

ABSTRACT
Antifungal treatment is often ineffectual, partly because of biofilm formation. In this study, by using a combined forward and reverse genetic strategy, we identified that nucleus-localized AfSsn3 and its partner AfSsn8, which constitute a Cdk8-cyclin pair, are required for azole resistance in Aspergillus fumigatus. Deletion of Afssn3 led to increased absorption and utilization of glucose and amino acids. Interestingly, absorption and utilization of glucose accelerated the extracellular polysaccharide formation, while utilization of the amino acids serine, threonine, and glycine increased sphingolipid pathway intermediate accumulation. In addition, the absence of Afssn3 induced the activity of the efflux pump proteins. These factors indicate the mature biofilm is responsible for the major mechanisms of A. fumigatus resistance to azoles in the ΔAfssn3 mutant. Collectively, the loss of Afssn3 led to two “barrier” layers between the intracellular and extracellular spaces, which consequently decreased drug penetration into the cell.
KEYWORDS: biofilm, Afssn3, extracellular polysaccharide, sphingolipid pathway intermediates, efflux pump, Aspergillus fumigatus
INTRODUCTION
Aspergillus fumigatus is a notorious opportunistic pathogen that causes invasive aspergillosis (IA) with high mortality (1, 2). At present, azoles are the first line of antifungal drugs for treating IA (3). Members of the azole class primarily include fluconazole, itraconazole (ITC), voriconazole (VRC), and posaconazole. Despite the fact that the widespread utilization of azole drugs has significantly increased the survival rate of patients with IA, the continued use of antifungal drugs has increased the emergence of drug-resistant strains over the years, which has resulted in less effective agents and high therapeutic costs (4–6). Even worse, the approved antifungals for IA are quite limited (7). Consequently, there is an urgent need to understand the molecular mechanisms of azole resistance, which may provide theoretical support for new antifungal drug design.
One important mechanism of azole resistance in A. fumigatus was mediated by the modification of the azole target protein Cyp51A, which encodes 14-α-demethylase and functions in ergosterol biosynthesis (8, 9). The inhibition of 14-α-demethylase caused by azoles results in the reduction of ergosterol biosynthesis and toxic sterol accumulation, which inhibits cell growth (8, 9). At present, more than 30 different Cyp51A modification sites have been described as being responsible for azole cross-resistance in clinical isolates and laboratory-derived mutants (5, 9).
Another important mechanism of azole resistance is mediated by the overexpression of multidrug resistance (MDR) pumps (9). The MDR efflux transporter genes of the ATP-binding cassette (ABC) and the major facilitator superfamily (MFS) classes have been shown to be clinically important in different pathogenic fungi (9). Under increased efflux pump activity, azoles might be easy to remove, leading to azole resistance (9–12). A. fumigatus is believed to harbor at least 49 genes that encode the efflux transporters of the ABC family and 278 genes of the MFS family (13, 14). Among them, the overexpression of AfuMDR1, AfuMDR2, AfuMDR3, AfuMDR4, and atrF has resulted in azole resistance (11).
In fungi, biofilm formation is complex and is also considered to play therapeutic roles in drug resistance. Biofilms are reportedly able to inhibit the penetration of antifungals and cause inefficacy in antifungal treatment, including the movement of fluconazole, amphotericin B, and flucytosine into cells to treat Candida albicans (9, 15, 16). Reports also showed that A. fumigatus can form biofilms, which were characterized by aggregates of multicellular hyphae enclosed within the extracellular matrix (ECM), and they are resistant to antifungal drugs, especially caspofungin and itraconazole (13, 17). In addition, biofilms are also partly associated with efflux pump activity (13).
In Manchester, 64 azole-resistant A. fumigatus strains isolated from patients or other places were collected and analyzed. The results showed that only 57% were found to have Cyp51A mutations, suggesting that other possible resistance mechanisms exist (18). In fact, laboratory-derived mutants with novel mutations were reportedly resistant to azoles to various degrees (19, 20). In addition, the heat shock protein Hsp90 is also involved in azole resistance (9).
Although many efforts have been made to understand azole resistance, knowledge of this molecular mechanism is still limited. To understand the azole resistance mechanism, the Agrobacterium tumefaciens-mediated transformation (ATMT) method was used to find new genes related to azole resistance in this study. ATMT is an efficient tool for constructing mutant libraries, which has been widely used not only in fungi but also in plants (21, 22).
In the present study, through ATMT, we identified that A. fumigatus nucleus-localized Afssn3 (Afu3g13990), along with its partner Afssn8 (Afu2g15790), which constituted a Cdk8-cyclin pair (23), was required for azole resistance. In Saccharomyces cerevisiae and Candida albicans, SSN3 is a cyclin-dependent kinase and SSN8 is a cyclin-like protein; both proteins are part of the heterotetrameric Cdk8 module that forms a complex with the transcriptional coregulator, a mediator. It has been reported that Ssn3 in S. cerevisiae or C. albicans is involved in multiple pathways, such as repression of carbon source utilization, biofilm formation, and drug resistance (24, 25). In humans, the homolog of Afssn3 is cdk8, which is considered to play crucial roles in colon cancer and melanoma formation (26, 27). Inactivating Afssn3 led to the increased uptake and consumption of glucose and subsequently resulted in increased extracellular polysaccharide (EPS)/ECM biosynthesis. Furthermore, the increased consumption and utilization of amino acids caused a significant accumulation of sphingolipid pathway intermediates (long-chain bases [LCBs]). In addition, deletion of Afssn3 increased the expression of efflux transporter genes. Collectively, these results demonstrated that the Afssn3-Afssn8 pair-constituted Cdk8 module was required for the regulation of glucose and amino acid metabolism, which would affect the EPS/ECM formation, sphingolipid metabolism, and efflux pump activities and thus formed hyperbiofilm to affect the azole resistance of the ΔAfssn3 mutant.
