

ABSTRACT
Pediatric hydrocephalus is characterized by an abnormal accumulation of cerebrospinal fluid (CSF) and is one of the most common congenital brain abnormalities. However, little is known about the molecular and cellular mechanisms regulating CSF flow in the developing brain. Through whole-genome sequencing analysis, we report that a homozygous splice site mutation in coiled-coil domain containing 39 (Ccdc39) is responsible for early postnatal hydrocephalus in the progressive hydrocephalus (prh) mouse mutant. Ccdc39 is selectively expressed in embryonic choroid plexus and ependymal cells on the medial wall of the forebrain ventricle, and the protein is localized to the axoneme of motile cilia. The Ccdc39prh/prh ependymal cells develop shorter cilia with disorganized microtubules lacking the axonemal inner arm dynein. Using high-speed video microscopy, we show that an orchestrated ependymal ciliary beating pattern controls unidirectional CSF flow on the ventricular surface, which generates bulk CSF flow in the developing brain. Collectively, our data provide the first evidence for involvement of Ccdc39 in hydrocephalus and suggest that the proper development of medial wall ependymal cilia is crucial for normal mouse brain development.
KEY WORDS: Cilia, Hydrocephalus, Ependymal cells, Cerebrospinal fluid, Brain development
Summary: Ccdc39-deficient ependymal cells exhibit abundant short, thin cilia that are incapable of creating the rhythmic beating pattern necessary for cerebrospinal fluid flow in the perinatal mouse brain.
INTRODUCTION
Hydrocephalus in children is a life-threatening condition and develops from excess accumulation of cerebrospinal fluid (CSF) in the cerebral ventricles. It affects 1-3 children per 1000 live births and represents roughly one-third of all congenital central nervous system (CNS) malformations with a variety of etiologies (Haverkamp et al., 1999; Schrander-Stumpel and Fryns, 1998). The traditional CSF bulk flow theory suggests that unidirectional flow moves CSF from the lateral ventricles posteriorly through the third and fourth ventricles and into the cerebral and lumbar subarachnoid spaces (Del Bigio, 2010; Lee, 2013). Regardless of the initial cause of CSF accumulation, the resulting increased intracranial pressure leads to thinning of the cerebral mantle and periventricular white matter tract injury, which can affect CNS function. Furthermore, the risks related to malfunctions in infant shunt implantation can be as high as 50% in the first year of life, and multiple revisions are often necessary within 10 years (Kang and Lee, 1999; Notarianni et al., 2009). CSF is mainly produced by the choroid plexus, and transports nutrients, signaling molecules and waste products throughout life (Faubel et al., 2016; Feliciano et al., 2014; Lehtinen et al., 2013). A better understanding of the embryonic development and mechanisms of CSF flow, production and absorption is needed to pave the way for improved congenital hydrocephalus treatments.
Cilia are microtubule-based organelles located on the cell surface that facilitate cell motility, extracellular fluid transport, mechanosensation and chemosensation. The typical motile ciliary axoneme has a ring of nine outer microtubules and two central microtubules. This ʻ9+2' axoneme contains the dynein motor protein that generates forces by sliding against adjacent peripheral microtubules and controls rhythmic wave movement of cilia in an ATP-dependent manner (Lodish et al., 2000). To date, over 2000 genes have been identified as ciliary genes. Disrupted cilia motility leads to primary ciliary dyskinesia, which presents with chronic bronchitis, situs inversus, congenital heart disease, infertility, and hydrocephalus (Davis et al., 2015; Praveen et al., 2015). Mouse models of ciliopathy have been reported to develop neonatal hydrocephalus (Banizs et al., 2005; Ha et al., 2016; Ibanez-Tallon et al., 2004; Lee, 2013; Lee et al., 2008). Although this is not a common symptom in human cases, there are several reports of hydrocephalus in primary ciliary dyskinesia patients (Ibanez-Tallon et al., 2004; Lee, 2013; Praveen et al., 2015; Vieira et al., 2012).
Each CNS ependymal cell has 10-100 multiple 9+2 motile cilia that exhibit fast planar back-and-forth motion to generate CSF flow in the cerebral ventricles (Del Bigio, 2010; Lee, 2013; Narita et al., 2012). The choroid plexus cells possess both 9+0 and 9+2 motile cilia that show variable patterns of ciliary beating during the perinatal period (Narita et al., 2012; Narita and Takeda, 2015; Nonami et al., 2013). However, the spatiotemporal pattern of ciliogenesis within the brain and the contribution of different types of motile cilia to the formation of hydrocephalus have not been elucidated.
The progressive hydrocephalus (prh) mouse allele was isolated in a forward genetic screen and causes a dramatic and highly penetrant early postnatal hydrocephalus phenotype (Stottmann et al., 2011). Here, we show that prh mice carry a splice site mutation in a motile cilia gene, coiled-coil domain containing 39 (Ccdc39), and have neural motile cilia defects that lead to impaired CSF flow in the perinatal brain. This ciliary immotility leads to abnormal local bulk CSF flow and accumulation in the forebrain. Our data show for the first time that motile cilia fully develop prenatally on the ependymal cells located in the ventromedial ventricular wall of the mouse brain, including the walls of the foramen of Monro. Ccdc39 is expressed as early as embryonic day (E) 14, coincident with development of motile cilia in these ependymal and choroid plexus cells. Homozygous or compound heterozygous mutations in CCDC39 have been reported in primary ciliary dyskinesia human patients and dogs (Antony et al., 2013; Merveille et al., 2011; Praveen et al., 2015), but the CNS function of the gene has not yet been fully determined. Our study of the prh mutant reveals a role for Ccdc39 in ependymal cell maturation that is essential for proper CSF circulation in the developing mouse brain.
RESULTS
Identification of a recessive splice donor site mutation in Ccdc39 in the prh mouse mutant
We initially mapped the prh mutation to a 76 Mb region on chromosome 3 with a genome-wide SNP scan (Stottmann et al., 2011). We further narrowed the critical region to a 9.4 Mb interval with microsatellite markers on 28 affected animals. The refined minimal interval was between D3Mit271 and D3Mit307 (30.02-39.6 Mb on bld37; Fig. 1A) and contained 57 genes. To identify the causal mutation, we performed whole genome sequencing of an affected mouse and analyzed high quality, unique, homozygous, single base-pair changes within the minimal interval. We found two novel homozygous single-nucleotide variants (mm9 chr3:g.33731448A>T and chr3:g.33715005_8delinsCTTTACCCG) within introns 7 and 14 of Ccdc39. Sanger sequencing and TaqMan probe-based SNP genotyping of affected mice (n=51) indicated complete concordance between homozygosity for the chr3:g.33731448A>T variant and the hydrocephalus phenotype. Carriers were heterozygous for this mutation (Fig. 1B). The homozygous mutation was not found in any of the unaffected mice (n>300) and is not reported in publicly available mouse SNP databases of 21 different mouse strains (Adams et al., 2015).
Fig. 1.
