

ABSTRACT
Ceftazidime-avibactam (CAZ-AVI) is a promising novel treatment for infections caused by carbapenem-resistant Enterobacteriaceae (CRE). Despite improved treatment outcomes compared to those achieved with aminoglycoside- and colistin-based regimens, the rapid evolution of CAZ-AVI resistance during treatment has previously been reported in Klebsiella pneumoniae sequence type 258 (ST258) blaKPC-3-harboring isolates. Here, we report the stepwise evolution and isolation of two phenotypically distinct CAZ-AVI-resistant Klebsiella pneumoniae isolates from a patient with pancreatitis. All susceptible (n = 3) and resistant (n = 5) isolates were of the ST307 clonal background, a rapidly emerging clone. Taking advantage of short-read Illumina and long-read Oxford Nanopore sequencing and full-length assembly of the core chromosome and plasmids, we demonstrate that CAZ-AVI resistance first occurred through a 532G → T blaKPC-2 point mutation in blaKPC-2 (D179Y protein substitution) following only 12 days of CAZ-AVI exposure. While subsequent isolates exhibited substantially decreased meropenem (MEM) MICs (≤2 μg/ml), later cultures demonstrated a second CAZ-AVI resistance phenotype with a lower CAZ-AVI MIC (12 μg/ml) but also MEM resistance (MIC > 128 μg/ml). These CAZ-AVI- and MEM-resistant isolates showed evidence of multiple genomic adaptations, mainly through insertions and deletions. This included amplification and transposition of wild-type blaKPC-2 into a novel plasmid, an IS1 insertion upstream of ompK36, and disruption of the rfb gene locus in these isolates. Our findings illustrate the potential of CAZ-AVI resistance to emerge in non-K. pneumoniae ST258 clonal backgrounds and alternative blaKPC variants. These results raise concerns about the strong selective pressures incurred by novel carbapenemase inhibitors, such as avibactam, on isolates previously considered invulnerable to CAZ-AVI resistance. There is an urgent need to further characterize non-KPC-mediated modes of carbapenem resistance and the intrinsic bacterial factors that facilitate the rapid emergence of resistance during treatment.
KEYWORDS: ceftazidime-avibactam, carbapenem-resistant Enterobacteriaceae, Klebsiella pneumoniae, long-read sequencing, antimicrobial resistance, bacterial genomics
INTRODUCTION
Ceftazidime-avibactam (CAZ-AVI) is a novel β-lactam–β-lactamase inhibitor combination with activity against serine β-lactamases, including Klebsiella pneumoniae carbapenemases (KPCs) (1). Since its approval by the U.S. Food and Drug Administration (FDA) in February 2015, CAZ-AVI has been considered a promising treatment for multidrug-resistant Gram-negative bacterial (MDR-GNB) infections, in particular, infections caused by carbapenem-resistant Enterobacteriaceae (CRE) expressing class A, class C, and some class D carbapenemases (2). However, resistance has been documented in several patients after short periods of CAZ-AVI exposure (3, 4).
Most isolates causing documented cases of CAZ-AVI-resistant K. pneumoniae infections reported to date harbored mutant blaKPC-3 (3, 5). Amplification of the blaKPC gene has also been postulated to be a potential mechanism of CAZ-AVI resistance (5, 6). However, the molecular processes mediating the in vivo evolution of CAZ-AVI resistance remain incompletely defined. Furthermore, the majority of blaKPC-producing CAZ-AVI-resistant infections occurred in the K. pneumoniae sequence type 258 (ST258) clonal background (3, 5, 6), while the emergence of KPC-mediated resistance in other clonal backgrounds has not been noted.
Here we report the development of CAZ-AVI resistance in the emerging K. pneumoniae clone ST307 harboring blaKPC-2 during CAZ-AVI treatment. Using short- and long-read sequencing, we describe the phenotypic and genomic evolution of these isolates and the independent evolution of high-level and low-level CAZ-AVI resistance.
RESULTS
Evolution of resistance during treatment.