RESULTS
Screening and identification of Afssn3.
Azole resistance in fungi has resulted in severe problems during clinical treatment. To further investigate the azole resistance mechanism, we constructed a randomly inserted mutant library of A. fumigatus (Af293) that was mediated by A. tumefaciens. Of the 2,000 tested transformants, eight azole-resistant transformants and one azole-susceptible, caspofungin-susceptible transformant were screened on plates containing ITC and caspofungin at 0.1 and 2 μg/ml, respectively (Fig. 1A and B). Most of the nine transformants displayed similar growth rates and colony morphology compared to the wild-type strain. These nine transformants were then selected for further analysis. Among the eight azole-resistant transformants, the transfer DNA (T-DNA) flanking sequence of mutant T637 (i.e., transformant 637) was first identified by thermal asymmetric interlaced PCR (TAIL-PCR). As Fig. 1C shows, T-DNA was inserted into the upstream (−290 bp) of Afu3g13990 initiation codon of the T637 mutant. To identify the homologs of Afu3g13990 in S. cerevisiae, the putative amino acid sequence of Afu3g13990 was used as a query to perform BLASTP analysis in the website of the National Center for Biotechnology Information (NCBI). The result showed that SSN3 of S. cerevisiae (NP_015283.1, 58% identity, E value = 3e−135), which encodes a serine threonine kinase, was a possible homolog of Afu3g13990 in A. fumigatus, referred to here as Afssn3 (Fig. 1D).
FIG 1.

Screening and identification of novel genes that confer altered antifungal susceptibility to A. fumigatus. (A and B) Comparison of wild-type (Af293) colonies and a representative colony of T-DNA insertion mutants in two different categories grown on YUU agar plates supplemented with ITC, VRC, and caspofungin (CS) at the indicated concentrations. Plates were incubated at 37°C for 2 days. (C) Schematic of T-DNA insertion sites in the genomic regions of A. fumigatus. The gene maps were adapted from pages on the CADRE website (https://bio.tools/cadre_genomes_browser). The insertion site determined by TAIL-PCR was located at bp −290. Italic and boldface letters indicate the right border (RB) sequence of T-DNA. The underlined sequence represents the 5′ noncoding region of AFUA_3G13990. (D) Schematic diagram of conserved domains of AfSsn3 in A. fumigatus. AfSsn3 contains an STKc_CDK8_like domain and a PKC_like domain with its superfamilies.
Disruption of Afssn3 in A. fumigatus increased azole resistance.
Given the important functions of SSN3 as reported in S. cerevisiae and humans, we deleted Afssn3 in the A1160 background strain (Fig. 2A), which was a ku80-null mutant. Diagnostic PCR showed that the open reading frame (ORF) of Afssn3 was fully replaced by the selection marker pyr4, indicating that the Afssn3-null mutant was constructed successfully (see Fig. S1A in the supplemental material). As shown in Fig. 2B, the Afssn3-null mutant displayed an attenuated conidiation ca. 40% that of the parental wild type per unit area when inoculated on YUU-rich medium (for the composition of YUU, see Materials and Methods), which is somewhat different from the observation for T637. This result indicated that Afssn3 is partly required for conidiation. To investigate whether Afssn3 was involved in azole resistance, the Afssn3-null mutant and the wild-type strain were first tested on YUU plates containing ITC at 2 μg/ml. As shown in Fig. 2A, the ΔAfssn3 and T637 mutants both displayed increased growth, whereas the growth of the wild-type strain was completely arrested in the presence of ITC or VRC, suggesting that Afssn3 is involved in azole resistance. However, when the functional Afssn3 gene was complemented at the locus of the ΔAfssn3 mutant, the strain (named ΔAfssn3C) showed similar azole susceptibility to that of the wild-type strain, indicating that the aberrant ΔAfssn3 azole resistance phenotype was specific to the loss of Afssn3 rather than other unexpected mutations (Fig. 2A).
FIG 2.
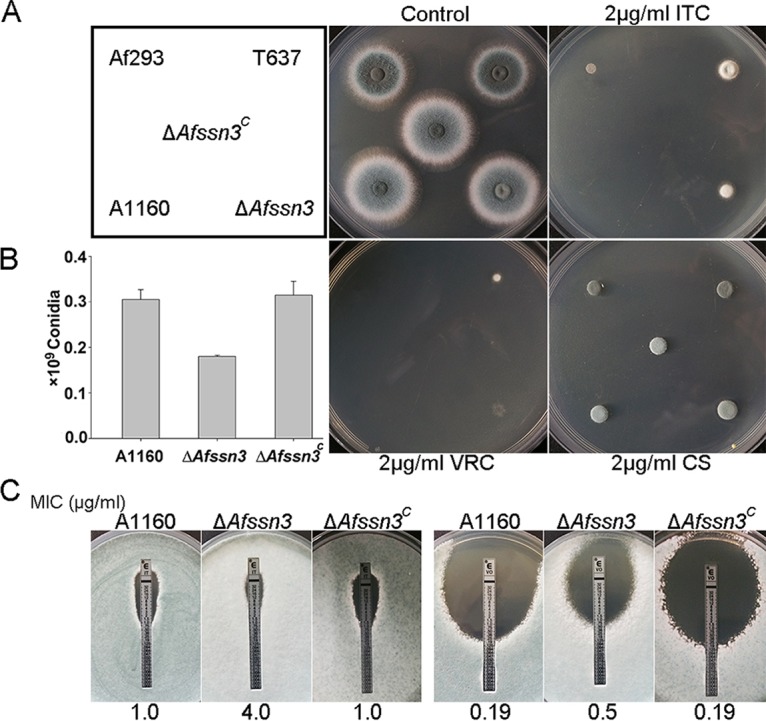
ΔAfssn3 mutants displayed azole-resistant phenotypes. (A) Colony phenotype comparison of the indicated strains. Portions (2 μl) of ddH2O containing 2 × 104 conidia of each strain were inoculated in YUU medium with or without 2 μg/ml ITC, VRC, and caspofungin (CS). Plates were cultured at 37°C for 2 days. (B) Quantitative data for the numbers of conidia in related strains cultured at 37°C for 2 days. Error bars represent the standard deviations from three replicates. (C) For each strain, 105 conidia were mixed in YUU, and Etest strips impregnated with an ITC or VRC gradient were placed on YUU agar plates and cultured for 2 days at 37°C before observation.
