are at some distance away from the Lhx2 transcription start site (TSS; Refseq Accession number: NM_010710), at −32 kb and +51 kb (Fig. 2G, peaks ‘a’ and ‘c’, respectively). One site is closer to the TSS, at −3.5 kb (Fig. 2G, peak ‘b’). To test the hypothesis that FOXG1 regulates LHX2 directly, we performed a luciferase assay using fragments corresponding to each of the a, b and c peaks individually, upstream of a minimal promoter that drives the luciferase reporter construct. Each fragment gave a significant induction of the luciferase reporter in the presence of Foxg1 (Fig. 2H). These results indicate that the occupancy regions we identified on the Lhx2 locus are indeed sites via which FOXG1 is able to positively regulate its target. Although the ChIP-seq was performed in E14.5 tissue, the accompanying luciferase assay also demonstrates the ability of Foxg1 to regulate Lhx2 in the context of a heterologous system. Taken together, these finding indicate that the Foxg1-binding sites we identified may also be relevant to the regulation of Lhx2 at earlier stages such as E9.5.
We further tested for changes in histone modification marks at the Lhx2 TSS upon loss of Foxg1. Chromatin was isolated from control and Foxg1lox/lox dorsal telencephalic tissue harvested at E12.5. In the absence of Foxg1, the Lhx2 TSS displays a fourfold increase in levels of the repressive H3K27me3 mark and an apparent reduction in levels of the activatory H3K27 acetyl mark compared with controls (Fig. 2I). Together, these data indicate that FOXG1 regulates Lhx2 directly. This is the first evidence of a direct upstream regulator for Lhx2 in the mammalian brain.
We next examined whether the apparent regulation of Lhx2 by FOXG1 is dependent on the stage of Foxg1 removal. Upon administration of tamoxifen at E9.5 to Foxg1lox/lox animals, which results in near-complete recombination of Foxg1, Lhx2 expression levels appear much weaker than in control brains, and some regions display no detectable Lhx2 expression (Fig. 3D,E; additional embryos in Fig. S5). It is noteworthy that only Lhx2-negative regions display hem markers, whereas Foxg1-negative regions do not do so as long as there is a detectable level of Lhx2 expression. Surprisingly, this is also seen when Foxg1 is disrupted at E11.5. Although the overall Lhx2 levels seem less affected (Fig. 3G-I; additional embryos in Fig. S6), loss of Foxg1 from E11.5 nevertheless causes some Lhx2-negative patches to form. Therefore, the ability of FOXG1 to regulate Lhx2 levels is strongest when Foxg1 is removed from E9.5. Intriguingly, regardless of the stage of Foxg1 removal, discrete Lhx2-negative domains are seen in the neuroepithelium, and these display hem markers. The presence of Lhx2-negative patches of tissue, regardless of when Foxg1 is removed, indicates that FOXG1 regulates Lhx2 expression, and its loss reliably causes downregulation of Lhx2 in some populations.
Fig. 3.