A man in his late 40s with a history of type II diabetes and hypertension presented with recurrent abdominal pain following a recent admission for acute pancreatitis in Puerto Rico. The patient's clinical details and treatments and the timeline of his 3-month hospitalization are summarized in Table 1. Empirical treatment with meropenem (MEM) began on hospital day 5, but carbapenem-resistant K. pneumoniae (CRKP) was cultured from pancreatic fluid collected on that day. Treatment was therefore changed to polymyxin B (PMB) and MEM, with the subsequent addition of tigecycline, given the persistently positive cultures. PMB and MEM were ceased due to nephrotoxicity, and CAZ-AVI was commenced on hospital day 37 with the continued addition of tigecycline. Within 24 h of initiation of CAZ-AVI, the patient required the initiation of continuous veno-venous hemofiltration, for which the dose was increased to 2.5 g every 8 h. Following 12 days of CAZ-AVI treatment, K. pneumoniae was cultured from the patient's bronchoalveolar lavage fluid on hospital day 49 and again on day 58 and from a tracheal aspirate on day 59. These isolates had lower MEM MICs (Table 1), and a CAZ-AVI Etest revealed MICs of >256 μg/ml, consistent with CAZ-AVI resistance. Once susceptibility results became available, CAZ-AVI treatment was discontinued, and PMB-MEM treatment was reinitiated. Tigecycline was held from this final regimen, given concerns for hepatotoxicity. Subsequently, blood cultures were positive for CRKP on hospital day 68 while on PMB-MEM therapy and remained positive until hospital day 72, when care was withdrawn.
TABLE 1.
Phenotypic and molecular characterization of K. pneumoniae isolatesa

Abbreviations: CAZ-AVI, ceftazidime-avibactam; MEM, meropenem; BMD, broth microdilution; PMB, polymyxin B; TGC, tigecycline; BAL, bronchoalveolar lavage.
Phenotype 3 isolates were characterized by an ompK35 C1049T mutation, resulting in a premature stop codon following amino acid 349, and an IS1 insertion sequence 49 bp upstream of ompK36.
Characterization of isolates.
Further testing for susceptibility to CAZ-AVI and MEM showed three distinct susceptibility phenotypes (Table 1): phenotype 1 isolates, collected at the baseline during MEM and PMB treatment, were MEM resistant (MIC by the broth microdilution [BMD] method, 128 μg/ml) and CAZ-AVI susceptible (MIC, 3 μg/ml). Phenotype 2 isolates, collected during CAZ-AVI exposure, displayed CAZ-AVI resistance and a decreased MEM MIC (CAZ-AVI MIC > 256 μg/ml; MEM MIC ≤ 2 μg/ml, considered intermediate resistance by Clinical and Laboratory Standards Institute [CLSI] interpretative criteria). However, phenotype 3 isolates, collected after CAZ-AVI discontinuation, reverted to MEM resistance (MIC by the BMD method, ≥128 μg/ml) but again harbored CAZ-AVI resistance, albeit with a lower MIC (12 μg/ml).
All isolates were typed as K. pneumoniae ST307 using in silico multilocus sequence typing (MLST). The pairwise single nucleotide polymorphism (SNP) distances ranged from 0 to 7 SNPs between isolates, showing a high degree of relatedness (Fig. 1A). Phenotype 2 isolates (CAZ-AVI resistant, MEM susceptible) harbored a novel nonsynonymous mutation in an ATP-dependent protease, ATP-binding subunit HslU, a heat shock protein expressed in many bacteria in response to cell stress (7). Phenotype 3 isolates lacked this SNP, suggesting that they emerged from the original phenotype 1 isolates. Phenotype 3 isolates were also distinguished by a nonsynonymous mutation in cra-1, a locus involved in the activation and repression of cellular catabolism, and a novel ompK35 C1049T mutation, resulting in a premature stop codon following amino acid 349 and 2 amino acids before the end of the protein.
FIG 1.
