

Abstract
Background
The objective of this study was to investigate the incidence, demographics, tumor characteristics, treatment, and survival of patients with hilar cholangiocarcinoma.
Methods
Data on Klatskin tumors between 2004 and 2013 was extracted from the Surveillance, Epidemiology and End Results Registry. The epidemiology of these tumors was then analyzed.
Results
A total of 254 patients with Klatskin tumors were identified. The overall age-adjusted incidence of Klatskin tumors between 2004 and 2013 was 0.38 per 1,000,000 per year. A gradual decline in the incidence was noted, with the highest (0.44) in 2005 and lowest (0.24) in 2010. Males had a higher incidence of Klatskin tumors compared to females (0.47 vs. 0.25 per 1,000,000 per year). These tumors were more common among Asian and Pacific islanders, who had an age-adjusted incidence rate of 0.48 per 1,000,000. Incidence increased with age, with the peak incidence between the ages of 80 and 84 years. The majority of the tumors were extrahepatic (67.3%). Approximately one-fourth (22.4%) of these patients had metastatic disease at presentation. Only 26.8% of patients had surgically resectable disease at presentation. One- and 5-year cause-specific survival for Klatskin tumors was 41% and 10.4%, respectively, with a median survival of 7 months. On Cox proportional hazard regression analysis, extrahepatic tumors (hazard ratio [HR] 0.54, 95% confidence interval [CI] 0.37-0.80, P=0.02) and those treated surgically (HR 0.47, 95%CI 0.29-0.77, P=0.003) had significantly better outcomes.
Conclusions
Klatskin tumors are rare and have a very poor prognosis with low survival rate. Among these tumors, extrahepatic and surgically treated tumors tend to have better outcomes.
Keywords: Hilar cholangiocarcinoma, Klatskin tumors, SEER, survival, incidence
Introduction
Cholangiocarcinomas (CCAs) are rare tumors, comprising 3% of all gastrointestinal tumors originating from the intra- or extrahepatic bile duct epithelium. They represent the second most common primary hepatic malignancy [1]. Approximately 50% of CCAs are perihilar or hilar, 40% are distal extrahepatic, and only 10% or less are intrahepatic [2]. In 1965, Klatskin identified CCAs originating from the hepatic duct bifurcation and named these hilar tumors as Klatskin tumors [3]. Klatskin tumors, or hilar CCAs, are bile duct tumors that involve the common hepatic duct bifurcation but may arise from the intrahepatic (IHCCA) or extrahepatic (EHCCA) portion of the biliary tree [4]. It has also been suggested that EHCCA and IHCCA should be combined as perihilar CCA, given the similarities in their behavior and treatment methods [5]. A comparison of the 10-year prevalence of these tumors between 1990 and 2000 in the United States showed a high age-adjusted prevalence of IHCCA in Hispanics, with the lowest prevalence among African Americans and Asian Pacific islanders [1].
Our current knowledge of the pathogenesis of CCA in general, in comparison to other gastrointestinal tumors, is limited. Genetic abnormalities and molecular defects in various oncogenes may play a role. A role for chronic inflammation and cholestasis in carcinogenesis has been also stipulated [1]. Various risk factors have been identified: primary sclerosing cholangitis [6-8], parasitic infestation (Clonorchis [9], Opisthorchis [10]), cholelithiasis [11], fibrocystic liver disease [12], toxins (thorotrast, rubber and chemical industries) [13], chronic liver disease [14-17], Lynch syndrome and biliary papillomatosis [18]. Histologically, the majority of these tumors are adenocarcinomas followed by squamous cell carcinomas.
International Classification of Disease – Oncology (ICD-O) consists of two coding systems to describe a tumor: i) the topographical code, which describes the anatomical site of origin of the tumor; and ii) the morphological code, which describes the histology of the tumor along with the behavior (malignant or benign). ICD-O-3 has been available since 2000 and was last revised in 2013. In the prior version, i.e. ICD-O-2, Klatskin tumors were assigned to the topography code for intrahepatic biliary duct tumors only, whereas in ICD-O-3, these hilar tumors were reassigned topographical codes for either intrahepatic or extrahepatic biliary duct tumors. Previous analysis of Surveillance, Epidemiology and End Results (SEER) data [19] noted that, before ICD-O-3, 91% of the Klatskin tumors were incorrectly coded as intrahepatic biliary duct (IHBD) tumors, leading to an overestimation of the latter’s incidence by 13% and an underestimation of extrahepatic biliary duct (EHBD) tumors. The switch to ICD-O-3 in 2001 resulted in Klatskin tumors being coded as EHBD, with a subsequent rise in their incidence [19,20]. In addition, ICD-O-3 referenced hilar CCA histology code 8162/3 to topographies for both IHCCA and EHCCA.
