

ABSTRACT
We defined how blood-derived vitronectin (VTN) rapidly and potently activates leukemia inhibitory factor (LIF) and pro-inflammatory interleukin 6 (IL-6) in vitro and after vascular injury in the brain. Treatment with VTN (but not fibrinogen, fibronectin, laminin-111 or collagen-I) substantially increased LIF and IL-6 within 4 h in C6-astroglioma cells, while VTN−/− mouse plasma was less effective than that from wild-type mice. LIF and IL-6 were induced by intracerebral injection of recombinant human (rh)VTN in mice, but induction seen upon intracerebral hemorrhage was less in VTN−/− mice than in wild-type littermates. In vitro, VTN effects were inhibited by RGD, αvβ3 and αvβ5 integrin-blocking peptides and antibodies. VTN activated focal adhesion kinase (FAK; also known as PTK2), whereas pharmacological- or siRNA-mediated inhibition of FAK, but not PYK2, reduced the expression of LIF and IL-6 in C6 and endothelial cells and after traumatic cell injury. Dominant-negative FAK (Y397F) reduced the amount of injury-induced LIF and IL-6. Pharmacological inhibition or knockdown of uPAR (also known as PLAUR), which binds VTN, also reduced cytokine expression, possibly through a common target of uPAR and integrins. We propose that VTN leakage into tissues promotes inflammation. Integrin–FAK signaling is therefore a novel IL-6 and LIF regulation mechanism relevant to the inflammation and stem cell fields.
KEY WORDS: FAK, IL-6, Integrin, LIF, Vitronectin, uPAR
Summary: Blood vitronectin is a unique and key inducer of pro-inflammatory IL-6 and LIF, through integrin and uPAR signaling, which had not been previously indicated as having a role in cytokine expression.
INTRODUCTION
Vitronectin (VTN) is mainly produced in the liver by hepatocytes and endothelial cells (Seiffert et al., 1991, 1995) and is found at high concentrations in blood serum (Hayman et al., 1983). VTN−/− mice display no overt phenotype (Zheng et al., 1995) but exhibit delayed angiogenesis and poor wound healing (Jang et al., 2000), and defective vascular repair, causing persistent leakiness after injury (Li et al., 2012). Thus, leaked VTN may induce tissue repair responses. VTN has been detected in tissues in inflammatory rheumatoid arthritis (Tomasini-Johansson et al., 1998) and lung, liver and kidney fibrosis (Jang et al., 2000). Both VTN and fibronectin (another enriched plasma protein) leak into the brain after stroke (del Zoppo et al., 2012) and can activate microglia in vitro (Milner and Campbell, 2003; Welser-Alves et al., 2011).
Microglia and astrocytes express the VTN receptors αvβ3 and αvβ5 integrin (Herrera-Molina et al., 2012; Kang et al., 2008; Milner, 2009; Welser-Alves et al., 2011). Microglia and astrocytes, as well as endothelial cells, are major producers of pro-inflammatory cytokines, such as IL-6 and TNFα, in vitro and after traumatic or ischemic injury to the brain (Banner et al., 1997; Erta et al., 2012; Lau and Yu, 2001) or upon self-induction by IL-6 (Van Wagoner and Benveniste, 1999). IL-6 is a major regulator of a variety of inflammatory disorders and a target for therapies (Hunter and Jones, 2015). Its levels are almost non-existent in the normal brain but increase rapidly and greatly after acute injuries, such as stroke (Kang et al., 2013; Suzuki et al., 2009; Van Wagoner and Benveniste, 1999). The initial trigger(s) for IL-6 induction in the brain remains largely unresolved (Suzuki et al., 2009), but might include leakage of blood proteins upon blood–brain barrier disruption, which occurs rapidly after stroke (Krueger et al., 2015).
LIF is a GP130 (also known as IL6ST) receptor-activating cytokine, and as such related to the IL-6 family of cytokines (Zigmond, 2012). LIF is well known for playing a role during development and for promoting stem cell self-renewal in vitro and in vivo (Bauer and Patterson, 2006; Cartwright et al., 2005). LIF is also expressed by astrocytes (Banner et al., 1997), microglia (Nakanishi et al., 2007) and endothelial cells (Mi et al., 2001). It can also be pro-inflammatory (Kerr and Patterson, 2004; Pan et al., 2008; Suzuki et al., 2009), facilitating neutrophil activation (Borish et al., 1986) and macrophage infiltration, as demonstrated by conditioned medium experiments from LIF−/− and IL-6−/− Schwann cell preparations from denervated mouse sciatic nerves (Tofaris et al., 2002). LIF is expressed at very low levels throughout the body, but increases following brain injury (Banner et al., 1997) and stroke (Kang et al., 2013). Its expression in injured peripheral nerves is decreased again after repair (Dowsing et al., 2001), perhaps coincident with re-establishment of vascular integrity. The mechanisms regulating LIF expression are not well understood, but may include stimulation by IL-1β, possibly through mRNA stabilization (Carlson et al., 1996).
VTN has an RGD motif (Suzuki et al., 1985) with which it binds to the VTN receptors αvβ3 and αvβ5 integrin (Plow et al., 2000). It also interacts with several other proteins (Leavesley et al., 2013). Besides its cell adhesive properties, VTN activates integrin intracellular signaling molecules (Giancotti and Ruoslahti, 1999), including FAK (also known as PTK2), one of the major integrin transducers. Phosphorylation of Y397 is critical to FAK activation (Liu et al., 2003) and induces a number of signaling cascades (Keasey et al., 2013). Phosphorylation of FAK at Y397 is critical for TNFα-stimulated expression of IL-6 (Schlaepfer et al., 2007), suggesting that it might be a signaling node for cytokine regulation. VTN is unique among extracellular matrix (ECM) molecules because it also binds to urokinase-type plasminogen activator (uPA) receptor (uPAR; also known as PLAUR) (Madsen et al., 2007), a membrane-bound glycoprotein that serves as the receptor for uPA.
Here, we determined whether blood-derived proteins such as VTN regulate LIF and IL-6 expression through integrin–FAK and/or uPAR signaling, by using cultured astroglioma and endothelial cell, and adult mouse models.
RESULTS
VTN uniquely increases LIF and IL-6 expression in vitro
We had previously shown that some ECM molecules, including VTN, inhibit CNTF through integrin signaling (Keasey et al., 2013). To determine whether the related cytokines LIF and IL-6 are regulated by such blood proteins we first used serum, which has very high levels of ECM proteins such as VTN, fibronectin (FN1) and fibrinogen (Hayman et al., 1985). We cultured C6 astroglioma cells for 24 h in 10% fetal bovine serum (FBS), then changed the medium to low 1% (v/v) or regular 10% serum medium for an additional 24 h. In 1% serum, LIF and IL-6 mRNA levels were only ∼33 and 45% (Fig. 1A,B), respectively, of that found when cells were in 10% serum. CNTF was upregulated in 1% serum (Fig. 1C), consistent with our previous study (Keasey et al., 2013), and suggesting that the decreases in LIF and IL-6 were not a general cellular response to serum withdrawal. We followed up this broad approach by testing specific ECM proteins. C6 cells were seeded onto plastic culture plates and maintained for 24 h and serum-deprived for a further 24 h. VTN, fibronectin, fibrinogen, laminin-111 (the isoform comprising α1, β1 and γ1 chains) or collagen-I were then added directly to the culture medium. Remarkably, after 4 h, only VTN produced a significant ∼8-fold increase in LIF (Fig. 1D) and ∼4-fold increase in IL-6 mRNA expression (Fig. 1E). VTN treatment for 24 h showed no discernable differences relative to vehicle controls (data not shown, n=2), suggesting that LIF and IL-6 are rapidly induced but then return to baseline levels. This could be due to VTN endocytosis and subsequent degradation (Memmo and McKeown-Longo, 1998). VTN effects were dose dependent (Fig. 1F,G) and resulted in increased LIF and IL-6 protein release from cells as confirmed with a dot blot assay of conditioned medium (Fig. 1H,I). In addition, C6 cells were grown in a medium containing 1% (v/v) plasma from VTN−/− or VTN+/+ littermate mice, but without fetal bovine serum. After 4 h, LIF and IL-6 mRNA increases with VTN−/− plasma were only ∼30% and 50%, respectively, of that seen with VTN+/+ plasma (Fig. 1J,K). This suggests that native mouse VTN can increase LIF and IL-6 expression, and that an additional plasma molecule(s) also has such effects.
Fig. 1.

Among ECM proteins, VTN uniquely activates LIF and IL-6 expression in C6 astroglioma cells. Serum contains a variety and various abundances of integrin-binding ECM proteins. We plated C6 cells for 24 h in 10% serum, then replaced the medium with a low serum formulation (1% v/v; LS) for 24 h, which reduced LIF (A) and IL-6 (B) mRNA expression, as measured by RT-qPCR, relative to that seen in control 10% serum (Ctrl). (C) In the same experiment as in A and B, CNTF mRNA was increased, likely due to inhibitory activity of serum ECMs, such as VTN, on CNTF expression which is negatively regulated by integrin–FAK signaling. In further experiments, we seeded C6 cells and maintained them for 24 h before serum was removed for a further 24 h. Then, VTN was ‘spiked’ into the medium (10 μg/ml concentration), where it rapidly (within 4 h) and robustly induced LIF (D) and IL-6 (E) mRNA relative to what was seen upon addition of PBS control (vehicle, no ECM substrate). Fibronectin (FN1), fibrinogen (FGN), laminin-111 (LM) or collagen-I (COL) addition had no effect. VTN increased LIF (F) and IL-6 (G) in a dose-dependent fashion. NS, no serum; concentrations of VTN are μg/ml. VTN treatment for 4 h caused increased release of LIF (H) and IL-6 (I) protein in conditioned medium, as shown by a dot blot assay (representative of three independent experiments). Plasma from VTN−/− mice induced LIF (J) and IL-6 (K) gene expression by less than plasma from VTN+/+ littermates when added for 4 h to C6 cells that had been serum-deprived for 24 h. Data are means±s.e.m. of three or four independent experiments (as denoted in columns). **P<0.01, ***P<0.001.
Recombinant VTN and VTN from blood leaked into the brain upregulate brain LIF and IL-6 in adult mice
Recombinant human (rh)VTN was injected into the striatum of the brains of young adult mice. In VTN+/+ (wild-type) mice, striatal LIF (2.8-fold) and IL-6 (2.5-fold) mRNA levels were increased 24 h later, relative to that seen after PBS control injections (Fig. 2A,C). In VTN−/− littermates (used to avoid the confounding effects of endogenous VTN), LIF (∼5-fold) and IL-6 (∼11-fold) were upregulated to higher levels than in the VTN+/+ mice (Fig. 2A,C). This may represent an adaptive response to VTN deficiency. Heat inactivated (denatured) rhVTN injected into the striatum had no effect on either LIF or IL-6 mRNA expression (Fig. 2A,C), suggesting that the response to VTN is not due to an immune response against human protein. LIF (∼4-fold) and IL-6 (∼10-fold) protein expression was also induced in the striatum by VTN, relative to PBS injections, as measured by an ELISA in the contralateral side of these mice (Fig. 2B,D).
Fig. 2.