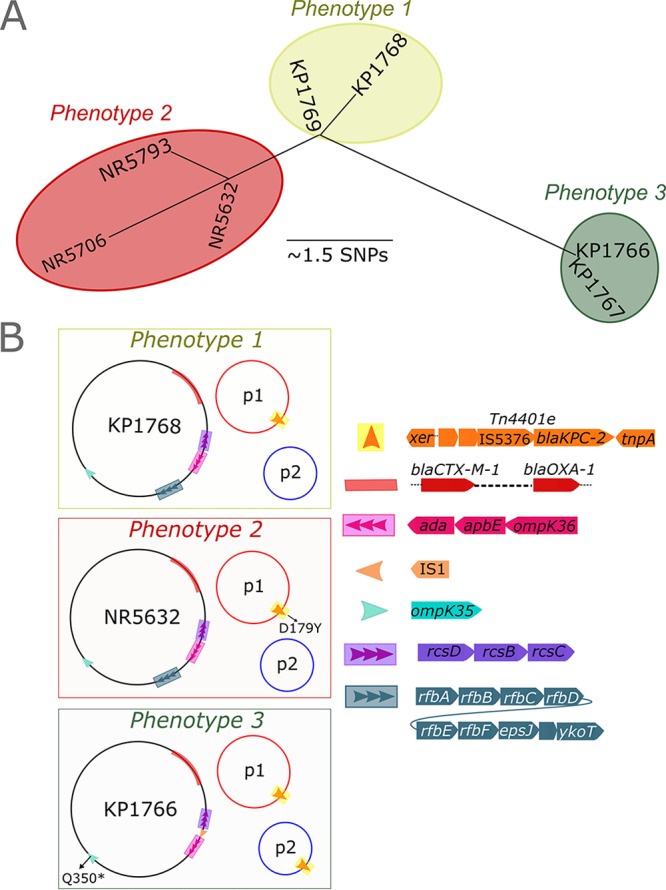
Schematic representation of genomic adaptations during evolution of CAZ-AVI resistance in K. pneumoniae ST307. (A) Neighbor-joining phylogenetic tree illustrating that all three phenotypes had closely related core chromosomes. Phenotype 2 and 3 isolates arose independently from the phenotype 1 isolate. (B) Phenotype 2 isolates (CAZ-AVI resistant, MER susceptible) acquired a D179Y mutation in KPC-2. Phenotype 3 isolates harbored wild-type blaKPC-2 in plasmid 1 as well as a second copy that was translocated as part of the Tn4401e cassette to the smaller plasmid. Additional adaptations included deletion of the rfb gene locus (green) and an insertion of IS1 between the rcs locus and ompK36 at position −48 bp.
Analysis of resistance genes showed that phenotype 1 and 3 isolates carried wild-type blaKPC-2. Phenotype 2 isolates, however, harbored mutant blaKPC-2 encoding a tyrosine-for-aspartic acid substitution at Ambler amino acid position 179 (D179Y), in the KPC Ω-loop region encompassing amino acid positions 165 to 179 (3, 8). The high read depth noted through short-read whole-genome sequencing (WGS) suggested a possible duplication of the KPC gene in phenotype 3 isolates compared to phenotype 1 and 2 isolates, which was confirmed by quantitative PCR.
Genomic insights through long-read sequencing.
Long-read sequencing and hybrid assembly produced assembled chromosomal and plasmid regions for one representative isolate from each phenotype, KP1768 (phenotype 1), NR5632 (phenotype 2), and KP1766 (phenotype 3). The chromosomes of KP1768 (phenotype 1) and NR5632 (phenotype 2) were each assembled into two contigs totaling 5.39 Mbp with an average coverage of 27 times and 14 times, respectively. The KP1766 (phenotype 3) chromosome, on the other hand, was assembled into a single 5.38-Mbp contig at 30 times coverage.
All three isolates harbored nearly identical versions of a novel IncA/C plasmid (∼205 kb) and IncFIBK plasmid (∼150 kb), which were each assembled into single contigs (Fig. 1B). We also detected a ColRNA plasmid-like region containing blaOXA-1 and blaCTX-M-1 integrated into the core chromosomes of all three isolates. In isolates KP1768 (phenotype 1) and NR5632 (phenotype 2), blaKPC-2 was present on the IncA/C group plasmid only and associated with a Tn4401e transposon (∼10 kb). In KP1766 (phenotype 3), there were copies of blaKPC-2 present on the IncA/C and IncFIBK plasmids. Further inspection of this region revealed that the 10-kb Tn4401e transposon had migrated to the IncFIBK plasmid as an intact unit.
In addition to the premature stop codon in ompK35, we assessed if porin genes were further altered in phenotype 3. While we did not detect mutations in ompK36 across the three phenotypes, we identified an IS1 insertion sequence 49 bp upstream of ompK36 in both phenotype 3 isolates. The functional impact of differences in ompK35 and ompK36 in phenotype 3 isolates remains to be determined. Phenotype 3 isolates also harbored a deletion of the rfb cluster implicated in the O-antigen biosynthetic pathway (9). This deletion occurred in the context of disruption of the rfb gene locus by an IS5 family transposase.