Subsequently, we tested the MICs of the ΔAfssn3 strain and the wild-type strain in response to ITC based on the CLSI-M38-A2 method. The MICs of the ΔAfssn3 mutant were 2-fold (4 μg/ml in the Afssn3-null mutant versus 2 μg/ml in the wild-type strain) and 4-fold (4 μg/ml in the Afssn3-null mutant versus 1 μg/ml in the wild-type strain) higher than that of the wild-type strain for ITC and VRC, respectively. This finding is consistent with the results for the T637 mutant and its parental stain Af293 (8 μg/ml in the T637 mutant versus 4 μg/ml in the parental strain in response to ITC and 8 μg/ml in the T637 mutant versus 2 μg/ml in the parental strain to VRC). Etest strips further confirmed that the ΔAfssn3 strain exhibited increased resistance to azoles (Fig. 2C). Taken together, these results strongly demonstrated that AfSsn3 is involved in azole resistance in A. fumigatus.
AfSsn3 is located in the nucleus.
In S. cerevisiae and C. albicans, Ssn3 has been shown to function as a transcriptional coactivator of RNA polymerase II (23–25). For this reason, we hypothesize that AfSsn3 could be located in the nucleus in A. fumigatus. To gain insight into the location of AfSsn3, we first tried to label AfSsn3 with green fluorescent protein (GFP) at its C terminus under the control of its native promoter through homologous recombination. However, we failed to detect the AfSsn3-GFP signals. We supposed that maybe the AfSsn3-GFP signal is too weak to detect. Therefore, we labeled AfSsn3 at its C terminus under the control of the gpdA promoter through ectopic integration. Western blot data showed that the molecular mass of AfSsn3-GFP was ∼75 kDa (including GFP, 27 kDa), which was in accordance with the predicted size, implying that the GFP-labeled strain we constructed was successful (Fig. 3A). Using fluorescent light microscopy observation, we found that in YUU media with or without ITC (1 μg/ml) stimulation, AfSsn3-GFP was always accumulated at the nucleus (Fig. 3B). This result suggests that AfSssn3 under our test conditions was located in the nucleus, and its subcellular location was not responsive to the ITC stimulation in the AfSsn3-GFP overexpression strain.
FIG 3.

AfSsn3 is located in the nucleus. (A) Western blotting of AfSsn3-GFP fusion with an anti-GFP antibody. The predicted size of AfSsn3-GFP is ∼78 kDa. (B) Colocalization of AfSsn3-GFP with or without ITC (1 μg/ml) stimulation (1 h). DAPI was used to visualize the nucleus. Scale bar, 10 μm.
AfSsn3 deficiency results in increased glucose absorption and hyperbiofilm.
Because the homolog of Ssn3 in humans is involved in colon cancer and melanoma formation and in C. albicans is involved in biofilm formation (25–27), we were interested in whether the Ssn3 deficiency would lead to some characteristics of tumor cells or biofilm formation in A. fumigatus. To this end, we first investigated the biofilm formation of the ΔAfssn3 mutant and the wild type. As expected, compared to the wild type, the ΔAfssn3 mutant showed significantly increased biofilm biomass (Fig. 4A), implying that Afssn3 is concerned with biofilm formation. Because glucose is the carbon source of biofilm formation, we then tested the glucose consumption of the ΔAfssn3, wild-type, and ΔAfssn3C strains under minimal media. As shown in Fig. 4B, the extracellular glucose levels of the ΔAfssn3 mutant were significantly lower than those of the wild-type and ΔAfssn3C strains at growth times of 16 and 20 h. These data suggest that, compared to the wild type, the glucose absorption activity of the ΔAfssn3 mutant was significantly increased.
FIG 4.

The absence of Afssn3 results in increased glucose uptake, biofilm formation, and abnormal nuclear morphology. (A) Quantification of A. fumigatus biofilms. The indicated strains (106 CFU) were cultured on serum-soaked silicone squares submerged in liquid YUU medium at 37°C for 48 h. The biofilms were then dried and weighed. The error bars represent the standard errors of the mean (n = 3). (B) The wild-type (A1160), ΔAfssn3, and ΔAfssn3C strains were cultured at 37°C 220 rpm for 24 h in liquid minimal medium (MM). The supernatants at the indicated times were collected for HPLC analysis of residual glucose. The extracellular glucose content was normalized to the biomass of each strain. *, P < 0.05; **, P < 0.01. (C) ECM structures of the indicated strains. The in vitro biofilm of each strain (106 CFU) incubated in 37°C for 48 h was observed by SEM. Arrows indicate ECM areas. (D) Phenotypic comparison of nuclei in the wild-type, ΔAfssn3, and ΔAfssn3C strains. All strains were grown in liquid YUU medium at 37°C for 12 h. The nuclei were visualized using DAPI. Arrows indicate the locations of the nuclei. Bars, 10 μm. (E) Quantitative data for cell nucleus diameters in related strains (**, P < 0.01).
Based on the above data that the loss of Afssn3 accelerated glucose consumption, we wondered whether the consumed glucose would cause an increase in ECM produced from the EPS. To test this possibility, we observed the extracellular polysaccharide matrix by using scanning electron microscopy (SEM). Under the same culture conditions, the extracellular polysaccharide matrix produced by the ΔAfssn3 strain was obviously greater than that of the wild type (Fig. 4C). Nevertheless, the components of the cell wall polysaccharide we tested, such as the glucan and chitin between the ΔAfssn3 and wild-type strains, were not changed (see Fig. S2A in the supplemental material).