Although several studies have reported on CCAs and their extrahepatic, intrahepatic and hilar types, studies focusing on Klatskin tumors are lacking. In this study, we present the epidemiology, tumor characteristics, treatment, and survival profile of Klatskin tumors diagnosed between 2004 and 2013. We further evaluate differences between extra- and intrahepatic Klatskin tumors, as prior reports suggest that they tend to present and behave differently.
Patients and methods
Data were extracted from the National Cancer Institute’s SEER registry. The SEER registry is an extensive data center of cancer incidence, staging, treatment and survival from population-based cancer registries of the United States. The SEER Program registries collect data on patient demographics, primary tumor site, tumor morphology, and stage at diagnosis, treatment, and follow up, and publish cancer incidence and survival data from cancer registries covering approximately 30% of the US population. SEER includes 26% of African Americans, 38% of Hispanics, 44% of American Indians and Alaska Natives, 50% of Asians, and 67% of Hawaiian/Pacific islanders. It comprises 18 cancer registries that gather data from 18 geographical areas of United States [21].
Data collection
Between 2004 and 2013, all cases of Klatskin tumors were extracted from the population-based cancer registries of the program. These dates were selected to avoid inclusion of the incorrectly coded tumors prior to the implementation of ICD-O-3 [22]. To identify the cases, various combinations of topographical and histology codes based on ICD-O-3 were used [23]. The topographical codes used for Klatskin tumors were: C22.0, C22.1, and C24.0. The ICD-O-3 histological code used for Klatskin tumors was 8162/3. Only malignant and microscopically confirmed cases were included. American Joint Committee on Cancer (AJCC) 2004 staging was available for the majority of cases and this was used. Survival was calculated for first primary Klatskin tumors only. Cases in which survival duration was unknown were excluded.
Statistical analysis
The incidence rates were calculated as the number of new cases per 1,000,000 person-years, age-adjusted to the 2000 US population. Significance for a trend in incidence was evaluated by testing the hypothesis that the annual percentage change was equal to zero. SEER*Stat 8.3.2 [23] was used to calculate age-adjusted incidence, age-wise distribution, and annual percentage change in incidence. For all other demographic and survival data, cases were copied to Excel and then analyzed using SPSS (IBM Corp. Released 2012. IBM SPSS Statistics for Windows, Version 21.0. Armonk, NY: IBM Corp). Pearson’s chi-square test was used for all categorical variables, while the Mann-Whitney U test was used to compare medians between continuous variables. Kaplan-Meier curves were plotted for survival analysis and 1- and 5- year survival was calculated by the Kaplan-Meier method. A Cox proportional hazard regression model was then used to determine predictors of survival.
Results
Incidence
The overall age-adjusted incidence of Klatskin tumors between 2004 and 2013 was 0.38 per 1,000,000 per year (Table 1). There was a higher incidence of Klatskin tumors in males compared to females (0.47 vs. 0.25 per 1,000,000 per year). These tumors were more common among Asian and Pacific islanders, who had an age-adjusted incidence rate of 0.48 per 1,000,000. A gradual decline in the incidence was noted, with the highest incidence (0.44) in 2005 and the lowest (0.24) in 2010 (Fig. 1). Incidence increased with age, with peak incidence between the ages of 80 to 84 years. No cases were reported in patients younger than 24 years of age (Fig. 2).
Table 1.
Incidence of Klatskin tumors

Figure 1.
