

ABSTRACT
The peroxide bond of the artemisinins inspired the development of a class of fully synthetic 1,2,4-trioxolane-based antimalarials, collectively known as the ozonides. Similar to the artemisinins, heme-mediated degradation of the ozonides generates highly reactive radical species that are thought to mediate parasite killing by damaging critical parasite biomolecules. We examined the relationship between parasite dependent degradation and antimalarial activity for two ozonides, OZ277 (arterolane) and OZ439 (artefenomel), using a combination of in vitro drug stability and pulsed-exposure activity assays. Our results showed that drug degradation is parasite stage dependent and positively correlates with parasite load. Increasing trophozoite-stage parasitemia leads to substantially higher rates of degradation for both OZ277 and OZ439, and this is associated with a reduction in in vitro antimalarial activity. Under conditions of very high parasitemia (∼90%), OZ277 and OZ439 were rapidly degraded and completely devoid of activity in trophozoite-stage parasite cultures exposed to a 3-h drug pulse. This study highlights the impact of increasing parasite load on ozonide stability and in vitro antimalarial activity and should be considered when investigating the antimalarial mode of action of the ozonide antimalarials under conditions of high parasitemia.
KEYWORDS: Plasmodium falciparum, antimalarial activity, artefenomel, arterolane, ozonide, peroxide antimalarial, stability
INTRODUCTION
Malaria remains a significant threat to human health in many parts of the developing world and was responsible for approximately 429,000 deaths in 2015 (1). The greatest morbidity and mortality in humans is caused by the parasite species Plasmodium falciparum and, in the absence of a reliable vaccine, management is heavily reliant on effective chemotherapeutic intervention. Rapid acting semisynthetic peroxide antimalarials based on artemisinin are the cornerstone of the current drug treatment strategy and are recommended in combination with a suitable long acting partner drug (artemisinin combination therapy [ACT]) as the first-line option for uncomplicated malaria. In the case of severe malaria, parenteral artesunate followed by ACT is the recommended course of treatment (2). Unfortunately, the artemisinins have short in vivo half-lives (1 to 2 h) that dictate the need for multiple doses over a 3-day treatment course, and this prevents their use in malaria prophylaxis. Of major concern is the emergence and spread of artemisinin resistant malaria in the Greater Mekong Subregion (3, 4), highlighting the urgent need for new and effective therapies.
OZ439 (artefenomel) is one of the most advanced antimalarial drug candidates currently in clinical development (5–8), and its first-generation predecessor, OZ277 (arterolane), is approved for use in India as a fixed-dose combination with piperaquine (9–12). Both of these ozonides are fully synthetic, and like the artemisinins (13), contain a peroxide bond that is required for activity (14–16). The artemisinins (17, 18) and the ozonides (19, 20) act on all blood stages of the parasite, including sexual-stage gametocytes (21). Similar to the artemisinins, OZ277 has a relatively short in vivo half-life that is only two to three times longer than that for dihydroartemisinin (DHA). In contrast, OZ439 has a greatly extended in vivo half-life and exposure profile and is currently undergoing phase II clinical trials in combination with ferroquine (ClinicalTrials.gov registration no. NCT02497612).
Although the mechanism of artemisinin action has not been completely resolved, it is widely accepted that artemisinins must be activated by iron-mediated reduction of the peroxide bond for antimalarial activity (22–25). Iron-mediated activation gives rise to highly reactive radical species that are thought to modify crucial parasite biomolecules resulting in cellular damage and parasite death (26). Recent work has confirmed that the most likely source of the iron-based activator is heme released through the process of parasite hemoglobin digestion (27–29). Blocking hemoglobin digestion with falcipain hemoglobinase inhibitors antagonized artemisinin activity up to 100-fold in trophozoite-stage parasites when a short pulse of drug exposure was used (28). Consistent with this hypothesis, the potency of artemisinin antimalarials is significantly lower during the mid-ring stage when hemoglobin digestion is less active (27, 30). Other potential mechanisms for artemisinin activity, including mitochondrial activation (31) and iron-independent pathways, such as direct inhibition of P. falciparum phosphatidylinositol-3-kinase (32) and the cofactor model (33), have also been proposed.
Recent in vitro studies have used drug pulse activity assays to examine the exposure time dependence of parasite response to selected peroxide antimalarials (34–36) across the asexual parasite life cycle. The extent of parasite killing was found to be dependent on parasite strain and life cycle stage, drug concentration, and drug exposure time. Similar to the artemisinins (30), short pulse treatment with the ozonides was less effective against mid-ring-stage parasites than against trophozoites, an observation consistent with enhanced hemoglobin degradation during the trophozoite stage. Accordingly, the iron chelator bipyridyl (BiPy) had little effect on ozonide potency, whereas inhibition of hemoglobin digestion using the falcipain inhibitor E64d significantly antagonized ozonide action. These results indicate that, like the artemisinins, the ozonides require active hemoglobin digestion for potent antimalarial activity. Moreover, extending the ozonide exposure time to mimic in vivo exposure led to enhanced activity and more effective killing than for the clinically used artemisinin derivative DHA (34).
Previous studies have also demonstrated that the ozonides are susceptible to chemical degradation in noninfected red blood cells (RBCs) and degrade at a higher rate in parasitized RBCs (5). The rate of degradation depends on the chemical structure; first-generation ozonides such as OZ277 have a more exposed peroxide bond and degrade more quickly than next-generation compounds such as OZ439 that contain a more sterically hindered peroxide bond (16, 37, 38). The increased rate of degradation of OZ277 in infected RBCs has been linked to the reduced plasma exposure seen with OZ277 in malaria-infected patients (9) relative to healthy volunteers (12). Notably, a similar decreased exposure was not evident when malaria patients were treated with OZ439 (7).
Understanding the mechanism of action of both the artemisinins and the ozonides is an active area of research. Various biochemical and biomimetic approaches have been utilized to identify both the molecular targets of peroxide antimalarials and parasite biochemical pathways perturbed in response to drug treatment (29, 39–42). Many of these in vitro techniques require the use of specific conditions such as high parasitemia (39, 40, 42, 43) for optimal assay performance, and these conditions differ considerably from those used in typical in vitro drug activity measurements. Given that the reactivity of the peroxide bond is essential for antimalarial activity, accurate interpretation of the biological relevance of results from in vitro experiments relies on a clear understanding of the interplay between parasite-mediated drug degradation and biological activity under various experimental conditions, such as varying hematocrit (Hct), parasitemia, or both.
The aim of the present study was to quantitatively assess both parasite-mediated drug degradation and the in vitro antimalarial activity of the ozonides at various levels of parasitemia, at various life cycle stages, and in the presence of selected inhibitors. We show that reduced ozonide potency at high parasitemia correlates with a significantly enhanced rate of drug degradation in vitro. Our results indicate that parasite load and drug degradation are important variables to consider when investigating the antimalarial mode of action of peroxides in vitro.
RESULTS
Assessment of ozonide washing procedure for drug pulse assays.
Removal of the ozonides from in vitro culture in the pulse activity assay format was recently shown to be insufficient if a standard washing method (three washes with culture medium) was used (34). Under the conditions tested by Yang et al. (200 μl, 0.2% Hct), effective removal of drug was achieved by washing cells with complete RPMI medium (containing 5% serum and 0.25% Albumax II) and incorporating a plate change as part of the washing method. In light of this, the suitability of different washing procedures was assessed for our specific in vitro conditions using 2% Hct and 0.5% Albumax II in RPMI. Using a method similar to that described previously (34), noninfected RBCs were exposed to drug, the drug was washed out, infected RBCs were added to the culture, and parasite growth was assessed after 48 h relative to an untreated control. DHA was effectively removed from RBCs according to the standard wash procedure, and parasite growth was equivalent to that of the untreated controls (data not shown). In comparison, when RBCs were incubated with either ozonide and washing was performed using the standard drug washout procedure, residual antimalarial activity was detected (see Fig. S1 in the supplemental material), and significant residual drug concentrations were measured by liquid chromatography-mass spectrometry (LC-MS) (Fig. 1, blue bars). When a plate transfer was incorporated into the standard wash method, OZ277 was effectively removed up to the maximum concentration tested (4.8 μM; see Fig. S2A in the supplemental material); however, a concentration-dependent reduction in parasite growth was still evident with OZ439-treated RBCs (see Fig. S2B in the supplemental material). Incorporating both the plate transfer and a modified washing medium containing 5% Albumax II led to efficient removal of OZ439 at concentrations up to 2.4 μM (see Fig. S2B, gray, in the supplemental material). At an intermediate Albumax II concentration of 2%, removal of OZ439 was effective up to a concentration of 1.2 μM (see Fig. S2B, red, in the supplemental material). Quantitative analysis confirmed that the optimized washing procedure (four washes containing 2% Albumax II and a plate change) reduced residual drug levels to below their limit of quantitation (Fig. 1, red bars).
