

ABSTRACT
Fungal infections frequently affect immunodeficient individuals and are estimated to kill 1.35 million people per annum. Azole antifungals target the membrane-bound cytochrome P450 monooxygenase lanosterol 14α-demethylase (CYP51; Erg11p). Mutations in CYP51 can render the widely used triazole drugs less effective. The Candida albicans CYP51 mutation G464S and the double mutation Y132F G464S (Y140F and G464S by Saccharomyces cerevisiae numbering) as well as the CYP51A G54E/R/W mutations of Aspergillus fumigatus (G73E/R/W by S. cerevisiae numbering) have been reproduced in a recombinant C-terminal hexahistidine-tagged version of S. cerevisiae CYP51 (ScErg11p6×His). Phenotypes and X-ray crystal structures were determined for the mutant enzymes. Liquid microdilution assays showed that the G464S mutation in ScErg11p6×His conferred no difference in the susceptibility of yeast to triazole drugs but in combination with the Y140F mutation gave a 4-fold reduction in susceptibility to the short-tailed triazole fluconazole. The ScErg11p6×His Y140F G464S mutant was unstable during purification and was not crystallized. The ScErg11p6×His G73E/R/W mutations conferred increased susceptibly to all triazoles tested in liquid microdilution assays. High-resolution X-ray crystal structures reveal two different conformations of the ligand itraconazole, including a previously unseen conformation, as well as interactions between the tail of this triazole and the E/W73 residue. This study shows that S. cerevisiae CYP51 adequately represents some but not all mutations in CYP51s of pathogenic fungi. Insight into the molecular mechanisms of resistance mutations in CYP51 will assist the development of inhibitors that will overcome antifungal resistance.
KEYWORDS: CYP51, X-ray crystallography, antifungal resistance, drug resistance mechanisms, fluconazole, fungal infections, itraconazole, lanosterol 14α-demethylase, voriconazole
INTRODUCTION
Fungal infections cause a variety of debilitating conditions in humans, animals, and plants. While superficial fungal infections are usually readily treated, invasive fungal infections that afflict the immunocompromised and individuals with a range of comorbidities have been estimated to kill about 1.35 million people annually (1). Candida and Aspergillus species are among the most common causes of fungal infection in humans. Aspergillus infections that cause chronic pulmonary aspergillosis and allergic bronchopulmonary aspergillosis in immunocompetent individuals affect millions of people worldwide (2). Immunosuppressed patients suffer potentially lethal infections such as invasive aspergillosis (IA), which affects >300,000 people worldwide (Global Action Fund for Fungal Infections [http://www.gaffi.org/why/fungal-disease-frequency/]). Candida albicans is a commensal that causes superficial infections on mucous membranes and lethal disseminated infections in hosts with impaired physical or immune defenses. This pathogen is a major cause of hospital-acquired bloodstream infections in vulnerable individuals, with mortality rates of 30 to 55% (3, 4).
The azole antifungals are a class of commonly used medications and agrochemicals. They target the membrane-bound enzyme lanosterol 14α-demethylase (CYP51; Erg11p), a member of the CYP51 class of monooxygenases in the cytochrome P450 superfamily, which catalyzes the rate-limiting step in ergosterol biosynthesis. The resistance of fungal pathogens to triazoles due to mutations in CYP51 reduces therapeutic options and makes treatment problematic. Furthermore, the widespread use of these azoles as fungicides in agriculture has led to cross-resistance between medical and agricultural triazoles (5, 6).
Aspergillus fumigatus is the most frequent cause of IA. A. fumigatus harbors two isoforms of lanosterol 14α-demethylase (CYP51A and CYP51B) (7). The pathogen's intrinsic resistance to the widely used triazole fluconazole (FLC) is thought to be due to the CYP51A isoform (8), but the molecular basis for resistance has yet to be elucidated. Voriconazole (VCZ) is not affected by this intrinsic resistance mechanism and has been the drug of choice for the treatment of IA (9). However, significant toxicity and pharmacokinetic issues require therapeutic monitoring during treatment with VCZ (10). The long-tailed triazoles itraconazole (ITC) and posaconazole (PCZ) are used as prophylactic treatments for aspergillosis. The long-term prophylactic use of ITC leads to the pathogen acquiring resistance affecting glycine 54 (G54) in CYP51 (11), which confers cross-resistance to the long-tailed triazole PCZ (12) but not to the short-tailed triazole VCZ (13, 14) or the medium-tail-length triazole isavuconazole (ISA) (15). The recent discovery of G54 mutations in environmental isolates of A. fumigatus (16) suggests that agricultural fungicides may also drive selection for this mutation. Early studies that expressed A. fumigatus CYP51A (AfCYP51A) G54 mutants in Saccharomyces cerevisiae, with native CYP51 repressed by using a tetracycline-regulated promoter, confirmed the reduced susceptibility of these strains to ITC and PCZ and the retention of susceptibility to VCZ (14). Structure-based sequence alignment using the full-length structure of His-tagged S. cerevisiae CYP51 in complex with ITC (ScErg11p6×His) (PDB accession number 5EQB) (17) as a template shows that A. fumigatus CYP51A G54 (G73 in S. cerevisiae) is located at the entry to the substrate channel. This suggests that mutation to a bulky residue might disrupt the interaction of long-tailed azoles with the mouth of the substrate channel, while the binding of short- and medium-tailed azoles that interact with only the active site is less likely to be affected.
The A. fumigatus CYP51A G448S mutation is located closer to the active site on the proximal side of the heme. It has an effect opposite that of the G73 mutations and confers resistance to the short-tailed triazole VCZ but not the long-tailed triazole ITC or PCZ (18, 19). The equivalent mutation is also found in C. albicans CYP51 (CaCYP51). The CaCYP51 G464S mutation confers resistance to FLC alone or in combination with other mutations (20–22). Enzyme kinetic studies using microsomal preparations from S. cerevisiae cells expressing the CaCYP51 G464S mutant found that the mutant enzyme has a reduced affinity for FLC and lower catalytic activity than the wild-type enzyme (23). It was suggested that the position of the heme within the active site could be altered due to the mutation.
The G464S mutation in CaCYP51 has been found in combination with the Y132H and R467K mutations as well as with the Y132H and H283R mutations (20, 21). The Y132H and G464S mutations each conferred a 4-fold increase in resistance to FLC compared to the wild-type enzyme. When both mutations were present in CaCYP51, the resistance of the strain to FLC was increased 32-fold (20). The residues equivalent to Y132H and G464S in ScCYP51 (Y140H and G464S) are located on the opposite sides of the heme (Fig. 1) (24). We previously demonstrated that the Y140F/H mutations in ScErg11p6×His eliminate the hydrogen bond between the heme ring C propionate group and the hydroxyl group of Y140 as well as water-mediated hydrogen bonds to the short-tailed azoles FLC and VCZ (25). Based on the structure of ScErg11p6×His in complex with FLC (PDB accession number 4WMZ) (24), we propose that the mutant G464S hydroxyl group replaces a water molecule that makes a hydrogen bond to the heme ring D propionate.
FIG 1.