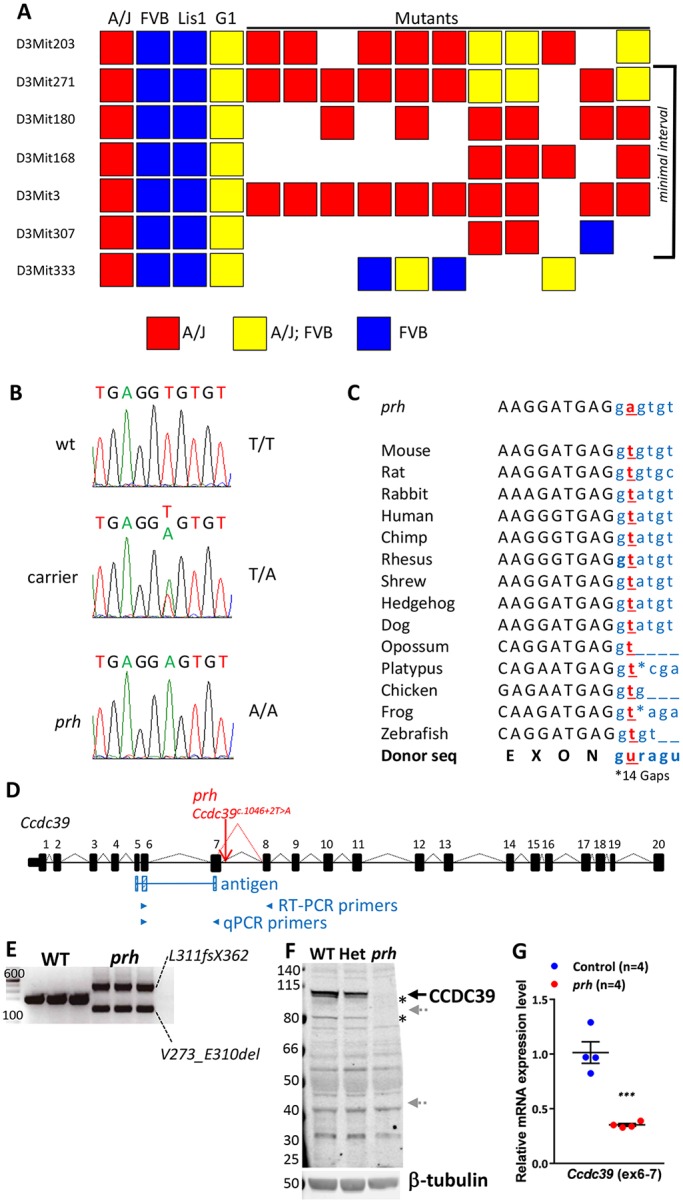
The prh mutation (chr3:g.33731448A>T) is at a conserved mRNA splicing donor site within Ccdc39 intron 7 (Ccdc39c.930+2T>A). (A) Genetic map of the region containing prh bounded proximally by D3Mit271 and distally by D3Mit307. (B) Sanger sequencing traces of the genomic DNA showing a homozygous chr3:g.33731448A>T change in the prh sequence. (C) The conserved splice donor site sequence (AGguragu) within Ccdc39 intron 7 in 14 different species from the UCSC genome database and nucleotide change found in prh (Ccdc39c.930+2T>A, red). Protein coding and intronic sequences are shown in black upper and blue lower case, respectively. (D) The wild-type Ccdc39 gene (from the UCSC genome database) and with the prh mutation. (E) RT-PCR on P1 brain showing two abnormal Ccdc39 transcripts, but no native isoform of Ccdc39, in the prh mutants. DNA size marker (left, bp). (F) Western blotting with CCDC39 antibody on P8 brain lysate from wild type (WT), heterozygous (Het) and prh mutant. CCDC39 protein was not found in the prh mutants and was reduced in the heterozygotes. No shorter protein products are detected of the size predicted to be encoded by the abnormal Ccdc39 mRNAs (∼99 kDa and ∼40 kDa, gray arrows). The CCDC39 isoforms are also missing in the prh mutant (asterisks). β-tubulin, loading control. (G) Quantitative RT-PCR on P1 brain RNA showing reduced Ccdc39 mRNA levels in the prh mutants. The locations of qPCR primers are indicated in D. ***P<0.001.
The candidate variant had a highly conserved thymidine residue 2 bp downstream of Ccdc39 exon 7 within an mRNA splicing donor site (Ccdc39c.930+2T) on the negative strand (Fig. 1C) (Reed, 1989). RT-PCR analysis of the Ccdc39 transcript revealed that the prh mutants expressed two atypical transcripts but no wild-type transcript (Fig. 1D,E). Sanger sequencing of these RT-PCR products showed that the abnormal Ccdc39 mRNAs are predicted to encode ∼40 kDa (L311fsS362) and ∼99 kDa (V273_E310del) protein products as a result of a failure in splicing intron 7 and cryptic alternative splicing in exon 7, respectively (Fig. 1E, Fig. S1A). Immunoblotting of wild-type and prh brain lysates indicated no CCDC39 protein in the mutant (Fig. 1F). Therefore, we concluded that the prh mutation results in loss of CCDC39 protein due to abnormal mRNA splicing. In fact, total Ccdc39 mRNA levels in the prh mouse brain were reduced to 30% of that in wild-type brains (Fig. 1G). The nucleotide insertion in intron 14 did not affect splicing of Ccdc39 (Fig. S1B). Together, our findings strongly suggest that the homozygous c.930+2T>A variant in the Ccdc39 gene is the causal mutation in the prh mouse allele. Hereafter, we refer to the prh allele as Ccdc39prh.
Genetic complementation assay for Ccdc39
We next performed a complementation test with an independently developed targeted conditional gene trap allele for Ccdc39 (Ccdc39tm1a). The Ccdc39tm1a allele is designed to generate a null allele for Ccdc39 with a lacZ gene trap cassette inserted into intron 7 (Skarnes et al., 2011). We crossed the Ccdc39tm1a allele with Ccdc3wt/prh heterozygous mice. The compound heterozygous Ccdc39tm1a/prh progeny were born with mild ventriculomegaly (Fig. 2A,B,D,E) and developed severe hydrocephalus by postnatal day (P) 9 (Fig. 2C,F). Moreover, these Ccdc39tm1a/prh mutants showed a similar failure to grow normally (Fig. 2G) and survive to the weaning phase (Fig. 2H) as observed in Ccdc39prh/prh mice. CCDC39 protein was not detected in Ccdc39tm1a/prh brain lysate (Fig. 2I). Thus, the failure of the Ccdc39tm1a allele to complement the Ccdc39prh mutation is consistent with our conclusion that the prh mutation is an allele of Ccdc39.
Fig. 2.

Ccdc39tm1a/prh mice phenocopy Ccdc39prh/prh mice. (A-F) Nissl staining of sections of P0 control (A,B) and Ccdc39tm1a/prh (D,E) mutant mouse brains. The Ccdc39tm1a/prh mice show moderate ventriculomegaly (asterisk in E), reproducing the brain phenotype of Ccdc39prh/prh mice at P0. Nissl staining of sections of P9 Ccdc39tm1a/prh (F) and control (C) mouse brains. Ccdc39tm1a/prh mice developed severe hydrocephalus with substantially enlarged lateral ventricles (asterisk in F). Body weight (G) and survival rate (H) of postnatal Ccdc39tm1a/prh mice recapitulated the phenotypes of Ccdc39prh/prh mice. The failure of the Ccdc39tm1a allele to complement the Ccdc39prh mutation is consistent with prh being an allele of Ccdc39. P values are generated in the comparisons of null mutant with wild type. (I) Western blotting with CCDC39 antibody on P8 brain lysate from Ccdc39wt/wt (WT), Ccdc39wt/tm1a and Ccdc39tm1a/prh mice. β-Tubulin, loading control. CP, choroid plexus. Scale bars: 2 mm (A,C,D,F), 0.5 mm (B,E).
Abnormal left-right patterning of visceral organs in the Ccdc39prh/prh mutant
Ccdc39 is expressed in the mouse node, and homozygous mutations are responsible for primary dyskinesia with situs inversus in humans and dogs (Merveille et al., 2011). Dextrocardia was also previously reported in a null allele for Ccdc39 (Li et al., 2015). Consistent with these previous reports, we found situs anomalies in early postnatal Ccdc39prh/prh pups with cases of situs inversus totalis (3/15) and heterotaxy (2/15) observed in the Ccdc39prh/prh mutants but not in control mice (10/10) (Fig. S2).
Hydrocephalus perturbs perinatal brain development in Ccdc39prh/prh mice
The Ccdc39prh/prh mutants appeared grossly normal at birth, but all of the homozygous mutant mice showed developmental growth defects and progressive hydrocephalus in the early postnatal period and died within 3 weeks (Stottmann et al., 2011) (Fig. 2F-H). Ccdc39prh/prh embryos had normal size ventricles at E18, but exhibited mild ventriculomegaly shortly after birth (Fig. 3I, Fig. S3A). Histological analysis revealed that embryonic brain patterning appears normal in the P1 mutant brain (Fig. 3A-F). Interestingly, the dense Nissl-stained germinal zones, including the ventricular zone (VZ) and subventricular zone (SVZ) regions of the telencephalic ventricular wall, were the most affected areas in the entire brain of mutants at P1 (Fig. 3C,F,J). This phenotype was even more pronounced at P5, when nearly all of the Nissl-stained germinal zone was lost (Fig. 3G,H). Neural progenitor cells in the postnatal VZ/SVZ mainly produce forebrain glia and olfactory neurons (Cayre et al., 2009). The levels of myelin basic protein (MBP) were reduced in the Ccdc39prh/prh mutant, whereas glial fibrillary acidic protein (GFAP) levels were increased, indicating impaired oligodendrogenesis and reactive gliosis, respectively (Fig. 3K). In addition, olfactory bulbs were smaller (data not shown). Thus, hydrocephalus caused by perturbation of ependymal and/or choroid plexus cilia may indirectly affect normal postnatal gliogenesis.
Fig. 3.