FIG 1.

Assessment of drug washout protocol for pulse activity assays with OZ277 and OZ439. LC-MS quantification of the amount of OZ277 (A) and OZ439 (B) remaining in RBC cultures (2% Hct) after washing using either the standard (blue) or the optimized (red) washing protocol, as defined in Materials and Methods. Values represent the means of two independent experiments (one experiment for 2,400 nM).
Ozonide stability in noninfected RBCs.
The extent of ozonide degradation under our standard in vitro culture conditions (2% Hct in RPMI containing 0.5% Albumax II) was examined following a 6 h incubation with noninfected RBCs. For OZ277, there was approximately 40% loss over this time period, which was reduced to <20% loss in the presence of the iron chelator deferiprone (DFP). Under the same conditions, minimal loss of OZ439 was observed over the incubation period (>85% remaining after 6 h), either in the absence or in the presence of DFP. Degradation rate constants for OZ277 and OZ439 in noninfected RBCs in the presence and absence of DFP are presented in Table 1. The stability of both compounds was confirmed in complete culture medium without RBCs (no significant degradation observed).
TABLE 1.
Activity, degradation half-lives, and rate constants of OZ277 and OZ439 in noninfected and P. falciparum-infected RBCsa
| Parasite stage | Condition(s) | OZ277 |
OZ439 |
||||
|---|---|---|---|---|---|---|---|
| IC50_3h (nM) | Half-life (h) | k (h−1) | IC50_3h (nM) | Half-life (h) | k (h−1) | ||
| Noninfected | 2% Hct | 8.9 ± 2.1 | 0.078 ± 0.018 | 41 ± 17 | 0.017 ± 0.007 | ||
| 2% Hct + DFP | 27 ± 4.1 | 0.026 ± 0.004 | 63 ± 34 | 0.011 ± 0.006 | |||
| Mid-rings | 1% P | NA | 7.5 ± 0.9 | 0.092 ± 0.011 | NA | 25 ± 3.5 | 0.028 ± 0.004 |
| 10% P | NA | 4.1 ± 0.5 | 0.17 ± 0.022 | NA | 18 ± 6.7 | 0.038 ± 0.014 | |
| 10% P + DFP | NA | 4.8 ± 0.7 | 0.15 ± 0.022 | NA | 22 ± 4.3 | 0.031 ± 0.006 | |
| 10% P + E64d | NA | 5.8 ± 0.6 | 0.12 ± 0.012 | NA | 20 ± 6.6 | 0.034 ± 0.011 | |
| Trophozoites | 1% P | 31 ± 23 | 3.5 ± 0.8 | 0.20 ± 0.047 | 31 ± 11 | 19 ± 2.5 | 0.037 ± 0.005 |
| 10% P | 295 ± 119 | 1.3 ± 0.2 | 0.51 ± 0.089 | 398 ± 272 | 3.7 ± 0.5 | 0.19 ± 0.024 | |
| 10% P + DFP | 377 ± 223 | 1.2 ± 0.2 | 0.57 ± 0.11 | 469 ± 271 | 3.7 ± 0.2 | 0.19 ± 0.011 | |
| 10% P + E64d | NA | 1.8 ± 0.4 | 0.39 ± 0.080 | NA | 6.4 ± 3.70 | 0.11 ± 0.063 | |
| >90% P | NA | 0.060, 0.10 | 12, 6.9 | NA | 0.30, 0.20 | 2.3, 3.5 | |
Values represent the means ± the standard deviations of three to five independent experiments (individual values from two independent experiments for >90% P condition). IC50_3h, 50% inhibitory concentration to a 3-h drug pulse; P, parasitemia; NA, not active. For the IC50_3h determinations, parasites were exposed to drug for 3-h pulse, washed using an optimized washing method to remove drug, diluted to 0.5% parasitemia, and incubated for 48 to 72 h before parasite growth was assessed. For the half-life and k determinations, the starting concentration of drug was 100 nM.
Parasite-dependent drug degradation and ozonide antimalarial activity at high parasitemia.
The effect of increasing parasitemia on the apparent rate of ozonide degradation was examined at different parasite stages. For OZ277, there was a significant increase in the apparent rate of drug degradation in 1% trophozoite-infected cells compared to noninfected RBCs, both at 2% Hct (Fig. 2A). In comparison, OZ439 stability was not significantly affected by the presence of trophozoites at 1% parasitemia (Fig. 2B). When the parasitemia was increased from 1 to 10% trophozoites, a marked increase in the apparent rate of degradation of both OZ277 and OZ439 was observed (Fig. 2A and B). When mid-trophozoite-stage cultures were substantially enriched to >90% parasitemia, there was a further increase in the apparent rate of degradation for both compounds, displaying an almost 100-fold higher degradation rate constant compared to that for a 1% trophozoite culture. At low parasitemia (1%), mid-ring-stage degradation of both ozonides was not significantly different from the noninfected RBC controls over the 6 h incubation period (see Fig. S3A and B in the supplemental material). Increasing the parasitemia from 1 to 10% mid-rings had only a moderate effect on the rate of degradation of OZ277 and no detectable effect on the degradation of OZ439 (see Fig. S3A and B in the supplemental material). At 10% ring-stage parasitemia, the rate of ozonide degradation was significantly lower than that for trophozoite-stage cultures at the same parasitemia, and rate constants were comparable to those for trophozoites at 1% parasitemia (Table 1).
FIG 2.

Parasite-dependent increase in ozonide degradation rate at increasing levels of trophozoite-stage parasitemia and the corresponding loss of in vitro activity. (A and B) Degradation profiles of OZ277 (A) and OZ439 (B) in noninfected RBCs (gray) and synchronized trophozoite-stage parasites at 1% (blue), 10% (red), and >90% (black) parasitemia. The starting concentration of both drugs was 100 nM. Values represent the means of two independent experiments or the means ± the standard errors of the mean (SEM) of three to five independent experiments. (C and D) Representative growth inhibition profiles of 3-h pulse treatment with OZ277 (C) and OZ439 (D) in 1% (blue), 10% (red), and >90% (black) trophozoite-stage cultures. Values represent the means of two technical replicates or the means ± the SEM of three technical replicates.
The parasite-dependent degradation of OZ277 was also assessed under more physiologically relevant conditions using asynchronous parasite cultures. Human RBCs were suspended in human plasma at a physiological Hct of 45%, and the parasitemia was varied between 0.2 and 3.8%. A similar progressive increase in the rate of degradation was apparent under these conditions (see Table S1 in the supplemental material). Increasing parasite load by altering the Hct between 15 and 45%, while maintaining a constant 1% parasitemia also resulted in increased degradation of OZ277 (see Table S2 in the supplemental material).
To determine whether a relationship existed between the number of parasites in a culture and the degradation rate constant (k), the number of parasites per μl of culture was plotted against the respective degradation rate constant under each condition (Fig. 3). A positive correlation was apparent for both OZ277 and OZ439 at standard in vitro conditions of 2% Hct. The same trend was also apparent for OZ277 when incubated under the more physiologically relevant conditions, although we acknowledge that these conditions are not directly comparable due to differences in parasite strain, medium, and drug concentration.
FIG 3.