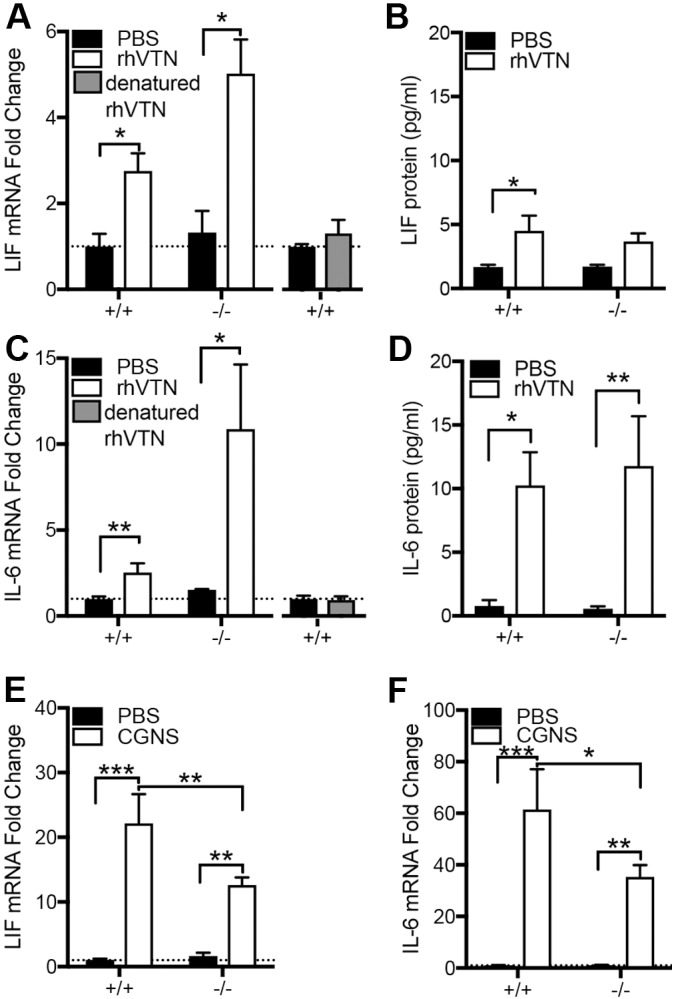
VTN regulates LIF and IL-6 expression in mouse brain. Injection of 1 µg rhVTN into the striatum of adult VTN+/+ or VTN−/− littermate mice leads to increased LIF mRNA (A), with no induction observed with heat denatured rhVTN (B). (C) rhVTN also induced IL-6 mRNA expression at 24 h relative to that seen upon PBS injections (VTN+/+ mice: PBS n=7, VTN n=7; VTN−/− mice: PBS, n=5, VTN, n=5) with no difference seen when using heat denatured rhVTN (D). Total LIF (E) and IL-6 (F) protein were induced by rhVTN injection into the striatum of the same mice as measured by ELISA. Hemorrhagic leakage induced through collagenase injection into the striatum caused a greater increase in LIF (G) and IL-6 (H) mRNA in VTN+/+ than in VTN−/− littermate mice after 24 h (n=6–7/group). Data are means±s.e.m. *P<0.05, **P<0.01, ***P<0.001.
To test the effects of leaked endogenous VTN from the blood, we induced a hemorrhage by injecting collagenase into the striatum of adult VTN mice. After 24 h, LIF expression was increased ∼22-fold relative to PBS control injections in VTN+/+ mice but only by half as much in VTN−/− littermates (Fig. 2E). IL-6 expression was increased ∼62-fold in VTN+/+ mice but by a lower amount (∼35-fold) in VTN−/− littermates (Fig. 2F). This confirms that endogenous VTN leakage into the brain contributed to increases in LIF and IL6, and that the rhVTN data are not an artefact of or an immune response to human protein.
VTN induces LIF and IL-6 expression through integrins and uPAR
To test whether VTN acts through integrins, C6 cells were co-incubated with a broad-spectrum inhibitor (RGDS) of RGD-dependent integrin ligands and a more potent integrin (P11) non-RGD-dependent blocking peptide (HSDVHK), thought to be αvβ3 specific (Choi et al., 2010, but see Fig. 6B,C). RGDS attenuated VTN-induced LIF and IL-6 mRNA expression at 4 h relative to control RAD peptide (Arg-Ala-Asp-D-Phe-Val) peptide (Fig. 3A,B), and to the same extent as P11. No cell detachment was observed after 4 h of treatment with P11 or RGDS. Expression of LIF and IL-6 was still high relative to that seen without VTN, suggesting that non-integrin mechanisms contributed to the VTN-mediated effects. VTN is known to bind to uPAR as well as affecting uPA binding to trigger ligand-independent integrin signaling via FAK (Madsen et al., 2007). The uPAR inhibitor BC-11, which functions through blocking the uPA N-terminal binding to uPAR (Longo et al., 2015; Magnussen et al., 2014) attenuated VTN-induced LIF and IL-6 induction in C6 cells, but had no effect by itself (Fig. 3C,D). Co-incubation of both P11 and BC-11 did not further decrease the VTN-mediated LIF and IL-6 induction (Fig. 3C,D), suggesting that both inhibitors function by inhibiting a common downstream pathway. Knockdown of uPAR in C6 cells significantly attenuated the VTN-mediated expression of LIF and IL-6 (Fig. 3E,F), while uPAR knockdown was confirmed by reverse transcription real-time quantitative PCR (RT-qPCR) (Fig. 3G).
Fig. 6.

FAK, but not PYK2, regulates LIF and IL-6 expression in human and mouse brain endothelial cells. Human CMEC cells were passaged and resuspended in serum-free medium and maintained in suspension for 1 h. Cells were then plated onto plastic (PBS; Ctrl), or VTN-, laminin- (LN) or fibronectin (FN1)-treated plates for 1 h. (A) VTN caused a much greater increase in pFAK Y397 and STAT3 Y705 over laminin and fibronectin. CMEC cells were pre-incubated with integrin β3- or β5-blocking antibodies before plating onto VTN. At 4 h after plating, induction of LIF (B) and IL-6 (C) by VTN were reduced when CMEC cells were pre-incubated with P11, β3- or β5-blocking antibodies (mean±s.e.m.; n=4–6, ANOVA with Fisher LSD test). To determine the role and specifity of FAK in regulating baseline LIF and IL-6 mRNA expression, we performed knockdown of FAK (siFAK) or PYK2 (siPYK2) using siRNAs in human CMEC cells over 6 days. siFAK reduced LIF (A) and IL-6 (B), while increasing CNTF (C) gene expression relative to non-targeting control siRNA. siPYK2 had no effect. In bEnd5 cells, siFAK also diminished both LIF (D) and IL-6 (E) expression while increasing CNTF (F) (mean±s.e.m.; n=3). Pharmacological FAK inhibition for 4 h with PF573228 (PF228) also suppressed LIF (G) and IL-6 (H) while upregulating CNTF (I) mRNA expression relative to vehicle-treated controls (Ctrl), as measured by RT-qPCR (n=4). Data are means±s.e.m. from n=4 independent experiments each. *P<0.05, **P<0.01, ***P<0.001; NS, not significant.
Fig. 3.
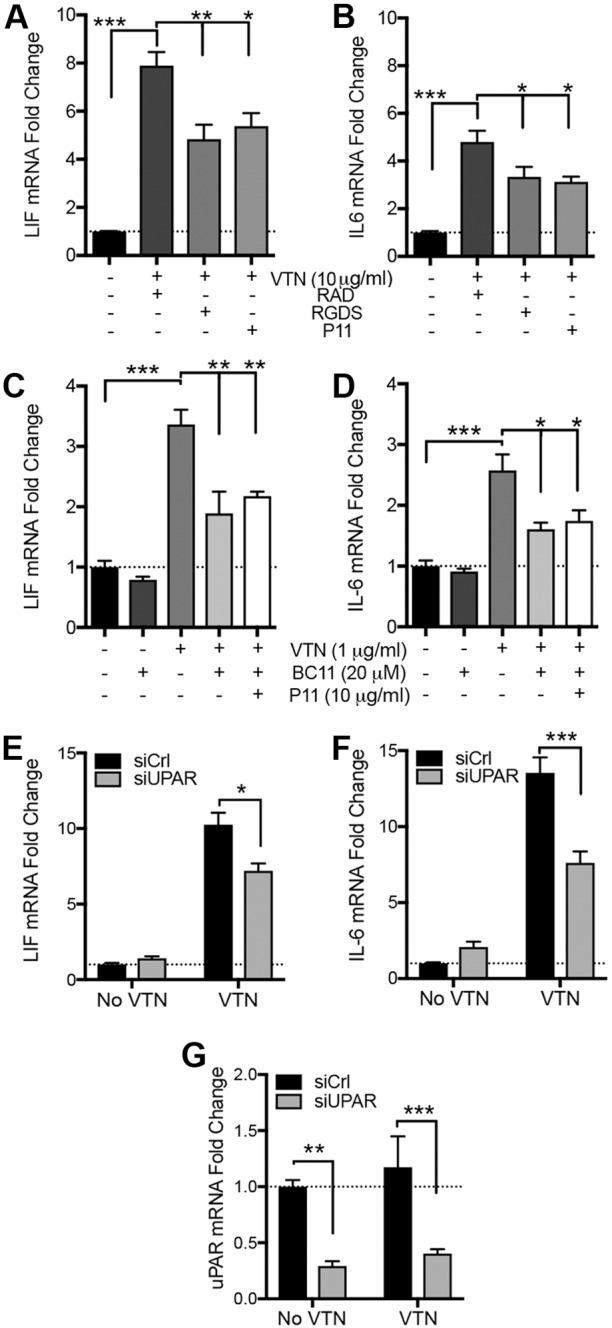
Integrins mediate VTN-induced LIF and IL-6 in C6 cells. C6 cells were seeded onto plastic for 24 h in serum-containing medium, then serum starved for 24 h before addition of VTN to the medium for 4 h. Co-treatment with RGD or αvβ3 (P11) integrin-blocking peptides reduced the effects of VTN on LIF (A) and IL-6 (B) mRNA expression. RAD is a non-RGD control peptide. LIF and IL-6 expression were not completely abolished in these experiments, suggesting that there is an additional VTN-activated mechanism. Indeed, BC-11, a uPA–uPAR inhibitor, decreased VTN-induced LIF (C) and IL-6 (D) mRNA expression, but did not have any effect alone, after 4 h. Data are means±s.e.m. of four independent experiments. Since 1 µg/ml VTN was effective in inducing LIF and IL-6, we only chose this dosage for these experiments. Co-incubation of both P11 and BC-11 could not further decrease the VTN-mediated LIF and IL-6 induction. Finally, knockdown of uPAR by means of siRNA (siUPAR) reduced by LIF (E) and IL-6 (F) induction by VTN (10 µg/ml). uPAR knockdown was confirmed by RT-qPCR (G, n=3). *P<0.05, **P<0.01, ***P<0.001.
VTN- or injury-mediated LIF and IL-6 expression is blocked by FAK inhibition in vitro
FAK is a major transducer of integrin signaling (Mitra et al., 2005). C6 cells were seeded onto plates pre-coated with VTN or plastic only (controls). VTN activated FAK (as judged by measuring Y397 phosphorylation, denoted pFAK-Y397) but not the closely related PYK2 (also known as PTK2B) at Y402, in C6 cells 4 h after seeding (Fig. 4A). The FAK inhibitor PF573228 added to the culture medium immediately after seeding completely blocked VTN-induced FAK phosphorylation (Fig. 4B), and also completely abolished the induction of LIF and IL-6 (Fig. 4C,D). Plating C6 cells onto culture plates pre-coated with rhVTN led to a similar induction of LIF and IL-6 to that seen when proteins were added directly to medium (Figs 1D,E and 4C,D). In separate experiments, other FAK inhibitors PF562271 and PND-1186, but surprisingly not Y11, also decreased pFAK-Y397 (Fig. S1A) and dose-dependently reduced baseline (without VTN) LIF and IL-6 expression after 4 h (Fig. S1B,C). We also found that FAK inhibition suppresses VTN-induced LIF and IL-6 expression when VTN was added directly to medium (Fig. 4E,F). Together, this shows that FAK signaling is a major regulator of VTN-mediated LIF and IL6 expression. Integrin blockade by P11, β3- or β5-blocking antibodies did not attenuate this induction (data not shown), suggesting that other mechanisms play a role.