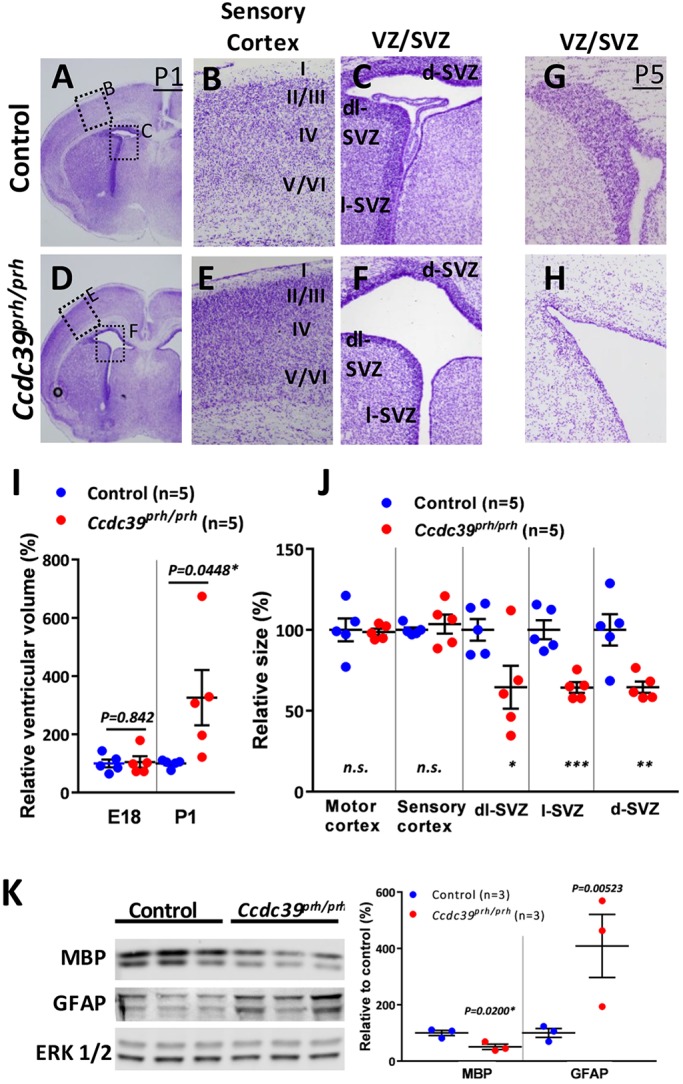
Postnatal hydrocephalus in Ccdc39prh/prh mice. (A-H) Nissl staining of P1 (A-F) and P5 (G,H) mutant and control brains. Development of the cerebral cortical layers was comparable between Ccdc39wt/wt (B) and Ccdc39prh/prh mutant (E) brain, whereas postnatal neural VZ/SVZ progenitors were significantly reduced in the Ccdc39prh/prh mutant at P1 (C,F) and nearly absent at P5 (G,H). (I) Cerebral ventricular size in E18 and P1 brains. (J) P1 mutant brain had significantly smaller VZ/SVZ than control littermate. *P<0.05, **P<0.01 and ***P<0.001. (K) Western blotting of whole brain lysate from P10 brains. Reduced MBP and increased GFAP proteins indicated hypomyelination and enhanced gliosis in the mutant brains as compared with control littermates. ERK1/2, loading control. d-, dorsal; l-, lateral; dl-, dorsolateral.
Ccdc39 is expressed by ciliated neural cells during ciliogenesis in the developing mouse forebrain
In order to identify the primarily affected neural cell types responsible for development of hydrocephalus in the Ccdc39prh/prh mutant, we examined the spatial and temporal distribution of Ccdc39 gene products in the developing brain. Ccdc39 mRNA was detected in E14 mouse choroid plexus tissues in all three (lateral, third and fourth) ventricles and caudal and ventral ependymal cell layers (Fig. 4A-C) (Diez-Roux et al., 2011). Immunohistochemistry confirmed the presence of CCDC39 protein in the E14 wild-type brain and its loss in Ccdc39prh/prh mutants (Fig. 4D-F). We found that the expression of CCDC39 coincides with that of FOXJ1, a transcription factor known to be required for motile multiciliogenesis, in ependymal cells on ventromedial walls as early as E16 and continuing through P0 (Fig. 4G). Axonemal extension starts at E16 with cytoplasmic and nascent ciliary expression of DNALI1 and is more robust by P0 (Fig. S4). To our surprise, scanning electron microscopy (SEM) of the P0/P1 mouse brain revealed well-developed multiciliated ependymal cells on the ventromedial wall of the lateral ventricle (Fig. 4H). Previous studies have shown that most ependymal cells on the lateral wall of the telencephalic ventricle are born between E14 and E16 (Spassky et al., 2005) and lack mature motile cilia at P1 (Banizs et al., 2005). Our SEM analyses support the lack of mature cilia on the lateral wall at birth but show, for the first time, the presence of motile cilia on the medial wall of the lateral ventricle.
Fig. 4.

Ccdc39 expression in choroid plexus and ependymal epithelial cells of the prenatal mouse brain, and fully ciliated ependymal cells in the P0 mouse forebrain. (A-C) RNA in situ hybridization for Ccdc39 mRNA at E145 (adapted from Eurexpress.org). Ccdc39 is expressed (arrowheads) in the choroid plexus (CP) of the lateral ventricle (LV) and fourth ventricle (4V), as well as in ependymal cells (EP) and ependymal wall of the ventral fourth ventricle. (D-F) Immunohistochemistry with CCDC39 antibody of E14.5 wild-type (D,E) and prh mutant (F) brains. The CCDC39 protein is reduced to nearly undetectable levels in the prh mutant. (G) Immunohistochemistry with CCDC39 (green) and FOXJ1 (red) antibodies in E16 and P0 mouse brain. M, medial; L, lateral. Arrows indicate the location of the high-magnification images in the insets. (H) SEM images of ventromedial walls of P0 mouse forebrain showing fully ciliated ependymal cells. Magnifications of boxed areas are shown to each side. Scale bars: 1 mm in G (top left); 500 µm in G (bottom left); 5 µm in G (top right); 10 µm in G (bottom left); 50 µm in H.
Axonemal and cytoplasmic localization of CCDC39 in ciliated neural cells
Consistent with its functions in ciliogenesis, CCDC39 protein was found in multiciliated choroid plexus cells (Fig. 5A,B,F,G) and ependymal cells on the medial wall of foramen of Monro (Fig. 5C,H), the subcommissural organ (Fig. 5D,I), and the central aqueduct (Fig. 5E,J, Fig. S3B). CCDC39 was localized within the axoneme as well as in the cytoplasm (Fig. 5K). These data suggest that the Ccdc39 gene has specific functions in motile ciliogenesis in choroid plexus and ependymal cells in the perinatal mouse brain. Therefore, loss of functional motile cilia on these cell types might be the primary cause of the hydrocephalus phenotype seen in the Ccdc39prh/prh mutant.
Fig. 5.

Axonemal and cytoplasmic localization of CCDC39 in the E18 mouse brain. (A-J) Immunohistochemistry with CCDC39 (green) and acetylated tubulin (red) antibodies in the E18 mouse brain. Ccdc39 is expressed in multiciliated choroid plexus epithelium cells, ependymal cells in the medial walls of the foramen of Monro, subcommissural organ, and central aqueducts. (K) SRRF images of CCDC39 in the axoneme. Solid arrowheads indicate expression of CCDC39 within cilia. Open arrowheads indicate CCDC39 expression within developing ependymal cells that are not yet ciliated. LV, lateral ventricle; 3V, third ventricle; FM, foramen of Monro; SCO, subcommissural organ; CA, central aqueduct; CP, choroid plexus; Ep/EP, ependymal cells. Scale bars: 100 µm (A-E), 10 µm (F-J), 1 µm (K).
Aberrant development of cilia motility structures in Ccdc39prh/prh ependymal cells
CCDC39 protein has previously been found to be necessary for the formation of inner dynein arms and nexin links of airway motile cilia (Merveille et al., 2011) and the flagella of Chlamydomonas (Lin et al., 2015; Oda et al., 2014). To examine the consequences of loss of CCDC39 in neural cilia, we used immunohistochemistry to examine the inner dynein arms with DNALI1 (Rashid et al., 2006) and the nexin-dynein regulatory complex (N-DRC) (Bekker et al., 2007) with GAS8, together with acetylated α-tubulin to label the axoneme (Fig. 6A,B). Wild-type ependymal cell cilia on the medial ventricular wall were immunopositive for both DNALI1 (Fig. 6A) and GAS8 (Fig. 6B), indicative of mature and motile cilia. In the Ccdc39prh/prh mutants, DNALI1 protein was not found in the ependymal cilia axoneme, whereas cytoplasm accumulation appeared normal or slightly enhanced (Fig. 6A). This phenotype became more pronounced at P9 (Fig. S5). Similarly, GAS8 was only rarely detected near the apical cytoplasm of the P0/P1 mutant ependymal cells and never localized to the mutant axoneme (Fig. 6B). These data indicate that Ccdc39-deficient ependymal cells have defective motile ciliogenesis due to a lack of crucial molecular components for motile cilia. Interestingly, the choroid plexus cilia did not show any differences in DNALI1 or GAS8 immunostaining (Fig. S6). We also found a lack of DNALI1 and normal expression of GAS8 in mutant respiratory cilia (Fig. S7). These data indicate that the molecular characteristics of motile cilia and their development vary from one tissue to another. The mutant mice did not show sinusitis or bronchitis in histology, probably owing to their early lethality after birth.