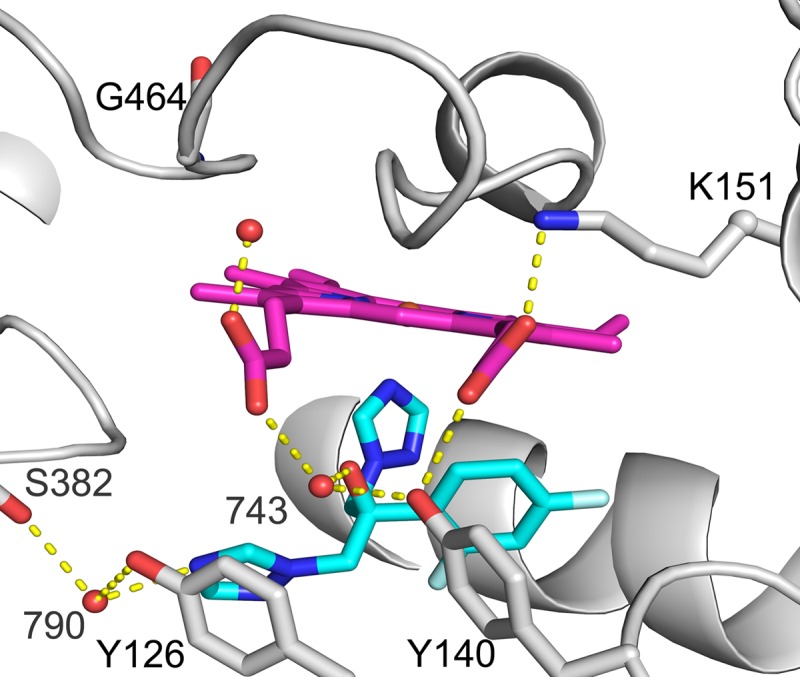
Fluconazole bound to the active site of wild-type ScErg11p6×His. Shown is a cartoon representation of ScErg11p6×His (PDB accession number 4WMZ) with heme (magenta), fluconazole (cyan), and residues Y140, Y126, G464, S382, and K151 shown as sticks. Water molecules are shown as red spheres, and hydrogen bonds are shown as yellow dotted lines.
To date, the crystal structures of CYP51s from S. cerevisiae, A. fumigatus (CYP51B), Candida glabrata (CgCYP51), and C. albicans have been reported. The full-length structures for ScCYP51 have been obtained in complex with the substrate lanosterol; the triazoles ITC, VCZ (17), and FLC (24); the tetrazole VT-1161 (PDB accession number 5UL0), and several agrochemical antifungals (26). Structures of N-terminally truncated C. albicans CYP51 in complex with PCZ and VT-1161 (27) and full-length C. glabrata CYP51 and CaCYP51 in complex with ITC (PDB accession numbers 5JLC and 5V5Z) were released in 2017. Crystal structures of N-terminally truncated A. fumigatus CYP51B in complex with VCZ and VNI {(R)-N-(1-[2,4-dichlorophenyl]-2-[1H-imidazol-1-yl]ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide} (28) have also been reported. At this time, there is no crystal structure of AfCYP51A. The enzyme expressed in Escherichia coli appears unstable with purification, as it gave a diagnostic inactive P420 complex when reduced in the presence of carbon monoxide (29). In order to assess the effects of mutations that confer significant azole resistance in C. albicans and A. fumigatus, we present structural and functional analyses of these CYP51 mutations recreated in ScErg11p6×His.
RESULTS
Quantitation of ScErg11p6×His mutant expression levels.
Table S1 in the supplemental material lists the strains used in this study. Recombinant ScERG11, including a C-terminal hexahistidine tag, was constitutively overexpressed at the PDR5 locus of these strains. Native ERG11 was retained in the AD2Δ background and deleted in the AD3Δ background. All mutations were confirmed by mass spectrometry of Ni-nitrilotriacetic acid (NTA) affinity- and size exclusion chromatography (SEC)-purified 62-kDa protein bands separated by SDS-polyacrylamide gel electrophoresis (see Fig. S1 to S5 in the supplemental material). Quantitation of protein levels in crude membrane preparations from strains of the AD3Δ background was performed for all mutants and the wild-type enzyme apart from the G73W mutant, which did not have a viable phenotype when endogenous ERG11 was deleted. Coomassie-stained SDS-PAGE gels were used to visualize the protein in the crude membranes, and Western blots were used to determine the relative content of ScErg11p6×His in each preparation (Fig. S6). The Western blots showed that the ScErg11p6×His G73R and G73W mutant enzymes were expressed at 93% and 120% of the level of the wild-type recombinant enzyme, respectively. The ScErg11p6×His G73E mutant, G464S mutant, and Y140F G464S double mutant enzymes were expressed at 40%, 30%, and 65% of the level of the wild-type enzyme, respectively.
Triazole susceptibilities of mutant strains.
MIC80 determinations were performed by using a modified liquid microdilution method using buffered synthetic defined (SD) medium for the yeast host and recombinant strains overexpressing wild-type and mutant ScERG11 from the PDR5 locus (see Table S1 in the supplemental material). We previously reported that the removal of endogenous ScCYP51 from the wild-type or mutant strains overexpressing ScErg11p6×His did not significantly alter susceptibilities to the triazole drugs tested (24, 25). Surprisingly, the G73 series of CYP51 mutants showed increased susceptibilities to all the triazole drugs tested compared to the strain overexpressing the wild-type enzyme (Table 1). In the AD2Δ background (with endogenous ERG11 intact), the MIC80s of the G73 mutants were reduced by up to 2.5-fold for FLC and VCZ but not ITC compared to the strain overexpressing the wild-type enzyme. The native enzyme appeared to make a significant contribution to susceptibility because the deletion of endogenous ERG11 reduced MIC80s 1.5- to 3.5-fold further against all azoles (Table 1). The comparable susceptibility patterns of the G73E and G73R mutants, in comparison with the wild-type strain, suggest that the 2.3-fold-lower level of expression of the G73E enzyme had little effect on susceptibility to the azoles. The deletion of native ScERG11 from AD2ΔScErg11p_G73W gave a nonviable phenotype. This indicated that the CYP51 G73W enzyme has insufficient activity to support cell growth despite expression at levels higher than those of the wild-type enzyme (Fig. S6).
TABLE 1.
MIC80 values for strains overexpressing wild-type or mutant ScErg11p6×His
| Strain | Mean MIC80 (μg/ml) (SEM)a |
||
|---|---|---|---|
| FLC | VCZ | ITC | |
| AD2ΔScErg11p | 1.90 (±0.10) | 0.239 (±0.017) | 0.106 (±0.006) |
| AD3ΔScErg11p | 1.90 (±0.02) | 0.248 (±0.010) | 0.105 (±0.004) |
| AD2ΔScErg11p_G73E | 0.83 (±0.02) | 0.136 (±0.039) | 0.09 (±0.007) |
| AD3ΔScErg11p_G73E | 0.50 (±0.06) | 0.038 (±0.006) | 0.05 (±0.001) |
| AD2ΔScErg11p_G73R | 0.91 (±0.03) | 0.082 (±0.010) | 0.097 (±0.007) |
| AD3ΔScErg11p_G73R | 0.50 (±0.01) | 0.047 (±0.004) | 0.062 (±0.006) |
| AD2ΔScErg11p_G73W | 0.95 (±0.05) | 0.089 (±0.010) | 0.103 (±0.006) |
| AD2ΔScErg11p_G464S | 1.82 (±0.10) | 0.247 (±0.014) | 0.076 (±0.009) |
| AD3ΔScErg11p_G464S | 1.80 (±0.10) | 0.241 (±0.025) | 0.112 (±0.008) |
| AD2ΔScErg11p_DM | 8.20 (±0.40) | 0.585 (±0.020) | 0.089 (±0.001) |
| AD3ΔScErg11p_DM | 7.50 (±0.08) | 0.570 (±0.019) | 0.063 (±0.007) |
Standard errors of the means are shown in parentheses. Cells were grown in SD medium. MIC80s were determined after 48 h at 30°C.
The recombinant strain overexpressing the ScErg11p6×His G464S enzyme gave susceptibilities comparable to those of the wild-type strain for each triazole drug tested (FLC, VCZ, and ITC) (Table 1). Compared to the AD2ΔScErg11p_G464S strain, the deletion of native ERG11 to obtain the AD3ΔScErg11p_G464S strain did not significantly alter susceptibility to these triazoles. The 70% lower level of expression of the mutant than of the wild-type enzyme in the AD3Δ background may be why this strain does not show resistance to triazoles. The AD2Δ and AD3Δ strains expressing the ScErg11p6×His Y140F enzyme from the PDR5 locus were described previously (25). They show 2-fold greater resistance to FLC, 1.7-fold greater resistance to VCZ, and similar susceptibility to ITC compared to control strains overexpressing wild-type ScErg11p6×His. The MIC80s of the AD2ΔScErg11p_DM strain overexpressing the ScErg11p6×His Y140F G464S mutant were not altered significantly with the deletion of native ERG11. It gave similar or slightly increased susceptibility to all three triazoles. The MIC80 of AD3ΔScErg11p_DM for FLC (7.50 μg/ml) was ∼4-fold higher than that of the wild-type ScErg11p6×His strain (1.90 μg/ml), despite the G464S mutation alone having no effect on FLC resistance. Similarly, the MIC80 of AD3ΔScErg11p_DM for VCZ (0.570 μg/ml) was ∼2-fold higher than the wild-type value of 0.248 μg/ml. In contrast, susceptibility to ITC was essentially unchanged by this pair of mutations in ScErg11p6×His.