We then observed the nuclear morphology, a characteristic phenotype of tumor cells, of the ΔAfssn3 and wild-type strains. Interestingly, we found that the nucleus of the ΔAfssn3 mutant was obviously bigger than that of the wild type and ΔAfssn3C (Fig. 4D and E). These data suggest that AfSsn3 is essential to keep the natural morphology of nucleus.
As it turns out, biofilm formation is a crucial factor in azole resistance (9, 13, 17). To understand the function of Afssn3 in azole resistance and to elucidate whether the increased biofilm formation of the Afssn3-null mutant resulted from the increased activity of glucose absorption, we tested the content of glucose and other small molecules in the ΔAfssn3 and wild-type strains during the early growth stage using untargeted metabolomics. To this end, we performed gas chromatography-mass spectrometry (GC-MS). After data pretreatment and multivariate statistics, including principal-component analysis (PCA) and partial least-squares discriminant analysis (PLS-DA), significantly changed metabolites were identified for further study (see Fig. S3A and B in the supplemental material). Approximately 72 metabolites (variable importance in projection [VIP] > 1; P < 0.05) were reported and 7 and 38 small molecules were up- and downregulated by >1.2- and <0.65-fold, respectively, in the ΔAfssn3 strain compared to the wild type (see Data Set S1 in the supplemental material). Representative differential metabolites can be divided into four categories: monosaccharides, amino acids, nucleotides, and organic acids. Surprisingly, monosaccharides and amino acids in the mutant made up the largest number of compounds at a lower level compared to that of the wild type.
Given that losing Afssn3 resulted in the increased assimilation of glucose and thus promoted biofilm formation, we questioned whether the increased glucose assimilation could be attributed to azole resistance. For this purpose, extra glucose at a different concentration was added to the media to test whether the azole resistance of the ΔAfssn3 mutant occurred in a glucose dose-dependent manner. As expected, with increased glucose concentrations, the azole resistance of ΔAfssn3 exhibited an enhanced trend (see Fig. S4 in the supplemental material). When glucose was added up to 8%, the azole resistance of ΔAfssn3 reached the highest level, and it decreased when the glucose concentration was more than 8%. In contrast, addition of the disaccharide trehalose, added as a control, was unable to increase azole resistance (see Fig. S4 in the supplemental material). Consequently, we concluded that AfSsn3 deficiency resulting in hyperbiofilm formation is partly due to increased glucose absorption and consumption.
Cellular amino acid category and concentration are involved in azole resistance.
Considering the aforementioned information obtained by GC-MS analysis that deletion of Afssn3 resulted in decreased amino acid accumulation in the cells, we sought to find whether there was some relationship between amino acid utilization and azole resistance. To this end, the azole resistances of ΔAfssn3 and wild-type strains were tested with different amino acid supplementation in the media. The results showed that the amino acids we tested can be divided into four categories according to the contribution to the azole resistance of the ΔAfssn3 strain (Fig. 5 and Fig. S5 in the supplemental material). Category I amino acids decrease the azole resistance of ΔAfssn3, such as Lys and His, and category II amino acids make no obvious or very little contribution to the azole resistance of ΔAfssn3, such as Arg, Met, Asp, Pro, Hse, Glu, Cys, Ala, Leu, Val, and Ile. Category III amino acids, such as Ser, Thr, and Gly, significantly increased the azole resistance of ΔAfssn3, and category IV amino acids confer azole resistance to both ΔAfssn3 and the wild type (most of these were aromatic amino acids such as Tyr, Phe, and Try). In conclusion, these data strongly suggest that Afssn3-involved amino acid metabolism is required for azole resistance.
FIG 5.

The addition of extra amino acids affected the resistance of ΔAfssn3 to ITC. The ITC resistance of the indicated strains was tested in YUU plates with 2% amino acid addition (pH 6.5 to 7.0). Plates were cultured at 37°C for 2 days.
ΔAfssn3 accumulates sphingolipid pathway intermediates.
Given the aforementioned fact that losing Afssn3 resulted in increased glucose and amino acid consumption, which subsequently led to azole resistance, we sought to determine how consumption of the glucose or amino acid affects azole resistance. Are there unknown macromolecular metabolites involved in azole resistance after glucose or amino acid consumption? To find out, the ΔAfssn3 and wild-type strains were subjected to cellular macromolecular metabolite analysis by ultrahigh performance liquid chromatography-quadrupole time-of-flight mass spectrometry (UPLC/Q-TOF MS) based on the untargeted intracellular metabolite metabolomics during the late growth stage. PCA, PLS-DA, hierarchical clustering analysis, and Kyoto encyclopedia of genes and genome (KEGG) pathway mapper analysis were performed to investigate the two strains. The PCA and PLS-DA score plots revealed that the specimens from the two strains were distinguished from one another (see Fig. S3C and D in the supplemental material). A hierarchical clustering analysis and KEGG pathway mapper analysis of the metabolites from the two strains is shown in Fig. 6A and Data Set S1 in the supplemental material. The contents of the 57 molecules (VIP > 2; P < 0.01) with high or low molecular masses that we tested, including the amino acids, lipids, and polysaccharides in ΔAfssn3, were changed compared to those of the wild type. Interestingly, sphingolipid pathway intermediates and polysaccharides were significantly accumulated in the ΔAfssn3 strain relative to the wild type. Compared to the wild-type strain, the ΔAfssn3 mutant exhibited a 3.4-fold increase in 3-keto-dihydrosphingosine (3-keto-DHS), a 4.6-fold increase in dihydrosphingosine (DHS), a 7-fold increase of phytosphingosine (PHS), and a 6-fold increase in dihydroceramide (DHC) (Fig. 6A and B and see Data Set S1 in the supplemental material). Coincidentally, the biosynthetic pathway of sphingolipids and ceramides starts with the condensation of serine and palmitoyl coenzyme A (palmitoyl-CoA) and yields 3-keto-DHS under the catalysis of serine palmitoyl-CoA transferase (SPT) (Fig. 6C) (28). Combined with the aforementioned information that ΔAfssn3 increases Ser, Thr, and Gly utilization (Thr and Gly can be converted into Ser during metabolism) and accumulates large amounts of sphingolipid pathway intermediates, we conclude that the derepression of Ser-, Thr-, and Gly-mediated sphingolipid biosynthesis in ΔAfssn3 is crucial for azole resistance.