Fig. 6.

Ciliary structural abnormalities in ependymal cells of the Ccdc39prh/prh mutant mouse. (A) Immunofluorescence of the P1 brain for acetylated α-tubulin (red) and DNALI1 (green). Boxed regions are shown at higher magnification on the right. DNALI1 staining appears to colocalize with the acetylated α-tubulin staining within the axoneme of wild-type ependymal cells, but not in mutant (arrows). (B) Immunofluorescence of the P1 brain for acetylated α-tubulin (red) and GAS8 (green). An abnormal punctate staining pattern of GAS8 was detected in the apical side of the ependymal cells (red arrows), and no localization of GAS8 within the axoneme (white arrows) was found in the mutant. (C) TEM images of ependymal cilia in Ccdc39prh/prh and wild-type mice. A variety of microtubule disorganizations was found in the mutant, including absence of inner dynein arm (red arrows), mislocalization of one or two microtubule peripheral doublets (MPD), and absence of the central pair (CP). Outer dynein arms were preserved in the mutant (black arrows). Ectopic abnormal ciliary membrane inclusions were occasionally found in the mutant (white arrow). (D) About 27% (7/26) of mutant cilia lost the CP and showed MPD (green), ∼23% (5/26) lost MPD (purple), and ∼50% (13/26) showed normal CP and MPD (red). (E) Cross-sectional diameter of ependymal cilia was significantly reduced in the Ccdc39prh/prh mutant. ***P<0.0001. (F) P1 ependymal cilia stained for γ-tubulin (red) and β-catenin (green). Note the increased number of γ-tubulin-stained basal bodies in the Ccdc39prh/prh ependymal cells. Four sections from two animals each; ***P<0.00001. Scale bars: 1 mm in A (left); 100 µm in A (middle); 10 µm in A (right), B and F (right); 100 nm in C; 1 μm in F (left).
We continued cilia microstructural analysis with transmission electron microscopy (TEM) and found that Ccdc39-deficient ependymal cilia were smaller in diameter, devoid of inner dynein arms, and exhibited various types of ciliary axonemal microtubule disorganization (Fig. 6C-E). In contrast to the mature motile ciliary 9+2 microtubule arrangement in wild-type mice (n=45), all the evaluated mutant cells showed the abnormal appearance of 8+2 and 9+0 arrangements of microtubule doublets due to mislocalization of one or two of the peripheral doublets into the central compartment of the axoneme (n=26), some of which also showed a lack of the central pair of microtubules (7/26) (Fig. 6C,D). Also, Ccdc39prh/prh peripheral microtubules lacked inner dynein arms (Fig. 6C, red arrows), although the outer dynein structure was preserved (Fig. 6C, black arrows). Frequently, the Ccdc39prh/prh mutant axonemes also had abnormal ectopic ciliary membrane inclusions (Fig. 6C, white arrow). The ependymal cilia of Ccdc39prh/prh mutants were thinner (298.7±7.3 nm, versus 372.6±4.8 nm in wild type; P<0.0001; Fig. 6C,E). We found a higher density of mutant ependymal cilia by immunohistochemistry against γ-tubulin, a cilia basal body marker (29.2±2.4 per cell, versus 16.4±1.1 per cell in wild type; P<0.0001; Fig. 6F).
Lack of ependymal cilia beating in neonatal mouse brain results in impaired CSF flow and circulation
CCDC39 mutations have previously been shown to alter ciliary beating amplitude and patterns (Merveille et al., 2011). We therefore analyzed cilia beating pattern and frequency ex vivo with high-speed video microscopy at P1 and P6. Based on strong expression of CCDC39 and the presence of motile cilia, we imaged ventromedial walls of the lateral ventricles (Movies 1-4) and the central aqueduct (Movies 5, 6). A strong forward power stroke and a backward recovery stroke (at a beat frequency of 15.50±0.40 Hz at P1 and 10.42±0.67 Hz at P6/P7) were found in the wild-type ependymal cilia (Fig. 7B). A representative kymograph of P6 wild-type cilia is shown in Fig. 7A. This speed is similar to that reported for third ventricle ependymal cell cilia (Liu et al., 2014), and indicated the full maturation of mouse ependymal cilia at P1. By contrast, many of the ependymal cilia in the Ccdc39prh/prh mutant appeared largely immobile with virtually undetectable ciliary beat pattern at P1 and P6 (Movies 2, 4, 6). Only 12% of the mutant cilia showed rigid and subtle movements (Fig. 7C), which resulted in a significantly reduced ciliary beating frequency (0.85±0.42 Hz, P<0.001; Fig. 7A,B).
Fig. 7.

Abnormalities in ependymal cilia beating and CSF flow in Ccdc39prh/prh mice. (A) Representative kymograph of P7 wild-type and Ccdc39prh/prh mutant ependymal cilia from the high-speed video microscopy study. (B) Ciliary beat frequency was significantly reduced in the Ccdc39prh/prh mutant ependymal cilia. ***P<0.0001. (C) Ependymal ciliary beat patterns analyzed in the slowed down videos showed that mutant cilia were unable to generate a repetitive beating pattern (green) and only few (∼12%) showed dyskinetic movement (red). (D) The velocity of the fluorescent micro-beads analyzed from tracks in ex vivo P7 mouse brain ventricles of control and Ccdc39prh/prh mutants (see also Movies 7, 8). All micro-beads tested in the wild-type brain showed consistent speed at 68±0.7 µm/s and moved out of the field by 5 s of imaging. The speed of floating beads was significantly reduced in the Ccdc39prh/prh mice to 11±0.07 µm/s. More than 350 micro-beads were analyzed and are displayed in different colors in the graph. Two animals from each genotype were tested. Data represent mean±s.e.m. ***P<0.0001. (E) SEM images of multiciliated ependymal cells on the ventromedial forebrain wall and the central aqueduct wall of P0 mutant and wild-type mice. The Ccdc39prh/prh ependymal cells have shorter, thinner, but more abundant cilia. Scale bars: 5 µm. (F) Ependymal cilia length is significantly reduced in the Ccdc39prh/prh mutant. ***P<0.0001.
The orchestrated beating of ependymal motile cilia generates localized CSF flow in each ventricle (Roth et al., 1985; Siyahhan et al., 2014). We detected unidirectional CSF flow generated by the beating motile cilia in the wild-type samples in the forebrain (Movies 1, 3) and the central aqueduct (Movie 5). By contrast, the Ccdc39prh/prh ependymal cells with immobile cilia showed no evidence of directional CSF movement in the ventricles (Movies 2, 4, 6). By adding fluorescent micro-beads to the adjacent ventricular lumen, we were able to quantify CSF flow velocity in each genotype (Fig. 7D, Movies 7, 8). At both the level of the ventromedial wall of the forebrain and the central aqueduct ependymal cells, the Ccdc39prh/prh mutant showed an ∼70% decrease in CSF flow speed compared with the wild-type brain (68±0.7 µm/s wild type versus 11±0.07 µm/s Ccdc39prh/prh; P≤0.0001) (Fig. 7D, Movies 7, 8). The presence of motile cilia was confirmed in SEM, although the mutant cilia were shorter (3.4±0.08 μm versus 6.5±1.1 μm in wild type; P<0.001; Fig. 7E,F) in both the forebrain and central aqueduct. These data indicate that loss of Ccdc39 results in a significant CSF flow retardation, especially locally at the narrow aqueducts: the foramen of Monro, which connects the lateral with the third ventricles, and the central aqueduct, which links third and fourth ventricles.
To investigate the effect on CSF bulk flow of loss of Ccdc39, we injected Evans Blue dye into the lateral ventricle on one side of 8-day-old wild-type or Ccdc39prh/prh mutant mice and evaluated the pattern of CSF flow by determining the distribution of the dye throughout the cerebral ventricles (Fig. 8A). In wild-type mice (n=5), the dye injected into the lateral ventricle was able to travel to the fourth ventricle within 10 min after the injection (Fig. 8A, arrowheads). By contrast, in the Ccdc39prh/prh mutants, both the third ventricle and the central aqueduct contained less of the dye-labeled CSF, and it was not detected at all in the fourth ventricle (n=7/8). Importantly, histological analysis showed that the size and shape of both the central aqueduct (Fig. 8B) and the foramen of Monro (data not shown) in the P8 Ccdc39prh/prh mutant were indistinguishable from those of wild-type controls, suggesting that reduced flow, and not blockage, is responsible for the lack of dye in the fourth ventricle. We also investigated aqueduct structure in P1 Ccdc39prh/prh mutants at the beginning of ventriculomegaly and in P14 mutants after severe hydrocephalus development. The mutants showed a normal or slightly enlarged central aqueduct at P1 and misaligned ependymal cells that left a wider aqueduct opening at P14 (Fig. S8). Moreover, we never observed a blood or tumor mass that could physically block CSF flow in the Ccdc39prh/prh brains.