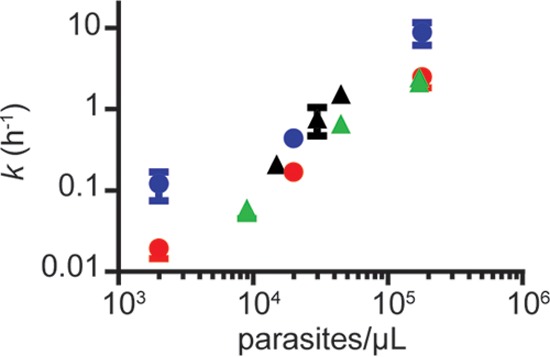
Relationship between degradation rate constant (k) and parasite load for OZ277 and OZ439. The degradation rate constants for 100 nM OZ277 (blue circles) and OZ439 (red circles) were determined when incubated with 3D7 strain P. falciparum parasites at different parasite loads achieved by increasing parasitemia and maintaining 2% Hct. To represent conditions comparable to human blood, degradation rate constants were also determined for 360 nM OZ277 when incubated with K1 strain-infected human RBCs suspended in human plasma. Different parasite loads were achieved by either altering parasitemia and maintaining 45% Hct (green triangles) or altering Hct (15, 30, and 45%) and maintaining constant 1% parasitemia (black triangles). Rate constants represent the means of two independent experiments or the means ± the SEM of three or more independent experiments minus the average degradation rate constant observed in noninfected RBCs.
The dependence of the apparent ozonide degradation rate on parasitemia and asexual parasite stage led us to assess how the differences in stability affected the in vitro potency (i.e., the 50% inhibitory concentration [IC50]). The sensitivity of synchronized mid-ring- and mid-trophozoite-stage cultures to a 3-h pulse with OZ277 or OZ439 was determined under low (1%)- and high (10%)-parasitemia conditions. In mid-ring-stage parasites, both ozonides showed poor activity and no significant change in the in vitro potency of either drug was apparent when parasitemia was increased to 10% (see Fig. S3C and D in the supplemental material). Incubation with a culture of high trophozoite parasitemia (10%) was associated with an ∼10-fold reduction in antimalarial activity compared to a low (1%) trophozoite infected culture in response to a 3-h pulse with either OZ277 or OZ439 (Fig. 2C and D). Enriched mid-trophozoite-stage cultures (>90% parasitemia) displayed a further significant reduction in potency (Fig. 2C and D), such that parasite growth was unaffected by a 3-h ozonide pulse up to a concentration of 2 μM.
Iron source responsible for increased ozonide degradation at high parasitemia.
The roles of chelatable iron and hemoglobin-derived heme in ozonide degradation and antimalarial activity at high parasitemia (10%) were assessed using sublethal concentrations of the iron chelator DFP and the falcipain inhibitor E64d, respectively. In mid-ring-stage parasites, the addition of either E64d or DFP had a negligible effect on the apparent rate of drug degradation of OZ277 and OZ439 (see Fig. S4A and B in the supplemental material). Under these conditions E64D, but not DFP, reduced OZ277 activity in mid-rings (see Fig. S4C in the supplemental material), but no effect was observed on the limited ring-stage activity of OZ439 under these conditions (see Fig. S4D in the supplemental material).
In trophozoites (10% parasitemia), E64d substantially reduced both OZ277 activity in the 3-h pulse assay (Fig. 4A) and the apparent rate of degradation (Fig. 4B). When OZ277 was coincubated with DFP, activity was unaffected (Fig. 4C), and there was minimal impact on the rate of degradation (Fig. 4B). Similarly, at 10% parasitemia, the addition of E64d also caused a marked reduction in OZ439 activity (Fig. 4D) and rate of degradation (Fig. 4E), whereas DFP had minimal effect on either activity or the rate of degradation (Fig. 4E and F).
FIG 4.

Activity and degradation profiles of OZ277 and OZ439 in the presence or absence of sublethal concentrations of DFP and E64d. (A) Representative 3-h activity profile of OZ277 in trophozoite-stage parasites in the absence (red) or presence (green) of 10 μM E64d. Values represent the means ± the SEM of three technical replicates. (B) Stability profile of OZ277 (100 nM) in trophozoite-infected RBCs at 10% parasitemia in the absence (red) or presence (green) of 10 μM E64d or 250 μM DFP (black). Values represent the means of two independent experiments or the means ± the SEM of three to four independent experiments. (C) Representative 3-h activity profile of OZ277 in trophozoite-stage parasites in the absence (red) or presence (black) of 250 μM DFP. Values represent the means ± the SEM of three technical replicates. (D) Representative 3-h activity profile of OZ439 in trophozoite-stage parasites in the absence (red) or presence (green) of 10 μM E64d. Values represent the means ± the SEM of three technical replicates. (E) Stability profile of OZ439 (100 nM) in trophozoite-infected RBCs at 10% parasitemia in the absence (red) or presence (green) of 10 μM E64d or 250 μM DFP (black). Values represent the means of two independent experiments or the means ± the SEM of three to four independent experiments. (F) Representative 3-h activity profile of OZ439 in trophozoite-stage parasites in the absence (red) or presence of 250 μM DFP (black). Values represent the means ± the SEM of three technical replicates.
DISCUSSION
The existing literature strongly supports the hypothesis that, like the artemisinins, ozonide antimalarial activity requires activation of the peroxide bond by a reduced iron source (16, 38, 44, 45), inextricably linking compound degradation and antimalarial activity. The requirement for hemoglobin digestion as a prerequisite for antimalarial activity has previously been demonstrated for DHA (27, 28, 30) and has recently also been reported for selected ozonides (34). These studies showed that OZ277 and OZ439 display stage-specific differences in antimalarial activity that correlate with periods of active hemoglobin digestion. The relative lack of sensitivity to a 3-h pulse with an ozonide during the mid-ring stage is also in agreement with previous reports that show ozonides exhibit increased activity at longer exposure times (19, 20, 34). Although the exact molecular target(s) responsible for parasite death after ozonide treatment is incompletely understood, activated peroxides are likely capable of covalently modifying parasite proteins (42), which is thought to ultimately result in parasite death. In the current investigations, we quantified the apparent rate of ozonide degradation in parallel to measuring ozonide activity under conditions of increased parasitemia as commonly used in mechanistic biochemical investigations. Understanding the relationship between degradation, activity, and stage specificity is essential to enable the informed interpretation of mechanistic data generated under these in vitro incubation conditions.
Consistent with previous results in noninfected blood (5), OZ277 degraded in the presence of noninfected RBCs under our standard culture conditions using 2% Hct, whereas OZ439 demonstrated minimal degradation. Most of the iron in noninfected RBCs is bound in the form of hemoglobin and concentrations of free heme are low (46). Noninfected erythrocytes also contain a pool of free iron known as the labile iron pool (LIP) (47) in which iron exists in a chelatable form (48) that is at least partially reduced (49). DFP, a membrane-permeable iron chelator, selectively chelates ferric iron, which in turn shifts the equilibrium of iron species from the ferrous to the ferric form. In the presence of DFP, the rate of OZ277 degradation was significantly reduced (Table 1) consistent with the hypothesis that free, chelatable iron, most likely as part of the LIP, is responsible for the degradation of OZ277 in noninfected RBCs. Previous studies have shown that ferric iron has no impact on the degradation of OZ277 (38). Unlike OZ277, OZ439 displays limited reactivity toward free Fe2+ (5), likely explaining the relative stability observed for OZ439 in noninfected RBCs.