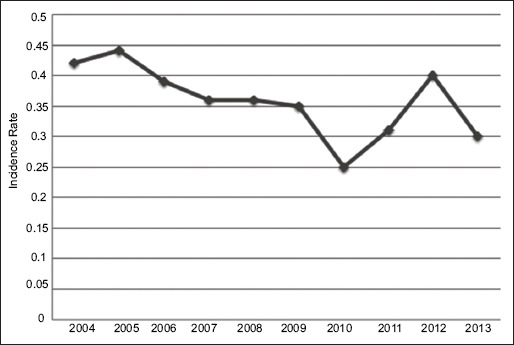
Annual incidence of Klatskin tumors
Figure 2.

Age-wise incidence of Klatskin tumors
Demographics and tumor characteristics
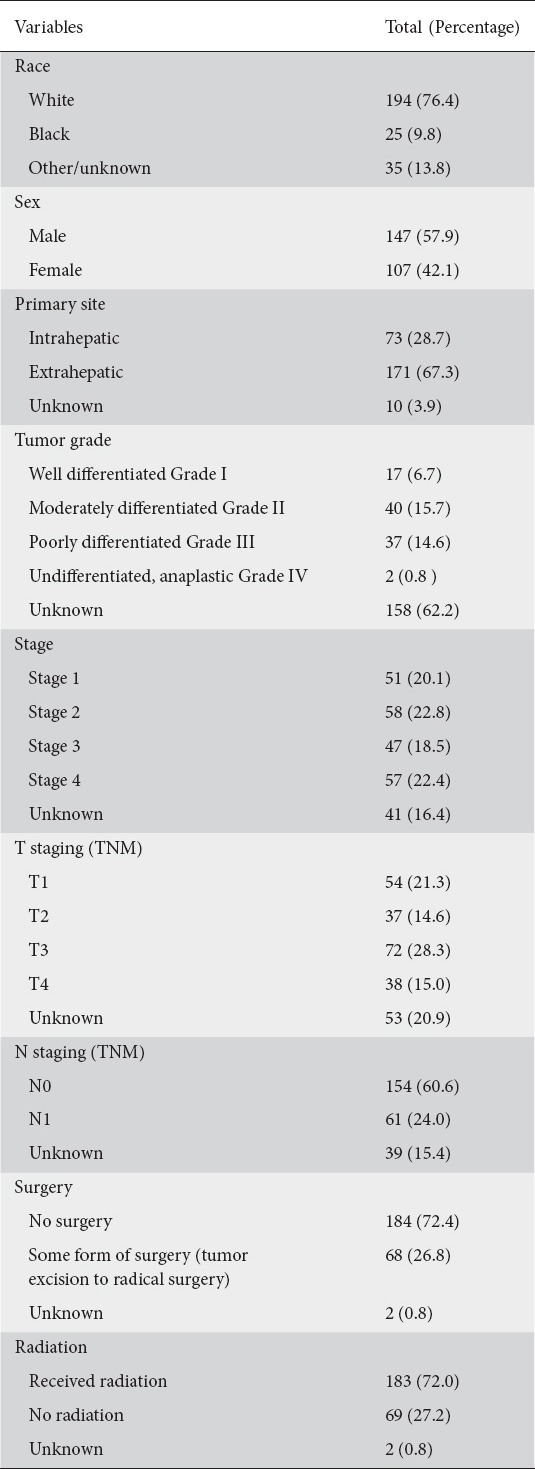
A total of 254 patients with Klatskin tumors who met our inclusion criteria were identified from the SEER database. The demographic and tumor characteristics are shown in Table 2. The majority of the tumors were extrahepatic (67.3%) and moderately differentiated (15.7%). The most common stage at presentation was Stage II (22.8%), followed by Stage IV (22.4%). The majority (72.0%) of these tumors received radiation therapy, whereas only 26.8% were amenable to surgical resection.
Table 2.