Fig. 4.
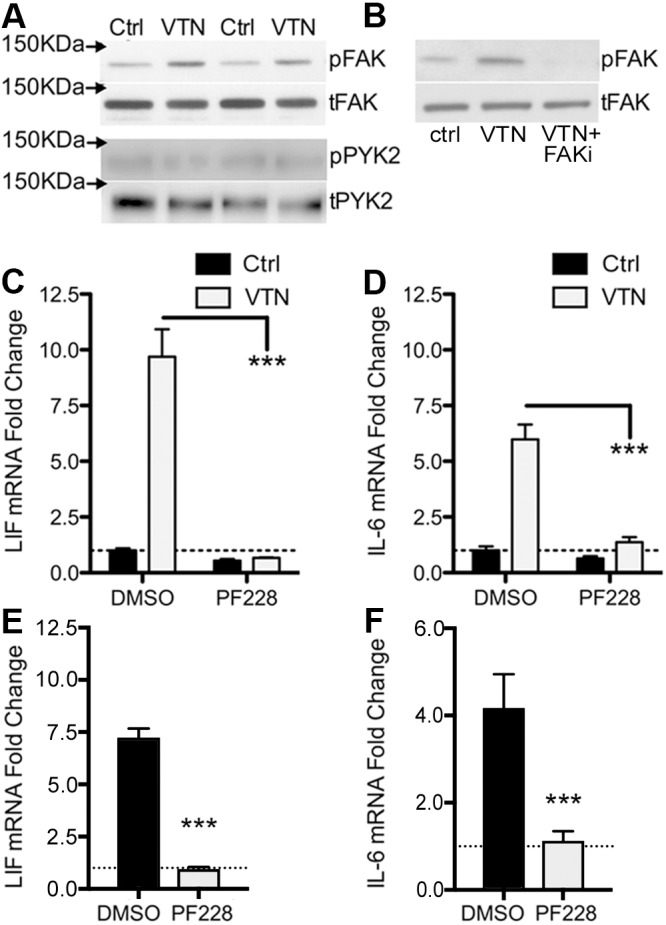
FAK inhibition abolishes VTN mediated LIF and Il-6 induction. C6 cells were seeded onto VTN-coated tissue culture plates (50 µg/ml) or non-treated control (Ctrl) plates and maintained for 4 h. (A) In these conditions, VTN stimulates phosphorylation of Y397-FAK (pFAK) but not of PYK2 at the corresponding Y402 residue (pPYK2), as shown in western blots. Loading controls are total FAK (tFAK) and PYK2 (tPYK2). Two representative lanes per group from three independent experiments are shown (n=3). (B) FAK antagonist PF573228 (10 µM, FAKi) completely blocked the VTN-stimulated pY397-FAK formation when added immediately after cells were seeded (n=3). In addition, the FAK inhibitor PF573228 (PF228) completely abolished VTN-induced LIF (C) and IL-6 (D) gene expression as measured by RT-qPCR. Control (Ctrl) vehicle is 0.1% DMSO (mean±s.e.m.; n=4 independent experiments). In further experiments, C6 cells were cultured in serum-free medium for 24 h before VTN was added directly to medium with or without FAK inhibition (PF228), (E) LIF and IL-6 (F) induction by VTN was also completely abolished by FAK inhibition in these conditions (mean±s.e.m.; n=3).
Both LIF and IL-6 are activated after injury to the brain (Banner et al., 1997; Lau and Yu, 2001), and we therefore sought to define the role of FAK in their injury-induced upregulation in vitro. C6 cells were seeded onto tissue culture plates (no coating) and maintained for 48 h before monolayers were removed and mechanically dissociated (swipe injury). Cells were allowed to re-adhere for 4 h in 10% (v/v) serum-containing medium, before RNA isolation. The injury caused ∼50% cell loss by 2 and 6 h as measured by MTT and Trypan Blue counts (Fig. S2A,B). LIF and IL-6 mRNA expression were increased ∼4- and 8-fold, respectively, in surviving cells relative to uninjured controls (Fig. 5A,B). Incubation of the validated FAK inhibitors PF573228, PND-1186 or PF562271 (Fig. S1) during the 4 h recovery abolished the injury-induced upregulation of LIF and IL-6 mRNA expression (Fig. 5A,B). The FAK inhibitor Y11, which had no effect on pY397-FAK (Fig. S1A) or LIF and IL-6 mRNA expression (Fig. S1B,C) in uninjured C6 cells, did not alter injury-induced LIF expression (Fig. 5A). Surprisingly, Y11 increased IL-6 expression above that caused by injury, suggesting that it was biologically active but had off-target effects (Fig. 5B). LIF and IL-6 proteins released into culture medium were induced by injury and were suppressed by FAK inhibition, as shown by dot blot assays (Fig. 5C,D). The larger increases seen in dot blots relative to mRNA induction could be explained by an accumulative effect of stable protein in the medium, while LIF and IL-6 mRNA are being actively turned over. LIF and IL-6 mRNA half-life is typically 30–45 min (Derigs and Boswell, 1993; Iwasaki et al., 2011). Also, it is possible that the uninjured controls retain much more of the IL-6 within the cytoplasm. The effects of the FAK inhibitors was not due to cell loss as they did not appear to affect cell viability after injury (data not shown). We also confirmed the importance of FAK Y397 phosphorylation, which is key to FAK activation (Sieg et al., 2000), as transfection of mutated functionally dead FAK Y397F plasmids into C6 cells significantly reduced LIF (Fig. 5E) and IL-6 (Fig. 5F) mRNA levels at 4 h following cell injury.
Fig. 5.

FAK mediates injury-induced LIF and IL-6 induction in C6 cells. C6 cells were seeded and maintained for 48 h in serum-containing medium without added VTN. Cells were then injured in an in vitro trauma model (swipe injury) with or without FAK inhibitors added at the time of injury. LIF (A) and IL-6 (B) mRNA expression were strongly induced (Ctrl Inj) at 4 h after injury compared to no injury controls (Ctrl NI), but were abolished by treatment with FAK antagonists, PND-1186 (PND), PF573228 (PF228), PF562271 (PF271), but not Y11. Surprisingly, Y11 further increased IL-6 expression after injury. Data are means±s.e.m. of three independent experiments and expressed as a fold change relative to uninjured controls, first normalized to GAPDH to account for differences in cell numbers. *P<0.05; **P<0.01; ***P<0.001; NS, not significant. In the same conditions as described for A and B, LIF (C) and IL-6 (D) protein expression and release was increased after injury, and attenuated by all inhibitors, except Y11, as shown by dot blots of conditioned medium (representative of three independent experiments). In addition, we found that C6 cells overexpressing wild-type (WT) FAK expressed significantly more LIF (E) and IL-6 (F) than cells overexpressing dominant negative Y397F mutant FAK following injury (mean±s.e.m.; n=3).
FAK also regulates LIF and IL-6 expression in endothelial cells
To test whether FAK had a similar role in other cell types, we chose human (hCMEC) and mouse (bEnd5) brain endothelial cell lines. They were chosen due to their importance, together with microglia and astrocytes, in the acute response of the neurovascular unit to insults (Hawkins, 2005). Furthermore, IL-6 is highly expressed by endothelial cells (Gertz et al., 2012) as well as in our cultures, making it easier to test reductions in baseline expression levels after FAK inhibition than in C6 cells. We first tested endothelial cell responsiveness to ECM proteins. After passaging, cells were re-suspended in serum-free medium and maintained in suspension for 1 h, before being seeded onto culture plates coated with vehicle (PBS), VTN, laminin-111 or fibronectin. Western blot analysis showed that there was a marked increase in pFAK-Y397 in cells maintained on VTN over control (vehicle), laminin-111 or fibronectin (Fig. 6A, n=3). Intriguingly, phosphorylation of STAT3 Y705, which positively regulates IL-6 production (Peruzzi et al., 2012), was also increased (Fig. 6A) in VTN-treated cells. To confirm the role of integrins in VTN-induced LIF and IL-6, we used function-blocking integrin antibodies on CMEC cells plated onto VTN. P11 peptide and anti-β5 antibody reduced the effects of VTN on LIF expression (P<0.05, n=4–6) with the anti-β3 antibody block not being significant (P=0.08, Fig. 6B). VTN-induced IL-6 expression was blocked by P11, and antibodies against β3 and β5 (P<0.05, n=4–6, Fig. 6C). In addition, we tested the specificity of P11 by co-incubation with the β3- and β5-blocking antibodies. No further effect was found on LIF and IL-6, suggesting that P11 acts through β3 and β5 integrins.
Some FAK antagonists reportedly also inhibit PYK2 (Slack-Davis et al., 2007). Therefore, we performed siRNA knockdown of FAK and PYK2 in CMEC cells over 6 days, with specificity confirmed by qPCR (Fig. S3A,B) and western blotting (Fig. S3C). ILK, another integrin signaling mediator, was not altered (Fig. S3C). Baseline LIF and IL-6 expression were decreased to ∼70% in FAK-knockdown CMECs relative to non-targeting siRNA controls (Fig. 6D,E). Cells were maintained in 5% serum as it can induce these cytokines (Fig. 1A,B). Knockdown of PYK2 had no effect (Fig. 6D,E), revealing the specificity of FAK signaling in LIF and IL-6 regulation. CNTF mRNA expression was increased upon treatment with siRNA against FAK (siFAK) but not against PYK2 (siPYK2) (Fig. 6F), consistent with our previous findings that FAK inhibition induces CNTF (Keasey et al., 2013).
We also tested for FAK specificity in the bEnd5 mouse endothelial cells. Targeted knockdown of FAK with siRNA was confirmed by decreased total FAK protein (Fig. S3D), with no effect on ILK expression (Fig. S3E). siFAK reduced LIF and IL-6 mRNA expression to ∼60% of controls (Fig. 6G,H) but increased CNTF by ∼30% (Fig. 6I). Pharmacological FAK inhibition in bEnd5 cells cultured in 10% serum with PF573228 for 4 h suppressed LIF and IL-6 expression to ∼25% and ∼50% that of controls, respectively (Fig. 6J,K). CNTF mRNA was dramatically induced by PF573228 in the same cells (Fig. 6L). The changes in LIF and IL-6 after the 6-day siRNA treatment were smaller than that seen with the 4 h treatment with the pharmacological inhibitor, perhaps due to adaptive responses independent of FAK signaling.
FAK antagonists suppress LIF and IL-6 specifically through inhibition of FAK
To further confirm the specificity of the FAK antagonists, siFAK pre-treated CMEC cells were incubated with PF573228 or PND-1186. PF573228 has been used successfully in vitro and in vivo (Keasey et al., 2013) PND-1186 suppressed LIF expression at lower concentrations (Fig. S1B) and were selected for these experiments. Quantitative capillary western blots confirmed that total FAK protein (Fig. 7A–C) and pFAK-Y397 (Fig. 7D–F) were reduced by siFAK (DMSO vehicle) but, as expected, were not further reduced by PF573228 or PND-1186. The reduction in LIF and IL-6 mRNA expression caused by PF573228 and PND-1186 was not significantly different when inhibitors are combined with a non-targeting siRNA control or siFAK, (Fig. 7G,H), suggesting that these inhibitors acted specifically through FAK and did not have off-target effects.
Fig. 7.