FIG 6.

Afssn3 deficiency accumulates sphingolipid pathway intermediates. (A) Hierarchical clustering dendrogram of differential metabolites from the wild-type (A1160) and ΔAfssn3 strains. Red, upregulation; blue, downregulation. Six biological replicates were tested for each sample. Arrows indicate the accumulation of the LCBs in ΔAfssn3. (B) Histogram of the different pathway −log(P = 10) values enriched in the ΔAfssn3 strain according to KEGG pathway analysis. The horizontal line at 1.3 indicates P < 0.05; the horizontal line at 2 indicates P < 0.01. (C) Sphingolipid biosynthesis pathway in yeast. Circles indicate the predicted yeast TSC10 and AUR1 orthologs in A. fumigatus: KsrA and AurA.
In addition, we also identified some other accumulated compounds, namely, 2-O-(β-d-galactopyranosyl-(1→6)-β-d-galactopyranosyl)2S,3R-dihydroxytridecanoic acid (1.6-fold) and O-β-d-Gal-(1→3)-O-[O-β-d-Gal-(1→4)-2-(acetylamino)-2-deoxy-β-d-Glc-(1→6)]-2-(acetylamino)-2-deoxy-d-galactose (1.5-fold) in ΔAfssn3, which may be intermediate products of the extracellular polysaccharide matrix, glycolipids, and/or glycoproteins.
LCB accumulation is an important factor contributing to Afssn3 deficiency leading to azole resistance.
In addition to the findings described above that ΔAfssn3 accumulated LCBs, we hypothesized that sphingolipid pathway proteins may also have functions in azole resistance. For this reason, two sole and essential genes—ksrA, which encodes a 3-keto-dihdrosphingosine reductase (3KSR) whose substrate is 3-keto-dihydrosphingosine, and aurA, which encodes an inositol phosphorylceramide synthase, whose substrate is phytoceramide—were selected for further investigation (Fig. 6C) (28). ksrA and aurA were then overexpressed in a wild-type background using the gpdA promoter, and the strains were named WTOE::ksrA and WTOE::aurA (Fig. 7A). The expression levels of ksrA and aurA in the two OE strains were determined by quantitative reverse transcription-PCR (qRT-PCR) analysis (Fig. 7B), and the results indicated that the mRNA levels of ksrA and aurA were sharply increased in the two OE strains. It should be noted that all of the strains contained their native alleles. From the colony growth phenotypes observed on the plates, we can see that WTOE::ksrA presented a clear phenotype of resistance to ITC (Fig. 7A). In contrast, the overexpression of aurA had little impact on ITC sensitivity (Fig. 7A).
FIG 7.

LCB accumulation is an important factor contributing to azole resistance in the ΔAfssn3 strain. (A) Susceptibility comparison of different strains on YAG plates supplemented with or without ITC (0.1 or 2 μg/ml) and serine (2%). (B) The transcription of ksrA and aurA in wild-type and ksrA- and aurA-overexpressing strains was detected by real-time PCR. (C) Transmission electron micrographs of the wild-type and ΔAfssn3 strains. (D) Intracellular ITC accumulation of the wild-type and ΔAfssn3 strains quantified by HPLC. The ITC content of the wild type was normalized to the level of ΔAfssn3 (wild type = 1).
To further confirm these phenomena, WTOE::ksrA, WTOE::aurA, and the wild-type strains were subjected to sphingolipid analysis by UPLC/Q-TOF MS based on the untargeted intracellular metabolite metabolomics during the late growth stage, as mentioned above. The PCA and PLS-DA score plots revealed that the specimens of the three strains were distinguished from each other (see Fig. S3E and F in the supplemental material). As expected, the LCBs accumulated in WTOE::ksrA so that the content levels of 3-keto-DHS, DHS, PHS, and DHC increased 1.12-, 1.5-, 1.38-, and 1.28-fold, respectively, compared to the wild-type strain (Table 1). However, the LCBs were diminished in WTOE::aurA; as a result, the levels of 3-keto-DHS, DHS, PHS, and DHC increased 0.67-, 0.83-, 0.79-, and 0.67-fold, respectively, compared to the wild-type strain (Table 1). These results suggested that ksrA and aurA overexpression would affect azole resistance or susceptibility, which were mediated by the content of LCBs.
TABLE 1.
Differential sphingolipid pathway intermediates identified in WTOE::ksrA versus WT and in WTOE::aurA versus WT experiments
| No. | Metabolite | m/z (Da) | Δppm | VIPa |
P value |
Fold change |
||
|---|---|---|---|---|---|---|---|---|
| WTOE::ksrA/WT | WTOE::aurA/WT | WTOE::ksrA/WT | WTOE::aurA/WT | |||||
| 1 | 3-Ketodihydrosphingosine | 299.28245 | 0.05 | 1.84011 | 0.00282 | 0.02127 | 1.124168 | 0.673864 |
| 2 | Dihydrospingosine | 301.2978 | 0.94 | 1.83252 | 0.00256 | 0.02216 | 1.507481 | 0.826465 |
| 3 | Phytospingosine | 317.29252 | 1.48 | 1.07507 | 0.04329 | 0.03383 | 1.376037 | 0.794281 |
| 4 | Dihydroceramide | 329.29297 | 0.09 | 2.1331 | 0.00057 | 3.77E-05 | 1.277921 | 0.670821 |
VIP, variable importance in projection.