Demographic and tumor characteristics
Cause-specific survival and predictors of survival
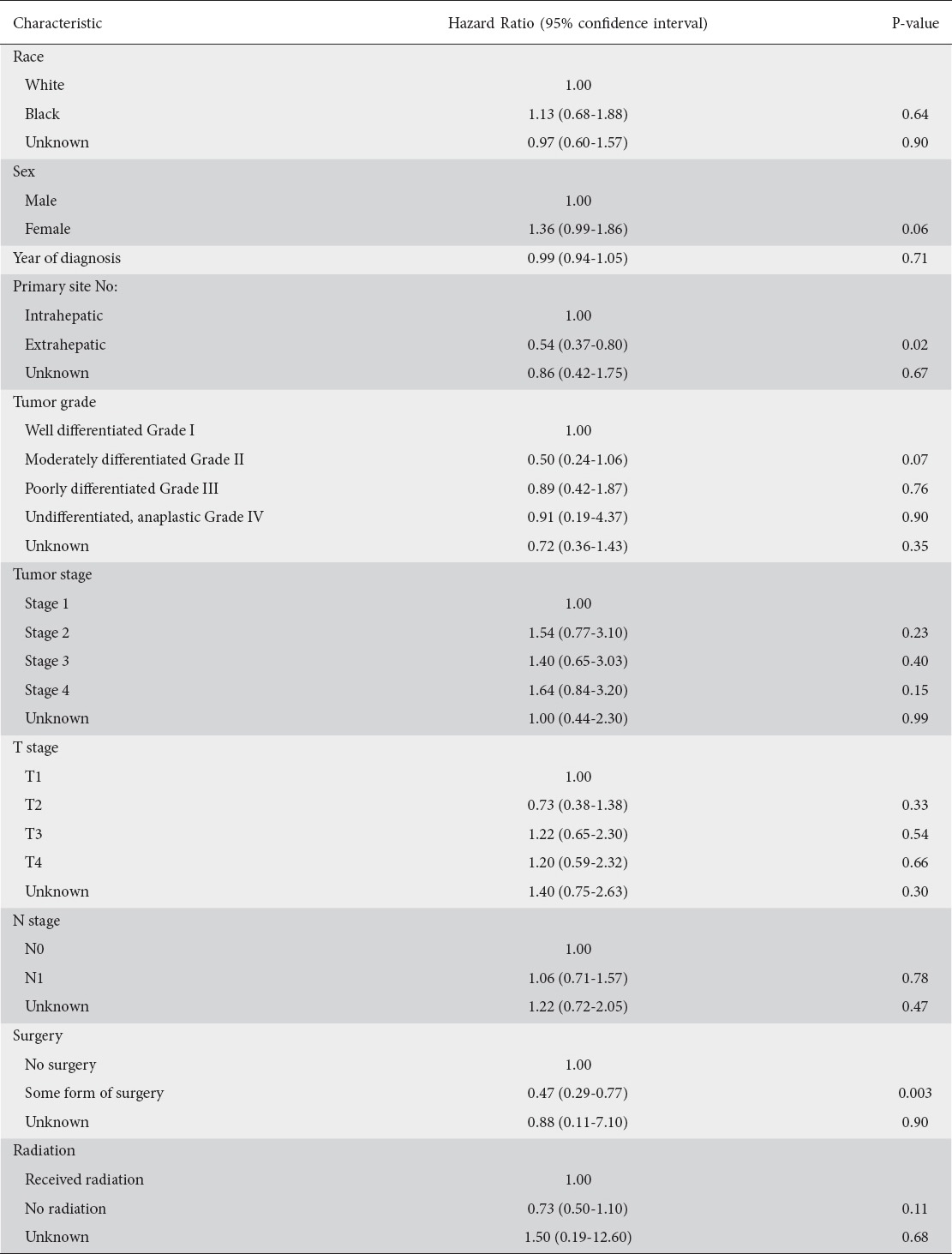
One- and 5-year cause-specific survival for Klatskin tumors was 41% and 10.4%, respectively, with a median survival of 7 months (Fig. 3, Table 3). On Cox proportional hazard regression analysis, extrahepatic tumors (hazard ratio [HR] 0.54, 95% confidence interval [CI] 0.37-0.80, P=0.02) and those treated surgically (HR 0.47, 95%CI 0.29-0.77, P=0.003) were noted to have better outcomes (Table 4). Patients’ age, sex, race, year of diagnosis, tumor grade, staging, and radiation exposure did not affect outcomes.
Figure 3.

Kaplan-Meier curve for survival analysis
Table 3.
Survival of patients with Klatskin tumors

Table 4.