Fig. 8.
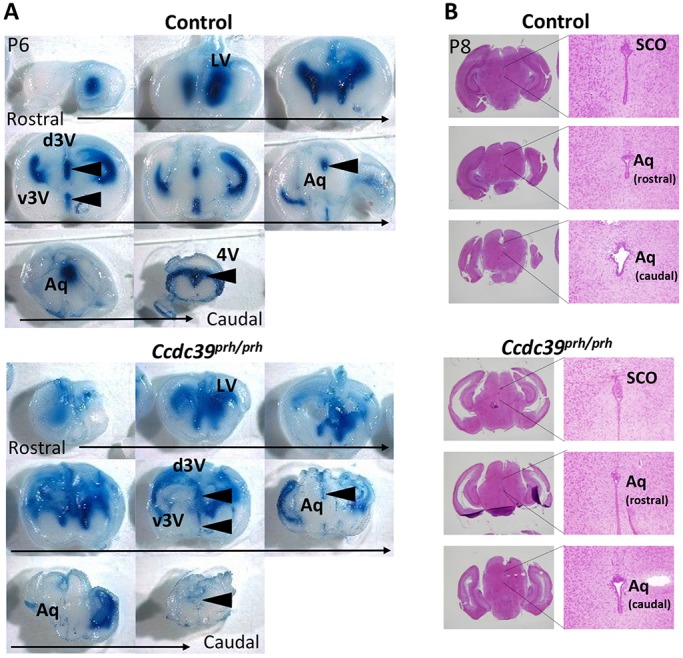
Retardation in CSF flow is not due to a physical obstruction in the central aqueduct of early postnatal prh mice. (A) CSF flow analysis in P6 Ccdc39prh/prh and control mice. Evans Blue dye injected into an anterior horn of the lateral ventricle (LV) traveled through the ventricular system and was detected in the third (3V) and fourth (4V) ventricles within 10 min in control mice (5/5) but not in the mutant (7/8) (arrowheads). (B) Histology of the central aqueduct (Aq) was comparable in mutant and littermate control mice at P8. SCO, subcommissural organ; d, dorsal; v, ventral.
Taken together, all of these data lead us to conclude that the coordinated beating of ependymal cilia requires Ccdc39 gene function. Our data demonstrate that disruption of the cilia beating pattern in the perinatal mouse brain (starting on the embryonic ventromedial ventricular wall) results in a reduced local CSF flow rate and retardation of bulk CSF flow without physical obstruction at the level of the foramen of Monro and the central aqueduct. The Ccdc39prh/prh mouse model mimics many features of congenital hydrocephalus cases, suggesting that the altered formation and/or function of ependymal cilia may contribute to the development of this congenital brain condition.
DISCUSSION
Loss of Ccdc39 causes hydrocephalus in the prh mouse mutant
In this study, we show that the prh mouse has a mutant allele of Ccdc39 that leads to disrupted motile ciliogenesis in the perinatal brain. This mouse model constitutes the first demonstration of a hydrocephalus phenotype due to loss of function of Ccdc39. We demonstrate the earliest appearance of motile cilia on forebrain ependymal cells of the ventromedial ventricular walls, which is complete by P1. Our findings suggest that the early postnatal hydrocephalus phenotype in the prh mouse model arises as a result of defective motile cilia already at embryonic stages, due to the Ccdc39 mutation.
The homozygous loss of the CCDC39 gene is one of more than 30 gene mutations causing primary ciliary dyskinesia, in which patients commonly develop sinusitis, bronchitis, laterality defects (heterotaxy and situs inversus) and infertility (Antony et al., 2013; Merveille et al., 2011; Olcese et al., 2017; Praveen et al., 2015; Vieira et al., 2012). Prenatal or neonatal hydrocephalus has been reported in some cases (Praveen et al., 2015; Vieira et al., 2012). Our study, along with that of other primary ciliary dyskinesia mouse models, suggests that ciliary function is also essential for CNS ventricular homeostasis. Consistent with previous findings of laterality defects in mouse mutants and patients (Li et al., 2015; Merveille et al., 2011), the Ccdc39prh/prh mice showed situs inversus and heterotaxy phenotypes, which are commonly seen in other ciliopathy mutants with motile cilia deficits. Although hydrocephalus is not a prevalent phenotype in primary ciliary dyskinesia patients, rodent models of the orthologous gene commonly develop hydrocephalus (Lee, 2013). This discrepancy is likely to be due to species-specific differences. The minimum width of the mouse foramen of Monro (<10 nm) is 100,000-fold smaller than that in humans (>1 mm) (Lang, 1992). These structural differences might make the rodent CSF system more dependent on motile cilia and predispose the ciliopathy animal models to hydrocephalus. Hydrocephalus is a heterogeneous condition, and different mechanisms are involved at multiple levels of its pathogenesis. The known mutations for congenital hydrocephalus (L1CAM, MPDZ and CCDC88C) are in different classes of proteins and, presumably, the cellular mechanisms differ.
Motile cilia development in the prenatal mouse brain
We found direct evidence in video microscopy experiments that the motile function of ependymal cilia at both the foramen of Monro and the central aqueduct is imperative for generating local CSF flow in the prenatal mouse brain. We show that Ccdc39 is almost exclusively expressed in ependymal walls of the foramen of Monro and the central aqueduct along with the subcommissural organ and the choroid plexus in the mouse brain as early as E14 (Fig. 4). Despite previous reports that ependymal motile cilia develop postnatally (Banizs et al., 2005), we show, for the first time, that medial walls of the foramen of Monro develop mature, functional, multiciliated ependymal cells at birth (Movie 1). This was confirmed by detection of mature motile cilia markers on medial ventricular wall cilia at birth (Fig. 6). Therefore, we provide previously undocumented and underappreciated functions of ependymal cilia in the prenatal mouse forebrain. Our data show that motile cilia defects and perturbation of hydrodynamic forces in the early developing brain result in severe hydrocephalus with impaired glial and neuronal cell development, white matter damage, and slowed CSF bulk flow. The cilia-mediated CSF directional flow might also be crucial to avoid exposure of the vulnerable developing brain to malnutrition, lack of signaling molecules, or toxic materials that can have detrimental consequences for the brain.
Consistent with previous studies in lung and flagella (Merveille et al., 2011; Oda et al., 2014), our TEM study showed that Ccdc39-deficient ependymal cells lack inner dynein arms, proper 9+2 microtubule structures, and have shorter and thinner cilia. Mutant ependymal cells did not have the axonemal dynein protein DNALI1 and were incapable of creating a rhythmic beating pattern necessary for CSF to flow. Cryo-electron tomography data in algae flagella showed that FAP59 (CCDC39 homolog) and FAP172 (CCDC40 homolog) form a complex and are crucial for forming 96 nm microtubule structural repeats of the motile axoneme (Oda et al., 2014). Therefore, a CCDC39/40 protein complex might define the binding site of radial spokes along the long axes of the nine peripheral microtubules and provide anchoring sites for inner dynein arms and N-DRCs in the mammalian ependymal cilia. Our results showing increased cilia in Ccdc39prh/prh ependymal cells also suggested a possible regulatory role of CCDC39 in centriole/deuterosome amplification and multiciliogenesis. Future functional studies of CCDC39 in centriole, fibrous granules, or deuterosome production and molecular interaction with known pathways might provide further insights into the molecular mechanisms regulating the initiation of multiciliogenesis (Mori et al., 2017; Stubbs et al., 2012; Zhao et al., 2013).
The choroid plexus epithelial cells exhibit both 9+2 and 9+0 cilia and are transiently motile in late embryonic development to early postnatal time points in mice (Narita et al., 2010, 2012; Narita and Takeda, 2015; Nonami et al., 2013). They have a very slow beat frequency and variable ciliary beating forms. Thus, their contribution to generating CSF flow is considered to be minimal. A sensory function of choroid plexus cilia has been suggested in transcytosis-mediated water transport (Narita et al., 2010), and ciliary function in intracellular pH control in choroid plexus epithelial cells has been studied in mouse mutants with an Ift88 mutation (Tg737orpk mice) (Banizs et al., 2007, 2005). Interestingly, our immunohistochemical study in the wild-type developing choroid plexus cilia showed the presence of the nexin-dynein regulatory complex protein GAS8 within the axoneme, but not the axonemal inner dynein protein DNAL1. Moreover, the prh mutation did not change this choroid plexus cilia-specific subcellular localization of DNAL1 and GAS8 (Fig. S6). This suggests that the role of Ccdc39 in the development and function of motile cilia in the choroid plexus differs from that in ependymal cells. Further analysis using a choroid plexus-specific deletion of the Ccdc39 gene might help to elucidate the development and function of these motile cilia during normal brain development.