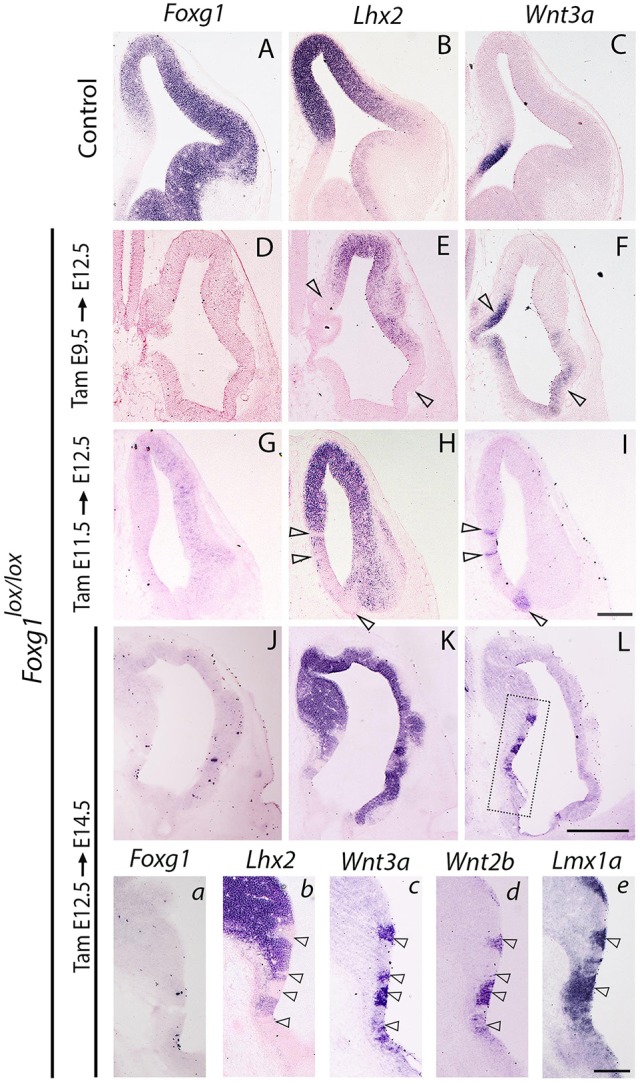
Lhx2 restricts the hem domain in a Foxg1 mutant background. (A-C) Expression of Foxg1, Lhx2 and Wnt3a in control brains. (D-I) Tamoxifen (Tam) was administered at E9.5 (D-F) or E11.5 (G-I) to control and CreERT2; Foxg1lox/lox embryos, and brains were harvested at E12.5. Regions devoid of Lhx2 expression correspond to ectopic hem patches marked by Wnt3a expression (open arrowheads, E,F,H,I). (J-L) Tamoxifen was administered to CreERT2; Foxg1lox/lox at E12.5 and the embryos were harvested at E14.5. (a-e) Magnified views of serial sections corresponding to the area within the box in L. When Foxg1 is removed at E12.5 (J,a), Lhx2 expression is lost in patches (K; b, arrowheads). These patches express the hem markers Wnt3a (L; c, arrowheads), Wnt2b (d, arrowheads) and Lmx1a (e, arrowheads). Sections a-e are in a continuous series, so a comparison between b (Lhx2) and c (Wnt3a) shows the alignment of hem patches in immediately adjacent sections. Scale bars: 200 μm for A-I; 400 µm for J-L; 100 µm for a-e.
It appears that, in the absence of FOXG1, the critical period in which LHX2 suppresses hem fate is expanded beyond E9.5, suggesting that FOXG1 in itself is crucial for suppressing cortical plasticity (Hanashima et al., 2004). We tested this hypothesis further, and administered tamoxifen to Foxg1lox/lox animals at E12.5, well after the hem has formed, and examined the embryos at E14.5. Surprisingly, we discovered ectopic patches of hem (Fig. 3J-L). These patches appeared only at very rostral levels of sectioning, and once again correlated with patches in which Lhx2-negative cells had accumulated. We ascertained that these patches were transformed to hem fate by examining three hem markers, Wnt3a, Wnt2b and Lmx1a, in serial sections. All three markers identify similar territories as ectopic hem (Fig. 3C-E and Fig. S7). Thus, FOXG1 appears to limit the critical period during which LHX2 suppresses hem fate; upon loss of Foxg1, this time window is expanded to as late as E12.5. These data are summarized in Table S1. It is important to note that although CreER is known mediate recombination within 6 h of tamoxifen administration, the actual loss of functional FOXG1 or LHX2 protein would depend on the half-life of both the mRNA and the stability of the protein that has already accumulated. For multifunctional factors such as these, we expect that the turnover of mRNA and protein would be comparable with the cell cycle duration of ∼8 h at E10.5 (Quinn et al., 2007) so as to ensure tight regulation of downstream targets during subsequent cell divisions. Consistent with this interpretation, administration of tamoxifen to Foxg1lox/lox at E11.5 results in Lhx2-negative patches by E12.5 that have transformed into ectopic hems (Fig. 3G,H). This indicates that 24 h is sufficient for: (1) CRE-mediated recombination of the Foxg1 locus; (2) the resulting drop in Foxg1 mRNA and protein levels to occur; (3) a consequential reduction in Lhx2 mRNA transcription to occur that results in LHX2 protein reaching sub-threshold levels; and (4) the transcription of hem markers to finally reach levels that are detectable by in situ hybridization.