Purification of the mutant enzymes.
Mutant ScErg11p6×His enzymes were purified from all strains of the AD3Δ background except the G73W mutant. The AD2ΔScErg11p_G73W mutant strain was used instead for protein purification, as the deletion of its native ERG11 gave a nonviable phenotype. Affinity chromatography and SEC gave comparable chromatograms and Coomassie blue-stained SDS-PAGE profiles for wild-type and ScErg11p6×His G73E/R/W mutant enzymes (see Fig. S7 in the supplemental material). In contrast, the ScErg11p6×His G464S and ScErg11p6×His Y140F G464S preparations gave additional peaks during SEC of the affinity-purified enzyme, which is suggestive of denaturation or degradation (Fig. S8 and S9). No degradation products were detected by SDS-PAGE in the SEC elution profile of the ScErg11p6×His G464S enzyme (Fig. S8). SDS-PAGE detected an additional protein band at ∼55 kDa in the Ni-NTA elution profile of the ScErg11p6×His Y140F G464S enzyme (Fig. S9). The preparation appeared yellow rather than the red color expected of a functional enzyme.
CYP450 concentration estimation and characterization by CO binding.
The ScErg11p6×His G73E/R/W enzymes prepared by affinity chromatography gave carbon monoxide difference spectrum profiles similar to that of the wild-type enzyme. For ScErg11p6×His, the reduced carbon monoxide-bound protein gave a dominant peak at ∼445 nm, as expected for a functional fungal CYP51 protein. The wild-type and the G73R mutant enzymes gave similar profiles, with a minor peak at 417 nm that indicated some denatured CYP450 enzyme in the preparation (Fig. 2). The majority of the purified Y140F G464S enzyme appeared nonfunctional, with a large peak at 417 nm and a slight shoulder at 445 nm. The ScErg11p6×His G464S enzyme had a slightly larger shoulder at 445 nm than the double mutant, but the majority of these proteins appeared nonfunctional (Fig. 2).
FIG 2.

Wild-type and mutant ScErg11p6×His carbon monoxide difference spectra. Difference spectra are shown for the wild-type ScErg11p6×His protein (a) and the Y140F G464S (b), G73R (as a representative of the G73W/R/E mutants, as all 3 profiles were similar) (c), and G464S (d) mutants. The difference spectra were obtained by using equal concentrations of the ScErg11p6×His protein in a control sample, which was reduced by sodium dithionite, and a test sample, which was reduced by sodium dithionite after the solution was saturated with carbon monoxide. The peak at 445 nm represents functional CYP450, and the peak at 417 nm represents nonfunctional CYP450.
Spectral characterization by type II binding studies.
The type II spectral shift is used to identify azole binding to the P450 enzymes. It occurs when the water molecule bound as an axial ligand at the heme iron is displaced by the nitrogen of the azole ring. The binding of the triazole drugs FLC, VCZ, ITC, and PCZ to the ScErg11p6×His G73E/R/W enzymes gave type II difference spectra (Fig. 3). In contrast, the ScErg11p6×His G464S and the Y140F G464S enzymes were not sufficiently stable to be purified without a ligand present, and thus, the type II difference spectra for triazole binding were not determined. The G73E/W ScErg11p6×His mutants gave ΔAmax values with peak (428 nm) and the trough (410 nm) wavelengths comparable to those of wild-type ScErg11p6×His. The ScErg11p6×His G73R mutant had a slightly lower overall ΔAmax, with the peak and the trough being shifted to 413 nm and 430 nm, respectively, for FLC binding and with the trough being shifted to 415 nm only for VCZ binding. The ΔAmax of the G73E mutant was comparable to that of the wild-type enzyme (Table 2). The G73W mutant gave the highest ΔAmax values for all triazoles tested, and the G73R mutant had significantly lower ΔAmax values. The resultant Kd (dissociation constant) values are presented in Table 2. The Kd values obtained are in the nanomolar range, which is indicative of tight binding. The majority of the data were best fitted by using the Hill equation. The Hill coefficients were between 1.5 and 2.6. The use of a derivative of the Morrison quadratic equation for tight binding gave lower Kd values than when data were fitted to the Hill equation. The Kd values were mostly in the range of 40 to 120 nM for the G73E/R mutants, which is comparable to those of the wild-type enzyme. A Kd value of 8 nM was calculated for FLC binding by the G73E mutant. All triazoles tested showed a high binding affinity for the G73W mutant enzyme (5 to 23 nM, best fit with the quadratic equation) except for FLC, which had a Kd value of 470 nM based on a best fit with the Hill equation.
FIG 3.

Type II difference spectra and FLC binding curves for ScErg11p6×His G73E/R/W enzymes. (a to c) The difference spectra were obtained by incremental additions of FLC dissolved in DMSO to 1 μM the ScErg11p6×His G73R (a), G73E (b), and G73W (c) mutants in the sample cuvette and a corresponding amount of DMSO added to 1 μM enzyme in the reference cuvette. (d) The absorbance difference between the trough and the peak was plotted against the triazole concentration to generate binding curves. The wild-type enzyme is represented by circles, the G73E mutant is represented by squares, the G73R mutant is represented by triangles, and the G73W mutant is represented by diamonds. The inset shows the type II difference spectra for the wild-type enzyme.
TABLE 2.
Type II binding of triazole drugs to wild-type and mutant ScErg11p6×His enzymesa
| ScErg11p6×His enzyme | Triazole | ΔAmax | Equation | Mean Kd (μM) (SE) | Hill coefficient | IC50 (µM) |
|---|---|---|---|---|---|---|
| WT | FLC | 0.044 | Hill | 0.141 (±0.028) | 1.5 | 0.27 |
| VCZ | 0.036 | Hill | 0.051 (±0.019) | 2 | 0.23 | |
| ITC | 0.033 | Hill | 0.123 (±0.027) | 1.6 | 0.28 | |
| PCZ | 0.034 | Hill | 0.078 (±0.023) | 2.2 | 0.32 | |
| G73E | FLC | 0.043 | Morrison | 0.008 (±0.007) | NA | 0.45 |
| VCZ | 0.043 | Hill | 0.08 (±0.025) | 2.5 | 0.36 | |
| ITC | 0.034 | Hill | 0.07 (±0.04) | 1.9 | 0.25 | |
| PCZ | 0.042 | Hill | 0.10 (±0.04) | 2.1 | 0.36 | |
| G73R | FLC | 0.030 | Hill | 0.08 (±0.02) | 2.6 | 0.37 |
| VCZ | 0.028 | Hill | 0.12 (±0.1) | 1.7 | 0.29 | |
| ITC | 0.030 | Hill | 0.08 (±0.04) | 2.2 | 0.31 | |
| PCZ | 0.031 | Hill | 0.04 (±0.02) | 2.3 | 0.25 | |
| G73W | FLC | 0.051 | Hill | 0.47 (±0.097) | 2.3 | 0.71 |
| VCZ | 0.053 | Morrison | 0.014 (±0.007) | NA | 0.51 | |
| ITC | 0.047 | Morrison | 0.023 (±0.009) | NA | 0.52 | |
| PCZ | 0.047 | Morrison | 0.005 (±0.006) | NA | 0.44 |
The preferred equation to fit the data was chosen by using the Akaike information criterion (47). Values in parentheses indicate standard errors. WT, wild type; NA, not applicable.
Structures of ScErg11p6×His G73E and G73W enzymes in complex with ITC.