Prenatal abnormal motile cilia development leads to impaired CSF bulk flow in the neonatal mouse brain
We note that the tubular structure of the central aqueduct as well as the size of the fourth ventricle were comparable in prh and wild-type mice (Fig. S8), but CSF bulk flow was slower or lacking in these regions in the mutant (Fig. 7). Clinically, aqueductal stenosis/obstructive hydrocephalus is often defined by just the dilated lateral and third ventricles with a normally shaped fourth ventricle. This is frequently used as evidence for structural collapse or physical blockage at the aqueduct. Our ex vivo video microscopy study demonstrates that defective ependymal cilia can lead to disrupted local CSF flow at the central aqueduct without physical obstruction of the aqueduct in mice. Therefore, our data suggest that clinical obstructive hydrocephalus cases [22-49% of patients (Haverkamp et al., 1999)] might include instances of partially disrupted motile cilia function in addition to other acquired etiologies.
In summary, we have identified the genetic mutation in Ccdc39 responsible for the prh mouse mutant allele, thereby revealing an essential motile ciliary function in perinatal mouse hydrocephalus development. It is especially noteworthy that this model closely recapitulates the developmental timing observed in the majority of congenital hydrocephalus cases. Our study indicates that motile cilia function is crucial for directional CSF flow as well as systemic CSF circulation within the perinatal mouse brain. A further understanding of CSF circulation mechanisms and their temporal relationships with normal human brain development could lead to new therapeutic strategies for hydrocephalus by altering CSF circulation and/or absorption.
MATERIALS AND METHODS
Mice
The Ccdc39prh allele (Stottmann et al., 2011) was maintained on a mixed strain background after the initial linkage analysis identified the causal mutation. The Ccdc39tm1a [Ccdc39tm1a(KOMP)Wtsi] mice were obtained from the UC Davis KOMP repository (project ID CSD36312) and were also maintained on a mixed genetic background. Mice were housed in specific pathogen-free conditions, and all experiments were performed according to the Institutional Animal Care and Use Committee guidelines of the Cincinnati Children's Hospital Medical Center.
Genetic mapping and sequencing of the prh mutation
Microsatellite markers were tested on at least 30 affected animals to narrow the critical interval using standard methods. Whole genome sequencing was performed with whole brain genomic DNA isolated from an affected mouse by 125 bp paired-end sequencing on the Illumina HiSeq2500 platform. The unique, homozygous, single-nucleotide changes within the minimal genetic interval were selected with the following filtering: (1) within 50 bp of the nearest exon; and (2) not reported as polymorphisms in publicly available sequence variation databases – Sanger Institute Mouse Genome Project (Keane et al., 2011), NCBI dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/), and Celera whole genome shotgun sequence (Mural et al., 2002). Finally, Sanger sequencing was also performed in unaffected siblings to exclude nucleotide changes not associated with the hydrocephalus phenotype. The prh mice were genotyped using the TaqMan Sample-to-SNP Kit (Applied Biosystems) for a single-nucleotide change at chr3:g.33731448A>T (assay ID: AH6R6X7).
RT-PCR and real-time PCR
Total RNA treated with DNase was extracted from whole brains of P10 mice using the RNeasy Plus Mini Kit (Qiagen). cDNA was synthesized using the SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific), and RT-PCR and cDNA sequencing were performed following standard methods. Quantitative real-time PCR was performed with QuantStudio 6 using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) according to the manufacturer's instructions. Primer sequences are listed in Table S1.
Western blotting
P10 whole brain lysates in RIPA buffer [50 mM Tris-Cl pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, 1% proteinase inhibitor cocktail (Thermo Fisher Scientific)] were used in fluorescence western blotting with standard procedures and minor modifications using 7% Tris-acetate PAGE gels (Thermo Fisher Scientific), PVDF membrane (Bio-Rad), Odyssey blocking buffer (Licor Bioscience), Odyssey CLx Imaging System, and the antibodies listed in Table S2.
Histology, immunohistochemistry, and fluorescence confocal microscopy
For Hematoxylin and Eosin staining, tissues were fixed in formalin for 24 h and embedded in paraffin after ethanol dehydration, and 5 µm-thick serial microtome sections were prepared. For immunofluorescence microscopy and Nissl staining, mouse brains fixed in 4% paraformaldehyde (PFA)/PBS overnight were washed and cryoprotected in 30% sucrose/PBS then immersed in NEG50 freezing medium (Thermo Fisher Scientific). 12 µm-thick cryosections were prepared and 0.1% Cresyl Violet solution was used for Nissl staining. Antigen retrieval was performed in citrate buffer (pH 6) for 45 min, as needed (see Table S2 for antibody list). After rehydration and blocking in 10% normal donkey serum/0.1% Triton X-100/PBS for 1 h, the sections were incubated with primary antibodies diluted in blocking buffer for 12-18 h at 4°C. After stringent washings and subsequent incubation with fluorophore-conjugated secondary antibodies for 1 h, sections were counterstained with DAPI (Sigma-Aldrich) for 5 min and mounted with DAPI Fluoromount-G mounting medium (Southern Biotech). For immunofluorescence, either a Nikon Ti-E widefield SpectraX inverted fluorescence microscope or a Nikon A1R LUN-V inverted laser scanning confocal microscope was used. Images were processed and analyzed using NIS Elements software (Nikon). For super-resolution radial fluctuation (SRRF) imaging, sections were imaged using the Nikon A1R LUN-V inverted confocal microscope. Time-lapses were generated using Galvano scanning, in which 200 frames were obtained over a period of 212 s (average of 0.94 frames/s). Time-lapses were then aligned with the Nikon Elements super-resolution alignment tool. Aligned images were analyzed with the Fiji NanoJ-Srrf plug-in, as previously described (Gustafsson et al., 2016). For whole-mount staining of the P1 brain, 500 µm coronal sections were obtained from PFA-fixed and agarose-embedded brains using a Leica VTS 1000 vibratome. After careful removal from agarose, sections were processed for immunohistochemistry with mouse anti-γ-tubulin and rabbit anti-β-catenin antibodies (Sigma). Tissues were imaged using the Nikon A1R LUN-V inverted confocal microscope, 100× objective with Nyquist sampling, and 10-12 steps (0.5 μm each step); z-stacks were obtained from each slice. Basal bodies were counted using NIS Elements AR4.40.00 software. In situ Ccdc39 mRNA images are adapted from and available at the Eurexpress expression database (http://www.eurexpress.org/ee/) (Diez-Roux et al., 2011).
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM)
The medial walls of P0 forebrains (for SEM) or P6 forebrains (for TEM) were fixed as previously described (Inoue et al., 2015). Briefly, samples were fixed in electron microscopy grade 2% PFA and 2.5% glutaraldehyde in 0.1 M Na cacodylate buffer (pH 7.4) at 4°C overnight. Tissues were washed thoroughly in 0.1 M Na cacodylate buffer (pH 7.4) then post-fixed in 1% osmium oxide (diluted in 0.1 M Na cacodylate buffer) for 1 h. Samples were washed thoroughly in 0.1 M Na cacodylate buffer and dehydrated prior to critical point drying in 100% ethanol. For SEM, tissues were gold palladium coated using a sputter coater (Leica EM ACE600) and scanned with a Hitachi SU8O1O scanning electron microscope. For TEM, samples were washed twice in propylene oxide and passed through ascending grades of LX-112 embedding resin in propylene oxide before embedding and polymerization in LX-112 embedding resin (Ladd Research Scientific). Ultra-thin sections were obtained using a Leica EMUC7 ultramicrotome and sections were placed on 200 mesh grids. The sections were stained with uranyl acetate and lead citrate to control for the background, then imaged using a Hitachi 7650 transmission electron microscope. Cilia length in SEM images was measured using NIS Elements AR4.40.00 software using its length measurement tool.