Regardless of the stage or extent of Foxg1 disruption, one striking result is that hem only forms where Lhx2 is completely undetectable. Regions of even very weak Lhx2 expression are complementary and completely non-overlapping with regions where hem is formed. This suggests that LHX2 is a proximal and highly effective suppressor of hem fate. Furthermore, hem does not form where Foxg1 is undetectable but Lhx2 is present, indicating that LHX2 is downstream of FOXG1 in restricting the hem to its location. However, it is not completely clear why only some cells entirely lose Lhx2 expression after Foxg1 deletion. It is possible that there is a threshold effect, such that LHX2 levels falling below a certain level triggers hem fate, which could then cause suppression of all remaining Lhx2 expression in those cells.
In earlier work, we demonstrated that Lhx2-null cells ‘prefer’ to aggregate with each other and exclude wild-type cells, and vice versa (Mangale et al., 2008). To our knowledge, this phenomenon appears to be unique to loss of LHX2 and limited to the embryonic dorsal telencephalon. In contrast, the ventral telencephalon in Lhx2lox/lox brains appears completely floxed when the dorsal telencephalon displays Lhx2+ and Lhx2− patches (three embryos in Fig. S3). In our current experiments, it appears that cells that have downregulated Lhx2 as a result of loss of Foxg1 behave similarly, and form clusters separated by those that express Lhx2. As LHX2 suppresses hem fate, clusters of cells that have downregulated Lhx2 transform into hem. The apparent homotypic aggregation of Lhx2− cells could be a mechanism that defines and maintains a key boundary between the hem and the hippocampal primordium: the organizer and the responsive tissue, respectively (Mangale et al., 2008). Indeed, when we examined whether hippocampal markers arise adjacent to the hem patches seen in Foxg1 single conditional mutants, Prox1, Lef1 and Ephb1 all appeared in ectopic locations adjacent to the ectopic hem patches (Fig. 4). Hippocampal markers appear only in tissue that expresses Lhx2, and it does not seem to matter whether Foxg1 is present or not (Fig. 4 shows two embryos displaying different extents of loss of Foxg1 due to some variability in recombination of the Foxg1lox/lox allele). Therefore, LHX2, but not FOXG1, is crucial for hippocampal fate specification. It is surprising that weak Lhx2 expression supports hippocampal specification given the normally high levels of LHX2 expression seen in the hippocampal primordium (Bulchand et al., 2001) (Fig. S1). Together, the results suggest a single threshold for LHX2 levels, below which hem fate is triggered, and above which hippocampal specification is possible.
Fig. 4.

Ectopic hippocampal markers appear adjacent to the ectopic hem. (A-F) Control brains at E12.5 display hippocampal primordium markers Lef1, Prox1 and Ephb1 in neuroepithelium that also expresses Foxg1 and Lhx2. (G-P) Tamoxifen was administered at E9.5 to CreERT2; Foxg1lox/lox embryos, and brains were harvested at E12.5. (G) Foxg1 expression is present in dorsal regions that escaped recombination, but not in ventral regions. (H) Lhx2 displays a similar pattern to FOXG1. (I) Ectopic hem identified by Lmx1a expression is seen in regions devoid of Foxg1 and Lhx2. (J-L) The hippocampal primordium markers Lef1, Prox1 and Ephb1 are seen in tissue that excludes the ectopic hem and expresses Foxg1 and Lhx2. (M-P) Another brain with complete recombination of Foxg1 (M) and reduced expression of Lhx2 (N). In the complete absence of Foxg1, ectopic hem is seen only regions lacking Lhx2 expression (arrowheads, N,O) and hippocampal marker Prox1 appears only in regions displaying Lhx2 expression. Scale bars: 200 µm. (Q) Summary schematic. (a) Cortical neuroepithelial cells express Foxg1 (pink) and Lhx2 (blue). (b-d) Removal of Foxg1 by tamoxifen administration at E9.5 causes a decrease in Lhx2 expression in some cells (light blue) and complete loss of Lhx2 in some cells (open circles). Lhx2-negative cells cluster together (red arrows, b) and transform into hem (yellow, d), whereas Lhx2-expressing cells also cluster together, and express hippocampal markers when they lie adjacent to the hem patches (green, d). This provides a molecular basis for the Foxg1 mutant phenotype in which only hem and hippocampal fates are seen. (R) Model of interactions. Solid red and green lines indicate interactions based on data in this study. Broken red and green lines represent regulatory effects hypothesized based on data in this study, and the dashed black line represents a predicted regulation of Foxg1 by LHX2 in the medial telencephalon, based on data from Mangale et al. (2008). Black lines indicate inductive effects.