The structures of ScErg11p6×His G73E and G73W in complex with ITC were obtained to resolutions of 1.98 Å and 2.15 Å, respectively (PDB accession numbers 5ESG and 5ESH, respectively) (see Table S2 in the supplemental material). Molecular replacement showed that these complexes were essentially identical to the wild-type structure but with evidence for the presence of mutant residues, the ligand, and changes in the conformation of some residues, as discussed below. Both mutant structures showed a conformation of ITC different from those reported for the structures of wild-type ScErg11p6×His (PDB accession number 5EQB) (17) and ScErg11p6×His Y140F/H in complex with ITC (PDB accession numbers 4ZDY and 4ZE3) (25). The piperazine ring of ITC, which has been modeled as either a chair or a twisted boat conformation, accommodated this difference by acting as a hinge (Fig. 4). ITC in the wild-type and Y140F/H mutant structures has a chair conformation of the six-membered piperazine ring, which allows for the extended conformation of the drug. In the structure of the ScErg11p6×His G73E mutant in complex with ITC, the twisted boat shape of the piperazine ring facilitated the bending of the ITC tail away from E73 (Fig. 4a). There are potential π-anion interactions between the carboxylate of E73 and the 1,2,4-triazolin-3-one group of ITC (30, 31). Chen et al. observed the same conformation for PCZ in complex with Trypanosoma brucei CYP51 (PDB accession numbers 2X2N and 2WV2) (32). The scattered Fo − Fc electron density maps obtained with two of their structures suggested that PCZ has two conformers in a dynamic equilibrium. In the structure of the ScErg11p6×His G73E mutant in complex with ITC, no density was detected initially following phase solution for the β or γ carbons for E73, but there was some density for the carboxyl group. After refinement, the 2Fo − Fc density at the mutation site detected all the atoms of the glutamic acid residue.
FIG 4.
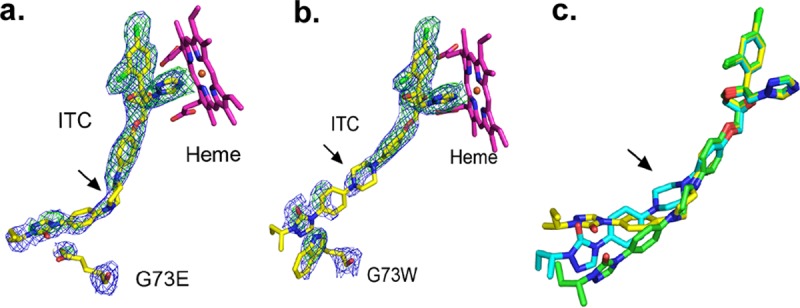
Itraconazole bound to wild-type and ScErg11p6×His G73E/W enzymes. (a and b) OMIT maps for ITC in complex with ScErg11p6×His G73E (a) and G73W (b) mutants. Electron density is shown for ITC and the site of the mutations G73E and G73W immediately following phasing and prior to modeling of the inhibitor. The final modeled ITC and the mutated residues are shown as sticks, with C atoms represented in yellow, N atoms in blue, O atoms in red, and Cl atoms in green. The heme is shown with C atoms in magenta. The 2Fo − Fc electron density map (blue) is contoured at 1σ, and the Fo − Fc map is contoured at 3σ (green). Maps (ccp4) were generated by Phenix for visualization in PyMOL. Both maps were calculated by using Fcalc refined from coordinates with no ligand or mutant residues present at the active site. Arrows indicate the piperazine ring. (c) The three conformations of ITC detected in three different structures are overlaid. The extended ITC conformation found in wild-type ScErg11p6×His (PDB accession number 5EQB) is shown with green carbon atoms, with the piperazine ring in a chair conformation. The piperazine ring of ITC (C atoms in yellow) is in a twisted boat conformation in the ScErg11p6×His G73E structure. In the ScErg11p6×His G73W structure, the piperazine ring of ITC (C atoms in cyan) remains in the chair conformation as in the wild type (C atoms in green), but the tail of the ligand is twisted to accommodate the mutation.
ITC in complex with the ScErg11p6×His G73W mutant adopted a conformation different from that seen with the ScErg11p6×His G73E mutant (Fig. 4b). The piperazine ring was modeled as the chair conformation, but the tail of the ligand was slightly twisted in order to accommodate the W73 residue. There are π-stacking interactions between the W73 and the triazolin-5-one group of ITC.
In the ScErg11p6×His Y140F ITC structure (PDB accession number 4ZDY), a hydrogen-bonding network was identified in a hydrophilic pocket. Residues P379, H381, S382, D504, S508, and M509 formed this hydrogen bond network with three water molecules, including one hydrogen bonded to the piperazine ring of ITC. This network was retained in the G73E ITC mutant structure. However, in the G73W structure, only one of these water molecules is retained, and it forms a hydrogen bond with the main-chain carbonyl and amide groups of S382. Another water molecule, not found in the Y140F or G73E ITC structures, occurs closer to the substrate entry channel in the ScErg11p6×His G73W structure. It forms hydrogen bonds to the side chain of H382 and main-chain carbonyls of Y72, W73, and F506.
Structures of the ScErg11p6×His G73W and G73E mutants in complex with ITC showed no electron density in the putative product exit channel (PPEC) detected in wild-type CYP51 (17), but some residues around the PPEC had different conformations compared to those of the wild-type structure complexed with ITC (PDB accession number 5EQB). These residues had different rotamers and positions (Fig. 5), particularly the side chains of residues F241 and F384. In addition, helix αB′ is slightly shifted to accommodate the movement of F241. In the G73E/W ITC structures, these residues point into the PPEC, and the F241 side chain occupies space within the channel, closing it off (Fig. 5). The same residues in the wild-type ITC structure point away from the channel to accommodate the ligand.
FIG 5.

Substrate and putative egress channels of wild-type and ScErg11p6×His G73W enzymes. The surface map (solvent-accessible surface) cutaway shows the substrate entry channel and the putative product egress channel of the wild-type enzyme complexed with ITC (PDB accession number 5EQB) (salmon), and the substrate channel is shown in a cutaway of the G73W mutant enzyme in complex with ITC (PDB accession number 5ESH) (lilac). The heme, ITC, and residues surrounding the putative product egress channel, R98, H128, L129, F241, and F384, are overlaid in the middle panel and represented as sticks, with C atoms in salmon for the wild-type enzyme and lilac for the G73W mutant, O atoms in red, N atoms in blue, and Cl atoms in green. The C atoms of ITC in the substrate channel of the G73W structure are shown in cyan and green for the wild type.
Structures of the ScErg11p6×His G73E and G73R mutants in complex with FLC.
Structures of the ScErg11p6×His G73E and G73R mutants in complex with FLC were obtained at resolutions of 2.25 Å and 2.20 Å, respectively (PDB accession numbers 5ESF and 5ESE) (see Table S2 in the supplemental material). The electron density maps showed good density for the ligand following molecular replacement in both structures (Fig. S10). The E73 and R73 side chains in both structures showed limited density after refinement. E73 had no density immediately after molecular replacement, and the 2Fo − Fc density covered only the β-carbon of the residue after initial refinement. The R73 residue lacks density for the β and γ carbons (Fig. S10). As previously described, the binding of the short-tailed ligand FLC to wild-type ScErg11p6×His (PDB accession number 4WMZ) (24) involves a network of water-mediated hydrogen bonds in the active site (Fig. 1). Both waters found in the wild-type structure are present in these mutant structures in the same positions; i.e., water molecules 743 and 790 form hydrogen bonds with FLC in both mutant structures.
ScErg11p6×His G73W apo structure.
The ScErg11p6×His G73W apo structure was determined to a resolution of 2.10 Å (PDB accession number 5ESI) (see Table S3 in the supplemental material). There was clear crystallographic evidence for the presence of the mutation, which confirmed data obtained by mass spectrometry. Although the mutant enzyme was purified without the addition of any ligand, some density was observed in the active site. It is difficult to identify what this density represents. The density at the heme iron could represent molecular oxygen, which was modeled into the ScErg11p6×His structure in complex with lanosterol (PDB accession number 4LXJ) (17). In the ScErg11p6×His G73W structure, M509 occupies space in the substrate entry channel pointing toward the PPEC (Fig. 6). This closes the substrate channel but leaves the PPEC open. In the rest of the mutant structures, M509 does not occupy space in the channel. However, a similar conformation of M509 is seen in the wild-type enzyme structure in complex with VCZ (PDB accession number 5HS1). This indicates the flexibility of M509 and surrounding residues in the substrate channel.