Both OZ277 and OZ439 displayed a higher rate of degradation when incubated with trophozoite-stage parasites compared to either ring stages or noninfected RBCs (Fig. 2A and B; see also Fig. S2A and B in the supplemental material). Although OZ439 was generally more stable than OZ277, the apparent rate of degradation of both compounds was significantly higher under conditions of high parasitemia (10 and >90%) for trophozoite-stage parasites, but less so for the mid-ring stage (see Fig. S2A and B in the supplemental material). Parasitic hemoglobin digestion is most active during the trophozoite stage of the parasite asexual life cycle (50) and liberates a large amount of redox-active free heme. The cell-permeable falcipain inhibitor E64d can be used to block hemoglobin degradation (leading to an accumulation of undigested hemoglobin) and subsequent heme release in P. falciparum (27, 51), while DFP is capable of depleting free Fe2+ in the parasite LIP (52). Prolonged exposure to either compound alone may be detrimental to the parasite. However, under the conditions tested, short incubations of E64d or DFP alone had no detectable effect on parasite growth, a finding consistent with previous reports (27, 34). Stabilization of both ozonides in parasitized RBCs (10% parasitemia) by E64d, but not by DFP (Fig. 4), is consistent with a parasite-mediated hemoglobin degradation product, most likely free heme, contributing to the parasitemia-dependent degradation of the parent ozonides in trophozoites. Previous in vitro studies have shown that the ozonides are stable in the presence of intact hemoglobin (53) but susceptible to heme-catalyzed degradation (37), further demonstrating the role of hemoglobin digestion in promoting ozonide degradation. The lack of a measurable effect of DFP on ozonide stability indicates free ferrous iron does not have a significant role in ozonide degradation under high parasitemia conditions (Fig. 4 and Table 1). For OZ439, the rate of degradation in the presence of E64d returned to levels similar to that seen in noninfected RBCs. Although the rate of OZ277 degradation was significantly reduced after the addition of E64d, it did not return to the noninfected control level, suggesting other factors may be contributing to in vitro degradation of OZ277 that are unrelated to antimalarial activity. This could potentially include aqueous mediated degradation pathways, common to first generation ozonides like OZ277 (54) or interaction with intraparasitic flavin cofactors, similar to that described for the artemisinins (55).
The measured rate of ozonide degradation in this experimental system reflects the contributions from each of the individual degradation processes that occur simultaneously in the different cell populations (i.e., noninfected and infected RBCs; Fig. 5). Ozonides within a P. falciparum infected RBC culture environment are in equilibrium between protein- (or lipid)-bound and free (unbound) drug, the latter of which can diffuse across membranes and enter infected and noninfected RBCs. The apparent rate of ozonide degradation in infected RBCs is substantially greater than that in noninfected RBCs (Table 1). Therefore, parasite load (the number of infected RBCs per μl), which is dependent on both parasitemia and Hct, is the primary determinant of the overall rate of degradation.
FIG 5.

Proposed pathways for ozonide degradation in an asynchronous P. falciparum culture. In an in vitro system, an equilibrium exists between ozonide (OZ) that is bound to proteins or lipids (OZBound) and free (unbound) drug (OZFree) that is available to permeate into an infected or noninfected RBC. The parasite stage (ring or trophozoite) and the relative proportion of each cell population (ring infected, trophozoite infected, and noninfected cells) differentially contribute to ozonide degradation. Hemoglobin-derived free heme (shown as heme) is the most significant catalyst, leading to degraded ozonide (OZ*) and is most abundant in trophozoite-infected RBC when hemoglobin digestion is most active. Trophozoite-mediated degradation is therefore the dominant ozonide degradation pathway (dark bold arrows), whereas the relative contribution of ring-stage parasites is less substantial (bold arrows) since hemoglobin digestion is not as active. In all cell populations, free ferrous iron as part of the labile iron pool (Fe2+) likely plays a minor role in degradation. Paired and single arrows represent reversible and irreversible processes, respectively, and bolder arrows represent more dominant pathways.
Similar to previous results that used the Fe2+-specific iron chelator bipyridyl (34), iron chelation with DFP had no appreciable affect on ozonide antimalarial activity (Fig. 4 and Table 1). However, inhibition of in vitro antiparasitic activity by coincubation with E64d supports the prevailing hypothesis that heme-mediated activation of the parent ozonide, rather than free iron-mediated activation, is essential for antimalarial activity. In this context, it is uncertain how a higher rate of degradation (or activation) observed with increased parasite load, might affect antimalarial potency. Our results show that at high trophozoite-stage parasitemia, the increased apparent rate of ozonide degradation parallels a significant and progressive reduction in in vitro antimalarial activity. Indeed, at the extreme conditions of >90% parasitemia, a 3-h pulse with either ozonide had no detectable effect on parasite growth (Fig. 2C and D). It is noted that the 3 h incubation at high parasitemia alone does not detrimentally affect parasite viability, since the enriched culture (>90% parasitemia) drug-free controls demonstrated the same growth as untreated 1% parasitemia culture when each condition was diluted to 0.5% parasitemia and incubated over 48 h. Interestingly, the rate of degradation of DHA also increases in the presence of P. falciparum infection (45% Hct) compared to noninfected RBCs (see Table S3 in the supplemental material) and antimalarial activity to a 3-h pulse is reduced when trophozoite-stage parasitemia increases from 1 to 10% (see Fig. S5 in the supplemental material). Together with the current data for OZ277 and OZ439, this indicates that parasite-mediated degradation and reduced activity under in vitro conditions of high trophozoite parasitemia could be a property of all peroxide antimalarials.
The activity of peroxide antimalarials is governed by parasite stage, drug concentration, and exposure time (34), while artemisinin resistance is associated with multiple mutations in the parasite protein K13 (56, 57). Recent evidence suggests that the duration of exposure necessary for effective killing is greater in artemisinin-resistant parasites compared to the wild type (34) and that the degree of K13 mutant susceptibility to OZ277 and OZ439 is dependent on the type of K13 mutation (35). Although the mechanism of artemisinin resistance remains elusive, some reports have suggested that it may involve an elevated stress response that enables the parasite to withstand the impact of short-term radical-mediated cellular damage (58). In our studies, the increase in trophozoite parasitemia results in a substantially reduced duration of parasite exposure to intact drug. Under conditions of very high parasitemia (>90%) where the duration of exposure and antimalarial activity are both limited, it is plausible that wild-type P. falciparum stress response mechanisms are able to overcome the short-lived radical-mediated damage until the drug is effectively depleted, resulting in an apparent loss of antimalarial activity.
Studies recently reported by Ismail et al. described a number of alkylated parasite proteins following treatment of high parasitemia, trophozoite-stage cultures with modified ozonide probes (42). Although the activity of the modified ozonide probes was not reported under the same conditions, our observations would suggest the possibility for limited antimalarial activity under these conditions used, assuming of course that the ozonide probes displayed similar stability properties to OZ277 and OZ439. Our results indicate that rapid peroxide activation is not sufficient to induce substantial antiparasitic activity when the duration of exposure to parent drug is limited. Therefore, the biochemical impact of peroxide antimalarials observed under in vitro conditions of high parasitemia should be interpreted with caution when investigating mechanisms of antimalarial activity.
The reduced activity of ozonide antimalarials at higher levels of in vitro parasitemia could also be related to the increased number of parasite targets that need to be engaged for growth inhibition to occur. This phenomenon is similar to the previously described inoculum effect, where increased concentrations of drug are required to inhibit growth as the concentration of microorganisms is increased. The inoculum effect has been described for various antimalarials, including chloroquine (59), quinine, mefloquine, halofantrine, and DHA (60). Antimalarials, such as chloroquine and mefloquine, that do not undergo parasite-mediated degradation, exhibit only a 2- to 3-fold reduction in activity in sensitive parasites for a 10-fold increase in parasitemia and the mechanism has been attributed to depletion of drug from the extracellular medium (60). For peroxides that mediate parasite killing by irreversible target modification, we hypothesize that the amount of activated drug per parasite, rather than the initial drug concentration in the culture system, may be the primary determinant of drug action. Therefore, increasing the parasite load effectively decreases the amount of drug available to inhibit each parasite, resulting in the observed decrease in antiparasitic activity. The pronounced decrease in potency of >10-fold in going from 1% parasitemia to 10% parasitemia or higher, and the corresponding progressive increase in the rate of drug degradation, may suggest that a combination of this modified inoculum effect and limited duration of exposure due to rapid degradation of the parent drug could be the basis for reduced ozonide potency at high in vitro parasitemia.