DISCUSSION
We identified CAZ-AVI resistance in the newly emerging K. pneumoniae ST307 clonal background through a 532G → T mutation in the blaKPC-2 D179Y protein substitution as the putative initial mechanism of CAZ-AVI resistance following only 12 days of CAZ-AVI exposure. These results are unique in the context of previous clinical reports of CAZ-AVI resistance, which note mutations in blaKPC-3 in K. pneumoniae ST258. Our findings demonstrate that CAZ-AVI resistance can rapidly emerge in non-ST258 K. pneumoniae clonal backgrounds and different blaKPC variants, raising concerns about the strong selective pressures incurred by CAZ-AVI treatment. Moreover, we documented a highly concerning additional CAZ-AVI resistance phenotype with dual CAZ-AVI and MEM resistance. Taking advantage of long-read sequencing, we identified transposition of wild-type blaKPC-2 from one plasmid into another plasmid harbored by the same isolate (resulting in a doubled blaKPC-2 copy number), an IS1 insertion upstream of ompK36, and disruption of the tag gene locus in these isolates as likely mechanisms of CAZ-AVI resistance in these isolates. While the MEM MIC was significantly decreased in isolates harboring KPC-2 D179Y, consistent with previous reports in KPC-3, phenotype 3 isolates were resistant to both CAZ-AVI and MEM. These discrepancies highlight inherent differences in CAZ-AVI resistance-conferring mechanisms between phenotypes. It remains unclear if the successive or combined pressure of CAZ-AVI and PMB-MEM led to these genomic adaptations.
Shields et al. first reported the evolution of CAZ-AVI resistance during treatment of K. pneumoniae infections in three patients (3). All isolates belonged to ST258 clade II and had three different blaKPC-3 mutations, all of which promoted increased CAZ-AVI MICs. These mutations matched those previously documented during CAZ-AVI in vitro passage experiments (10). The KPC-2 D179Y substitution observed in our phenotype 2 isolates matches a previously described blaKPC-3 Ω-loop subregion mutation (3) and refutes the hypothesis that blaKPC-2 lacks a predisposition to CAZ-AVI resistance (3, 11). Mutant blaKPC-2 enzymes conferring CAZ-AVI resistance have been described in vitro (12) but had not been noted clinically prior to the present report. While CAZ-AVI resistance has emerged in blaKPC-2-carrying K. pneumoniae isolates, this was not due to blaKPC-2 mutations (13). Our results stress the importance of mutations in the Ω-loop region in hindering avibactam binding, regardless of the KPC type. This is of concern at clinical sites where blaKPC-2-harboring K. pneumoniae isolates predominate, such as at the Columbia University Medical Center (14). Further investigations of the mechanisms underlying blaKPC-2-conferred CAZ-AVI resistance in other clonal backgrounds are needed.
Additional mechanisms of resistance to CAZ-AVI have been proposed. In a previous study, an increased copy number of a blaKPC-3 gene in combination with mutations in the ompK35 and ompK36 genes was thought to be responsible for CAZ-AVI resistance (6). Of note, the isolate from the prior report was from a patient without CAZ-AVI exposure. In the CAZ-AVI- and MEM-resistant phenotype 3 isolates described herein, resistance was likely driven by insertion sequence-mediated amplification of the blaKPC gene, in addition to porin disruption and transposon insertions upstream of ompK36 and in the O-antigen pathway gene cluster. The O antigen makes up the outer layer of the lipopolysaccharide and has been shown to play a role in both virulence and modulating the host response through its antigenic variability. Further studies are needed to determine the impact of the detected mutations and their impact on virulence or hindered host immune detection of affected isolates, explaining their persistence. We note that our phenotype 3 isolates were recovered after the cessation of CAZ-AVI and during PMB and MEM exposure, raising the possibility that PMB and MEM exposure or the host response rather than CAZ-AVI alone can lead to genetic modifications producing broad-spectrum antibiotic resistance. This claim is supported by a recent in vitro study of KPC-3 variants, which showed that passage of CAZ-AVI-resistant isolates through sublethal meropenem concentrations brought about mutations in blaKPC-3 and ompK36 and correlated with meropenem resistance (15).