FIG 6.

Conformation of the M509 residue in the wild-type and ScErg11p6×His G73W apo structures. The surface cutaway of the ScErg11p6×His G73W apo structure (PDB accession number 5ESI) (cyan) shows the altered substrate channel due to a different conformation of the M509 residue. The G73W mutant structure was aligned to the structure of wild-type ScErg11p6×His in complex with the FLC structure (PDB accession number 4WMZ) (inset). The M509 and S508 residues are shown as sticks, with C atoms in cyan in the G73W apo substrate channel and in yellow in the wild-type FLC structure.
Structures of the ScErg11p6×His G464S mutant.
Structures of the ScErg11p6×His G464S mutant in complex with ITC and FLC were obtained at resolutions of 2.24 Å and 2.15 Å, respectively (PDB accession numbers 5ESK and 5ESJ) (see Table S3 in the supplemental material). Both structures showed clear evidence for the presence of the mutation and the ligand (Fig. 7). The binding of ITC and FLC appeared similar to the binding of these ligands in the structures of wild-type ScErg11p6×His (PDB accession numbers 4WMZ and 5EQB).
FIG 7.
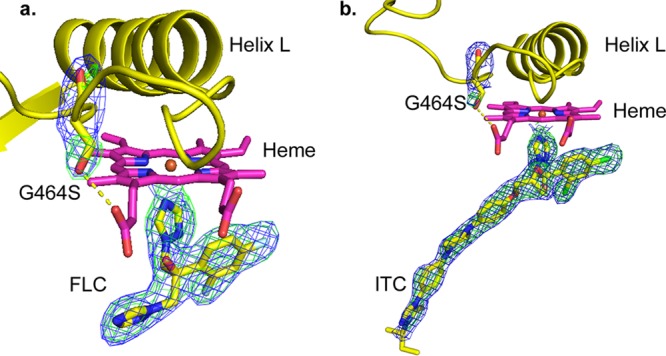
OMIT maps for mutation site and ligands in ScErg11p6×His G464S structures. (a) FLC; (b) ITC. The 2Fo − Fc electron density map (blue) is contoured at 1σ, and the Fo − Fc map is contoured at 3σ (green). Maps (ccp4) were generated by Phenix for visualization in PyMOL. Both maps were calculated by using Fcalc refined from coordinates with no ligand or mutant residue present at the active site. C atoms are shown in yellow for S464, ITC, and FLC. Helix L and the K′L loop are presented in a yellow cartoon form. The yellow dashed line represents a hydrogen bond.
Residue S464 is located on the K′L loop several residues away from the conserved cysteine 470 in the heme bulge. As predicted, the side-chain hydroxyl of S464 forms a hydrogen bond with the heme ring D propionate, replacing the water present in the structure of wild-type ScErg11p6×His (PDB accession number 4WMZ). The hydrogen bonding of S464 to the heme propionate did not affect the tilt of the heme.
DISCUSSION
This study has reproduced and characterized resistance-conferring CYP51 mutations from clinical isolates of fungal pathogens by using S. cerevisiae CYP51 as a surrogate. The introduction of G73R/W/E and G464S single mutations into ScErg11p6×His did not produce strains with triazole-resistant phenotypes found in their pathogenic counterparts. The increased susceptibility of ScErg11p6×His G73E/R/W mutant-overexpressing strains to the triazoles FLC, VCZ, and ITC suggests that the catalytic activity of the enzyme is affected by these mutations at the mouth of the substrate entry channel. A reduction in catalytic activity may explain why the deletion of the native ERG11 open reading frame (ORF) from AD2ΔScErg11p_G73W results in a nonviable strain; i.e., the mutant enzyme is insufficiently active to support growth. The contribution of native CYP51 is more apparent in resistance levels of the G73E/R mutant strains than in other mutants (Table 1). The possibility of the G73W mutant enzyme being misfolded or insufficiently expressed is excluded, as the hexahistidine-tagged enzyme was purified from crude membranes of the AD2ΔScErg11p_G73W strain, subjected to carbon monoxide and type II binding studies, and structurally resolved by X-ray crystallography. The crystallographic data detected extra interactions between the site of the G73E/W mutations and the 1,2,4-triazolin-3-one ring at the end of the long-tailed triazole ITC (Fig. 4). The ITC molecule is observed in a bent and slightly twisted conformation when bound to the ScErg11p×His G73W mutant, which has not been observed previously. PCZ has a structure very similar to that of ITC and may bind in a similar manner to the ScErg11p6×His G73W mutant.
The interactions of the long-tailed triazoles and the E/W73 mutation sites might enable the drug to dissociate more slowly after it binds. However, no significant differences were detected in the Kds for the equilibrium binding of short- and long-tailed triazole drugs by these mutant enzymes (Table 2). A. fumigatus has two CYP51 homologues, and as far as we are aware, the G54 mutation has been found only in CYP51A (13). An assay of the activity of AfCYP51A reconstituted with its cognate reductase demonstrated that the G54W mutation reduces the catalytic activity of the enzyme 3-fold compared to that of the wild-type enzyme (29). In addition, this mutation conferred 11- and 34-fold-higher 50% inhibitory concentrations (IC50s) for ITC and PCZ, respectively, compared to a moderate 2-fold increase in the IC50 for VCZ.
Homology modeling of AfCYP51A, based on the human CYP51 structure (PDB accession number 3JUS) (33), was used to suggest that G54 mutations might block the substrate entry channel or directly interact with the long-tailed triazoles (34). One explanation for the differential effect of mutations in the mouth of the substrate channel on ScCYP51 and AfCYP51A is that the AfCYP51A substrate channel opening is narrower than it is in ScCYP51. The presence of a bulky residue at the G54 mutation site of AfCYP51A could provide insufficient space to accommodate the tail of a long-tailed triazole. The result would be reduced binding of long-tailed triazoles without affecting the binding of short-tailed triazoles. However, homology models of AfCYP51A based on ScErg11p6×His provide no convincing evidence for this hypothesis (J. D. A. Tyndall et al., unpublished data). The N-terminally truncated structures of AfCYP51B show that the substrate entry channel differs when the short-tailed triazole is bound (PDB accession number 4UYM) compared to the binding of a long-tailed VNI (PDB accession number 4UYL) (28). With VNI at the active site, the substrate entry channel opening is narrower than when VCZ is in the active site. The integrity and rigidity of the substrate entry channel may be maintained by the presence of the transmembrane helix. CaCYP51 has been crystallized with the transmembrane helix truncated (PDB accession number 5FSA) (27) and intact (PDB accession number 5V5Z). The substrate channel opening appears narrower in the structure with the transmembrane helix intact. Thus, an intact transmembrane helix may be needed to better understand mutations at the substrate entry channel opening.
The G464S mutation alone had no effect on triazole susceptibility. This could be explained by the 70% lower expression levels of the mutant enzyme. In addition, the CaCYP51 G464S and AfCYP51A G448S mutants could be heterologously expressed in our S. cerevisiae system to gain more insight into the effect of this mutation on resistance to triazoles. The introduction of the second mutation, ScErg11p6×His Y140F G464S, reduced susceptibility to short-tailed but not long-tailed triazoles more than did the single Y140F mutation. The G464S mutation deleteriously affected the function of the purified enzyme preparation, and the Y140F mutation reduced stability further, with heme being retained poorly within the protein. As described previously for the CaCYP51 G464S mutant (23), microsomal preparations may give more stable samples of the single and double mutants for enzyme assays and type II difference spectrum measurements to assess the affinity of triazole drugs.
The G464S mutation creates a hydrogen bond to the heme ring D propionate (Fig. 7), and the Y140F mutation abolishes a hydrogen bond to the heme ring C propionate. In addition to effects on protein stability, an altered tilt of the heme might produce a resistant phenotype. Our finding that the tilt of the heme was unaffected in all the single mutant structures in complex with triazole drugs implies that the effects of the mutations on the electronic environment of the heme is a significantly more important factor in the conferral of azole resistance.