All measurements of ozonide stability in the present study were determined using an initial ozonide concentration of 100 nM. Since we have not explored whether degradation rates are concentration dependent, these degradation rate constants are specific for this particular starting concentration. Although it is not possible to quantitatively extrapolate this in vitro data to what might be seen in vivo, the results do raise interesting questions regarding possible clinical implications under conditions of hyperparasitemia. In the case of arterolane (OZ277), reduced plasma concentrations are seen in malaria patients compared to healthy volunteers (9), which is consistent with the increased rate of peroxide degradation in the presence of parasites as detected in vitro, although this effect is somewhat attenuated when OZ277 is combined with piperaquine (9). It was also noted that as patient parasitemia levels were reduced there was a corresponding increase in plasma concentrations of arterolane after daily dosing (9), providing further support for the role of parasites in peroxide clearance. Additional clinical studies have also shown malaria-infected patients with a high parasite burden (either increased parasitemia, Hct, or both) have increased rates of recrudescence compared to patients with lower parasitemias, when treated with the peroxide antimalarial, artesunate (61), so it is interesting to speculate whether enhanced drug degradation at high parasite load could have impacted peroxide exposure and antimalarial efficacy. The undertreatment of hyperparasitemic malaria patients is a contributing factor to drug resistance in the field (62) and, as such, the potential influence of parasite load on heme-mediated clearance of peroxide antimalarials clearly warrants further investigation.
Conclusion.
This study systematically demonstrates that parasite-mediated degradation of selected ozonide antimalarials is associated with a significant reduction in activity under in vitro conditions of high trophozoite parasitemia. In addition, ozonide degradation is more rapid when exposed to parasites in the trophozoite stage compared to the ring stage of the asexual life cycle, is positively correlated with parasite load, and can be stabilized in the presence of the falcipain inhibitor E64d. These results indicate a balance between heme-mediated activation and degradation appears to be critical for optimal in vitro activity of these drugs. This has implications for in vitro mechanistic studies that are conducted under conditions that substantially reduce ozonide stability and antimalarial activity and, as such, parasite load should be considered when making associations between peroxide antimalarial activity and biochemical perturbations in vitro.
MATERIALS AND METHODS
Reagents.
SYBR green I nucleic acid staining dye (10,000× in dimethyl sulfoxide [DMSO]) was purchased from Life Technologies. E64d and potassium dichromate (K2Cr2O7) were from Sigma-Aldrich. Acetonitrile (ACN) was LC-MS grade (B&J Brand). OZ277 and OZ439 were provided by the Medicines for Malaria Venture (Geneva). Human RBCs and human plasma were obtained from the Australian Red Cross Blood Service in Melbourne (Victoria, Australia).
Culturing of parasites and synchronization.
The 3D7 strain of P. falciparum was maintained in continuous culture in human RBCs suspended with RPMI 1640 medium (10.4 g/liter), HEPES (5.94 g/liter), hypoxanthine (50 mg/liter), NaHCO3 (2.1 g/liter), and Albumax II (5 g/liter) at 37°C under a gas environment of 94% N2, 5% CO2, and 1% O2, as previously described (63). To examine the effect of parasite stage on drug activity and rate of drug degradation, synchronized cultures were achieved using multiple treatments with 5% sorbitol (64). Synchronization and parasitemia were confirmed by light-microscopic evaluation of Giemsa-stained thin blood smears (>600 parasites counted per slide), and the Hct was determined by counting cells with a hemocytometer.
Washing procedure and drug pulse activity assays.
Two different washing procedures were explored using in vitro culture conditions of 2% Hct and varying parasitemia in 200 μl of culture medium. The standard procedure involved four washes with 200 μl of complete RPMI culture medium supplemented with 0.5% Albumax II. The optimized method included four washes with 200 μl of complete RPMI culture medium supplemented with 2 to 5% Albumax II and involved transferring cultures to a fresh plate after the last wash. The effectiveness of each method was assessed by incubating various concentrations of OZ277 or OZ439 with noninfected RBCs for 3 h and then removing the drug using either the standard or optimized washing procedure and testing for residual drug activity by adding infected RBCs to the washed cells. Parasite growth was then determined after 48 h relative to an untreated control. Quantification of the concentration of drug remaining in the wells after washing was performed by LC-MS as described below.
Parasite sensitivity to the ozonides was assessed using drug pulse activity assays as described previously (65). Briefly, parasites of a specific age were exposed to drug for a defined period of time and parasite viability determined in the next asexual cycle after the drug was removed by washing. Drug stock solutions in DMSO were first diluted with 50% ACN/H2O and then serially diluted with 50% ACN/H2O in V-bottom microplates to provide the desired concentration range for spiking into the infected RBC culture. Spiking solutions (5 μl) were then added to the appropriate wells of a flat-bottom microplate containing the synchronized mid-ring- or mid-trophozoite-stage parasites so that a final Hct of 2% and either 1 or 10% parasitemia was achieved. Experiments using enriched trophozoite cultures used a commercially available magnetic cell fractionation system (Miltenyi Biotec) to magnetically separate synchronized mid-trophozoite-stage parasites from noninfected RBCs (66). After the magnetic harvest, the mid-trophozoite-stage culture was incubated at 37°C for 1 h, and the Hct was adjusted to 2%. Spiking solutions of drug were prepared as described above and added to the appropriate wells of a flat-bottom microplate containing the enriched culture so that a final Hct of 2% and enriched trophozoite parasitemia was achieved.
Cultures were then incubated with drug for 3 h at 37°C under a gas environment of 94% N2, 5% CO2, and 1% O2. At the end of the incubation period, the drug was removed by washing cultures in a V-bottom microplate using the optimized washing protocol described previously. For all levels of parasitemia tested (1%, 10%, and enriched), cultures were then adjusted to 0.5% parasitemia and 2% Hct (final volume, 200 μl) to support parasite reinvasion into the next asexual cycle and prevent collapse of high parasitemia cultures. This was achieved by resuspending a proportion of the infected culture from each well into a flat-bottom microplate containing complete RPMI medium and fresh noninfected RBCs so that the desired final culture conditions were obtained. Cultures were incubated under these conditions (2% final Hct; 0.5% final parasitemia) for 48 to 72 h at 37°C and a gas environment of 94% N2, 5% CO2, and 1% O2. Unwashed samples matching the 48 h growth conditions (2% final Hct; 0.5% final parasitemia) that were maintained under lethal drug pressure for 48 to 72 h acted as controls for 100% parasite killing, and infected erythrocytes that were incubated in the absence of drug served as controls for parasite growth. Assays were performed in duplicate or triplicate in at least two independent experiments using RBCs from different donors.
After 48 to 72 h of growth, parasite drug susceptibility was assessed by measurement of SYBR green I fluorescence as previously described (67). Briefly, lysis buffer containing 0.1 μl/ml SYBR green I was added to each well, and the contents were mixed until no visible erythrocyte sediment remained. After 1 to 2 h of incubation in the dark at room temperature, fluorescence was measured using an Enspire plate reader (Perkin-Elmer) with excitation and emission wavelengths of 485 and 530 nm, respectively. The fluorescence values were plotted against the logarithm of the drug concentration, and the data analyzed using Prism (v6.05; GraphPad Software, San Diego, CA) by nonlinear regression [log(inhibitor) versus normalized response] to yield the drug concentration that produced 50% growth inhibition relative to the drug-free controls (IC50).
Parasite- and RBC-mediated drug degradation.
Samples for assessment of OZ277 and OZ439 degradation were prepared in parallel to the parasite sensitivity samples using the same batch of noninfected and P. falciparum (3D7 strain)-infected RBCs. Parasite cultures (1 and 10% parasitemia, 2% Hct) and noninfected RBCs (2% Hct) were spiked with test compound to a final concentration of 100 nM in a flat-bottom microplate. The plate was incubated at 37°C under a gas environment of 94% N2, 5% CO2, and 1% O2. At each sampling point (between 0 and 6 h), 10 μl of 0.4 M K2Cr2O7 was added to the appropriate culture well in the flat-bottom plate to immediately oxidize Fe2+ to Fe3+. Proteins were then precipitated by addition of 300 μl of ACN (containing 150 ng/ml of diazepam as the internal standard), and the samples were transferred to microcentrifuge tubes, vortex mixed, and allowed to extract on ice for 10 min. After centrifugation for 5 min at 4°C, 150 μl of supernatant was transferred to an analytical vial and stored at 4°C until analysis within 8 h.