Pharmacological FAK inhibitors are specific in suppressing LIF and IL-6 in CMEC cells. To confirm the specificity of PF573228 (PF228) and PND-1186 (PND), FAK was first knocked down in CMECs with siRNA for 5 days followed by 4 h inhibitor treatments. Total FAK (tFAK) and pFAK protein knockdown was quantified by capillary western blots (A). For illustration purposes, synthetic bands were produced from chemiluminescence spectrograms (B). TUB, α-tubulin. (C) Quantification showing that siFAK decreases total FAK normalized to α-tubulin, which is not affected by the inhibitors PF228 or PND. (D–F) Similarly, siFAK decreased pY397-FAK levels, with the PF228 and PND decreasing pFAK only under control siRNA conditions (siCtrl). Critically, LIF (G) and IL-6 (H) gene expression (RT-qPCR) were not significantly different when the inhibitors were incubated with siCtrl or siFAK, and provided no additive suppression in the presence of siFAK, showing that their effects were entirely mediated through FAK. Data are means±s.e.m. of four independent experiments and presented relative to DMSO- and control (siCtrl)-treated cells. *P<0.05; **P<0.01; NS, not significant.
DISCUSSION
Our data has identified a VTN–integrin–FAK signaling pathway that rapidly and robustly induces expression of LIF and IL-6 in vitro and in adult mice after a cerebrovascular injury (Fig. 8A,B). This is consistent with the finding that FAK can mediate TNFα-induced IL-6 expression in cancer cells and myoblasts (Schlaepfer et al., 2007). Prior to this study, gp130–JAK–STAT3, p38 MAPK and nuclear factor (NF)-κB signaling was well known to regulate IL-6, which can also regulate its own expression (Kang et al., 2013). The finding that ligand–integrin binding induces LIF and IL-6 expression in concert with suppression of CNTF is novel. Others have reported that α2 laminins induce IL-6 (Delimont et al., 2014) but did not study involvement of integrins. Regulation of LIF through integrins or FAK was previously unknown. In the absence of LIF, laminin-111 and -511 (laminin-511 is composed of α5, β1 and γ1 chains) promote stem cell renewal in vitro through α6β1 integrin (Cattavarayane et al., 2015). This might involve induction of LIF, since it is widely used to maintain self-renewal. However, here, LIF was not affected by laminin-111.
Fig. 8.

Blood VTN leaks into the brain after stroke. (A) Schematic showing an overview of the signaling pathway by which VTN induces LIF and IL-6 gene expression, by binding to αvβ3 integrin and specifically activating downstream FAK. (B) Proposed model of VTN leakage after blood–brain barrier (BBB) breakdown. Under normal conditions, the intact BBB, characterized by tight junctions (TJ) between endothelial cells (EC), keeps VTN in blood from entering the central nervous system (CNS) tissue. BM, basement membrane. Under pathological conditions that cause BBB breakdown, such as stroke and hemorrhage, leakage of VTN in the brain parenchyma induces LIF and IL-6 expression by astrocytes (gray cells), microglia (blue cells) and endothelial cells (green cells).
Our results suggest that FAK is unique in regulating LIF and IL-6 compared to other ECM–integrin signaling transducers such as the highly related PYK2 (Mitra et al., 2005), which can also be activated by αvβ3 integrin (Butler and Blystone, 2005). The potential specificity of FAK would make it a good therapeutic target, for example, to downregulate LIF and/or IL-6. In fact, we have previously shown that systemic FAK inhibitor treatment promotes adult neurogenesis in mice (Keasey et al., 2013). Here, FAK inhibitors could completely attenuate injury-induced LIF and IL-6 in C6 glioma cells. FAK inhibitors also reduced baseline LIF and IL-6 in endothelial cells, suggesting that FAK is a major signal transducer for LIF and IL-6 signaling. Integrin blockade did not reduce cytokine induction, possibly due to the mechanical trauma and stretching of cells, which is known to activate pFAK Y397 (Delimont et al., 2014; Tornatore et al., 2011). Together, our data suggest that FAK inhibition would be beneficial in inflammatory diseases where IL-6 plays a major role, such as rheumatoid arthritis, diabetes, cancer and neurodegenerative diseases (Mauer et al., 2015; Rothaug et al., 2016). FAK inhibitors have been and are in clinical Phase I and II trials for solid tumors (www.clinicaltrials.gov) and seem to be well tolerated after systemic administration.
The rapid and potent effects of VTN in cultured C6 cells were mediated by αvβ3 and/or αvβ5 integrins, and by downstream FAK, as shown by pharmacological inhibitors and siRNA knockdown. Integrin αvβ3 is expressed by aorta endothelial cells where it mediates increased FAK phosphorylation and downstream ERK and JNK activation upon shear stress (Li et al., 1997). This is consistent with our findings showing that FAK signaling induces LIF and IL-6 in two types of brain endothelial cells. Others have shown that treatment with another αvβ3-blocking peptide reduces FAK phosphorylation and infarct volume after ischemic cerebral stroke in rats (Shimamura et al., 2006). It is possible that this was in part due to a reduction in LIF and IL-6, as suggested by our findings that P11 peptide, a reported antagonist of αvβ3 (Bang et al., 2011), and antibodies against β3 suppress LIF and IL-6 expression. Our data suggest that P11 acts both on β3 or β5 integrins because no additive effect of the antibodies was seen in reducing LIF or IL-6. The αvβ5 integrin likely also plays a role, as antibodies to the β5 subunit, which exists only in αvβ5 integrin, were similarly effective to β3 inhibition in blocking VTN-induced LIF and IL-6 expression. IL-6 is well known to promote inflammation after spinal cord injury (Lacroix et al., 2002). LIF mediates neutrophil activation and macrophage recruitment in culture (Borish et al., 1986; Tofaris et al., 2002), and microglia and macrophage activation in the injured spinal cord (Kerr and Patterson, 2004). If substantiated in animal models, such specificity may provide opportunities for more selective therapies in regulating detrimental inflammation.
In vitro, VTN was unique among the other ECM molecules, fibronectin, fibrinogen, laminin and collagen, in its ability to activate LIF and IL-6. This is consistent with VTN, but not fibronectin or laminin, promoting the formation of pFAK-Y397 in CMEC cells. This was surprising because fibrinogen and fibronectin also bind to αvβ3 integrin via RGD domains (Charo et al., 1990). Also, both VTN and fibronectin activate microglia (Milner et al., 2007) and support endothelial cell survival via α5β1 and αvβ3 integrins (Wang and Milner, 2006). Fibronectin enhances IL-1-mediated induction of IL-6 in vitro (Ostberg et al., 1995), but its effect on IL-6 alone has not been tested. Differential activation through the same integrins has been found, for example, fibronectin or osteopontin, but not VTN, activate FAK in osteoblast cells (Liu et al., 1997). Also, chronic exposure to VTN leads to greater upregulation of pro-MMP9 than fibronectin (Milner et al., 2007). Our integrin-blocking experiments suggested that there is an additional non-integrin mechanism mediating the effects of VTN, perhaps not shared by fibronectin or fibrinogen. VTN can be cleaved to produce a peptide that binds to the uPAR (Wei et al., 1994), which can boost VTN-mediated cell motility via interaction with αvβ3 (Degryse et al., 2005). In addition, uPAR has been implicated in FAK activation together with α5β1 integrin (Aguirre Ghiso, 2002). Indeed, here we found that pharmacological and siRNA uPAR inhibition reduced VTN-mediated LIF and IL-6 expression in vitro. However, it remains to be determined whether the unique effects of VTN compared to the other ECM molecules is due to its ability to bind uPAR. Inhibition of both αvβ3 integrin and uPAR did not attenuate VTN-induced LIF and IL-6 more than either alone, suggesting a common mechanism may exist in their signaling pathways. FAK inhibition alone completely blocked VTN-mediated LIF and IL-6 induction, suggesting that both mechanisms are dependent on FAK signaling. Others have shown that uPAR leads to integrin and FAK activation (Aguirre Ghiso, 2002). The possibility of uPAR mediating LIF and IL-6 expression through FAK is a new idea. The mechanism that uPAR uses to achieve this is likely associated with integrins, given that P11 did not further suppress VTN-mediated LIF and IL-6 induction in the presence of BC-11. The interaction between uPAR and integrins would be an intriguing area for further study. It remains to be determined how unique VTN is compared to other ECM molecules in its potential to regulate inflammatory signalling through integrin receptors in vivo. There probably are additional ECM proteins that regulate cytokine expression, including α2 laminin, which induces IL-6 in kidney cells (Delimont et al., 2014), while α1 laminin does not, consistent with our finding that laminin-111 was without effects. It also remains to be determined how VTN, fibronectin and laminin-111 can downregulate CNTF in C6 astroglioma cells (Keasey et al., 2013), while only VTN induces LIF and IL-6, possibly providing opportunities to develop pharmacological approaches for differential cytokine regulation.
Collectively, our data suggest that VTN leaks into the brain after trauma and induces LIF and IL-6 expression. It is perhaps not surprising that major blood proteins should trigger inflammation and possibly also subsequent repair. Consistent with our results is the finding that lipopolysaccharide (LPS) injections into lungs of mice lead to upregulation of VTN, which induces IL-1β, MIP-2 (also known as CXCL2) and IL-6 expression, as shown in VTN−/− mice (Tsuruta et al., 2007). Our data suggest that other leaked blood proteins could also play a pro-inflammatory role because hemorrhage in VTN−/− mice and plasma from VTN−/− mice in vitro induces about half as much LIF and IL-6 as was the case for the wild-type mice or plasma, respectively. This suggests that VTN is important but not the exclusive molecule that regulates LIF and IL-6 cytokine expression, and that other blood-derived molecules involved in this process remain to be identified. VTN and fibronectin leak into the brain after stroke (del Zoppo et al., 2012) or demyelinating injuries (Milner et al., 2007), where they have detrimental effects, and can activate microglia (Milner and Campbell, 2003). Here, fibronectin did not induce LIF or IL-6 in vitro, raising the possibility that leaked blood fibronectin regulates inflammation through different signaling mechanisms from VTN. VTN probably also contributes to induction of repair processes because VTN−/− mice have delayed and poor wound healing and angiogenesis (Jang et al., 2000), as well as persistent blood leakiness after injury (Li et al., 2012). Such defects are also seen in IL-6−/− mice (Gertz et al., 2012; Lin, 2003), probably because IL-6 is important for angiogenesis (Gertz et al., 2012).
Astrocytes, microglia, infiltrating macrophages and endothelial cells probably respond directly to VTN leakage to produce LIF and IL-6. All of these cells express αvβ3 integrin (Herrera-Molina et al., 2012; Milner, 2009; Wang and Milner, 2006), and can produce LIF and IL-6 after injury (Ishibashi et al., 2006; Van Wagoner and Benveniste, 1999). The astrocytes and microglia are in a prime location around microvessels to act as first-responders upon blood–brain barrier disruption (Fig. 8B). The luminal side of endothelial cells is always exposed to VTN but αvβ3 integrin–FAK clusters may be predominantly in an abluminal localization in endothelial cells (Li et al., 1997). LIF is a chemoattractant to macrophages (Tofaris et al., 2002) and their recruitment to ischemic injuries is reduced in VTN−/− mice (Li et al., 2012). This suggests that VTN-induced LIF attracts macrophages into injured tissue where they can respond to VTN to produce IL-6 (Antonov et al., 2011).
Compared to IL-6, much less is known about LIF gene regulation. LIF mRNA is increased by cAMP and MEK signaling in Schwann cells, astrocytes and non-neuronal lineages (Banner and Patterson, 1994; Nagamoto-Combs et al., 1999), as well as by TGFβ1 through PKCβ (Matsuoka et al., 1997). MEK signaling appears to be downstream of FAK in astrocytes (Schlaepfer et al., 2007), while PKCβ plays a role in regulating focal adhesion turnover in keratinocytes (Vandenberghe et al., 2013). Fibronectin promotes stem cell differentiation through increased FAK activation and focal adhesion formation (Hayashi et al., 2007). However, the role of FAK in regulating LIF expression had not been previously reported. This new insight may provide additional tools to influence biological processes where LIF plays an important role, including stem cell behavior (Bauer and Patterson, 2006), inflammatory processes (Kerr and Patterson, 2004; Sugiura et al., 2000) or trophoblast implantation (Vogiagis and Salamonsen, 1999).