Video microscopy of ependymal cilia and local CSF flow
P6/P7 wild-type and Ccdc39prh/prh brains were collected and dissected in warm Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F12) supplemented with L-glutamine (Gibco) and 1% N2 supplement (Gibco) at room temperature. 400 µm-thick coronal sections including either the ventromedial walls of the lateral ventricle or the central aqueduct were obtained using a Leica VTS1000 vibratome. The sections were subjected to video monitoring using an inverted IX50 Olympus microscope equipped with custom assembled EMCCD phase camera. Videos were obtained at 200 frames over a period of 7 s (28.6 frames/s). For concurrent imaging of the beating cilia with green fluorescent micro-beads we used an inverted Nikon Ti-E widefield microscope fitted with a Nikon 40× Plan Apo 0.95 N.A. air objective and an Andor Zyla 4.2 PLUS sCMOS monochromatic camera. For cilia beat frequency analysis, NIS Elements AR4 was used to generate the cilia kymograph. For local CSF flow analysis, 0.5 μm diameter latex microspheres (FluoSpheres, Thermo Fisher) were added to the medium immediately before imaging. The Lumencor Spectra X LED light source was used in tandem with the Prior LED white light source for fast switching between transmitted DIC and 470 nm excitation. Light was channeled through a custom quad-pass filter and 300 frames were collected at ∼40 frames/s. Tracking and quantitative analysis of the moving beads were performed using NIS Elements software. Briefly, the green fluorescent microspheres were detected with the bright spot detection tool, and then the movement of the green signal was tracked using the binary tracking tool of the software. The velocity of each microsphere track was then exported and data were subjected to statistical analysis using Student's t-test in GraphPad Prism software.
Cerebral ventricular injection of tracers (systemic CSF flow analysis)
The pattern of systemic CSF flow was assessed as described previously (Banizs et al., 2005; Liu et al., 2016). Briefly, P8 mice were deeply anesthetized using ketamine (100 mg/kg) and xylazine (10 mg/kg), administered intraperitoneally. Evans Blue dye (4% in PBS) was injected into the left lateral ventricle at 5 µl/min using the following coordinates: −1.8 mm deep, −0.8 mm left, and −1.7 mm posterior from the Bregma. Mice were then sacrificed 10 min after the injection, brains fixed in 4% PFA overnight and then cut into 2 mm-thick coronal slices.
Note added in proof
While we were revising our manuscript, we came across a recent paper mentioning a complementary finding of hydrocephalus in Ccdc39-null mice (Solomon et al., 2017), although no data in the brain were presented in this paper.
Supplementary Material
Acknowledgements
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: K.C., F.T.M., R.W.S., J.G.; Methodology: Z.A., S.M.V., L.H., R.W.S., J.G.; Software: R.W.S.; Formal analysis: Z.A., A.T., R.W.S., J.G.; Investigation: Z.A., R.W.S., J.G.; Resources: D.B., R.W.S., J.G.; Data curation: Z.A., S.M.V., L.H., C.S., A.T., J.G.; Writing - original draft: Z.A., J.G.; Writing - review & editing: Z.A., S.M.V., L.H., D.B., K.C., F.T.M., R.W.S., J.G.; Visualization: Z.A., J.G.; Supervision: K.C., F.T.M., R.W.S., J.G.; Project administration: F.T.M., R.W.S., J.G.; Funding acquisition: K.C., F.T.M., R.W.S., J.G.
Funding
This work was supported by Hydrocephalus Association Innovator Awards (to J.G.); by the Cincinnati Children's Research Foundation (CCRF) Trustee Grant (to J.G.); by the Center for Clinical and Translational Science University of Cincinnati JIT grant (to R.W.S. and J.G.); and by the National Institutes of Health (R01NS085023 to R.W.S.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.154500.supplemental
References
- Adams D. J., Doran A. G., Lilue J. and Keane T. M. (2015). The mouse genomes project: a repository of inbred laboratory mouse strain genomes. Mamm. Genome 26, 403-412. 10.1007/s00335-015-9579-6 [DOI] [PubMed] [Google Scholar]
- Antony D., Becker-Heck A., Zariwala M. A., Schmidts M., Onoufriadis A., Forouhan M., Wilson R., Taylor-Cox T., Dewar A., Jackson C. et al. (2013). Mutations in CCDC39 and CCDC40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Hum. Mutat. 34, 462-472. 10.1002/humu.22261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banizs B., Pike M. M., Millican C. L., Ferguson W. B., Komlosi P., Sheetz J., Bell P. D., Schwiebert E. M. and Yoder B. K. (2005). Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development 132, 5329-5339. 10.1242/dev.02153 [DOI] [PubMed] [Google Scholar]
- Banizs B., Komlosi P., Bevensee M. O., Schwiebert E. M., Bell P. D. and Yoder B. K. (2007). Altered pH(i) regulation and Na(+)/HCO3(−) transporter activity in choroid plexus of cilia-defective Tg737(orpk) mutant mouse. Am. J. Physiol. Cell Physiol. 292, C1409-C1416. 10.1152/ajpcell.00408.2006 [DOI] [PubMed] [Google Scholar]
- Bekker J. M., Colantonio J. R., Stephens A. D., Clarke W. T., King S. J., Hill K. L. and Crosbie R. H. (2007). Direct interaction of Gas11 with microtubules: implications for the dynein regulatory complex. Cell Motil. Cytoskelet. 64, 461-473. 10.1002/cm.20196 [DOI] [PubMed] [Google Scholar]
- Cayre M., Canoll P. and Goldman J. E. (2009). Cell migration in the normal and pathological postnatal mammalian brain. Prog. Neurobiol. 88, 41-63. 10.1016/j.pneurobio.2009.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S. D., Ferkol T. W., Rosenfeld M., Lee H.-S., Dell S. D., Sagel S. D., Milla C., Zariwala M. A., Pittman J. E., Shapiro A. J. et al. (2015). Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am. J. Respir. Crit. Care. Med. 191, 316-324. 10.1164/rccm.201409-1672OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bigio M. R. (2010). Ependymal cells: biology and pathology. Acta Neuropathol. 119, 55-73. 10.1007/s00401-009-0624-y [DOI] [PubMed] [Google Scholar]
- Diez-Roux G., Banfi S., Sultan M., Geffers L., Anand S., Rozado D., Magen A., Canidio E., Pagani M., Peluso I. et al. (2011). A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 9, e1000582 10.1371/journal.pbio.1000582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubel R., Westendorf C., Bodenschatz E. and Eichele G. (2016). Cilia-based flow network in the brain ventricles. Science 353, 176-178. 10.1126/science.aae0450 [DOI] [PubMed] [Google Scholar]
- Feliciano D. M., Zhang S., Nasrallah C. M., Lisgo S. N. and Bordey A. (2014). Embryonic cerebrospinal fluid nanovesicles carry evolutionarily conserved molecules and promote neural stem cell amplification. PLoS ONE 9, e88810 10.1371/journal.pone.0088810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson N., Culley S., Ashdown G., Owen D. M., Pereira P. M. and Henriques R. (2016). Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat. Commun. 7, 12471 10.1038/ncomms12471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S., Lindsay A. M., Timms A. E. and Beier D. R. (2016). Mutations in Dnaaf1 and Lrrc48 cause hydrocephalus, laterality defects, and sinusitis in mice. G3 (Bethesda) 6, 2479-2487. 10.1534/g3.116.030791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverkamp F., Wölfle J., Aretz M., Krämer A., Höhmann B., Fahnenstich H. and Zerres K. (1999). Congenital hydrocephalus internus and aqueduct stenosis: aetiology and implications for genetic counselling. Eur. J. Pediatr. 158, 474-478. 10.1007/s004310051123 [DOI] [PubMed] [Google Scholar]
- Ibanez-Tallon I., Pagenstecher A., Fliegauf M., Olbrich H., Kispert A., Ketelsen U.-P., North A., Heintz N. and Omran H. (2004). Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum. Mol. Genet. 13, 2133-2141. 10.1093/hmg/ddh219 [DOI] [PubMed] [Google Scholar]
- Inoue T., Narita K., Nonami Y., Nakamura H. and Takeda S. (2015). Observation of the ciliary movement of choroid plexus epithelial cells ex vivo. J. Vis. Exp. 101, e52991 10.3791/52991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J.-K. and Lee I. W. (1999). Long-term follow-up of shunting therapy. Childs Nerv. Syst. 15, 711-717. 10.1007/s003810050460 [DOI] [PubMed] [Google Scholar]
- Keane T. M., Goodstadt L., Danecek P., White M. A., Wong K., Yalcin B., Heger A., Agam A., Slater G., Goodson M. et al. (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289-294. 10.1038/nature10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang J. (1992). Topographic anatomy of preformed intracranial spaces. Acta Neurochir. Suppl. (Wien) 54, 1-10. 10.1007/978-3-7091-6687-1_1 [DOI] [PubMed] [Google Scholar]