For each LC-MS analytical run, calibration standards were freshly prepared in noninfected RBCs suspended in complete culture medium (2% Hct). Aliquots (100 μl) were first stabilized with 10 μl of 0.4 M K2CrO7 and then spiked with compound stock solution (prepared in 50% ACN/H2O) to achieve concentrations typically between 3 and 250 nM. Calibration standards were then vortex mixed, extracted on ice for 10 min, and centrifuged at 4°C for 5 min. Supernatant (150 μl) was then transferred to analytical vials for analysis.
LC-MS analysis.
Drug stability samples were analyzed using a Shimadzu 8050 triple quadrupole mass spectrometer coupled with a Shimadzu high-pressure liquid chromatography system. Analytical separation was performed on an Ascentis Express C18 reversed-phase column (50 by 2.1 mm, 2.7 μm) with a guard column of the same material (Sigma-Aldrich). Compounds were eluted using a binary gradient solvent system consisting of Milli-Q water with 0.05% formic acid (solvent A) and ACN (solvent B). The gradient profile was as follows: 0 to 0.2 min, 2% B; 0.2 to 0.3 min, 2 to 10% B; 0.3 to 2.7 min, 10 to 70% B; 2.7 to 2.8 min, 70 to 95% B; 2.8 to 3.3 min, 95% B; 3.3 to 3.5 min, 95 to 2% B; and 3.5 to 4 min, 2% B. The peaks of interest eluted between 2 and 3 min at a flow rate of 0.4 ml/min. Mass spectrometry was performed at heat block and desolvation temperatures of 250°C and a heating and drying gas flow of 10 liters/min. Nebulizing gas flow was set at 3 liters/min. The interface voltage and interface temperature were 2 kV and 300°C, respectively. Compounds were monitored by multiple reaction monitoring (MRM) employing electrospray ionization in positive mode. The m/z fragmentation transitions for MRM of OZ277, OZ439, and diazepam were m/z 393.2 to 227.1, m/z 470.1 to 304.1, and m/z 285 to 154.1, respectively. The cone voltage for the different compounds was optimized by infusing individual working stock solutions and ranged from 14 to 32 V. Data were acquired and processed using the Lab Solutions software.
Analyte concentrations were determined by comparison to calibration standards prepared in the same matrix. Quality control (QC) samples were also included in the analysis of the study samples. The validity of each analytical run was determined by ensuring the regression coefficient (R2) for the calibration curve was >0.999 and the measured concentration of the QC samples was within ±15% of the nominal concentration. The limits of quantitation for OZ277 and OZ439 were determined to be 8 and 4 nM, respectively. Experimental degradation data were fit to a first-order exponential decay function, based on initial rates.
Effect of iron chelation and cysteine protease inhibition on ozonide degradation and antimalarial activity.
A stock solution of the iron chelator DFP was prepared at 500 mM in water by dropwise addition of 1 M HCl until complete dissolution occurred. A stock solution of the cysteine protease inhibitor E64d (10 mM) was prepared in DMSO. Synchronized mid-ring- and mid-trophozoite-stage 3D7 parasites at 10% parasitemia were pretreated for 30 min with either DFP (250 μM) or E64d (10 μM), followed by incubation with OZ277 or OZ439 for an additional 3 h. Parasite sensitivity and the rate of drug degradation were determined using the SYBR green I and LC-MS assays, respectively, as described above.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Australian Red Cross Blood Service for their donation of human RBCs and plasma, Phil Wright and Kim Deadman of the Helen Macpherson Smith Trust lab from Monash Institute for Pharmaceutical Sciences (MIPS) for advice and technical assistance with the LC-MS instruments, and Rahul Patil, Francis Chiu, and Jessica Saunders from the Centre for Drug Candidate Optimization at MIPS for reagents, LC-MS advice, and technical assistance.
Partial financial support was provided by the National Health and Medical Research Council (grants APP1128003 and APP1088855).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01566-17.
REFERENCES
- 1.World Health Organization. 2016. World malaria report 2016. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.World Health Organization. 2015. Guidelines for the treatment of malaria, 3rd ed World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, et al. 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charman SA, Arbe-Barnes S, Bathurst IC, Brun R, Campbell M, Charman WN, Chiu FC, Chollet J, Craft JC, Creek DJ, Dong Y, Matile H, Maurer M, Morizzi J, Nguyen T, Papastogiannidis P, Scheurer C, Shackleford DM, Sriraghavan K, Stingelin L, Tang Y, Urwyler H, Wang X, White KL, Wittlin S, Zhou L, Vennerstrom JL. 2011. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci U S A 108:4400–4405. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moehrle JJ, Duparc S, Siethoff C, van Giersbergen PL, Craft JC, Arbe-Barnes S, Charman SA, Gutierrez M, Wittlin S, Vennerstrom JL. 2013. First-in-man safety and pharmacokinetics of synthetic ozonide OZ439 demonstrates an improved exposure profile relative to other peroxide antimalarials. Br J Clin Pharmacol 75:524–537. doi: 10.1111/j.1365-2125.2012.04368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phyo AP, Jittamala P, Nosten FH, Pukrittayakamee S, Imwong M, White NJ, Duparc S, Macintyre F, Baker M, Möhrle JJ. 2015. Antimalarial activity of artefenomel (OZ439), a novel synthetic antimalarial endoperoxide, in patients with Plasmodium falciparum and Plasmodium vivax malaria: an open-label phase 2 trial. Lancet Infect Dis 16:61–69. doi: 10.1016/S1473-3099(15)00320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCarthy JS, Baker M, O'Rourke P, Marquart L, Griffin P, van Huijsduijnen RH, Möhrle JJ. 2016. Efficacy of OZ439 (artefenomel) against early Plasmodium falciparum blood-stage malaria infection in healthy volunteers. J Antimicrob Chemother 71:2620–2627. doi: 10.1093/jac/dkw174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gautam A, Ahmed T, Sharma P, Varshney B, Kothari M, Saha N, Roy A, Moehrle JJ, Paliwal J. 2011. Pharmacokinetics and pharmacodynamics of arterolane maleate following multiple oral doses in adult patients with Plasmodium falciparum malaria. J Clin Pharmacol 51:1519–1528. doi: 10.1177/0091270010385578. [DOI] [PubMed] [Google Scholar]
- 10.Valecha N, Krudsood S, Tangpukdee N, Mohanty S, Sharma S, Tyagi P, Anvikar A, Mohanty R, Rao B, Jha A. 2012. Arterolane maleate plus piperaquine phosphate for treatment of uncomplicated Plasmodium falciparum malaria: a comparative, multicenter, randomized clinical trial. Clin Infect Dis 55:663–671. doi: 10.1093/cid/cis475. [DOI] [PubMed] [Google Scholar]
- 11.Valecha N, Looareesuwan S, Martensson A, Abdulla SM, Krudsood S, Tangpukdee N, Mohanty S, Mishra SK, Tyagi P, Sharma S. 2010. Arterolane, a new synthetic trioxolane for treatment of uncomplicated Plasmodium falciparum malaria: a phase II, multicenter, randomized, dose-finding clinical trial. Clin Infect Dis 51:684–691. doi: 10.1086/655831. [DOI] [PubMed] [Google Scholar]
- 12.Saha N, Moehrle JJ, Zutshi A, Sharma P, Kaur P, Iyer SS. 2014. Safety, tolerability and pharmacokinetic profile of single and multiple oral doses of arterolane (RBx11160) maleate in healthy subjects. J Clin Pharmacol 54:386–393. doi: 10.1002/jcph.232. [DOI] [PubMed] [Google Scholar]