In this study, resistance arose in K. pneumoniae ST307, a newly emerging clone (11, 16). Recently, infections caused by extended-spectrum β-lactamase (ESBL)-producing K. pneumoniae ST307 outnumbered ESBL K. pneumoniae ST258 infections at a Houston, TX, hospital (11). Our findings highlight that K. pneumoniae ST307 can serve as a background for extensive drug resistance through both plasmid uptake and chromosomal modifications. While our isolates had high carbapenem MICs at the baseline, the integration of two β-lactamases into the core chromosome may have accelerated the evolution of this clone. Further studies are needed to explore if isolates with higher carbapenem MICs are more adept at developing resistance to novel carbapenemase inhibitors.
When the findings are taken together, even though CAZ-AVI is a promising treatment for CRE infection, diverse mechanisms of CAZ-AVI resistance can emerge on therapy following short periods of treatment and may be further fueled by PMB and MEM exposure. Intrinsic bacterial factors may predispose select isolates to the more rapid evolution of CAZ-AVI resistance. While novel carbapenemase inhibitors fulfill an important need, they are unlikely to end the CRE epidemic. It remains to be determined if other carbapenemase inhibitors currently in trials will be equally vulnerable to the rapid evolution of resistance and which genetic backgrounds may be particularly problematic. This highlights the need to optimize the use of current agents to minimize the emergence of resistance and track the evolution of resistance with novel genomic tools, as well as the urgent need for novel treatments against MDR-GNB infections.
MATERIALS AND METHODS
Isolates and drug susceptibility testing.
We reviewed the clinical course of a patient who developed CAZ-AVI resistance during treatment. Seven K. pneumoniae isolates cultured from the patient over a 3-month period were available for further analysis (Table 1). Drug susceptibility testing was carried out by use of the Vitek-2 system (bioMérieux, Marcy l'Etoile, France) and broth microdilution (BMD) according to Clinical and Laboratory Standards Institute (CLSI) guidelines (17). CAZ-AVI susceptibility testing was carried out by Etest (bioMérieux, Marcy l'Etoile, France), and the FDA susceptible breakpoint of ≤8/4 μg/ml was applied.
Whole-genome sequencing (WGS).
DNA was extracted on an EpMotion liquid-handling workstation using a Qiagen UltraClean microbial DNA isolation kit (Hilden, Germany). Libraries were prepared using a Nextera XT DNA library preparation kit and sequenced on a MiSeq apparatus (Illumina, San Diego, CA). SRST2 analysis was performed for multilocus sequence typing (MLST) and characterization of resistance determinants (18).
For comparative sequence analyses, Illumina reads were mapped against a K. pneumoniae ST307 reference genome (GenBank assembly accession no. GCA_002148835.1). Variant calling was performed using the Snippy (version 3.1) program after exclusion of mobile genetic elements with the PHASTER and IslandViewer (version 3) programs (19–21). A neighbor-joining phylogenetic tree was generated in Geneious (version 10.1.3) software (22).
Three isolates were selected for single-molecule long-read sequencing of total DNA with the Oxford Nanopore MinION platform (Oxford Nanopore, Oxford, United Kingdom). Primary base calling was performed using the MinKNOW (version 1.7.10) program, and the Epi2Me (version 2.48.6) program was used for secondary base calling and demultiplexing of barcoded libraries (Oxford Nanopore, Oxford, United Kingdom). The resulting reads were filtered with the Poretools tool kit with a minimum length of 5,000 bp and the mothur (version 1.22.2) program with a maximum homopolymeric region length of 20 bp (23). Hybrid assembly was performed using quality-filtered Nanopore long reads and Illumina short reads with the SPAdes (version 3.10.1) program using the following k-mer parameters: −k 21, 33, 55, 77, 99, 127 (24). The Prokka (version 1.12) program was used for open reading frame prediction and annotation of the three resultant assemblies (25). The progressiveMauve plug-in in Geneious (version 10.1.3) software was used to align the three genomes, distinguish chromosomal and plasmid regions, reorder contigs, and identify possible recombination sites (22, 26).
We performed PCR sequencing of putative integration sites to validate assemblies (for the primers, see Table 2). Primers were strategically positioned on either side of presumed junctions to ensure exclusive amplification of target regions and rule out possible assembly errors. Reactions were performed on select isolates of all phenotypes, including those hypothesized to lack the cassettes of interest, which were considered negative controls. The Sanger sequencing output was aligned against the assemblies for confirmation. The blaKPC copy number was determined by quantitative PCR using SYBR green as previously described (6, 18).