Perhaps the most unexpected result in our study is that removing Lhx2 alone prior to E9.5 produces ectopic hem (Fig. 1) (Mangale et al., 2008), whereas removing Lhx2 after E9.5 does not do so unless Foxg1 is also removed (Figs 3 and 4). As schematized in Fig. 4R, we propose possible interactions that could explain this intriguing finding. (1) We suggest that LHX2 is required for Foxg1 expression in the medial telencephalon prior to E9.5, but it is redundant, i.e. substituted by other regulators of Foxg1, from E9.5 onwards. (2) It is also possible that, in addition to an indirect role via LHX2, FOXG1 is capable of suppressing hem directly. If this were true, then loss of both factors would be necessary to induce ectopic hem. The pre- and post-E9.5 scenarios would be as follows: loss of Lhx2 prior to E9.5 apparently causes loss of Foxg1 in medial tissue, and therefore produces ectopic hem medially, consistent with our previous findings (Mangale et al., 2008). In contrast, after E9.5, loss of Lhx2 is no longer sufficient to cause loss of Foxg1 (Fig. 2), therefore ectopic hems cannot form, as we demonstrate in Fig. 1. However, removal of Foxg1 causes loss of Lhx2 in some cells, and these double-negative cells then aggregate to form ectopic hem, which is consistent with our findings in Fig. 3.
In summary, our work suggests both hierarchy and synergy in the interactions between the fundamental regulators of cortical development, FOXG1 and LHX2, that is not obvious from examination of individual loss-of-function phenotypes. We also demonstrate that FOXG1 is a direct upstream regulator of Lhx2. Together with studies in the zebrafish, where LHX2 mediates the activity of SIX3 in regulating forebrain size (Ando et al., 2005), it appears that LHX2 functions at a key node in the network that controls the earliest developmental decisions required for telencephalic patterning. Our findings suggest avenues for future studies that could focus on this fascinating transcription factor biology at the earliest stages of telencephalic patterning that sets the stage for subsequent stages of cerebral cortical development.
MATERIALS AND METHODS
Mice
All animal protocols were approved by the Institutional Animal Ethics Committee (Tata Institute of Fundamental Research, Mumbai, India) according to regulations devised by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), India. The tamoxifen-inducible CreERT2 line [strain name: B6; 129-Gt(ROSA)26Sortm1(Cre/ERT)Nat/J; stock number: 004847] was obtained from the Jackson Laboratory. The Lhx2 lox/lox line used in this study was obtained from Edwin Monuki (University of California, Irvine, CA, USA) (Mangale et al., 2008). The Foxg1 lox/lox line has been described previously (Miyoshi and Fishell, 2012). Three or more embryos of each genotype were examined. For each embryo, the extent of recombination was examined in one series of sections. The stage of tamoxifen administration varied from E8.5 to E12.5; however, all embryos were examined at E12.5, except the E12.5 tamoxifen timepoint, which was analysed at E14.5.