- Lee L. (2013). Riding the wave of ependymal cilia: genetic susceptibility to hydrocephalus in primary ciliary dyskinesia. J. Neurosci. Res. 91, 1117-1132. 10.1002/jnr.23238 [DOI] [PubMed] [Google Scholar]
- Lee L., Campagna D. R., Pinkus J. L., Mulhern H., Wyatt T. A., Sisson J. H., Pavlik J. A., Pinkus G. S. and Fleming M. D. (2008). Primary ciliary dyskinesia in mice lacking the novel ciliary protein Pcdp1. Mol. Cell. Biol. 28, 949-957. 10.1128/MCB.00354-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen M. K., Bjornsson C. S., Dymecki S. M., Gilbertson R. J., Holtzman D. M. and Monuki E. S. (2013). The choroid plexus and cerebrospinal fluid: emerging roles in development, disease, and therapy. J. Neurosci. 33, 17553-17559. 10.1523/JNEUROSCI.3258-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Klena N. T., Gabriel G. C., Liu X., Kim A. J., Lemke K., Chen Y., Chatterjee B., Devine W., Damerla R. R. et al. (2015). Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 521, 520-524. 10.1038/nature14269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Zhang Z., Guo S., Chen F., Kessler J. M., Wang Y. M. and Dutcher S. K. (2015). A NIMA-related kinase suppresses the flagellar instability associated with the loss of multiple axonemal structures. PLoS Genet. 11, e1005508 10.1371/journal.pgen.1005508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Jin X., Prasad R. M., Sari Y. and Nauli S. M. (2014). Three types of ependymal cells with intracellular calcium oscillation are characterized by distinct cilia beating properties. J. Neurosci. Res. 92, 1199-1204. 10.1002/jnr.23405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M., Guan Z., Shen Q., Lalor P., Fitzgerald U., O'Brien T., Dockery P. and Shen S. (2016). Ulk4 is essential for ciliogenesis and CSF flow. J. Neurosci. 36, 7589-7600. 10.1523/JNEUROSCI.0621-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish H., Berk A., Zipursky L. S., Matsudaira P., Baltimore D. and Darnell J. (2000). Cilia and flagella: structure and movement. In Molecular Cell Biology, 4th edn. New York: W. H. Freeman and Co. [Google Scholar]
- Merveille A.-C., Davis E. E., Becker-Heck A., Legendre M., Amirav I., Bataille G., Belmont J., Beydon N., Billen F., Clément A. et al. (2011). CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 43, 72-78. 10.1038/ng.726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M., Hazan R., Danielian P. S., Mahoney J. E., Li H., Lu J., Miller E. S., Zhu X., Lees J. A. and Cardoso W. V. (2017). Cytoplasmic E2f4 forms organizing centres for initiation of centriole amplification during multiciliogenesis. Nat. Commun. 8, 15857 10.1038/ncomms15857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mural R. J., Adams M. D., Myers E. W., Smith H. O., Miklos G. L., Wides R., Halpern A., Li P. W., Sutton G. G., Nadeau J. et al. (2002). A comparison of whole-genome shotgun-derived mouse chromosome 16 and the human genome. Science 296, 1661-1671. 10.1126/science.1069193 [DOI] [PubMed] [Google Scholar]
- Narita K. and Takeda S. (2015). Cilia in the choroid plexus: their roles in hydrocephalus and beyond. Front. Cell Neurosci. 9, 39 10.3389/fncel.2015.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita K., Kawate T., Kakinuma N. and Takeda S. (2010). Multiple primary cilia modulate the fluid transcytosis in choroid plexus epithelium. Traffic 11, 287-301. 10.1111/j.1600-0854.2009.01016.x [DOI] [PubMed] [Google Scholar]
- Narita K., Kozuka-Hata H., Nonami Y., Ao-Kondo H., Suzuki T., Nakamura H., Yamakawa K., Oyama M., Inoue T. and Takeda S. (2012). Proteomic analysis of multiple primary cilia reveals a novel mode of ciliary development in mammals. Biol. Open 1, 815-825. 10.1242/bio.20121081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonami Y., Narita K., Nakamura H., Inoue T. and Takeda S. (2013). Developmental changes in ciliary motility on choroid plexus epithelial cells during the perinatal period. Cytoskeleton (Hoboken) 70, 797-803. 10.1002/cm.21132 [DOI] [PubMed] [Google Scholar]
- Notarianni C., Vannemreddy P., Caldito G., Bollam P., Wylen E., Willis B. and Nanda A. (2009). Congenital hydrocephalus and ventriculoperitoneal shunts: influence of etiology and programmable shunts on revisions. J. Neurosurg. Pediatr. 4, 547-552. 10.3171/2009.7.PEDS08371 [DOI] [PubMed] [Google Scholar]
- Oda T., Yanagisawa H., Kamiya R. and Kikkawa M. (2014). A molecular ruler determines the repeat length in eukaryotic cilia and flagella. Science 346, 857-860. 10.1126/science.1260214 [DOI] [PubMed] [Google Scholar]
- Olcese C., Patel M. P., Shoemark A., Kiviluoto S., Legendre M., Williams H. J., Vaughan C. K., Hayward J., Goldenberg A., Emes R. D. et al. (2017). X-linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nat. Commun. 8, 14279 10.1038/ncomms14279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praveen K., Davis E. E. and Katsanis N. (2015). Unique among ciliopathies: primary ciliary dyskinesia, a motile cilia disorder. F1000Prime Rep. 7, 36 10.12703/P7-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid S., Breckle R., Hupe M., Geisler S., Doerwald N. and Neesen J. (2006). The murine Dnali1 gene encodes a flagellar protein that interacts with the cytoplasmic dynein heavy chain 1. Mol. Reprod. Dev. 73, 784-794. 10.1002/mrd.20475 [DOI] [PubMed] [Google Scholar]
- Reed R. (1989). The organization of 3′ splice-site sequences in mammalian introns. Genes Dev. 3, 2113-2123. 10.1101/gad.3.12b.2113 [DOI] [PubMed] [Google Scholar]
- Roth Y., Kimhi Y., Edery H., Aharonson E. and Priel Z. (1985). Ciliary motility in brain ventricular system and trachea of hamster. Brain Res. 330, 291-297. 10.1016/0006-8993(85)90688-2 [DOI] [PubMed] [Google Scholar]
- Schrander-Stumpel C. and Fryns J.-P. (1998). Congenital hydrocephalus: nosology and guidelines for clinical approach and genetic counselling. Eur. J. Pediatr. 157, 355-362. 10.1007/s004310050830 [DOI] [PubMed] [Google Scholar]
- Siyahhan B., Knobloch V., de Zelicourt D., Asgari M., Schmid Daners M., Poulikakos D. and Kurtcuoglu V. (2014). Flow induced by ependymal cilia dominates near-wall cerebrospinal fluid dynamics in the lateral ventricles. J. R. Soc. Interface 11, 20131189 10.1098/rsif.2013.1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes W. C., Rosen B., West A. P., Koutsourakis M., Bushell W., Iyer V., Mujica A. O., Thomas M., Harrow J., Cox T. et al. (2011). A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337-342. 10.1038/nature10163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon G. M., Francis R., Chu K. K., Birket S. E., Gabriel G., Trombley J. E., Lemke K. L., Klena N., Turner B., Tearney G. J., (2017). Assessment of ciliary phenotype in primary ciliary dyskinesia by micro-optical coherence tomography. JCI Insight 2, e91702 10.1172/jci.insight.91702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassky N., Merkle F. T., Flames N., Tramontin A. D., Garcia-Verdugo J. M. and Alvarez-Buylla A. (2005). Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J. Neurosci. 25, 10-18. 10.1523/JNEUROSCI.1108-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stottmann R. W., Moran J. L., Turbe-Doan A., Driver E., Kelley M. and Beier D. R. (2011). Focusing forward genetics: a tripartite ENU screen for neurodevelopmental mutations in the mouse. Genetics 188, 615-624. 10.1534/genetics.111.126862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs J. L., Vladar E. K., Axelrod J. D. and Kintner C. (2012). Multicilin promotes centriole assembly and ciliogenesis during multiciliate cell differentiation. Nat. Cell Biol. 14, 140-147. 10.1038/ncb2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J. P., Lopes P. and Silva R. (2012). Primary ciliary dyskinesia and hydrocephalus with aqueductal stenosis. J. Child Neurol. 27, 938-941. 10.1177/0883073811429856 [DOI] [PubMed] [Google Scholar]
- Zhao H., Zhu L., Zhu Y., Cao J., Li S., Huang Q., Xu T., Huang X., Yan X. and Zhu X. (2013). The Cep63 paralogue Deup1 enables massive de novo centriole biogenesis for vertebrate multiciliogenesis. Nat. Cell Biol. 15, 1434-1444. 10.1038/ncb2880 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.