- 13.Posner GH, Oh CH, Wang D, Gerena L, Milhous WK, Meshnick SR, Asawamahasadka W. 1994. Mechanism-based design, synthesis, and in vitro antimalarial testing of new 4-methylated trioxanes structurally related to artemisinin: the importance of a carbon-centered radical for antimalarial activity. J Med Chem 37:1256–1258. doi: 10.1021/jm00035a003. [DOI] [PubMed] [Google Scholar]
- 14.Dong Y, Chollet J, Matile H, Charman SA, Chiu FC, Charman WN, Scorneaux B, Urwyler H, Santo Tomas J, Scheurer C. 2005. Spiro- and dispiro-1,2,4-trioxolanes as antimalarial peroxides: charting a workable structure-activity relationship using simple prototypes. J Med Chem 48:4953–4961. doi: 10.1021/jm049040u. [DOI] [PubMed] [Google Scholar]
- 15.Kaiser M, Wittlin S, Nehrbass-Stuedli A, Dong Y, Wang X, Hemphill A, Matile H, Brun R, Vennerstrom JL. 2007. Peroxide bond-dependent antiplasmodial specificity of artemisinin and OZ277 (RBx11160). Antimicrob Agents Chemother 51:2991–2993. doi: 10.1128/AAC.00225-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang Y, Dong Y, Wang X, Sriraghavan K, Wood JK, Vennerstrom JL. 2005. Dispiro-1,2,4-trioxane analogues of a prototype dispiro-1,2,4-trioxolane: mechanistic comparators for artemisinin in the context of reaction pathways with iron(II). J Org Chem 70:5103–5110. doi: 10.1021/jo050385+. [DOI] [PubMed] [Google Scholar]
- 17.Skinner TS, Manning LS, Johnston WA, Davis TM. 1996. In vitro stage-specific sensitivity of Plasmodium falciparum to quinine and artemisinin drugs. Int J Parasitol 26:519–525. doi: 10.1016/0020-7519(96)89380-5. [DOI] [PubMed] [Google Scholar]
- 18.Geary TG, Divo AA, Jensen JB. 1989. Stage-specific actions of antimalarial drugs on Plasmodium falciparum in culture. Am J Trop Med Hyg 40:240–244. doi: 10.4269/ajtmh.1989.40.240. [DOI] [PubMed] [Google Scholar]
- 19.Hofer S, Brun R, Maerki S, Matile H, Scheurer C, Wittlin S. 2008. In vitro assessment of the pharmacodynamic properties of DB75, piperaquine, OZ277, and OZ401 in cultures of Plasmodium falciparum. J Antimicrob Chemother 62:1061–1064. doi: 10.1093/jac/dkn315. [DOI] [PubMed] [Google Scholar]
- 20.Maerki S, Brun R, Charman SA, Dorn A, Matile H, Wittlin S. 2006. In vitro assessment of the pharmacodynamic properties and the partitioning of OZ277/RBx-11160 in cultures of Plasmodium falciparum. J Antimicrob Chemother 58:52–58. doi: 10.1093/jac/dkl209. [DOI] [PubMed] [Google Scholar]
- 21.Delves M, Plouffe D, Scheurer C, Meister S, Wittlin S, Winzeler EA, Sinden RE, Leroy D. 2012. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS Med 9:e1001169. doi: 10.1371/journal.pmed.1001169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meshnick SR, Thomas A, Ranz A, Xu CM, Pan HZ. 1991. Artemisinin (qinghaosu): the role of intracellular hemin in its mechanism of antimalarial action. Mol Biochem Parasitol 49:181–189. doi: 10.1016/0166-6851(91)90062-B. [DOI] [PubMed] [Google Scholar]
- 23.Meshnick SR, Yang YZ, Lima V, Kuypers F, Kamchonwongpaisan S, Yuthavong Y. 1993. Iron-dependent free radical generation from the antimalarial agent artemisinin (qinghaosu). Antimicrob Agents Chemother 37:1108–1114. doi: 10.1128/AAC.37.5.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Posner GH, Oh CH. 1992. Regiospecifically oxygen-18 labeled 1,2,4-trioxane: a simple chemical model system to probe the mechanism (s) for the antimalarial activity of artemisinin (qinghaosu). J Am Chem Soc 114:8328–8329. doi: 10.1021/ja00047a076. [DOI] [Google Scholar]
- 25.Zhang F, Gosser DK, Meshnick SR. 1992. Hemin-catalyzed decomposition of artemisinin (qinghaosu). Biochem Pharmacol 43:1805–1809. doi: 10.1016/0006-2952(92)90713-S. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Zhou B. 2010. Biological actions of artemisinin: insights from medicinal chemistry studies. Molecules 15:1378–1397. doi: 10.3390/molecules15031378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie SC, Dogovski C, Hanssen E, Chiu F, Yang T, Crespo MP, Stafford C, Batinovic S, Teguh S, Charman S. 2016. Haemoglobin degradation underpins the sensitivity of early ring stage Plasmodium falciparum to artemisinins. J Cell Sci 129:406–416. doi: 10.1242/jcs.178830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klonis N, Crespo-Ortiz MP, Bottova I, Abu-Bakar N, Kenny S, Rosenthal PJ, Tilley L. 2011. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci U S A 108:11405–11410. doi: 10.1073/pnas.1104063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Zhang C-J, Chia WN, Loh CC, Li Z, Lee YM, He Y, Yuan L-X, Lim TK, Liu M, Liew CX, Lee YQ, Zhang J, Lu N, Lim CT, Hua Z-C, Liu B, Shen H-M, Tan KSW, Lin Q. 2015. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat Commun 6:10111. doi: 10.1038/ncomms10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klonis N, Xie SC, McCaw JM, Crespo-Ortiz MP, Zaloumis SG, Simpson JA, Tilley L. 2013. Altered temporal response of malaria parasites determines differential sensitivity to artemisinin. Proc Natl Acad Sci U S A 110:5157–5162. doi: 10.1073/pnas.1217452110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Huang L, Li J, Fan Q, Long Y, Li Y, Zhou B. 2010. Artemisinin directly targets malarial mitochondria through its specific mitochondrial activation. PLoS One 5:e9582. doi: 10.1371/journal.pone.0009582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mbengue A, Bhattacharjee S, Pandharkar T, Liu HN, Estiu G, Stahelin RV, Rizk SS, Njimoh DL, Ryan Y, Chotivanich K, Nguon C, Ghorbal M, Lopez-Rubio JJ, Pfrender M, Emrich S, Mohandas N, Dondorp AM, Wiest O, Haldar K. 2015. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 520:683–687. doi: 10.1038/nature14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haynes RK, Cheu KW, Chan HW, Wong HN, Li KY, Tang MM, Chen MJ, Guo ZF, Guo ZH, Sinniah K, Witte AB, Coghi P, Monti D. 2012. Interactions between artemisinins and other antimalarial drugs in relation to the cofactor model-a unifying proposal for drug action. ChemMedChem 7:2204–2226. doi: 10.1002/cmdc.201200383. [DOI] [PubMed] [Google Scholar]
- 34.Yang T, Xie SC, Cao P, Giannangelo C, McCaw J, Creek DJ, Charman SA, Klonis N, Tilley L. 2016. A comparison of the exposure time-dependence of the activities of synthetic ozonide antimalarials and dihydroartemisinin against K13 wild-type and mutant Plasmodium falciparum. Antimicrob Agents Chemother 60:4501–4510. doi: 10.1128/AAC.00574-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Straimer J, Gnädig NF, Stokes BH, Ehrenberger M, Crane AA, Fidock DA. 2017. Plasmodium falciparum K13 mutations differentially impact ozonide susceptibility and parasite fitness in vitro. mBio 8:e00172-17. doi: 10.1128/mBio.00172-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baumgärtner F, Jourdan J, Scheurer C, Blasco B, Campo B, Mäser P, Wittlin S. 2017. In vitro activity of antimalarial ozonides against an artemisinin-resistant isolate. Malar J 16:45. doi: 10.1186/s12936-017-1696-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Creek DJ, Charman WN, Chiu FC, Prankerd RJ, Dong Y, Vennerstrom JL, Charman SA. 2008. Relationship between antimalarial activity and heme alkylation for spiro- and dispiro-1,2,4-trioxolane antimalarials. Antimicrob Agents Chemother 52:1291–1296. doi: 10.1128/AAC.01033-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Creek DJ, Charman WN, Chiu FC, Prankerd RJ, McCullough KJ, Dong Y, Vennerstrom JL, Charman SA. 2007. Iron-mediated degradation kinetics of substituted dispiro-1,2,4-trioxolane antimalarials. J Pharm Sci 96:2945–2956. doi: 10.1002/jps.20958. [DOI] [PubMed] [Google Scholar]