Noon of the day of vaginal plug was designated as embryonic day 0.5 (E0.5). Tamoxifen (Sigma) was administered to the pregnant dams at different time points, as mentioned in the text, and embryos were harvested at E12.5. Control embryos were littermates with one wild-type copy of the relevant gene. For Lhx2 lox/lox mice, the tamoxifen dose administered was 75 µg/gm body weight. Foxg1 lox/lox mice were found to be sensitive to tamoxifen toxicity, so lower doses were tested, and the tamoxifen dose used in the experiments was 28 µg/g body weight of the animal. We ascertained that extensive recombination was seen 6 h post tamoxifen administration using an Ai9 reporter (Fig. S8).
Sample preparation and In situ hybridization
Freshly harvested brains were fixed in 4% paraformaldehyde (PFA) (Sigma) overnight and equilibrated in 15% sucrose followed by 30% sucrose (SRL Chem). The brains were sectioned at 16 µm sections using a freezing microtome and mounted on Superfrost Plus slides (Electron Microscopy Sciences). Sections were post-fixed in 4% PFA, washed in phosphate-buffered Saline (PBS) and treated with proteinase K (Sigma) (1 µg/ml) at 37°C for 10 min. One more round of post-fixing and PBS washes was performed, and the sections were incubated in hybridization buffer (5×SSC, 50% formamide, 1% SDS) containing different antisense RNA probes at 70°C overnight. Probes were prepared using a kit according to the manufacturer's instructions (Roche). The next day, after washes with solution X (2×SSC, 50% formamide, 1% SDS) at 70°C, followed by stringent washes with 2×SSC and 0.2×SSC at room temperature, the sections were washed with TBST [25 mM Tris-HCl (pH 7.5) and 150 mM NaCl, 0.1% KCl, 0.5% Tween-20]. The slides were then incubated with alkaline phosphatase-conjugated anti-digoxigenin Fab fragments (1:5000, Roche) for 16 h at 4°C. The slides were then washed four times with TBST and then with developing buffer NTMT [100 mM NaCl, 100 mM Tris (pH 9.5), 50 mM MgCl, 1% Tween-20]. The colour reaction was performed using Nitro-blue tetrazolium chloride and 5-bromo-4-chloro-3-indolyl-phosphate (NBT-BCIP, Roche) according to the manufacturer's instructions. The incubation was performed for 10-40 h and terminated when the colour reaction had developed satisfactorily, as assessed by the intensity of signal and low background. The reaction was stopped using Tris-EDTA [10 mM Tris-HCl (pH 7.5), 10 mM EDTA (pH 8.0)] and fixed 3.7% formaldehyde [diluted in PBS from a 37% stock (Sigma)] for 1 h at room temperature. Finally, the slides were washed in PBS, dried and mounted in DPX mountant (S.D Fine Chem).
To identify unrecombined cells, RNA probes against Lhx2 exon2/3 were made by PCR followed by in vitro transcription (Roche) according to the manufacturer's instructions. All in situ hybridization experiments were performed on at least three brains of each genotype, for every time point of tamoxifen administration. For each embryo, the extent of recombination was examined by in situ hybridization for the recombined exon in one series of sections.
ChIP-seq
Chromatin immunoprecipitation (ChIP) followed by sequencing was performed on E14.5 cerebral cortices from wild-type embryos using FOXG1 antibody (Santa Cruz Biotechnology) as described previously (Kumamoto et al., 2013). Two independent ChIP experiments were sequenced on an Illumina Hiseq 2000 platform. FOXG1 binding peaks were identified using MACS (Model-based Analysis of ChIP-Seq) and the binding motif was identified using MEME-ChIP analysis (Dreme: discriminative regular expression motif elicitation) (Kumamoto et al., 2013). The Lhx2 transcriptional start site (TSS) was obtained using Refseq (accession number NM_010710).