The yeast expression system enables the robust overexpression of the ScCYP51 protein for phenotypic analysis and the routine preparation of high-resolution crystals of the full-length enzyme. These features provide an attractive model to study pathogenic mutations of homologous CYP51 enzymes. We conclude that ScCYP51 is a useful surrogate to investigate resistance mutations in cases such as the C. albicans CYP51 mutations Y132F and Y132F G464S. It is worth noting that residues G464 and Y140 in ScCYP51 overlay G464 and Y132 in CaCYP51 (PDB accession number 5V5Z) without much deviation. However, the phenotypes of other mutations may be less accurately represented, as is the case with the A. fumigatus CYP51A G54E/R/W mutants.
MATERIALS AND METHODS
Yeast strains and growth media.
The S. cerevisiae AD2Δ strain was used to overexpress mutant and wild-type ScErg11p6×His, as previously described (24). The AD2Δ strains lack 7 major ATP binding cassette transporters, and a pdr1-3 mutation in the PDR1 transcriptional regulator enables the constitutive overexpression of ScErg11p6×His from the PDR5 locus. Strains used and generated in this study are listed in Table S1 in the supplemental material. Liquid and solid yeast extract-peptone-dextrose (YPD) media were used for yeast strain growth and maintenance. YPD medium consisted of 1% (wt/vol) Bacto yeast extract (BD Difco Laboratories Inc., Franklin Lakes, NJ), 2% (wt/vol) Bacto peptone (BD Difco), and 2% (wt/vol) glucose. SD medium contained 2% (wt/vol) glucose, 0.67% (wt/vol) yeast nitrogen base without amino acids (BD Difco), 2% (wt/vol) agar (Oxoid Ltd., Hampshire, UK), and either uracil dropout (Qbiogene, Irvine, CA) or histidine dropout (Formedium, Norfolk, UK) complete supplement mixture. Solid SD medium was used for the selection of successful clones. Liquid SD medium, used for MIC80 determinations, contained complete supplement mixture (Formedium, Norfolk, UK) with 10 mM morpholineethanesulfonic acid (MES) and 20 mM HEPES buffered with Tris to pH 6.8.
Construction of yeast ScErg11p6×His mutant strains.
The G73E/R/W and G464S mutations in ScERG11 were introduced by recombinant PCR using genomic DNA of the ADΔ ScErg11p6×His-overexpressing strain as the template (35) (see Table S1 in the supplemental material). The outside primers PDR5F (GAACATGAACGTTCCTCAGCGCG) and PDR5DS (TATGAGAAGACGGTTCGCCATTCGGACAG) were used in combination with mutation primers (Table S4) to make fragments for recombinant PCR to introduce the G73E/R/W and G464S mutations. The ScERG11 Y140F G464S mutant was created by using template genomic DNA obtained from the strain overexpressing the ScErg11p6×His Y140F mutant (AD3ΔScErg11p_Y140F) (Table S1) (25), with the G464S mutation introduced by using primers specific for this mutation (Table S4). Mutant ScERG11 DNA cassettes were introduced into the PDR5 locus of the AD2Δ strain by homologous recombination. The ScERG11 DNA cassettes included a C-terminal hexahistidine tag and a URA3 selection marker plus bordering sequences from the PDR5 locus (35). Transformants were selected by using SD-Ura (synthetic defined uracil dropout medium) agar plates, and the correct integration of the cassette was confirmed by colony PCR. The ScERG11 open reading frame was sequenced, and the presence of the expected mutations was verified (Genetic Analysis Services, Department of Anatomy, University of Otago, Dunedin, New Zealand). The ScErg11p6×His mutant strains (Table S1) were further manipulated to remove endogenous ScERG11. Native ScERG11 was replaced with a disruption cassette containing the HIS1 marker by homologous recombination (24). Transformants were selected on SD-His (synthetic defined histidine dropout medium) agar plates. Colony PCR and DNA sequence analysis verified the correct integration and presence of the mutations. The resulting strains were designated AD3ΔScErg11_G73E, AD3ΔScErg11_G73R, AD3ΔScErg11p_DM, and AD3ΔScErg11p_G464S (Table S1). The deletion of native ScERG11 gave a nonviable phenotype for the AD2ΔScErg11_G73W strain.
Azole susceptibility assays of yeast mutant strains.
Broth microdilution assays were used to determine the triazole susceptibilities of the ScErg11p6×His-overexpressing mutant strains. The MICs of triazole drugs were defined at 80% growth inhibition (MIC80) compared to no-drug controls. Triazole drugs are not fungicidal and thus can give trailing growth at high concentrations. Broth microdilution assays were carried out as previously described (36), with the exception that SD liquid medium buffered to pH 6.8 was used (25). Cells were seeded at an optical density at 600 nm (OD600) of 0.005 in 96-well microtiter plates containing 1.4-fold serial dilutions of FLC, ITC, or VCZ. Plates were incubated at 30°C with shaking at 200 rpm for 48 h. Cell density was measured as the OD600 by using a BioTek Synergy 2 multimode plate reader (BioTek Instruments, VT, USA). Three separate experiments using three clones of each strain were carried to determine each MIC80 value.
Purification of ScErg11p.
ScErg11p6×His G73E/R/W, G464S, and G464S Y140F mutants were purified according to methods described previously by Monk et al. (17). Strains with endogenous ERG11 deleted were used for the purification of the mutant enzymes (AD3Δ background), except in the case of ScErg11p6×His G73W, where the AD2ΔScErg11_G73W strain was used. YPD cultures of yeast cells grown at 30°C with shaking at 200 rpm were harvested at an OD600 of ∼10. The cells were broken by bead beating, and crude membranes were isolated by differential centrifugation. The amount of protein in the crude membranes was quantitated by using the Lowry method (37), with bovine serum albumin (Thermo Fisher, Waltham, MA, USA) as the standard. The detergent n-decyl-β-d-maltoside (DM) was used to solubilize crude membranes at a 10× critical micelle concentration (CMC) in medium containing 5 mg/ml crude membranes in 10% (wt/vol) glycerol, 250 mM NaCl, 20 mM Tris (pH 7.5), 0.5 mM phenylmethanesulfonyl fluoride (PMSF), and 1 Roche EDTA-free protease inhibitor pill per 200 ml. The solubilized membranes were incubated with 2 ml of packed Ni-NTA-agarose matrix (Qiagen) per g of crude membranes for affinity purification of ScErg11p6×His. The Ni-NTA matrix was washed with affinity purification buffer containing 10% (wt/vol) glycerol, 250 mM NaCl, 20 mM Tris (pH 7.5), 0.5 mM PMSF, 6.4 mM DM (4× CMC), 20 mM imidazole, and 1 Roche EDTA-free protease inhibitor pill in 200 ml. The elution buffer was affinity purification buffer with the inclusion of 200 mM imidazole.
The ScErg11p6×His mutant enzymes were further purified by SEC with a Superdex 200 10/300 GL column (GE Healthcare Life Sciences, UK). SEC buffer consisted of 10% (wt/vol) glycerol, 150 mM NaCl, 20 mM HEPES, 0.5 mM PMSF, and 6.4 mM DM (4× CMC) at pH 7.5 at room temperature together with 2 Roche EDTA-free protease inhibitor pills per 400 ml. The appropriate triazole drug dissolved in dimethyl sulfoxide (DMSO) was added to the buffer for copurification: 10 μM FLC, 10 μM VCZ, 2 μM ITC, or 2 μM PCZ. The purified red fractions containing 62-kDa ScErg11p6×His were collected and centrifugally concentrated in 50-kDa-molecular-mass-cutoff Amicon Ultra-4 filters (Merck Millipore Ltd., Cork, Ireland).
Affinity purification of ScErg11p6×His for spectral assays.