- 39.Ismail HM, Barton V, Phanchana M, Charoensutthivarakul S, Wong MHL, Hemingway J, Biagini GA, O'Neill PM, Ward SA. 2016. Artemisinin activity-based probes identify multiple molecular targets within the asexual stage of the malaria parasites Plasmodium falciparum 3D7. Proc Natl Acad Sci U S A 113:2080–2085. doi: 10.1073/pnas.1600459113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cobbold SA, Chua HH, Nijagal B, Creek DJ, Ralph SA, McConville MJ. 2015. Metabolic dysregulation induced in Plasmodium falciparum by dihydroartemisinin and other front line antimalarial drugs. J Infect Dis 213:276–286. doi: 10.1093/infdis/jiv372. [DOI] [PubMed] [Google Scholar]
- 41.O'Neill PM, Barton VE, Ward SA. 2010. The molecular mechanism of action of artemisinin: the debate continues. Molecules 15:1705–1721. doi: 10.3390/molecules15031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ismail HM, Barton VE, Panchana M, Charoensutthivarakul S, Biagini GA, Ward SA, O'Neill PM. 2016. A click chemistry-based proteomic approach reveals that 1,2,4-trioxolane and artemisinin antimalarials share a common protein alkylation profile. Angew Chem Int Ed Engl 55:6401–6405. doi: 10.1002/anie.201512062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allman EL, Painter HJ, Samra J, Carrasquilla M, Llinás M. 2016. Metabolomic profiling of the malaria box reveals antimalarial target pathways. Antimicrob Agents Chemother 60:6635–6649. doi: 10.1128/AAC.01224-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Dong Y, Wittlin S, Creek D, Chollet J, Charman SA, Tomas JS, Scheurer C, Snyder C, Vennerstrom JL. 2007. Spiro- and dispiro-1,2-dioxolanes: contribution of iron(II)-mediated one-electron versus two-electron reduction to the activity of antimalarial peroxides. J Med Chem 50:5840–5847. doi: 10.1021/jm0707673. [DOI] [PubMed] [Google Scholar]
- 45.Fügi MA, Wittlin S, Dong Y, Vennerstrom JL. 2010. Probing the antimalarial mechanism of artemisinin and OZ277 (arterolane) with nonperoxidic isosteres and nitroxyl radicals. Antimicrob Agents Chemother 54:1042–1046. doi: 10.1128/AAC.01305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scholl PF, Tripathi AK, Sullivan DJ. 2005. Bioavailable iron and heme metabolism in Plasmodium falciparum. Curr Top Microbiol Immunol 295:293–324. [DOI] [PubMed] [Google Scholar]
- 47.Prus E, Fibach E. 2008. The labile iron pool in human erythroid cells. Br J Haematol 142:301–307. doi: 10.1111/j.1365-2141.2008.07192.x. [DOI] [PubMed] [Google Scholar]
- 48.Petrat F, Groot Hd, Sustmann R, Rauen U. 2002. The chelatable iron pool in living cells: a methodically defined quantity. Biol Chem 383:489–502. doi: 10.1515/BC.2002.051. [DOI] [PubMed] [Google Scholar]
- 49.Breuer W, Epsztejn S, Cabantchik ZI. 1995. Iron acquired from transferrin by K562 cells is delivered into a cytoplasmic pool of chelatable iron (II). J Biol Chem 270:24209–24215. doi: 10.1074/jbc.270.41.24209. [DOI] [PubMed] [Google Scholar]
- 50.Francis SE, Sullivan DJ Jr, Goldberg ED. 1997. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol 51:97–123. doi: 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- 51.Rosenthal PJ. 2004. Cysteine proteases of malaria parasites. Int J Parasitol 34:1489–1499. doi: 10.1016/j.ijpara.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 52.Clark M, Fisher NC, Kasthuri R, Cerami Hand C. 2013. Parasite maturation and host serum iron influence the labile iron pool of erythrocyte-stage Plasmodium falciparum. Br J Haematol 161:262–269. doi: 10.1111/bjh.12234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Creek DJ, Ryan E, Charman WN, Chiu FC, Prankerd RJ, Vennerstrom JL, Charman SA. 2009. Stability of peroxide antimalarials in the presence of human hemoglobin. Antimicrob Agents Chemother 53:3496–3500. doi: 10.1128/AAC.00363-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perry CS, Charman SA, Prankerd RJ, Chiu FCK, Dong Y, Vennerstrom JL, Charman WN. 2006. Chemical kinetics and aqueous degradation pathways of a new class of synthetic ozonide antimalarials. J Pharm Sci 95:737–747. doi: 10.1002/jps.20568. [DOI] [PubMed] [Google Scholar]
- 55.Haynes RK, Chan WC, Wong HN, Li KY, Wu WK, Fan KM, Sung HH, Williams ID, Prosperi D, Melato S, Coghi P, Monti D. 2010. Facile oxidation of leucomethylene blue and dihydroflavins by artemisinins: relationship with flavoenzyme function and antimalarial mechanism of action. ChemMedChem 5:1282–1299. doi: 10.1002/cmdc.201000225. [DOI] [PubMed] [Google Scholar]
- 56.Straimer J, Gnadig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Menard D, Fidock DA. 2015. Drug resistance: K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347:428–431. doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois A-C, Khim N, Kim S, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Menard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale J-C, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Menard D. 2014. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dogovski C, Xie SC, Burgio G, Bridgford J, Mok S, McCaw JM, Chotivanich K, Kenny S, Gnädig N, Straimer J, Bozdech Z, Fidock DA, Simpson JA, Dondorp AM, Foote S, Klonis N, Tilley L. 2015. Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol 13:e1002132. doi: 10.1371/journal.pbio.1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gluzman IY, Schlesinger PH, Krogstad DJ. 1987. Inoculum effect with chloroquine and Plasmodium falciparum. Antimicrob Agents Chemother 31:32–36. doi: 10.1128/AAC.31.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duraisingh MT, Jones P, Sambou I, Von Seidlein L, Pinder M, Warhurst DC. 1999. Inoculum effect leads to overestimation of in vitro resistance for artemisinin derivatives and standard antimalarials: a Gambian field study. Parasitology 119:435–440. doi: 10.1017/S0031182099004953. [DOI] [PubMed] [Google Scholar]
- 61.Ittarat W, Pickard AL, Rattanasinganchan P, Wilairatana P, Looareesuwan S, Emery K, Low J, Udomsangpetch R, Meshnick SR. 2003. Recrudescence in artesunate-treated patients with falciparum malaria is dependent on parasite burden not on parasite factors. Am J Trop Med Hyg 68:147–152. [PubMed] [Google Scholar]
- 62.White NJ, Pongtavornpinyo W, Maude RJ, Saralamba S, Aguas R, Stepniewska K, Lee SJ, Dondorp AM, White LJ, Day NP. 2009. Hyperparasitaemia and low dosing are an important source of anti-malarial drug resistance. Malar J 8:253. doi: 10.1186/1475-2875-8-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 64.Lambros C, Vanderberg JP. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65:418–420. doi: 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- 65.Xie SC, Dogovski C, Kenny S, Tilley L, Klonis N. 2014. Optimal assay design for determining the in vitro sensitivity of ring stage Plasmodium falciparum to artemisinins. Int J Parasitol 44:893–899. doi: 10.1016/j.ijpara.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 66.Mata-Cantero L, Lafuente M, Sanz L, Rodriguez M. 2014. Magnetic isolation of Plasmodium falciparum schizonts iRBCs to generate a high parasitaemia and synchronized in vitro culture. Malar J 13:112–120. doi: 10.1186/1475-2875-13-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. 2004. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother 48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.