FAK inhibition induced CNTF in vitro, as we reported previously (Keasey et al., 2013), while reducing LIF and IL-6 expression. This opposing effect seems consistent with the findings that endogenous CNTF is anti-inflammatory (Linker et al., 2002), and that LIF and CNTF have opposite effects on neural stem cell self-renewal and neurogenesis (Bauer and Patterson, 2006; Yang et al., 2008). It is conceivable that FAK signaling differentially regulates these cytokines to avoid contrasting gp130 signaling and biological outcomes. The involvement of cAMP in LIF expression in cultured astrocytes (Murphy et al., 1995) may explain why LIF and CNTF are differentially regulated, where cAMP downregulates CNTF (Yang et al., 2008), and may be downstream of integrin–FAK signaling.
In conclusion, we propose that leaked VTN plays a major and specific role in acutely and robustly activating cerebral and, possibly, other inflammatory processes. This provides new insight into potential pathophysiological mechanisms caused by the loss of vascular integrity that occurs in a variety of disorders. Furthermore, integrin–FAK signaling is a novel therapeutic target for regulating the inflammatory response following injury or disease, with pharmacological FAK inhibition representing a potent tool for attenuating inflammatory cytokine induction. We suggest that plasma VTN might act as a flagging molecule, directly triggering cytokine expression in injured tissues to recruit inflammatory cells (Fig. 8).
MATERIALS AND METHODS
Cell culture
We used rat astroglioma C6 cells (Cat #CCL-107, ATCC), mouse bEnd5 endothelial cells (a gift from Dr Engelhardt, University of Bern, Switzerland) and human CMEC/D3 endothelial cells (Cat# CLU512, Cellutions Biosystems). Cells were tested for contamination by fluorescent staining with DAPI. C6 cells were maintained in high serum (HS) medium [Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine and 1% penicillin-streptomycin] or low serum (LS) medium (advanced DMEM supplemented with 1% fetal bovine serum, 2 mM L-glutamine and 1% penicillin-streptomycin) where noted. BEnd5 cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 4 mM L-glutamine, 1% penicillin-streptomycin, 1 mM sodium pyruvate and 1× non-essential amino acids. CMEC cells were grown in fetal bovine serum (5%), penicillin-streptomycin (5%), hydrocortisone (1.4 μM, Sigma, Cat# H0135), ascorbic acid (5 μg/ml), chemically defined lipid concentrate (1%, Invitrogen Cat# 11905031), HEPES (10 mM), basic fibroblast growth factor (1 ng/ml, Sigma, Cat# F0291) in endothelial basal media-2 (Lonza, Cat# 00190860). Cells were maintained in a humidified CO2 (5%) incubator at 37°C. C6 cells were plated at 160,000 per ml, and bEnd5 and CMEC cells at 100,000 per ml. All cells were maintained for 24 h after plating before commencing experiments.
Cell cultures with ECM proteins and integrin blockade
C6 cells were serum-deprived for 24 h. Recombinant human VTN (1 or 10 µg/ml, Cat# SRP3186), fibronectin (10 µg/ml, Cat# F3667), fibrinogen (50 μg/ml, #F3879), collagen-I (10 µg/ml, Cat# C5533), or laminin-111 (EHS murine sarcoma basement membrane, 10 µg/ml, Cat# L2020), all from Sigma, were re-suspended in sterile water and added directly to the medium. Alternatively, cells treated with VTN (10 µg/ml) were co-incubated with RGDS (10 µg/ml, Cat# 3498, Tocris), αvβ3 peptide antagonist (P11, HSDVHK, 10 µg/ml, Cat# 4744, Tocris; Choi et al., 2010) or control RAD peptides (Arg-Ala-Asp-D-Phe-Val,10 µg/ml, Cat# BML-AM1010-0001, ENZO). P11 has been reported to function through binding to the metal ion-dependent adhesion site (MIDAS) and adjacent to MIDAS (ADMIDAS) (Bang et al., 2011; Choi et al., 2010), reducing ligand binding and αv and β3 subunit interaction (Xiong, 2002). P11 has been demonstrated to act with high potency on the αvβ3 integrin receptor with an IC50 of 1.74 pg/ml (Lee et al., 2004). Its specificity for αvβ3 has been demonstrated over α5β1 and α3β1 (Choi et al., 2010). After 4 h, total RNA or protein was isolated for RT-qPCR or western blotting, respectively. CMEC cells were passaged and resuspended in regular culture medium without serum (0% FBS) and maintained for 1 h in suspension (at 500,000/ml) using non-tissue culture-treated plates. Cells were then transferred to tissue culture plates pre-coated with vehicle (PBS), VTN, laminin or fibronectin (50 µg/ml, Keasey et al., 2013). Cells were lysed after 1 h and protein collected for analyses. Integrin-blocking antibodies used were against β3 (Biolegend, Cat# 304414, 10 µg/ml) and β5 (Ebioscience, Cat# 14-0497-82, 10 µg/ml) and added to the suspended CMEC cells. The cell suspension was then transferred to VTN-coated plates for 4 h before RNA isolation. C6 cells were seeded directly onto precoated plates without the 1 h suspension used for the CMEC cells and maintained with or without PF573228 (FAKi) added at the same time as seeding for 4 h.
In vitro drug treatments
Cells were incubated with pharmacological antagonists of FAK for 4 h before RNA or protein isolation. FAK antagonists were PF573228 (Cat# 3239, Tocris), PF562271 (Cat# S2890, Selleck), PND-1186 (Cat# S7653, Selleck) and Y11 (Cat# 4498, Tocris), used at concentrations between 0.1 µM to 10 µM as noted. BC-11 was used at 20 µM (Cat# 4372, Tocris). It has been shown that the N-terminal fragments of uPA preadsorbed with BC-11 limit the growth-suppressing activity of BC-11 (Longo et al., 2015). In addition, incubating high molecular mass uPA with plasminogen reduces plasmin formation, as determined by plasminogen zymography (Magnussen et al., 2014). This suggests that BC-11 functions through blockade of the enzymatic activity of uPA which may occur in addition to blockade of the binding uPA N-terminal fragments to uPAR. More recently, BC-11 was shown to reduce uPAR cleavage by uPA in a similar fashion to that seen for the serine protease inhibitor aprotinin (Magnussen et al., 2017). Drugs were dissolved in up to 0.1% DMSO of final volume in culture medium (vehicle).
Transfections
siRNAs against human FAK (PTK2, Cat# L-003164) or PYK2 (PTK2B, Cat# L-003165) or a non-targeting negative control (Cat# D-001810-10), all from Dharmacon, were transfected into CMEC cells 24 h after plating (200,000 cells/well) on six-well plates at 50 nM siRNA and 0.5% Lipofectamine-2000 (Cat#11668-019, Invitrogen). A second transfection was performed at 48 h due to FAK's long protein half-life (Ochel et al., 1999). Cells were maintained for a further 5 days before analysis or treatment with FAK antagonists. Transfection efficiency was ∼60–80% as visualized by performing fluorescent microscopy after co-transfection with siGLO RNA (Cat# D-001630-02, Dharmacon). Knockdown of uPAR in rat C6 glioma cells was performed with siRNA (Cat# L-1000500, Dharmacon, 50 nM). Transfection was performed with Lipofectamine-3000 [(0.25%) Cat# L3000015, Invitrogen] according to the manufacturer's instructions. Experiments were performed at ∼36 h post transfection.
FAK-Y397F mutant transfections
Wild-type pGFP-FAK (Addgene plasmid #50515) and pGFP-FAK Y397F (Addgene plasmid #50516) were obtained from Addgene and were both deposited by Kenneth Yamada (Gu et al., 1999). Transfections using Lipofectamine 3000 were performed according to the manufacturer's protocol, at a final concentration of 0.25% with 1 µg of plasmid DNA in a single well of a six-well plate. Western blotting with anti-phospho-Y397 antibody was used to confirm plasmids were expressed the wild-type and Y397F protein.
In vitro swipe injury model
C6 cells were plated at 320,000 cells/ml in 6-well plates with 10% fetal bovine serum medium, and grown to confluency for 48 h. No exogenous ECM protein was used in this assay. In the same growth medium, cells were removed by scraping the bottom of the well with an inverted sterile p1000 pipette tip and then triturated 10× with a p1000 pipette to mechanically dissociate them. Afterward, the cells were allowed to re-adhere in the same wells, and maintained for 2–6 h to assess cell viability or 4 h for RNA isolation. Cell survival was assessed by use of MTT (50 µg/ml, Cat# M6494, Invitrogen) added to culture medium for 2 h, before cells were washed with ice-cold PBS. Resulting formazan was dissolved with 500 µl acidified isopropanol, which was transferred to 96-well plates for spectrophotometric analysis at 570 nm. Trypan Blue (0.2%, Cat#15250061, Invitrogen) was added to cells dissociated with trypsin, and mixed 1:1 with culture medium to inactivate trypsin. Counts were performed using a hemocytometer.
Western blotting
Total protein was isolated from cell lysates using 1 ml of RIPA buffer supplemented with protease and phosphatase inhibitors as described (Keasey et al., 2013). Proteins were separated by SDS-PAGE and after transfer, PVDF membranes were incubated with antibodies for pY397-FAK (1:1000, Cat#3283, RRID AB_2173659), tFAK (1:1000, Cat#3285, RRID AB_2269034), pY402-PYK2 (1:1000, Cat#3291, RRID AB_2300530), PYK2 (1:1000, Cat# 3480, RRID AB_2174093), α-tubulin (1:2000; Cat#2125, RRID AB_2619646), STAT3 Y705 (1:1000, Cat#9145, RRID AB_2491009) and STAT3 (1:1000, Cat# 12640, RRID AB_2629499) all from Cell Signaling Technology, pS246-ILK (1:1000, Cat# AB1076, RRID AB_11211802, Millipore), tILK (1:1000, Cat# 04-1149, RRID AB_1977290, Millipore), followed by appropriate secondary antibodies (anti-mouse-IgG, Cat# 7076; anti-rabbit-IgG, Cat# 7074, Cell Signaling), as described previously (Keasey et al., 2013). Enhanced chemiluminescence (ECL; Cat# 34080, ThermoFisher) was used to visualize the antibody staining through use of a Li-Cor chemiluminescence imager (Li-Cor model Odyssey-FC 2800) or standard X-ray film.
Quantitative capillary ProteinSimple western blotting
Analysis of protein expression was performed according to the ProteinSimple protocol guide with reagents from the kit (Cat# PS-MK01, ProteinSimple) except where noted. Briefly, cell lysates were diluted to 0.2 µg/µl with 0.1× sample buffer supplemented with 1× fluorescent molecular mass markers and 40 mM DTT for a 5 µl reaction (1 µg protein/reaction). Samples were heated at 95°C for 5 min before loading into 24 single designated wells of a pre-filled plate along with blocking reagent, primary antibodies (1:50, 1:50 or 1:500 in antibody diluent, pFAK, FAK, α-tubulin, with pFAK and Tubulin or FAK and Tubulin mixed together for detection within the same capillary), horseradish peroxidase (HRP)-conjugated anti-rabbit-IgG secondary antibody, luminol-peroxide mix, to generate chemiluminescence, and washing buffer. Plates were loaded into the automated ProteinSimple ‘Wes’ for electrophoresis and imaged for fluorescence in real-time by means of a CCD camera for immunodetection in the capillary system at default settings (electrophoresis, 375 volts, 25 min; blocking, 5 min; primary antibody, 30 min; secondary antibody, 30 min). Data was analyzed by using Compass software (ProteinSimple) with data displayed as peak intensity or synthetic bands. Quantification was performed by normalizing areas under protein peaks to α-tubulin loading control.