It has been reported that sphingolipid and its intermediates function in cell membrane formation (28), so we speculated that the ΔAfssn3 strain probably has a differentiated cell membrane morphology from that of the wild type. To test our hypothesis, the cell membranes of ΔAfssn3 and the wild-type hyphae were examined by transmission electron microscopy (TEM). As shown in Fig. 7C, ΔAfssn3 cell membranes were more densely stained than those of the wild type. Similarly, the intracellular membrane of the ΔAfssn3 cells also showed a denser staining pattern, suggesting that Afssn3 is also essential for the architecture of cellular membranes.
RNA sequencing reveals that the loss of Afssn3 upregulates ABC and MFS multidrug transporter transcription.
Our metabolomics and ksrA overexpression data led us to propose that deleting Afssn3 probably increases the transcription of sphingolipid pathway-related genes, which are associated with azole resistance. To test this hypothesis, the whole-genome transcript profiles of the Afssn3-null mutant and the wild-type strain were comparatively analyzed. Among the 9,929 A. fumigatus transcripts, 572 were significantly differentially regulated (P ≤ 0.05, ∣log2FC∣ ≥ 1) in the ΔAfssn3 mutant compared to the wild-type strain (see Data Set S2 in the supplemental material). Of the 572 differentially expressed genes, 411 were upregulated, and 161 were downregulated, indicating that Afssn3 primarily functions as a transcriptional repressor. The gene ontology (GO) functional enrichment analysis showed that the differentially expressed genes were primarily involved in the alkaloid biosynthetic process, hydrogen peroxide catabolic process, hexose transmembrane transport, glucose import, ergosterol biosynthetic process, transmembrane transport, cell adhesion, and others (Fig. 8A and B). Interestingly, the transcription of genes related to the sphingolipid pathway was not affected. Four putative MDR efflux transporter genes, seven putative monosaccharide transporter genes, and five genes related to ergosterol biosynthesis were expressed at relatively higher levels in the absence of Afssn3 in A. fumigatus (Table 2).
FIG 8.

Afssn3 inactivation increased the activity of efflux pumps. (A and B) Histogram of the upregulated pathway (A) and the downregulated pathway (B) −log(P = 10) values enriched in ΔAfssn3 based on a GO functional enrichment analysis. (C) Rh123 accumulation is reduced in strain ΔAfssn3. Rh123 accumulation was measured by flow cytometry. The fluorescein isothiocyanate (FITC) at the x axis represents the relative value of fluorescence intensity and side scatter-high (SSC-H) at the y axis, which was the relative value of the light scattering intensity in A. fumigatus.
TABLE 2.
Differentially expressed genes selected in RNA-seq data
| Transcript ID | Gene product | Fold change (ΔAfssn3/WT) |
|---|---|---|
| Putative MDR efflux transporters | ||
| AFUB_009810/abcB | Putative ABC multidrug transporter | 4.14 |
| AFUB_030790/abcA | Multidrug transporter of ABC superfamily | 4.26 |
| AFUB_030800 | Putative MSF drug transporter | 2.46 |
| AFUB_053630/mdr1/abcA | ABC multidrug transporter | 2.47 |
| Putative glucose transporters | ||
| AFUB_003940 | Putative hexose transmembrane transporter | 4.25 |
| AFUB_021600 | Putative hexose transmembrane transporter | 4.93 |
| AFUB_048240 | Putative hexose transmembrane transporter | 2.53 |
| AFUB_051360 | Putative glucose transporter of the MFS | 1.67 |
| AFUB_071910 | Putative hexose transmembrane transporter | 2.47 |
| AFUB_091410 | Putative MFS glucose/myoinositol transporter | 13.75 |
| AFUB_095240/mstA | Putative MFS monosaccharide transporter | 1.87 |
| Ergosterol biosynthesis-related transcripts | ||
| AFUB_003560/erg24 | Putative C-14 sterol reductase with a predicted role in ergosterol biosynthesis | 2.66 |
| AFUB_017380/erg3 | Putative sterol delta-5,6-desaturase with a predicted role in ergosterol biosynthesis | 1.95 |
| AFUB_033070/erg26 | Putative C-3 sterol dehydrogenase/C-4 decarboxylase | 3.43 |
| AFUB_063960/erg11A | 14-Alpha sterol demethylase | 1.92 |
| AFUB_084150/erg25B | Putative C-4 methyl sterol oxidase with a predicted role in ergosterol biosynthesis | 2.42 |
The overexpression of MDR transporter genes in A. fumigatus is an important mechanism of azole resistance. To gain additional insight into the observed changes in transporter gene expression, we further compared the activity of efflux pumps by detecting the cellular accumulation of the rhodamine-123 (Rh123) fluorescent compound using flow cytometry in both the wild type and the Afssn3-null mutant. Consistent with the transcriptome sequencing (RNA-seq) data, the results in Fig. 8C showed that a striking reduction of Rh123 was observed in ΔAfssn3, indicating a relatively high activity by the efflux pumps in ΔAfssn3. Further, the intracellular ITC contents of the Afssn3-null mutant and the wild type were directly measured by high-pressure liquid chromatography (HPLC). HPLC analysis showed that the intracellular ITC accumulation in ΔAfssn3 was obviously lower than that in the wild type (Fig. 7D). This result also suggested that the efflux pump activity in ΔAfssn3 was higher than that in the wild type. Interestingly, Gal11p/MED15, a subunit of the mediator coactivator, has been demonstrated in yeasts to play a critical role in regulating drug efflux pumps and MDR (29). Because ergosterol biosynthesis is the drug target of ITC and VRC, the total ergosterol contents of the Afssn3-null mutant and the wild-type strain were then analyzed using HPLC. However, no significant differences were detected in the total ergosterol contents of the two strains (see Fig. S2B in the supplemental material).