Chromatin immunoprecipitation and qPCR
Tamoxifen was administered at E9.5 and cortices were dissected from E12.5 CreERT2; Foxg1 lox/lox and littermate control embryos. Cortices from two embryos were pooled for each sample. Three independent experiments were performed (biological replicates). The tissue was fixed for 5 min with 1% formaldehyde and quenched with 125 mM glycine. Cells were lysed and the chromatin was sheared into ≥200 bp fragments using a Covaris S220 sonicator (15 cycles: 60 s on/30 s off) in SDS lysis buffer. Immunoprecipitation was performed with Protein A and G Dynabeads (Invitrogen) mixed in equal proportions and using antibodies against H3K27acetyl (Diagenode), H3K27 trimethyl (Diagenode), rabbit IgG (Sigma) and panH3 control (Abcam) in the ratio 1 μg antibody for every 5 μg of chromatin. The immunoprecipitated DNA was purified using phenol-chloroform-isoamyl alcohol (Ambion). Equal amounts of input and immunoprecipitated DNA were used for quantitative PCR (qPCR) using SYBR Green (Light Cycler 480, Roche).
The primer sequences used were: LHX2 F, 5′ GATGTAGCTGCCCCCACGCC 3′; and LHX2 R, 5′TGTGGAACAGCATCGCGGC 3′. These primers span the region from −206 to +13 bps at the Lhx2 TSS. Each sample was run in duplicates for qPCR, and the average of these technical replicate Ct readings was used for further calculation. Wild-type control and Foxg1lox/lox Ct values were normalized to their respective panH3 Ct values. The fold enrichment was calculated as a ratio of 2-ΔCt. Data from control and Foxg1lox/lox samples from the three independent experiments was compared using Student's unpaired t-test to determine statistical significance. Statistical analyses were performed using Microsoft Excel and values are expressed as mean±s.e.m.
Cloning the FOXG1 occupancy sites on Lhx2 locus in the pGL4.23[luc2/minP] vector
Primers were designed for amplifying the regions corresponding to the three peaks seen in the ChIP-seq occupancy profile, marked a, b and c in Fig. 2. Genomic DNA was used as the template. The primer sequences are as mentioned in Table 1. For cloning regions ‘a’ and ‘c’, KpnI and XhoI were used; for cloning region ‘b’, SacI and XhoI were used to digest pGL4.23[luc2/minP] (Promega, E8411) at the MCS.
Table 1.
Primer sequences

Luciferase assay
U-87 MG cells (a kind gift from Neelam Shirsat, The Advanced Centre for Treatment, Research and Education in Cancer, Navi Mumbai, India) were cultured in DMEM supplemented with 10% (vol/vol) FBS, glutamax, penicillin (100 U/ml) and streptomycin (100 μg/ml). For transfections, 5×105 cells per well were seeded into 24-well plates. After an overnight incubation, the medium was removed and replaced with DMEM supplemented with 10% (vol/vol) FBS, glutamax without antibiotics for 2 h. After 2 h, the DMEM medium was replaced with transfection medium. For transfection, the pGL4.23[luc2/minP] vector with individually cloned FOXG1 occupancy regions (a, b and c) was added with either GFP (control) or Foxg1-GFP (pCAGGS-Foxg1) (500 ng/well for each construct) together with Renilla luciferase vector (100 ng/well) as a normalization control. Transfection was performed using 0.5 µl PLUS reagent and 2 μl of lipofectamine-LTX (Invitrogen) in 400 µl of optimem medium per well and incubated for 6 h. After 6 h, the transfection medium was replaced with fresh complete medium with antibiotics. Luciferase activity was measured using a commercial dual-Glo luciferase assay system (Promega, E2920) 48 h after transfection using Tecan Infinite Lumi Plate reader. The firefly luciferase readouts were normalized to their respective Renilla luciferase readouts obtained from the same cell lysate. The fold change in response to FOXG1 activity was calculated with respect to EGFP controls. All the values are expressed as the mean±s.e.m. of four biological replicates.
Supplementary Material
Acknowledgements
We thank A. Pierani (Dbx1 and Fgf17), C. Ragsdale (Wnt2b and Wnt3a), Y. Zhao (Lhx5), K. Millen (Lmx1a), E. Lai (Foxg1) for gifts of plasmid DNA used for generating RNA probes; N. Shirsat for the U87 cell line; S. Sakhalkar for technical assistance; S. Suryavanshi and the animal house staff of the Tata Institute for Fundamental Research (TIFR) for excellent support; and E. Monuki for the Lhx2lox/lox line.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: G.G., A.S.S., S.T.; Methodology: G.G., A.S.S., S.T.; Investigation: G.G., A.S.S., A.R., L.D.'S, B.C., G.M., G.F.; Resources: G.M., G.F., S.T.; Writing - original draft: S.T.; Writing - review & editing: G.G., A.S.S., A.R., B.C., G.M., G.F., S.T.; Visualization: S.T.; Supervision: S.T.; Project administration: S.T.; Funding acquisition: G.G., S.T.