Results of Cox proportional hazard regression
Discussion
Klatskin tumors, or hilar CCAs, are rare in the US population, but recent data suggests a rising incidence [24,25]. Our study provides the most recent comprehensive information on Klatskin tumors from the SEER cancer registry. The SEER database was used for this analysis as it is an extensive data center for cancer incidence, staging, treatment, and survival derived from population-based cancer registries of the United States. Contrary to prior reports [24], our study highlights a decline in the annual incidence of these tumors, with the highest incidence in 2005 and the lowest in 2010 (Fig. 1). The exact reason for this decline is not clearly understood. However, it may in part be explained by a possible decrease in incidence of the risk factors for Klatskin tumors in the US population. Further studies are warranted for a better understanding of this decreasing trend.
Klatskin tumors had the highest incidence among Asians and Pacific islanders, who had an age-adjusted incidence rate of 0.48 per 1,000,000. Factors that explain this racial predisposition for Asians to developing Klatskin tumors include the increased rate of hepatolithiasis and liver-fluke infections among these patients. The highest prevalence of CCA has been reported from Southeast Asia [5,19,24]. The reason behind the increased predisposition to these tumors among Asians in the United States who lack the usual risk factors may, however, suggest an alternative pathogenesis. Our analysis noted that the incidence of Klatskin tumors increases with age, with a peak between 60s-80s. It can affect all age groups, but the age distribution seen in our study is similar to prior reports [26]. We noted that no cases were reported in patients younger than 24 years, which suggests chronic long-standing inflammation with advancing age leading to hyperplasia as an underlying pathogenic mechanism.
In a SEER analysis between 1992-2000, 91% (246 of 269) of hilar CCAs were reported as IHCCA. In contrast, for the years 2001 and 2002, after the switch to ICD-O-3, 54% (22 of 41) of hilar CCAs were reported as IHCCA [19]. In our analysis (2004-2013), 67% (n=171) were reported as EHCCA while only 29% (n=73) were IHCCA, showing an increasing trend towards EHCCA. The rise in incidence can in part be explained by the change in coding system from ICD-O-2 to ICD-O-3. However, further studies are needed to understand the underlying factors for this trend.
Several classification systems have been published to classify hilar CCA, such as the Bismuth-Corlette system, TNM staging by the AJCC group, and the Memorial Sloan Kettering Cancer Center classification. The AJCC staging is the most widely used to determine prognosis and treatment [26-28]. Our analysis suggests that the majority of the patients present in advanced stages (Stages 3-4 and T3-T4) per this staging system.
The only curative treatment for hilar CCA is margin-negative surgical resection, which usually consists of en bloc resection of the bile duct, extended liver resection and portal vein resection [29]. There are no trials that demonstrate a therapeutic benefit for radiation, chemotherapy or combination therapy. The majority of the patients in our analysis were treated with radiation; however, those treated surgically fared better. Our study notes that, even though surgery is the preferred treatment for these tumors, only a minority (26.8%) of them were amenable to surgical resection. This is probably because most of these patients present in stages where the disease is too advanced for recovery by surgical resection.
We acknowledge several limitations in our study. First, it had an observational and retrospective design. Though the SEER database is large, our sample size of 254 Klatskin tumors is small. In addition, SEER does not provide data on various topics, including chemotherapeutic regimen and dose, local and distant recurrence data, and comorbidities. Another limitation of SEER is related to the ICD-O coding error of Klatskin tumors. We minimized this limitation somewhat by excluding cases before 2004; by doing this, we were able to avoid the coding error associated with ICD-O-2, where Klatskin tumors were assigned to the topography code for intrahepatic biliary duct tumors. However, ICD-O-3 uses the histology code 8162/3 for either intra- or extrahepatic bile duct tumors and does not use specific codes for intra- or extrahepatic, rendering our analysis imperfect given the possibility of coding misclassification. This issue can only be solved when specific histology codes are assigned to these tumors. Furthermore, the SEER database lacks data regarding risk factors, clinical presentation, duration of symptoms, diagnostic modalities employed, or the use of endoscopic ultrasonography, cholangioscopy, fluorescence in situ hybridization for tissue diagnosis, positron emission tomography scan data, or details regarding biliary drainage, which would have made our study even more robust and comprehensive.
In conclusion, Klatskin tumors, or hilar CCAs, are rare gastrointestinal tumors that have declined in incidence over the past decade in the United States. The majority of these tumors are diagnosed in their advanced stages with metastases. Surgery is regarded as the best treatment modality for these tumors. However, only a small number of patients are treated surgically, probably because they present in advanced stages. The tumors continue to carry a poor prognosis, with median survival duration measured in months.