ScErg11p6×His used for spectroscopic assays was affinity purified as described previously (38), using 50 mM l-histidine rather than imidazole for elution in affinity purification buffer. The eluted enzyme was washed by centrifugal filtration (Amicon Ultra; Millipore) in affinity purification buffer without the addition of l-histidine to obtain a ligand-free enzyme. The absence of l-histidine in the enzyme preparation was confirmed by using an Ultrospec 6300 pro UV-visible spectrophotometer. The heme peaks with l-histidine bound are at ∼420 nm for the wild-type protein and ∼417 nm with no ligand.
Determination of the concentration of cytochrome P450.
Carbon monoxide binding was used to determine the concentration of functional cytochrome P450 for drug binding studies as described previously by Guengerich et al. (39). The suitably diluted affinity-purified enzyme preparation was split into two 1-ml cuvettes. The sample cuvette was saturated with CO gas. About 1 mg of sodium dithionite was added to the sample and the reference cuvette. The P450 concentration was determined by measuring the difference in absorbances at 446 and 490 nm and using an extinction coefficient of 91 mM−1 cm−1 (40). Absorption spectra were recorded with a Cary 1 Bio UV-visible spectrophotometer using 10-mm-path-length UV-transparent plastic cuvettes (GE Healthcare Life Sciences, UK).
Type II difference spectra.
The affinity-purified ScErg11p6×His wild-type or G73E/R/W enzyme was used to determine type II difference spectra. The enzyme diluted to 1 μM was split into two cuvettes. Incremental additions of triazole drugs, dissolved in DMSO, were added to the sample cuvette, and corresponding amounts of DMSO were added to the reference cuvette. Titrations for FLC, VCZ, ITC, and PCZ were carried out, and the difference spectra were recorded at between 350 to 500 nm by using a Cary 1 Bio UV-visible spectrophotometer. Trough-peak absorbance differences were plotted against the triazole concentration to obtain binding curves. The Hill equation, ΔA = ΔAmax [azole]n/([azole]n + Kdn), or the quadratic equation for tight ligand binding (a derivative of the Morrison equation designed to take into account the amount of enzyme in the assay mixture) was used to determine the Kd using GraphPad Prism 6 software (GraphPad, San Diego, CA), where ΔAmax is the maximum change in the absorbance, [azole] is the azole concentration, n is the Hill coefficient, and Et is the total amount of the enzyme. The quadratic equation is ΔA = ΔAmax/2 × Et × {(Et + [azole] + Kd) − [(Et + [azole] + Kd)2 − (4 × Et × [azole])]0.5}.
Crystallization and data collection.
The ScErg11p6×His G73E/R/W and G464S mutants were either copurified with triazole drugs or purified without an added ligand. The crystals were grown by hanging-drop vapor diffusion in a 4-μl drop containing a 1:1 ratio of protein (20 mg/ml in SEC buffer) and the reservoir solution (45% polyethylene glycol 400 in 100 mM glycine at a pH range of 9.3 to 9.55), as previously described (17). The crystals grew after about 1 week at 18°C. Data sets were collected at a wavelength of 0.954 Å at the Australian Synchrotron (Melbourne, Australia). Data indexing and integration were done by using iMosflm (41) and scaled with Aimless (42). Molecular replacement was done with the ScErg11p6×His structure complexed with lanosterol (PDB accession number 4LXJ) (17) as the template, using Phaser-MR (43) from Phenix. Phenix.Refine (44) and Coot (45) were used for refinement and model building, respectively. The Grade Global Phasing online tool (Global Phasing Ltd.) was used to generate the crystallographic information (.cif) files for triazole drugs. The Fe-nitrogen and Fe-sulfur distances were constrained to 2.15 Å and 2.33 Å, based on the average coordinate bond distance of more than 80 known Fe-N (triazole) complexes and 4 heme Fe-S complexes in the Cambridge Structural Database (46), as previously described (24).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Marsden Fund of the Royal Society of New Zealand and the Health Research Council of New Zealand awarded to B.C.M. The New Zealand Synchrotron Group sponsored travel to the Australian Synchrotron.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02242-17.
REFERENCES
- 1.Brown GD, Denning DW, Levitz SM. 2012. Tackling human fungal infections. Science 336:647. doi: 10.1126/science.1222236. [DOI] [PubMed] [Google Scholar]
- 2.Denning DW, Perlin DS. 2011. Azole resistance in Aspergillus: a growing public health menace. Future Microbiol 6:1229–1232. doi: 10.2217/fmb.11.118. [DOI] [PubMed] [Google Scholar]
- 3.Horn DL, Fishman JA, Steinbach WJ, Anaissie EJ, Marr KA, Olyaei AJ, Pfaller MA, Weiss MA, Webster KM, Neofytos D. 2007. Presentation of the PATH Alliance registry for prospective data collection and analysis of the epidemiology, therapy, and outcomes of invasive fungal infections. Diagn Microbiol Infect Dis 59:407–414. doi: 10.1016/j.diagmicrobio.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 5.Snelders E, Camps SM, Karawajczyk A, Schaftenaar G, Kema GH, van der Lee HA, Klaassen CH, Melchers WJ, Verweij PE. 2012. Triazole fungicides can induce cross-resistance to medical triazoles in Aspergillus fumigatus. PLoS One 7:e31801. doi: 10.1371/journal.pone.0031801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faria-Ramos I, Tavares PR, Farinha S, Neves-Maia J, Miranda IM, Silva RM, Estevinho LM, Pina-Vaz C, Rodrigues AG. 2014. Environmental azole fungicide, prochloraz, can induce cross-resistance to medical triazoles in Candida glabrata. FEMS Yeast Res 14:1119–1123. doi: 10.1111/1567-1364.12193. [DOI] [PubMed] [Google Scholar]
- 7.Mellado E, Diaz-Guerra TM, Cuenca-Estrella M, Rodriguez-Tudela JL. 2001. Identification of two different 14-alpha sterol demethylase-related genes (cyp51A and cyp51B) in Aspergillus fumigatus and other Aspergillus species. J Clin Microbiol 39:2431–2438. doi: 10.1128/JCM.39.7.2431-2438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mellado E, Garcia-Effron G, Buitrago MJ, Alcazar-Fuoli L, Cuenca-Estrella M, Rodriguez-Tudela JL. 2005. Targeted gene disruption of the 14-alpha sterol demethylase (cyp51A) in Aspergillus fumigatus and its role in azole drug susceptibility. Antimicrob Agents Chemother 49:2536–2538. doi: 10.1128/AAC.49.6.2536-2538.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denning DW, Bromley MJ. 2015. How to bolster the antifungal pipeline. Science 347:1414–1416. doi: 10.1126/science.aaa6097. [DOI] [PubMed] [Google Scholar]
- 10.Ashbee HR, Barnes RA, Johnson EM, Richardson MD, Gorton R, Hope WW. 2014. Therapeutic drug monitoring (TDM) of antifungal agents: guidelines from the British Society for Medical Mycology. J Antimicrob Chemother 69:1162–1176. doi: 10.1093/jac/dkt508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tashiro M, Izumikawa K, Hirano K, Ide S, Mihara T, Hosogaya N, Takazono T, Morinaga Y, Nakamura S, Kurihara S, Imamura Y, Miyazaki T, Nishino T, Tsukamoto M, Kakeya H, Yamamoto Y, Yanagihara K, Yasuoka A, Tashiro T, Kohno S. 2012. Correlation between triazole treatment history and susceptibility in clinically isolated Aspergillus fumigatus. Antimicrob Agents Chemother 56:4870–4875. doi: 10.1128/AAC.00514-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez-Tudela JL, Alcazar-Fuoli L, Mellado E, Alastruey-Izquierdo A, Monzon A, Cuenca-Estrella M. 2008. Epidemiological cutoffs and cross-resistance to azole drugs in Aspergillus fumigatus. Antimicrob Agents Chemother 52:2468–2472. doi: 10.1128/AAC.00156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mann PA, Parmegiani RM, Wei SQ, Mendrick CA, Li X, Loebenberg D, DiDomenico B, Hare RS, Walker SS, McNicholas PM. 2003. Mutations in Aspergillus fumigatus resulting in reduced susceptibility to posaconazole appear to be restricted to a single amino acid in the cytochrome P450 14alpha-demethylase. Antimicrob Agents Chemother 47:577–581. doi: 10.1128/AAC.47.2.577-581.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alcazar-Fuoli L, Mellado E, Cuenca-Estrella M, Sanglard D. 2011. Probing the role of point mutations in the cyp51A gene from Aspergillus fumigatus in the model yeast Saccharomyces cerevisiae. Med Mycol 49:276–284. doi: 10.3109/13693786.2010.512926. [DOI] [PubMed] [Google Scholar]