Funding
This work was supported by a Wellcome Trust/DBT India Alliance Early Career Fellowship (IA/E/11/1/500402 to G.G.); a grant from the Department of Biotechnology, Ministry of Science and Technology, India (PR8681 to S.T.); intramural funds from Tata Institute of Fundamental Research-Department of Atomic Energy (S.T.); grants from the National Institutes of Health (R01 NS081297, P01 NS074972 and R01 MH095147 to G.F. and G.M.; R01 MH094589 and R01 NS089777 to B.C.; R01 MH094589 to G.F.); the Simons Foundation (G.F. and G.M.); the Takeda Science Foundation (G.M.); a Shanti Swarup Bhatnagar award from the Council of Scientific and Industrial Research (Government of India) (to S.T.); and the Infosys Prize for Life Sciences from the Infosys Science Foundation (to S.T.). Deposited in PMC for immediate release.
Data availability
ChIP-seq data have been deposited in Gene Expression Omnibus under accession number GSE96070.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.154583.supplemental
References
- Ando H., Kobayashi M., Tsubokawa T., Uyemura K., Furuta T. and Okamoto H. (2005). LHX2 mediates the activity of Six3 in zebrafish forebrain growth. Dev. Biol. 287, 456-468. 10.1016/j.ydbio.2005.09.023 [DOI] [PubMed] [Google Scholar]
- Bulchand S., Grove E. A., Porter F. D. and Tole S. (2001). LIM-homeodomain gene LHX2 regulates the formation of the cortical hem. Mech. Dev. 100, 165-175. 10.1016/S0925-4773(00)00515-3 [DOI] [PubMed] [Google Scholar]
- Caronia-Brown G., Yoshida M., Gulden F., Assimacopoulos S. and Grove E. A. (2014). The cortical hem regulates the size and patterning of neocortex. Development 141, 2855-2865. 10.1242/dev.106914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S.-J., Perez-Garcia C., Kroll T. T. and O'leary D. D. M. (2009). LHX2 specifies regional fate in Emx1 lineage of telencephalic progenitors generating cerebral cortex. Nat. Neurosci. 12, 1381-1389. 10.1038/nn.2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y., Piston D. W. and Hogan B. L. (1997). Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development 124, 2203-2212. [DOI] [PubMed] [Google Scholar]
- Grove E. A., Tole S., Limon J., Yip L. and Ragsdale C. W. (1998). The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development 125, 2315-2325. [DOI] [PubMed] [Google Scholar]
- Hanashima C., Li S. C., Shen L., Lai E. and Fishell G. (2004). FOXG1 suppresses early cortical cell fate. Science 303, 56-59. 10.1126/science.1090674 [DOI] [PubMed] [Google Scholar]
- Hébert J. M. and Mcconnell S. K. (2000). Targeting of cre to the FOXG1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev. Biol. 222, 296-306. 10.1006/dbio.2000.9732 [DOI] [PubMed] [Google Scholar]
- Huh S. O., Hatini V., Marcus R. C., Li S. C. and Lai E. (1999). Dorsal-ventral patterning defects in the eye of BF-1-deficient mice associated with a restricted loss of shh expression. Dev. Biol. 211, 53-63. 10.1006/dbio.1999.9303 [DOI] [PubMed] [Google Scholar]
- Kumamoto T., Toma K., Gunadi K., Mckenna W. L., Kasukawa T., Katzman S., Chen B. and Hanashima C. (2013). FOXG1 coordinates the switch from nonradially to radially migrating glutamatergic subtypes in the neocortex through spatiotemporal repression. Cell Rep. 3, 931-945. 10.1016/j.celrep.2013.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. M., Tole S., Grove E. and Mcmahon A. P. (2000). A local Wnt-3a signal is required for development of the mammalian hippocampus. Development 127, 457-467. [DOI] [PubMed] [Google Scholar]