TABLE 2.
Primers for amplification of integration and deletion sites
| Primer | Sequence (5′ → 3′) | Target description |
|---|---|---|
| 123728F | TCCAGCGCGGATTTAAAGTC | Chromosomal plasmid integration |
| 124869R | CGCACCTTCTTGATGACCTTC | |
| 126112R | TAATCAATGCCACACCCAGTC | |
| 171002F | TCAAGACGACGCGTACTATCC | |
| 172513R | GCCGTCTCGACTTTATGATGC | |
| 109141F | CTTTTGCAATATTGCTGGCCGCAC | Plasmid-borne Tn4401e |
| 96896R | GTGATCCCCTGGGCGAAATGCGCCTGGTAAGCA | |
| 106692F | GGCGGCGTTATCACTGTATT | |
| 105185F | GGCGAATGTGCGGGTCCACG | |
| 1884723F | TCCCTCGAGAGCCAGCGCAAG | Chromosomal O antigen |
| 1886053R | TGCTGCTGCAGGACCAGGGG | |
| 1874656F | GGTTTGAACGGATGTGAG | |
| 1875938R | CGTCCAATCAGGGCAACAG |
Accession number(s).
Sequence data are deposited in NCBI under BioProject no. PRJNA420753. This includes raw Illumina and Oxford Nanopore sequence files in the NCBI Sequence Read Archive (SRA; https://trace.ncbi.nlm.nih.gov/Traces/sra/) under accession no. SRP126176, as well as chromosomal and plasmid assemblies for KP1768 (GenBank accession no. CP025140 to CP025142), NR5632 (GenBank accession no. CP025143 to CP025145), and KP1766 (GenBank accession no. CP025146 to CP025148).
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (R01 AI116939 to A.-C.U.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
A.-C.U. has received research funding from Merck, unrelated to the current study. All other authors declare no conflict of interest.
REFERENCES
- 1.van Duin D, Bonomo RA. 2016. Ceftazidime/avibactam and ceftolozane/tazobactam: second-generation β-lactam/β-lactamase inhibitor combinations. Clin Infect Dis 63:234–241. doi: 10.1093/cid/ciw243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carmeli Y, Armstrong J, Laud PJ, Newell P, Stone G, Wardman A, Gasink LB. 2016. Ceftazidime-avibactam or best available therapy in patients with ceftazidime-resistant Enterobacteriaceae and Pseudomonas aeruginosa complicated urinary tract infections or complicated intra-abdominal infections (REPRISE): a randomised, pathogen-directed, phase 3 study. Lancet Infect Dis 16:661–673. doi: 10.1016/S1473-3099(16)30004-4. [DOI] [PubMed] [Google Scholar]
- 3.Shields RK, Chen L, Cheng S, Chavda KD, Press EG, Snyder A, Pandey R, Doi Y, Kreiswirth BN, Nguyen MH, Clancy CJ. 2017. Emergence of ceftazidime-avibactam resistance due to plasmid-borne blaKPC-3 mutations during treatment of carbapenem-resistant Klebsiella pneumoniae infections. Antimicrob Agents Chemother 61:e02097-16. doi: 10.1128/AAC.02097-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humphries RM, Yang S, Hemarajata P, Ward KW, Hindler JA, Miller SA, Gregson A. 2015. First report of ceftazidime-avibactam resistance in a KPC-3-expressing Klebsiella pneumoniae isolate. Antimicrob Agents Chemother 59:6605–6607. doi: 10.1128/AAC.01165-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humphries RM, Hemarajata P. 2017. Resistance to ceftazidime-avibactam in Klebsiella pneumoniae due to porin mutations and the increased expression of KPC-3. Antimicrob Agents Chemother 61:e00537-17. doi: 10.1128/AAC.00537-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson K, Hemarajata P, Sun D, Rubio-Aparicio D, Tsivkovski R, Yang S, Sebra R, Kasarskis A, Nguyen H, Hanson BM, Leopold S, Weinstock G, Lomovskaya O, Humphries RM. 2017. Resistance to ceftazidime-avibactam is due to transposition of KPC in a porin-deficient strain of Klebsiella pneumoniae with increased efflux activity. Antimicrob Agents Chemother 61:e00989-17. doi: 10.1128/AAC.00989-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramachandran R, Hartmann C, Song HK, Huber R, Bochtler M. 2002. Functional interactions of HslV (ClpQ) with the ATPase HslU (ClpY). Proc Natl Acad Sci U S A 99:7396–7401. doi: 10.1073/pnas.102188799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haidar G, Clancy CJ, Shields RK, Hao B, Cheng S, Nguyen MH. 2017. Mutations in blaKPC-3 that confer ceftazidime-avibactam resistance encode novel KPC-3 variants that function as extended-spectrum