Conditioned medium and dot blots
Conditioned cell culture medium (1.5 ml from one well of a six-well plate/condition) was collected from C6 cells, centrifuged at 5000 rpm for 10 min to remove cell debris and the supernatant frozen overnight at −80°C before lyophilization. The dehydrated proteins were resuspended in 500 μl ddH2O, and concentrated in protein concentration columns (Cat# UFC500324, Millipore) according to manufacturer's protocol. After centrifugation, the retained 50 μl volume was re-diluted ten-fold in fresh ddH2O to dilute salts from culture medium and then re-centrifuged. Western blotting revealed no visible bands for either LIF or IL-6. We therefore chose to run dot blots. 1 μl of the retained protein was pipetted onto a nitrocellulose membrane (Thermo Fisher, Cat# 88024) and allowed to dry at room temperature. Membranes were blocked in 5% BSA in TBS:Tween 20 (0.1%, v/v) for 1 h before overnight incubation with rabbit anti-IL-6 (1:1000, Cat# AF506, RRID AB_355398, R&D Systems), rabbit anti-LIF (1:200, Cat# SC-20087, RRID AB_2136098, Santa Cruz Biotechnology) antibodies in 5% BSA TBS:Tween. Membranes were washed 3×5 min with TBS:T then incubated with HRP-conjugated anti-rabbit-IgG or anti-mouse-IgG antibodies. Signal was visualized by ECL according to standard methods and visualized with a Li-Cor chemiluminescence imager.
RT-qPCR
Total RNA was collected from cells according to the manufacturer's protocol (Cat# 74106, Qiagen RNAeasy) and RT-qPCR was performed as described previously (Keasey et al., 2013). Briefly, 500 ng of RNA was reverse-transcribed and used at a final concentration of 20% in RT-qPCR reactions using TaqMan probes for rat C6 cells, GAPDH (Rn9999916_s1), LIF (Rn00573491_g1), IL-6 (Rn01410330_m1), CNTF (Rn00755092_m1), for human CMEC cells, GAPDH (Hs02758991_g1), LIF (Hs01055668_m1), IL-6 (Hs00174360_m1), CNTF (Hs00173456_m1), for mouse bEnd5 cells intracerebral injection brains, GAPDH (4352932E), LIF (Mm00434762_g1), IL-6 (Mm00446190_m1), CNTF (Mm00446373_m1), TNFα (Mm00443258_m1), CD45 (Mm01293575_m1) and CD68 (Mm03047340_m1). All reactions were performed in triplicate and normalized to the levels of GAPDH. Data were analyzed according to the ΔΔCt method.
Animals
Vitronectin-null (VTN−/−; JAX Stock 004371) were purchased from Jackson Laboratory (Bar Harbor, ME) and had been on a C57BL/6 background for 12 generations. Heterozygous mice were bred to produce VTN+/+ (wild type) and VTN−/− littermates for experiments. They were bred behind a barrier in our AALAC-accredited vivarium. Tail snips were collected and genotyping was performed with the protocol provided by The Jackson Laboratory. Males were used for the collagenase intracerebral injection hemorrhage experiments at 8–12 weeks of age. All mice were housed with food and water available ad libitum, and maintained on a 12 h light–12 h dark on/off cycle. All procedures were approved by the East Tennessee State University IACUC, in compliance with the NIH Guide on Care and Use of Animals.
Plasma isolation from mice
Whole blood (125 µl) collected retro-orbitally or vena cava blood (500 μl) from adult VTN+/+ and VTN−/− littermate mice was placed in EDTA-coated mini-capillary collection tubes (Cat# 07 6011, Ram Scientific) and centrifuged at 4000 g for 20 min at 4°C to separate plasma. Plasma from three mice was pooled and aliquots were stored at −80°C and used at the indicated concentrations in vitro and in vivo.
Stereotaxic intrastriatal injections in mice
Intrastriatal injections were performed as described previously (Kang et al., 2012). Briefly, mice were anesthetized with Avertin (2,2,2-tribromoethanol, 400 mg/kg body weight, Sigma-Aldrich) and placed in a Kopf stereotaxic apparatus. A 1 mm burr hole was drilled at coordinates 1 mm rostral and 1.5 mm lateral from Bregma. The needle of a 10 µl Hamilton syringe was clamped in an electrode holder and lowered 3.5 mm ventral to the dura to the center of the striatum. After 2 min, 1 µl PBS or 1 µl PBS plus active or denatured (60°C for 30 min) recombinant human VTN (1 µg/µl, Cat# SRP3186, Sigma-Aldrich) was injected over 3 min, followed by a 2 min pause to reduce backflow. After the needle was withdrawn, the skin was sutured and mice were kept on the heating pad until they fully recovered. The striatum was collected 24 h later from 2 mm brain slices and snap frozen in liquid nitrogen, and stored at −80°C for later analysis. To induce intracerebral hemorrhage (Mracsko et al., 2014), 0.5 µl collagenase (0.03 units, Cat# C2399, Sigma) or 0.5 µl control PBS was injected into the striatum. At 24 h, these mice were transcardially perfused with ice-cold PBS and the striatum tissue collected.
LIF and IL-6 ELISA from adult mouse brain
Protein levels were measured in extracts from the striatum of the hemorrhage mice and those injected with rhVTN by ELISA, using kits for mouse IL-6 (Cat # MLF00, R&D Systems) and LIF (Cat # MLF00, R&D Systems) and performed according to manufacturer's protocol.
Statistical analyses
Statistical analyses were performed in GraphPad Prism (version 5a or 7). A one-way ANOVA with Dunnett post hoc tests or a two-way ANOVA with multiple comparisons was used when comparing genotypes or siRNA treatments combined with pharmacological inhibitors. If only two groups were compared, a Student's t-test was used. Statistical analysis for Fig. 2A,C was performed with a non-parametric Kruskal–Wallis test, as the data were not normally distributed. ELISA data in Fig. 2B,D was analyzed by two-way ANOVA, which showed no difference in genotype or genotype treatment interaction but showed a treatment effect. We therefore performed a Fisher's least squares difference (LSD) test, which treats both groups as individual experiments and for the effect of treatment within each genotype. A value of P<0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgements
We express our gratitude to Hannah Malone for animal care, and Rhesa Dykes and Michelle Duffourc of the Microbiology Core Facility for guidance in the WES technology. Dr Engelhardt of the Theodor Kocher institute graciously provided the bEnd5 cells.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: M.P.K., T.H.; Methodology: M.P.K., C.J., L.F.P., T.H.; Validation: M.P.K., T.H.; Formal analysis: M.P.K., C.J., T.H.; Investigation: M.P.K., C.J., L.F.P., R.R.S., C.L.; Resources: T.H.; Data curation: M.P.K., T.H.; Writing - original draft: M.P.K., T.H.; Writing - review & editing: M.P.K., T.H.; Visualization: M.P.K., C.J., T.H.; Supervision: M.P.K., T.H.; Project administration: M.P.K., T.H.; Funding acquisition: T.H.
Funding
Funding was provided by National Institutes of Health (NIH) (R01 grant AG29493, and in part by C06RR0306551) and the Quillen College of Medicine at East Tennessee State University. L.F.P. was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (454077/2014-9), Fundação do Amparo a Ciência e Tecnologia (AMD-0291-2.00/14) and Coordenadoria de Aperfeiçoamento de Pessoal de Nível Superior (88881.133822/2016-01). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.202580.supplemental
References
- Aguirre Ghiso J. A. (2002). Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene 21, 2513-2524. 10.1038/sj.onc.1205342 [DOI] [PubMed] [Google Scholar]
- Antonov A. S., Antonova G. N., Munn D. H., Mivechi N., Lucas R., Catravas J. D. and Verin A. D. (2011). αVβ3 integrin regulates macrophage inflammatory responses via PI3 kinase/Akt-dependent NF-κB activation. J. Cell. Physiol. 226, 469-476. 10.1002/jcp.22356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang J.-Y., Kim E.-Y., Kang D.-K., Chang S.-I., Han M. H., Baek K.-H. and Kang I.-C. (2011). Pharmacoproteomic analysis of a novel cell-permeable peptide inhibitor of tumor-induced angiogenesis. Mol. Cell Proteomics 10, M110.005264 10.1074/mcp.M110.005264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banner L. R. and Patterson P. H. (1994). Major changes in the expression of the mRNAs for cholinergic differentiation factor/leukemia inhibitory factor and its receptor after injury to adult peripheral nerves and ganglia. Proc. Natl. Acad. Sci. USA 91, 7109-7113. 10.1073/pnas.91.15.7109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banner L. R., Moayeri N. N. and Patterson P. H. (1997). Leukemia inhibitory factor is expressed in astrocytes following cortical brain injury. Exp. Neurol. 147, 1-9. 10.1006/exnr.1997.6536 [DOI] [PubMed] [Google Scholar]
- Bauer S. and Patterson P. H. (2006). Leukemia inhibitory factor promotes neural stem cell self-renewal in the adult brain. J. Neurosci. 26, 12089-12099. 10.1523/JNEUROSCI.3047-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borish L., O'Reilly D., Klempner M. S. and Rocklin R. E. (1986). Leukocyte Inhibitory Factor (Lif) Potentiates Neutrophil Responses to Formyl-Methionyl-Leucyl-Phenylalanine. J. Immunol. 137, 1897-1903. [PubMed] [Google Scholar]
- Butler B. and Blystone S. D. (2005). Tyrosine phosphorylation of beta3 integrin provides a binding site for Pyk2. J. Biol. Chem. 280, 14556-14562. 10.1074/jbc.M411765200 [DOI] [PubMed] [Google Scholar]
- Carlson C. D., Bai Y., Jonakait G. M. and Hart R. P. (1996). Interleukin-1 beta increases leukemia inhibitory factor mRNA levels through transient stimulation of transcription rate. Glia 18, 141-151. [DOI] [PubMed] [Google Scholar]
- Cartwright P., McLean C., Sheppard A., Rivett D., Jones K. and Dalton S. (2005). LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 132, 885-896. 10.1242/dev.01670 [DOI] [PubMed] [Google Scholar]
- Cattavarayane S., Palovuori R., Tanjore Ramanathan J. and Manninen A. (2015). α6β1- and αV-integrins are required for long-term self-renewal of murine embryonic stem cells in the absence of LIF. BMC Cell Biol. 16, 3 10.1186/s12860-015-0051-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charo I. F., Nannizzi L., Smith J. W. and Cheresh D. A. (1990). The vitronectin receptor alpha v beta 3 binds fibronectin and acts in concert with alpha 5 beta 1 in promoting cellular attachment and spreading on fibronectin. J. Cell Biol. 111, 2795-2800. 10.1083/jcb.111.6.2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y., Kim E., Lee Y., Han M. H. and Kang I.-C. (2010). Site-specific inhibition of integrin alpha v beta 3-vitronectin association by a ser-asp-val sequence through an Arg-Gly-Asp-binding site of the integrin. Proteomics 10, 72-80. 10.1002/pmic.200900146 [DOI] [PubMed] [Google Scholar]