- Mangale V. S., Hirokawa K. E., Satyaki P. R. V., Gokulchandran N., Chikbire S., Subramanian L., Shetty A. S., Martynoga B., Paul J., Mai M. V. et al. (2008). LHX2 selector activity specifies cortical identity and suppresses hippocampal organizer fate. Science 319, 304-309. 10.1126/science.1151695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynoga B., Morrson H., Price D. J. and Mason J. O. (2005). FOXG1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev. Biol. 283, 113-127. 10.1016/j.ydbio.2005.04.005 [DOI] [PubMed] [Google Scholar]
- Miyoshi G. and Fishell G. (2012). Dynamic FoxG1 expression coordinates the integration of multipolar pyramidal neuron precursors into the cortical plate. Neuron 74, 1045-1058. 10.1016/j.neuron.2012.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monuki E. S., Porter F. D. and Walsh C. A. (2001). Patterning of the dorsal telencephalon and cerebral cortex by a roof plate-LHX2 pathway. Neuron 32, 591-604. 10.1016/S0896-6273(01)00504-9 [DOI] [PubMed] [Google Scholar]
- Muzio L. and Mallamaci A. (2005). FOXG1 confines Cajal-Retzius neuronogenesis and hippocampal morphogenesis to the dorsomedial pallium. J. Neurosci. 25, 4435-4441. 10.1523/JNEUROSCI.4804-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn J. C., Molinek M., Martynoga B. S., Zaki P. A., Faedo A., Bulfone A., Hevner R. F., West J. D. and Price D. J. (2007). Pax6 controls cerebral cortical cell number by regulating exit from the cell cycle and specifies cortical cell identity by a cell autonomous mechanism. Dev. Biol. 302, 50-65. 10.1016/j.ydbio.2006.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A., Gonzalez-Gomez M., Pierani A., Meyer G. and Tole S. (2014). Lhx2 regulates the development of the forebrain hem system. Cereb. Cortex 24, 1361-1372. 10.1093/cercor/bhs421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetreault N., Champagne M.-P. and Bernier G. (2009). The LIM homeobox transcription factor LHX2 is required to specify the retina field and synergistically cooperates with Pax6 for Six6 trans-activation. Dev. Biol. 327, 541-550. 10.1016/j.ydbio.2008.12.022 [DOI] [PubMed] [Google Scholar]
- Toresson H., Potter S. S. and Campbell K. (2000). Genetic control of dorsal-ventral identity in the telencephalon: opposing roles for Pax6 and Gsh2. Development 127, 4361-4371. [DOI] [PubMed] [Google Scholar]
- Vyas A., Saha B., Lai E. and Tole S. (2003). Paleocortex is specified in mice in which dorsal telencephalic patterning is severely disrupted. J. Comp. Neurol. 466, 545-553. 10.1002/cne.10900 [DOI] [PubMed] [Google Scholar]
- Walther C. and Gruss P. (1991). Pax-6, a murine paired box gene, is expressed in the developing Cns. Development 113, 1435-1449. [DOI] [PubMed] [Google Scholar]
- Xuan S., Baptista C. A., Balas G., Tao W., Soares V. C. and Lai E. (1995). Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron 14, 1141-1152. 10.1016/0896-6273(95)90262-7 [DOI] [PubMed] [Google Scholar]
- Yoshida M., Assimacopoulos S., Jones K. R. and Grove E. A. (2006). Massive loss of Cajal-Retzius cells does not disrupt neocortical layer order. Development 133, 537-545. 10.1242/dev.02209 [DOI] [PubMed] [Google Scholar]
- Yun K., Potter S. and Rubenstein J. L. (2001). Gsh2 and Pax6 play complementary roles in dorsoventral patterning of the mammalian telencephalon. Development 128, 193-205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.