- 15.Howard SJ, Lass-Florl C, Cuenca-Estrella M, Gomez-Lopez A, Arendrup MC. 2013. Determination of isavuconazole susceptibility of Aspergillus and Candida species by the EUCAST method. Antimicrob Agents Chemother 57:5426–5431. doi: 10.1128/AAC.01111-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma C, Hagen F, Moroti R, Meis JF, Chowdhary A. 2015. Triazole-resistant Aspergillus fumigatus harbouring G54 mutation: is it de novo or environmentally acquired? J Glob Antimicrob Resist 3:69–74. doi: 10.1016/j.jgar.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Monk BC, Tomasiak TM, Keniya MV, Huschmann FU, Tyndall JD, O'Connell JD, Cannon RD, McDonald JG, Rodriguez A, Finer-Moore JS, Stroud RM. 2014. Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc Natl Acad Sci U S A 111:3865–3870. doi: 10.1073/pnas.1324245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howard SJ, Cerar D, Anderson MJ, Albarrag A, Fisher MC, Pasqualotto AC, Laverdiere M, Arendrup MC, Perlin DS, Denning DW. 2009. Frequency and evolution of azole resistance in Aspergillus fumigatus associated with treatment failure. Emerg Infect Dis 15:1068–1076. doi: 10.3201/eid1507.090043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pelaez T, Gijon P, Bunsow E, Bouza E, Sanchez-Yebra W, Valerio M, Gama B, Cuenca-Estrella M, Mellado E. 2012. Resistance to voriconazole due to a G448S substitution in Aspergillus fumigatus in a patient with cerebral aspergillosis. J Clin Microbiol 50:2531–2534. doi: 10.1128/JCM.00329-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanglard D, Ischer F, Koymans L, Bille J. 1998. Amino acid substitutions in the cytochrome P-450 lanosterol 14alpha-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob Agents Chemother 42:241–253. doi: 10.1093/jac/42.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chau AS, Mendrick CA, Sabatelli FJ, Loebenberg D, McNicholas PM. 2004. Application of real-time quantitative PCR to molecular analysis of Candida albicans strains exhibiting reduced susceptibility to azoles. Antimicrob Agents Chemother 48:2124–2131. doi: 10.1128/AAC.48.6.2124-2131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flowers SA, Colon B, Whaley SG, Schuler MA, Rogers PD. 2015. Contribution of clinically derived mutations in ERG11 to azole resistance in Candida albicans. Antimicrob Agents Chemother 59:450–460. doi: 10.1128/AAC.03470-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelly SL, Lamb DC, Loeffler J, Einsele H, Kelly DE. 1999. The G464S amino acid substitution in Candida albicans sterol 14alpha-demethylase causes fluconazole resistance in the clinic through reduced affinity. Biochem Biophys Res Commun 262:174–179. doi: 10.1006/bbrc.1999.1136. [DOI] [PubMed] [Google Scholar]
- 24.Sagatova AA, Keniya MV, Wilson RK, Monk BC, Tyndall JD. 2015. Structural insights into binding of the antifungal drug fluconazole to Saccharomyces cerevisiae lanosterol 14alpha-demethylase. Antimicrob Agents Chemother 59:4982–4989. doi: 10.1128/AAC.00925-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sagatova AA, Keniya MV, Wilson RK, Sabherwal M, Tyndall JD, Monk BC. 2016. Triazole resistance mediated by mutations of a conserved active site tyrosine in fungal lanosterol 14alpha-demethylase. Sci Rep 6:26213. doi: 10.1038/srep26213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tyndall JD, Sabherwal M, Sagatova AA, Keniya MV, Negroni J, Wilson RK, Woods MA, Tietjen K, Monk BC. 2016. Structural and functional elucidation of yeast lanosterol 14alpha-demethylase in complex with agrochemical antifungals. PLoS One 11:e0167485. doi: 10.1371/journal.pone.0167485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hargrove TY, Friggeri L, Wawrzak Z, Qi A, Hoekstra WJ, Schotzinger RJ, York JD, Guengerich FP, Lepesheva GI. 2017. Structural analyses of Candida albicans sterol 14alpha-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J Biol Chem 292:6728–6743. doi: 10.1074/jbc.M117.778308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hargrove TY, Wawrzak Z, Lamb DC, Guengerich FP, Lepesheva GI. 2015. Structure-functional characterization of cytochrome P450 sterol 14alpha-demethylase (CYP51B) from Aspergillus fumigatus and molecular basis for the development of antifungal drugs. J Biol Chem 290:23916–23934. doi: 10.1074/jbc.M115.677310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warrilow AG, Parker JE, Price CL, Nes WD, Kelly SL, Kelly DE. 12 October 2015. CYP51-mediated azole resistance in Aspergillus fumigatus: an in vitro biochemical study. Antimicrob Agents Chemother doi: 10.1128/AAC.01806-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mascal M, Armstrong A, Bartberger MD. 2002. Anion-aromatic bonding: a case for anion recognition by pi-acidic rings. J Am Chem Soc 124:6274–6276. doi: 10.1021/ja017449s. [DOI] [PubMed] [Google Scholar]
- 31.Schwans JP, Sunden F, Lassila JK, Gonzalez A, Tsai Y, Herschlag D. 2013. Use of anion-aromatic interactions to position the general base in the ketosteroid isomerase active site. Proc Natl Acad Sci U S A 110:11308–11313. doi: 10.1073/pnas.1206710110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen CK, Leung SS, Guilbert C, Jacobson MP, McKerrow JH, Podust LM. 2010. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl Trop Dis 4:e651. doi: 10.1371/journal.pntd.0000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strushkevich N, Usanov SA, Park HW. 2010. Structural basis of human CYP51 inhibition by antifungal azoles. J Mol Biol 397:1067–1078. doi: 10.1016/j.jmb.2010.01.075. [DOI] [PubMed] [Google Scholar]
- 34.Fraczek MG, Bromley M, Bowyer P. 2011. An improved model of the Aspergillus fumigatus CYP51A protein. Antimicrob Agents Chemother 55:2483–2486. doi: 10.1128/AAC.01651-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamping E, Monk BC, Niimi K, Holmes AR, Tsao S, Tanabe K, Niimi M, Uehara Y, Cannon RD. 2007. Characterization of three classes of membrane proteins involved in fungal azole resistance by functional hyperexpression in Saccharomyces cerevisiae. Eukaryot Cell 6:1150–1165. doi: 10.1128/EC.00091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard, 3rd ed, M27-A3. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 37.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275. [PubMed] [Google Scholar]
- 38.Warrilow AG, Martel CM, Parker JE, Melo N, Lamb DC, Nes WD, Kelly DE, Kelly SL. 2010. Azole binding properties of Candida albicans sterol 14-alpha demethylase (CaCYP51). Antimicrob Agents Chemother 54:4235–4245. doi: 10.1128/AAC.00587-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guengerich FP, Martin MV, Sohl CD, Cheng Q. 2009. Measurement of cytochrome P450 and NADPH-cytochrome P450 reductase. Nat Protoc 4:1245–1251. doi: 10.1038/nprot.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Omura T, Sato R. 1964. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J Biol Chem 239:2370–2378. [PubMed] [Google Scholar]
- 41.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evans PR, Murshudov GN. 2013. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allen FH. 2002. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr B 58:380–388. doi: 10.1107/S0108768102003890. [DOI] [PubMed] [Google Scholar]
- 47.Akaike H. 1974. A new look at the statistical model identification. IEEE Trans Automat Contr 19:716–723. doi: 10.1109/TAC.1974.1100705. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.