- Degryse B., Resnati M., Czekay R.-P., Loskutoff D. J. and Blasi F. (2005). Domain 2 of the urokinase receptor contains an integrin-interacting epitope with intrinsic signaling activity: generation of a new integrin inhibitor. J. Biol. Chem. 280, 24792-24803. 10.1074/jbc.M413954200 [DOI] [PubMed] [Google Scholar]
- del Zoppo G. J., Frankowski H., Gu Y.-H., Osada T., Kanazawa M., Milner R., Wang X., Hosomi N., Mabuchi T. and Koziol J. A. (2012). Microglial cell activation is a source of metalloproteinase generation during hemorrhagic transformation. J. Cereb. Blood Flow Metab. 32, 919-932. 10.1038/jcbfm.2012.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delimont D., Dufek B. M., Meehan D. T., Zallocchi M., Gratton M. A., Phillips G. and Cosgrove D. (2014). Laminin α2-mediated focal adhesion kinase activation triggers Alport glomerular pathogenesis. PLoS ONE 9, e99083 10.1371/journal.pone.0099083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derigs H. G. and Boswell H. S. (1993). LIF mRNA expression is transcriptionally regulated in murine bone marrow stromal cells. Leukemia 7, 630-634. [PubMed] [Google Scholar]
- Dowsing B. J., Romeo R. and Morrison W. A. (2001). Expression of leukemia inhibitory factor in human nerve following injury. J. Neurotrauma 18, 1279-1287. 10.1089/089771501317095313 [DOI] [PubMed] [Google Scholar]
- Erta M. A., Quintana A. and Hidalgo J. (2012). Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 8, 1254-1266. 10.7150/ijbs.4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz K., Kronenberg G., Kälin R. E., Baldinger T., Werner C., Balkaya M., Eom G. D., Hellmann-Regen J., Kröber J., Miller K. R. et al. (2012). Essential role of interleukin-6 in post-stroke angiogenesis. Brain 135, 1964-1980. 10.1093/brain/aws075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti F. G. and Ruoslahti E. (1999). Integrin signaling. Science 285, 1028-1032. 10.1126/science.285.5430.1028 [DOI] [PubMed] [Google Scholar]
- Gu J., Tamura M., Pankov R., Danen E. H. J., Takino T., Matsumoto K. and Yamada K. M. (1999). Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J. Cell Biol. 146, 389-403. 10.1083/jcb.146.2.389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins B.T. (2005). The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 57, 173-185. 10.1124/pr.57.2.4 [DOI] [PubMed] [Google Scholar]
- Hayashi Y., Furue M. K., Okamoto T., Ohnuma K., Myoishi Y., Fukuhara Y., Abe T., Sato J. D., Hata R.-I. and Asashima M. (2007). Integrins regulate mouse embryonic stem cell self-renewal. Stem Cells 25, 3005-3015. 10.1634/stemcells.2007-0103 [DOI] [PubMed] [Google Scholar]
- Hayman E. G., Pierschbacher M. D., Ohgren Y. and Ruoslahti E. (1983). Serum spreading factor (vitronectin) is present at the cell surface and in tissues. Proc. Natl. Acad. Sci. USA 80, 4003-4007. 10.1073/pnas.80.13.4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman E. G., Pierschbacher M. D., Suzuki S. and Ruoslahti E. (1985). Vitronectin–a major cell attachment-promoting protein in fetal bovine serum. Exp. Cell Res. 160, 245-258. 10.1016/0014-4827(85)90173-9 [DOI] [PubMed] [Google Scholar]
- Herrera-Molina R., Frischknecht R., Maldonado H., Seidenbecher C. I., Gundelfinger E. D., Hetz C., Aylwin M. L., Schneider P., Quest A. F. G. and Leyton L. (2012). Astrocytic αVβ3 integrin inhibits neurite outgrowth and promotes retraction of neuronal processes by clustering Thy-1. PLoS ONE 7, e34295 10.1371/journal.pone.0034295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter C. A. and Jones S. A. (2015). IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 16, 448-457. 10.1038/ni.3153 [DOI] [PubMed] [Google Scholar]
- Ishibashi T., Dakin K. A., Stevens B., Lee P. R., Kozlov S. V., Stewart C. L. and Fields R. D. (2006). Astrocytes promote myelination in response to electrical impulses. Neuron 49, 823-832. 10.1016/j.neuron.2006.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki H., Takeuchi O., Teraguchi S., Matsushita K., Uehata T., Kuniyoshi K., Satoh T., Saitoh T., Matsushita M., Standley D. M. et al. (2011). The IκB kinase complex regulates the stability of cytokine-encoding mRNA induced by TLR–IL-1R by controlling degradation of regnase-1. Nat. Immunol. 12, 1167-1175. 10.1038/ni.2137 [DOI] [PubMed] [Google Scholar]
- Jang Y.-C., Tsou R., Gibran N. S. and Isik F. F. (2000). Vitronectin deficiency is associated with increased wound fibrinolysis and decreased microvascular angiogenesis in mice. Surgery 127, 696-704. 10.1067/msy.2000.105858 [DOI] [PubMed] [Google Scholar]
- Kang W.-S., Choi J.-S., Shin Y.-J., Kim H.-Y., Cha J.-H., Lee J.-Y., Chun M.-H. and Lee M.-Y. (2008). Differential regulation of osteopontin receptors, CD44 and the alpha(v) and beta(3) integrin subunits, in the rat hippocampus following transient forebrain ischemia. Brain Res. 1228, 208-216. 10.1016/j.brainres.2008.06.106 [DOI] [PubMed] [Google Scholar]
- Kang S. S., Keasey M. P., Cai J. and Hagg T. (2012). Loss of neuron-astroglial interaction rapidly induces protective CNTF expression after stroke in mice. J. Neurosci. 32, 9277-9287. 10.1523/JNEUROSCI.1746-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S. S., Keasey M. P. and Hagg T. (2013). P2X7 Receptor Inhibition Increases CNTF in the Subventricular Zone, But Not Neurogenesis or Neuroprotection After Stroke in Adult Mice. Transl. Stroke Res. 4, 533-545. 10.1007/s12975-013-0265-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keasey M. P., Kang S. S., Lovins C. and Hagg T. (2013). Inhibition of a novel specific neuroglial integrin signaling pathway increases STAT3-mediated CNTF expression. Cell Commun. Signal. 11, 35 10.1186/1478-811X-11-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr B. J. and Patterson P. H. (2004). Potent pro-inflammatory actions of leukemia inhibitory factor in the spinal cord of the adult mouse. Exp. Neurol. 188, 391-407. 10.1016/j.expneurol.2004.04.012 [DOI] [PubMed] [Google Scholar]
- Krueger M., Bechmann I., Immig K., Reichenbach A., Härtig W. and Michalski D. (2015). Blood-brain barrier breakdown involves four distinct stages of vascular damage in various models of experimental focal cerebral ischemia. J. Cereb. Blood Flow Metab. 35, 292-303. 10.1038/jcbfm.2014.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix S., Chang L., Rose-John S. and Tuszynski M. H. (2002). Delivery of hyper-interleukin-6 to the injured spinal cord increases neutrophil and macrophage infiltration and inhibits axonal growth. J. Comp. Neurol. 454, 213-228. 10.1002/cne.10407 [DOI] [PubMed] [Google Scholar]
- Lau L. T. and Yu A. C.-H. (2001). Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J. Neurotrauma 18, 351-359. 10.1089/08977150151071035 [DOI] [PubMed] [Google Scholar]
- Leavesley D. I., Kashyap A. S., Croll T., Sivaramakrishnan M., Shokoohmand A., Hollier B. G. and Upton Z. (2013). Vitronectin—Master controller or micromanager? IUBMB Life 65, 807-818. 10.1089/10.1002/iub.1203 [DOI] [PubMed] [Google Scholar]
- Lee Y., Kang D.-K., Chang S.-I., Han M. H. and Kang I.-C. (2004). High-throughput screening of novel peptide inhibitors of an integrin receptor from the hexapeptide library by using a protein microarray chip. J. Biomol. Screen. 9, 687-694. 10.1177/1087057104268125 [DOI] [PubMed] [Google Scholar]
- Li S., Kim M., Hu Y. L., Jalali S., Schlaepfer D. D., Hunter T., Chien S. and Shyy J. Y. (1997). Fluid shear stress activation of focal adhesion kinase. Linking to mitogen-activated protein kinases. J. Biol. Chem. 272, 30455-30462. [DOI] [PubMed] [Google Scholar]
- Li R., Ren M., Chen N., Luo M., Zhang Z. and Wu J. (2012). Vitronectin increases vascular permeability by promoting VE-cadherin internalization at cell junctions. PLoS ONE 7, e37195 10.1371/journal.pone.0037195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z.-Q. (2003). Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J. Leukoc. Biol. 73, 713-721. 10.1189/jlb.0802397 [DOI] [PubMed] [Google Scholar]
- Linker R. A., Mäurer M., Gaupp S., Martini R., Holtmann B., Giess R., Rieckmann P., Lassmann H., Toyka K. V., Sendtner M. et al. (2002). CNTF is a major protective factor in demyelinating CNS disease: a neurotrophic cytokine as modulator in neuroinflammation. Nat. Med. 8, 620-624. 10.1038/nm0602-620 [DOI] [PubMed] [Google Scholar]
- Liu E., Côté J.-F. and Vuori K. (2003). Negative regulation of FAK signaling by SOCS proteins. EMBO J. 22, 5036-5046. 10.1093/emboj/cdg503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.-K., Uemura T., Nemoto A., Yabe T., Fujii N., Ushida T. and Tateishi T. (1997). Osteopontin involvement in integrin-mediated cell signaling and regulation of expression of alkaline phosphatase during early differentiation of UMR cells. FEBS Lett. 420, 112-116. 10.1016/S0014-5793(97)01498-1 [DOI] [PubMed] [Google Scholar]
- Longo A., Librizzi M., Chuckowree I. S., Baltus C. B., Spencer J. and Luparello C. (2015). Cytotoxicity of the Urokinase-Plasminogen Activator Inhibitor Carbamimidothioic Acid (4-Boronophenyl) Methyl Ester Hydrobromide (BC-11) on Triple-Negative MDA-MB231 Breast Cancer Cells. Molecules 20, 9879-9889. 10.3390/molecules20069879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen C. D., Ferraris G. M. S., Andolfo A., Cunningham O. and Sidenius N. (2007). uPAR-induced cell adhesion and migration: vitronectin provides the key. J. Cell Biol. 177, 927-939. 10.1083/jcb.200612058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnussen S., Hadler-Olsen E., Latysheva N., Pirila E., Steigen S. E., Hanes R., Salo T., Winberg J.-O., Uhlin-Hansen L. and Svineng G. (2014). Tumour microenvironments induce expression of urokinase plasminogen activator receptor (uPAR) and concomitant activation of gelatinolytic enzymes. PLoS ONE 9, e105929 10.1371/journal.pone.0105929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnussen S. N., Hadler-Olsen E., Costea D. E., Berg E., Jacobsen C. C., Mortensen B., Salo T., Martinez-Zubiaurre I., Winberg J.-O., Uhlin-Hansen L. et al. (2017). Cleavage of the urokinase receptor (uPAR) on oral cancer cells: regulation by transforming growth factor - β1 (TGF-β1) and potential effects on migration and invasion. BMC Cancer 17, 350 10.1186/s12885-017-3349-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka I., Nakane A. and Kurihara K. (1997). Induction of LIF-mRNA by TGF-beta 1 in Schwann cells. Brain Res. 776, 170-180. 10.1016/S0006-8993(97)01015-9 [DOI] [PubMed] [Google Scholar]