AfSsn3 and AfSsn8 coordinately function in azole resistance.
In S. cerevisiae and C. albicans, Ssn3 and Ssn8 are components of the Cdk8 module of the mediator (23, 25). Therefore, we were interested in determining whether the deletion of Afssn8 would lead to a phenotype similar to that of ΔAfssn3 with azole resistance. A plate test directly demonstrated that AfSsn8 deficiency (see Fig. S1B in the supplemental material) also resulted in azole resistance and increased biofilm formation, but this deficiency was a bit weaker than that of ΔAfssn3 (Fig. 4A and C and see Fig. S6B in the supplemental material). To test whether overexpressing Afssn8 in the ΔAfssn3 strain would rescue the azole resistance phenotype, we overexpressed Afssn8 under the control of gpdA in a background of ΔAfssn3. As exhibited in Fig. S6B in the supplemental material, ΔAfssn3OE::Afssn8 showed a phenotype similar to that of ΔAfssn3 with azole resistance. However, the strains that overexpress Afssn3 or Afssn8 in the background of the wild type exhibited azole susceptibilities similar to that of the wild type (see Fig. S6A and B in the supplemental material). Furthermore, to address whether the deletions of Afssn3 and Afssn8 would accelerate the azole resistance of a single null mutant, a double-mutant strain (strain ΔAfssn3ΔAfssn8) was constructed in the background of ΔAfssn3. Unexpectedly, strain ΔAfssn3ΔAfssn8 displays an azole resistance similar to that of strain ΔAfssn3 (see Fig. S6B). Taken together, these data strongly demonstrate that AfSsn3 and AfSsn8 have overlapping but not compensatory roles toward azole resistance in A. fumigatus.
DISCUSSION
The invasive filamentous mold A. fumigatus is associated with life-threatening pulmonary infections in immunocompromised individuals (30, 31). In many of these cases, A. fumigatus has the capacity to construct a biofilm, which enhances its persistence and may result in significant morbidity and mortality in severe cases (13, 17, 32). The four stages of A. fumigatus biofilm formation include the following: (i) adhesion and secretion of an exopolymeric substance; (ii) the germination of conidia into hyphae; (iii) biofilm maturation, hyphae, and ECM, which form a complex structural arrangement; and finally, (iv) cell dispersion, dissemination, and propagation (33).
In this study, we identified Afssn3 was important for azole resistance in A. fumigatus. The evidence to explain how AfSsn3 deficiency causes azole resistance is as follows. First, ΔAfssn3 led to an increased expression of glucose transporter genes and thus resulted in the derepression of glucose uptake. The derepressed glucose uptake then stimulated EPS formation, one of the primary materials of ECM. Second, the increased consumption and utilization of amino acids, especially serine, resulted in the accumulation of LCBs. LCBs were essential components of the cell membrane, without which the composition and integrity of the cell membrane were affected. Finally, the absence of Afssn3 increased the MDR efflux pump activity. Taken together, these three factors construct a compact biofilm system and are responsible for major mechanisms of resistance to azoles in ΔAfssn3. They form a two-layer barrier between the intracellular and extracellular domains and consequently cause azoles to be difficult to take up (Fig. 7D) and easy to remove, causing increased azole resistance. A putative working model of this mechanism is presented in Fig. 9. Interestingly, we found that the azole resistance phenotype in the ΔAfssn3 strain can be suppressed by caspofungin (Fig. 2A), an echinocandin antifungal drug that targets β-1,3-glucan synthase and thus inhibits biofilm synthesis (34), indicating that Afssn3 inactivation leading to azole resistance partly depended on biofilm formation. Previous studies of A. fumigatus and C. albicans have demonstrated that the glucan and chitin biofilm can prevent echinocandin drugs from reaching the cell membrane (9, 17). However, other studies have shown that caspofungin is able to inhibit the formation of biofilm in C. albicans (35, 36). The opposing effects on susceptibility to the echinocandins could be explained by the fact that the different compositions of the biofilm altered its permeability to echinocandins. In this study, we hypothesize that the altered sphingolipid composition of the plasma membrane is the dominant mechanism of azole resistance and that caspofungin primarily functions outside the cell membrane so it can penetrate and vanquish the biofilm azole resistance of ΔAfssn3. Thus, our findings are the first to characterize Afssn3 in A. fumigatus and also provide new and systematic insights into its function in biofilm biosynthesis.
FIG 9.

Potential mechanism of Afssn3-mediated azole resistance. The increased EPS layer outside the cell wall and the elevated LCB levels within the plasma membrane may inhibit the penetration of azoles into the cells. Increased efflux pump activity also may contribute to azole resistance.