Summary Box.
What is already known:
Klatskin tumors, or hilar cholangiocarcinomas, are rare in the United States population but they may have a rising incidence
The incidence of Klatskin tumors increases with age and patients younger than 20 years of age are rare
The only curative treatment for hilar cholangiocarcinoma is margin-negative surgical resection
What the new findings are:
The annual incidence of Klatskin tumors has actually declined in the United States over the past decade
Even though surgery is the preferred treatment, only a minority (26.8%) of patients are amenable to surgical resection, as they present in advanced stages with metastases
These tumors continue to carry a poor prognosis, with median survival duration measured in months
Biography
Yale New Haven Health-Bridgeport Hospital; Mayo Clinic, Rochester, USA
Footnotes
Conflict of Interest: None
References
- 1.Blechacz BR, Gores GJ. Cholangiocarcinoma. Clin Liver Dis. 2008;12:131–150, ix. doi: 10.1016/j.cld.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 2.DeOliveira ML, Cunningham SC, Cameron JL, et al. Cholangiocarcinoma:thirty-one-year experience with 564 patients at a single institution. Ann Surg. 2007;245:755–762. doi: 10.1097/01.sla.0000251366.62632.d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okuda K, Kubo Y, Okazaki N, Arishima T, Hashimoto M. Clinical aspects of intrahepatic bile duct carcinoma including hilar carcinoma:a study of 57 autopsy-proven cases. Cancer. 1977;39:232–246. doi: 10.1002/1097-0142(197701)39:1<232::aid-cncr2820390137>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 4.Klatskin G. Adenocarcinoma of the hepatic duct at its bifurcation within theporta hepatis. An unusual tumor with distinctive clinical and pathological features. Am J Med. 1965;38:241–256. doi: 10.1016/0002-9343(65)90178-6. [DOI] [PubMed] [Google Scholar]
- 5.Ebata T, Kamiya J, Nishio H, Nagasaka T, Nimura Y, Nagino M. The concept of perihilar cholangiocarcinoma is valid. Br J Surg. 2009;96:926–934. doi: 10.1002/bjs.6655. [DOI] [PubMed] [Google Scholar]
- 6.deGroen PC. Cholangiocarcinoma in primary sclerosing cholangitis:who is at risk and how do we screen? Hepatology. 2000;31:247–248. doi: 10.1002/hep.510310137. [DOI] [PubMed] [Google Scholar]
- 7.Burak K, Angulo P, Pasha TM, Egan K, Petz J, Lindor KD. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol. 2004;99:523–526. doi: 10.1111/j.1572-0241.2004.04067.x. [DOI] [PubMed] [Google Scholar]
- 8.Claessen MM, Vleggaar FP, Tytgat KM, Siersema PD, van Buuren HR. High lifetime risk of cancer in primary sclerosing cholangitis. J Hepatol. 2009;50:158–164. doi: 10.1016/j.jhep.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 9.Shin HR, Lee CU, Park HJ, et al. Hepatitis B and C virusClonorchis sinensisfor the risk of liver cancer:a case-control study in Pusan, Korea. Int J Epidemiol. 1996;25:933–940. doi: 10.1093/ije/25.5.933. [DOI] [PubMed] [Google Scholar]
- 10.Honjo S, Srivatanakul P, Sriplung H, et al. Genetic and environmental determinants of risk for cholangiocarcinoma viaOpisthorchis viverriniin a densely infested area in Nakhon Phanom, northeast Thailand. Int J Cancer. 2005;117:854–860. doi: 10.1002/ijc.21146. [DOI] [PubMed] [Google Scholar]
- 11.Hsing AW, Gao YT, Han TQ, et al. Gallstones and the risk of biliary tract cancer:a population-based study in China. Br J Cancer. 2007;97:1577–1582. doi: 10.1038/sj.bjc.6604047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott J, Shousha S, Thomas HC, Sherlock S. Bile duct carcinoma:a late complication of congenital hepatic fibrosis. Case report and review of literature. Am J Gastroenterol. 1980;73:113–119. [PubMed] [Google Scholar]