- Mauer J., Denson J. L. and Brüning J. C. (2015). Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 36, 92-101. 10.1016/j.it.2014.12.008 [DOI] [PubMed] [Google Scholar]
- Memmo L. M. and McKeown-Longo P. (1998). The alphavbeta5 integrin functions as an endocytic receptor for vitronectin. J. Cell Sci. 111, 425-433. [DOI] [PubMed] [Google Scholar]
- Mi H., Haeberle H. and Barres B. A. (2001). Induction of astrocyte differentiation by endothelial cells. J. Neurosci. 21, 1538-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner R. (2009). Microglial expression of alphavbeta3 and alphavbeta5 integrins is regulated by cytokines and the extracellular matrix: beta5 integrin null microglia show no defects in adhesion or MMP-9 expression on vitronectin. Glia 57, 714-723. 10.1002/glia.20799 [DOI] [PubMed] [Google Scholar]
- Milner R. and Campbell I. L. (2003). The extracellular matrix and cytokines regulate microglial integrin expression and activation. J. Immunol. 170, 3850-3858. 10.4049/jimmunol.170.7.3850 [DOI] [PubMed] [Google Scholar]
- Milner R., Crocker S. J., Hung S., Wang X., Frausto R. F. and del Zoppo G. J. (2007). Fibronectin- and vitronectin-induced microglial activation and matrix metalloproteinase-9 expression is mediated by integrins alpha5beta1 and alphavbeta5. J. Immunol. 178, 8158-8167. 10.4049/jimmunol.178.12.8158 [DOI] [PubMed] [Google Scholar]
- Mitra S. K., Hanson D. A. and Schlaepfer D. D. (2005). Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56-68. 10.1038/nrm1549 [DOI] [PubMed] [Google Scholar]
- Mracsko E., Javidi E., Na S.-Y., Kahn A., Liesz A. and Veltkamp R. (2014). Leukocyte invasion of the brain after experimental intracerebral hemorrhage in mice. Stroke 45, 2107-2114. 10.1161/STROKEAHA.114.005801 [DOI] [PubMed] [Google Scholar]
- Murphy G. M., Song Y., Ong E., Lee Y. L., Schmidt K. G., Bocchini V. and Eng L. F. (1995). Leukemia inhibitory factor mRNA is expressed in cortical astrocyte cultures but not in an immortalized microglial cell line. Neurosci. Lett. 184, 48-51. 10.1016/0304-3940(94)11165-F [DOI] [PubMed] [Google Scholar]
- Nagamoto-Combs K., Vaccariello S. A. and Zigmond R. E. (1999). The levels of leukemia inhibitory factor mRNA in a Schwann cell line are regulated by multiple second messenger pathways. J. Neurochem. 72, 1871-1881. 10.1046/j.1471-4159.1999.0721871.x [DOI] [PubMed] [Google Scholar]
- Nakanishi M., Niidome T., Matsuda S., Akaike A., Kihara T. and Sugimoto H. (2007). Microglia-derived interleukin-6 and leukaemia inhibitory factor promote astrocytic differentiation of neural stem/progenitor cells. Eur. J. Neurosci. 25, 649-658. 10.1111/j.1460-9568.2007.05309.x [DOI] [PubMed] [Google Scholar]
- Ochel H.-J., Schulte T. W., Nguyen P., Trepel J. and Neckers L. (1999). The benzoquinone ansamycin geldanamycin stimulates proteolytic degradation of focal adhesion kinase. Mol. Genet. Metab. 66, 24-30. 10.1006/mgme.1998.2774 [DOI] [PubMed] [Google Scholar]
- Ostberg C. O., Zhu P., Wight T. N. and Qwarnstrom E. E. (1995). Fibronectin attachment is permissive for IL-1 mediated gene regulation. FEBS Lett. 367, 93-97. 10.1016/0014-5793(95)00509-8 [DOI] [PubMed] [Google Scholar]
- Pan W., Yu C., Hsuchou H., Zhang Y. and Kastin A. J. (2008). Neuroinflammation facilitates LIF entry into brain: role of TNF. Am. J. Physiol. Cell. Physiol. 294, C1436-C1442. 10.1152/ajpcell.00489.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzi B., Cappariello A., Del Fattore A., Rucci N., De Benedetti F. and Teti A. (2012). c-Src and IL-6 inhibit osteoblast differentiation and integrate IGFBP5 signalling. Nat. Comms. 3, 630 10.1038/ncomms1651 [DOI] [PubMed] [Google Scholar]
- Plow E. F., Haas T. K., Zhang L., Loftus J. and Smith J. W. (2000). Ligand binding to integrins. J. Biochem. 275, 21785-21788. 10.1074/jbc.R000003200 [DOI] [PubMed] [Google Scholar]
- Rothaug M., Becker-Pauly C. and Rose-John S. (2016). The role of interleukin-6 signaling in nervous tissue. Biochim. Biophys. Acta 1863, 1218-1227. 10.1016/j.bbamcr.2016.03.018 [DOI] [PubMed] [Google Scholar]
- Schlaepfer D. D., Hou S., Lim S.-T., Tomar A., Yu H., Lim Y., Hanson D. A., Uryu S. A., Molina J. and Mitra S. K. (2007). Tumor necrosis factor-alpha stimulates focal adhesion kinase activity required for mitogen-activated kinase-associated interleukin 6 expression. J. Biol. Chem. 282, 17450-17459. 10.1074/jbc.M610672200 [DOI] [PubMed] [Google Scholar]
- Seiffert D., Keeton M., Eguchi Y., Sawdey M. and Loskutoff D. J. (1991). Detection of vitronectin mRNA in tissues and cells of the mouse. Proc. Natl. Acad. Sci. USA 88, 9402-9406. 10.1073/pnas.88.21.9402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiffert D., Iruela-Arispe M. L., Sage E. H. and Loskutoff D. J. (1995). Distribution of vitronectin mRNA during murine development. Dev. Dyn. 203, 71-79. 10.1002/aja.1002030108 [DOI] [PubMed] [Google Scholar]
- Shimamura N., Matchett G., Yatsushige H., Calvert J. W., Ohkuma H. and Zhang J. (2006). Inhibition of integrin alphavbeta3 ameliorates focal cerebral ischemic damage in the rat middle cerebral artery occlusion model. Stroke 37, 1902-1909. 10.1161/01.STR.0000226991.27540.f2 [DOI] [PubMed] [Google Scholar]
- Sieg D. J., Hauck C. R., Ilić D., Klingbeil C. K., Schaefer E., Damsky C. H. and Schlaepfer D. D. (2000). FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2, 249-256. 10.1038/35010517 [DOI] [PubMed] [Google Scholar]
- Slack-Davis J. K., Martin K. H., Tilghman R. W., Iwanicki M., Ung E. J., Autry C., Luzzio M. J., Cooper B., Kath J. C., Roberts W. G. et al. (2007). Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 282, 14845-14852. 10.1074/jbc.M606695200 [DOI] [PubMed] [Google Scholar]
- Sugiura S., Lahav R., Han J., Kou S.-Y., Banner L. R., de Pablo F. and Patterson P. H. (2000). Leukaemia inhibitory factor is required for normal inflammatory responses to injury in the peripheral and central nervous systems in vivo and is chemotactic for macrophages in vitro. Eur. J. Neurosci. 12, 457-466. 10.1046/j.1460-9568.2000.00922.x [DOI] [PubMed] [Google Scholar]
- Suzuki S., Oldberg A., Hayman E. G., Pierschbacher M. D. and Ruoslahti E. (1985). Complete amino acid sequence of human vitronectin deduced from cDNA. Similarity of cell attachment sites in vitronectin and fibronectin. EMBO J. 4, 2519-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S., Tanaka K. and Suzuki N. (2009). Ambivalent aspects of interleukin-6 in cerebral ischemia: inflammatory versus neurotrophic aspects. J. Cereb. Blood Flow Metab. 29, 464-479. 10.1038/jcbfm.2008.141 [DOI] [PubMed] [Google Scholar]
- Tofaris G. K., Patterson P. H., Jessen K. R. and Mirsky R. (2002). Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. J. Neurosci. 22, 6696-6703. 10.1038/20026699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasini-Johansson B. R., Milbrink J. and Pejler G. (1998). Vitronectin expression in rheumatoid arthritic synovia–inhibition of plasmin generation by vitronectin produced in vitro. Br. J. Rheumatol. 37, 620-629. 10.1093/rheumatology/37.6.620 [DOI] [PubMed] [Google Scholar]
- Tornatore T. F., Dalla Costa A. P., Clemente C. F. M. Z., Judice C., Rocco S. A., Calegari V. C., Cardoso L., Cardoso A. C., Goncalves A. J. and Franchini K. G. (2011). A role for focal adhesion kinase in cardiac mitochondrial biogenesis induced by mechanical stress. Am. J. Physiol. Heart Circ. Physiol. 300, H902-H912. 10.1152/ajpheart.00319.2010 [DOI] [PubMed] [Google Scholar]
- Tsuruta Y., Park Y.-J., Siegal G. P., Liu G. and Abraham E. (2007). Involvement of vitronectin in lipopolysaccaride-induced acute lung injury. J. Immunol. 179, 7079-7086. 10.4049/jimmunol.179.10.7079 [DOI] [PubMed] [Google Scholar]
- Van Wagoner N. J. and Benveniste E. N. (1999). Interleukin-6 expression and regulation in astrocytes. J. Neuroimmunol. 100, 124-139. 10.1016/S0165-5728(99)00187-3 [DOI] [PubMed] [Google Scholar]
- Vandenberghe M., Raphaël M., Lehen'kyi V., Gordienko D., Hastie R., Oddos T., Rao A., Hogan P. G., Skryma R. and Prevarskaya N. (2013). ORAI1 calcium channel orchestrates skin homeostasis. Proc. Natl. Acad. Sci. USA 110, E4839-E4848. 10.1073/pnas.1310394110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiagis D. and Salamonsen L. A. (1999). Review: The role of leukaemia inhibitory factor in the establishment of pregnancy. J. Endocrinol. 160, 181-190. 10.1677/joe.0.1600181 [DOI] [PubMed] [Google Scholar]
- Wang J. and Milner R. (2006). Fibronectin promotes brain capillary endothelial cell survival and proliferation through alpha5beta1 and alphavbeta3 integrins via MAP kinase signalling. J. Neurochem. 96, 148-159. 10.1111/j.1471-4159.2005.03521.x [DOI] [PubMed] [Google Scholar]
- Wei Y., Waltz D. A., Rao N., Drummond R. J., Rosenberg S. and Chapman H. A. (1994). Identification of the urokinase receptor as an adhesion receptor for vitronectin. J. Biol. Chem. 269, 32380-32388. [PubMed] [Google Scholar]
- Welser-Alves J. V., Boroujerdi A., Tigges U. and Milner R. (2011). Microglia use multiple mechanisms to mediate interactions with vitronectin; non-essential roles for the highly-expressed αvβ3 and αvβ5 integrins. J. Neuroinflammation 8, 157 10.1186/1742-2094-8-157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J.-P. (2002). Crystal Structure of the Extracellular Segment of Integrin alpha Vbeta 3 in Complex with an Arg-Gly-Asp Ligand. Science 296, 151-155. 10.1126/science.1069040 [DOI] [PubMed] [Google Scholar]
- Yang P., Arnold S. A., Habas A., Hetman M. and Hagg T. (2008). Ciliary neurotrophic factor mediates dopamine D2 receptor-induced CNS neurogenesis in adult mice. J. Neurosci. 28, 2231-2241. 10.1523/JNEUROSCI.3574-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X., Saunders T. L., Camper S. A., Samuelson L. C. and Ginsburg D. (1995). Vitronectin is not essential for normal mammalian development and fertility. Proc. Natl. Acad. Sci. USA 92, 12426-12430. 10.1073/pnas.92.26.12426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond R. E. (2012). gp130 cytokines are positive signals triggering changes in gene expression and axon outgrowth in peripheral neurons following injury. Front. Mol. Neurosci. 4, 62 10.3389/fnmol.2011.00062 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.