beta-lactamases. Antimicrob Agents Chemother 61:e02534-16. doi: 10.1128/AAC.02534-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Follador R, Heinz E, Wyres KL, Ellington MJ, Kowarik M, Holt KE, Thomson NR. 2016. The diversity of Klebsiella pneumoniae surface polysaccharides. Microb Genom 2:e000073. doi: 10.1099/mgen.0.000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Livermore DM, Warner M, Jamrozy D, Mushtaq S, Nichols WW, Mustafa N, Woodford N. 2015. In vitro selection of ceftazidime-avibactam resistance in Enterobacteriaceae with KPC-3 carbapenemase. Antimicrob Agents Chemother 59:5324–5330. doi: 10.1128/AAC.00678-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Long SW, Olsen RJ, Eagar TN, Beres SB, Zhao P, Davis JJ, Brettin T, Xia F, Musser JM. 2017. Population genomic analysis of 1,777 extended-spectrum beta-lactamase-producing Klebsiella pneumoniae isolates, Houston, Texas: unexpected abundance of clonal group 307. mBio 8:e00489-17. doi: 10.1128/mBio.00489-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papp-Wallace KM, Winkler ML, Taracila MA, Bonomo RA. 2015. Variants of beta-lactamase KPC-2 that are resistant to inhibition by avibactam. Antimicrob Agents Chemother 59:3710–3717. doi: 10.1128/AAC.04406-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castanheira M, Mills JC, Costello SE, Jones RN, Sader HS. 2015. Ceftazidime-avibactam activity tested against Enterobacteriaceae isolates from U.S. hospitals (2011 to 2013) and characterization of beta-lactamase-producing strains. Antimicrob Agents Chemother 59:3509–3517. doi: 10.1128/AAC.00163-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomez-Simmonds A, Greenman M, Sullivan SB, Tanner JP, Sowash MG, Whittier S, Uhlemann AC. 2015. Population structure of Klebsiella pneumoniae causing bloodstream infections at a New York City tertiary care hospital: diversification of multidrug-resistant isolates. J Clin Microbiol 53:2060–2067. doi: 10.1128/JCM.03455-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shields RK, Nguyen MH, Press EG, Chen L, Kreiswirth BN, Clancy CJ. 2017. In vitro selection of meropenem resistance among ceftazidime-avibactam-resistant, meropenem-susceptible Klebsiella pneumoniae isolates with variant KPC-3 carbapenemases. Antimicrob Agents Chemother 61:e00079-17. doi: 10.1128/AAC.00079-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villa L, Feudi C, Fortini D, Brisse S, Passet V, Bonura C, Endimiani A, Mammina C, Ocampo AM, Jimenez JN, Doumith M, Woodford N, Hopkins K, Carattoli A. 2017. Diversity, virulence, and antimicrobial resistance of the KPC-producing Klebsiella pneumoniae ST307 clone. Microb Genom 3:e000110. doi: 10.1099/mgen.0.000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clinical and Laboratory Standards Institute. 2015. Performance standards for antimicrobial susceptibility testing, 25th informational supplement Document M100-S25. Clinical and Laboratory Standards Institute, Wayne, PA, [Google Scholar]
- 18.Inouye M, Dashnow H, Raven LA, Schultz MB, Pope BJ, Tomita T, Zobel J, Holt KE. 2014. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med 6:90. doi: 10.1186/s13073-014-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seemann T. 2017. Rapid haploid variant calling and core SNP phylogeny. https://github.com/tseemann/snippy Accessed 15 August 2017.
- 20.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. 2016. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44:W16–W21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhillon BK, Laird MR, Shay JA, Winsor GL, Lo R, Nizam F, Pereira SK, Waglechner N, McArthur AG, Langille MG, Brinkman FS. 2015. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res 43:W104–W108. doi: 10.1093/nar/gkv401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loman NJ, Quinlan AR. 2014. Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics 30:3399–3401. doi: 10.1093/bioinformatics/btu555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 26.Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]