- 13.Sahani D, Prasad SR, Tannabe KK, Hahn PF, Mueller PR, Saini S. Thorotrast-induced cholangiocarcinoma:case report. Abdom Imaging. 2003;28:72–74. doi: 10.1007/s00261-001-0148-y. [DOI] [PubMed] [Google Scholar]
- 14.Welzel TM, Graubard BI, El-Serag HB, et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States:a population-based case-control study. Clin Gastroenterol Hepatol. 2007;5:1221–1228. doi: 10.1016/j.cgh.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi M, Ikeda K, Saitoh S, et al. Incidence of primary cholangiocellular carcinoma of the liver in Japanese patients with hepatitis C virus-related cirrhosis. Cancer. 2000;88:2471–2477. doi: 10.1002/1097-0142(20000601)88:11<2471::aid-cncr7>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 16.Sorensen HT, Friis S, Olsen JH, et al. Risk of liver and other types of cancer in patients with cirrhosis:a nationwide cohort study in Denmark. Hepatology. 1998;28:921–925. doi: 10.1002/hep.510280404. [DOI] [PubMed] [Google Scholar]
- 17.Hsing AW, Zhang M, Rashid A, et al. Hepatitis B and C virus infection and the risk of biliary tract cancer:a population-based study in China. Int J Cancer. 2008;122:1849–1853. doi: 10.1002/ijc.23251. [DOI] [PubMed] [Google Scholar]
- 18.Lee SS, Kim MH, Lee SK, et al. Clinicopathologic review of 58 patients with biliary papillomatosis. Cancer. 2004;100:783–793. doi: 10.1002/cncr.20031. [DOI] [PubMed] [Google Scholar]
- 19.Khan SA, Emadossadaty S, Ladep NG, et al. Rising trends in cholangiocarcinoma:is the ICD classification system misleading us? J Hepatol. 2012;56:848–854. doi: 10.1016/j.jhep.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 20.Welzel TM, McGlynn KA, Hsing AW, O'Brien TR, Pfeiffer RM. Impact of classification of hilar cholangiocarcinomas (Klatskin tumors) on the incidence of intra- and extrahepatic cholangiocarcinoma in the United States. J Natl Cancer Inst. 2006;98:873–875. doi: 10.1093/jnci/djj234. [DOI] [PubMed] [Google Scholar]
- 21.Overview of the SEER program. [Accessed January 3 2018]. Available from: http://seer.cancer.gov/about/overview.html .
- 22.Fritz A, Percy C, Jack A, et al. International Classification of Diseases for Oncology. 3rd edition, 1st revision. Geneva: World Health Organization; 2013. [Google Scholar]
- 23.SEER*Stat Software. [Accessed January 3 2018]. Available from: http://seer.cancer.gov/seerstat/
- 24.Plentz RR, Malek NP. Clinical presentation, risk factors and staging systems of cholangiocarcinoma. Best Pract Res Clin Gastroenterol. 2015;29:245–252. doi: 10.1016/j.bpg.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Shaib YH, Davila JA, McGlynn K, El-Serag HB. Rising incidence of intrahepatic cholangiocarcinoma in the United States:a true increase? J Hepatol. 2004;40:472–477. doi: 10.1016/j.jhep.2003.11.030. [DOI] [PubMed] [Google Scholar]
- 26.Edge SB American Joint Committee on Cancer. AJCC cancer staging manual. 7th ed. New York: Springer; 2010. [DOI] [PubMed] [Google Scholar]
- 27.Deoliveira ML, Schulick RD, Nimura Y, et al. New staging system and a registry for perihilar cholangiocarcinoma. Hepatology. 2011;53:1363–1371. doi: 10.1002/hep.24227. [DOI] [PubMed] [Google Scholar]
- 28.Mansour JC, Aloia TA, Crane CH, Heimbach JK, Nagino M, Vauthey JN. Hilar cholangiocarcinoma:expert consensus statement. HPB (Oxford) 2015;17:691–699. doi: 10.1111/hpb.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ito F, Cho CS, Rikkers LF, Weber SM. Hilar cholangiocarcinoma:current management. Ann Surg. 2009;250:210–218. doi: 10.1097/SLA.0b013e3181afe0ab. [DOI] [PubMed] [Google Scholar]