Cellular metabolism and metabolic regulation have great importance during the elucidation of the biofilm formation mechanism. Previous results have confirmed that the amino acid metabolism was at a high level during C. albicans biofilm development (25, 37). In this study, the amino acid contents in strain ΔAfssn3 were assessed by GC-MS, and deletion of Afssn3 significantly increased the amino acid consumption and utilization (see Data Set S1 in the supplemental material). It is important to highlight that some categories of amino acids, such as serine, function as important ingredients in the biosynthesis of sphingolipids. Liquid chromatography-mass spectrometry (LC-MS) experiments further showed that the accumulation of the major LCBs (3-keto-DHS, DHS, PHS, and DHC) in the Afssn3-null mutant was significantly increased (Fig. 6A and B). Sphingolipids are essential components of cell membranes. For opportunistic fungal pathogens, the composition of the cell membrane is a significant factor in virulence and antifungal agent resistance because the changed composition of the cell membrane would affect the drug recognition and the permeation or activities of membrane-located azole pumps (38–41). In these studies, the accumulation of LCBs resulted in the increased susceptibility of C. albicans to azoles and the CRS-MIS (caspofungin reduced susceptibility but micafungin increased susceptibility) phenotype, which are different from our results. The conflicting results may be due to the different kinds and amounts of LCBs that exist in these different cells. In addition, our results showed that the addition of Ser, Gly, and Thr significantly influenced the resistance of ΔAfssn3 to azoles (Fig. 5). Gly and Thr were able to convert to Ser, which acts as the substrate of LCBs. That is why Ser, Gly, and Thr were able to increase azole resistance significantly. In addition, the overexpression of ksrA, which encodes the 3KSR that functions in sphingolipid biosynthesis, was also able to increase resistance to azoles to a great extent, and this overexpression was accompanied by a marked increase in LCBs (Table 1). Interestingly, the LCBs were reduced in WTOE::aurA, which was more sensitive than the wild-type strain in the presence of azoles. Therefore, our results indicate that the presence of high levels of LCB accumulation lead to more resistance to azoles and low levels of LCB accumulation lead to greater azole sensitivity. Further study by TEM showed that a much denser staining was present in the cell membrane in strain ΔAfssn3 than in the wild type (Fig. 7C), which suggests that the composition and integrity of the cell membrane were changed in ΔAfssn3. The changed cell membrane of ΔAfssn3 most probably inhibited azole permeation and thus led to azole resistance. Taken together, our findings suggest a close link between LCB accumulation and azole resistance in A. fumigatus.
A subgroup of Srb proteins—Med12/Srb8, Med13/Srb9, Cdk8/Srb10, and CycC/Srb11—forms a specific module (the Cdk8 module) (24). The mediator complex lacking the Cdk8 module has a stimulatory effect on basal transcription, whereas the mediator containing the Cdk8 module represses basal transcription (23, 24). Through RNA-seq analysis of the Afssn3 mutant (a homolog of cdk8/Srb10) and the wild type, we found that among 572 differentially expressed genes, 411 were upregulated and 161 were downregulated (see Data Set S2 in the supplemental material), indicating that Afssn3 exerts both positive and negative regulation effects but primarily negative regulation effects.
As a Cdk8-cyclin pair, the cyclin-dependent protein kinase Afssn3 and cyclin homolog Afssn8 both function in azole resistance, and Afssn3 probably plays a dominant role. The evidence for this conclusion is as follows. First, plate tests have demonstrated that ΔAfssn3 and ΔAfssn3ΔAfssn8 have almost the same azole resistance phenotype and biofilm biomass (Fig. 4A and C and see also Fig. S6B in the supplemental material). Afssn8 deficiency also resulted in azole resistance and increased biofilm biomass, which was a little weaker than that of strains ΔAfssn3 and ΔAfssn3ΔAfssn8. Second, the overexpression of Afssn8 in the ΔAfssn3 strain could not rescue the azole resistance phenotype (see Fig. S6B in the supplemental material). Third, when Afssn3 or Afssn8 was overexpressed in the background of the wild type, all of the strains exhibited azole susceptibilities similar to that of the wild type (see Fig. S6B in the supplemental material). Because of the importance of the mediator that regulates the basal RNA polymerase II transcription, microorganisms have evolved a subgroup of Cdk8 module proteins, namely, AfSsn3, AfSsn8, AfSrb8 and AfSrb9, in A. fumigatus. We plan to further examine the relationship between AfSrb8 and AfSrb9 and azole resistance in A. fumigatus in future studies.
An important cause of azole resistance in clinical A. fumigatus strains is the upregulation of the cyp51A gene (42, 43). In the present study, we found that an Afssn3 deficiency led to increased transcription of cyp51A (2-fold) (see Data Set S2 in the supplemental material and Table 1). We then sought to determine whether Afssn3 deficiency caused azole resistance because of increased cyp51A transcription. To address this hypothesis, we constructed a cyp51A single mutant and an Afssn3/cyp51A double mutant. If Afssn3 deficiency leading to azole resistance depended on increased cyp51A expression, Δcyp51A and ΔAfssn3 Δcyp51A mutants would show similar azole resistance. As shown in Fig. S7 in the supplemental material, compared to the wild type, Δcyp51A and ΔAfssn3 Δcyp51A strains both showed increased susceptibility to ITC. However, compared to the ΔAfssn3 Δcyp51A strain, the Δcyp51A strain was more susceptible to ITC (see Fig. S7 in the supplemental material), suggesting that the increased drug resistance of the ssn3-null mutant was probably not due to increased cyp51A transcription.
We systematically examined here Afssn3-mediated azole resistance. Nevertheless, some puzzles, such as how sphingolipid affects azole resistance, require further investigation.
MATERIALS AND METHODS
Strains, media, and culture condition.
All the A. fumigatus strains used in this study are shown in Table 3. The media used in this study included YAG (2% glucose, 0.5% yeast extract [1 ml/liter], 1,000× trace elements, 2% agar), YUU (YAG supplemented with 5 mM uridine and 10 mM uracil), and minimal medium (1% glucose [50 ml/liter], 1,000× trace elements, 20× salts [pH 6.5]) (44). For the transformant screening, generally, 300 μg of hygromycin B/ml was added to the corresponding medium. For the antifungal test, A. fumigatus strains were grown on YAG or YUU medium containing ITC, VRC, and caspofungin, respectively.
TABLE 3.
Aspergillus fumigatus strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| Af293 | Wild type | FGSCa |
| T637 | Afssn3 | This study |
| A1160 | ΔKU80 pyrG1 | FGSC |
| A1160C | ΔKU80 A1160::pyrG1 | This study |
| ΔAfssn3 | ΔKU80 pyrG1 ΔAfssn3::pyr4 | This study |
| ΔAfssn3C | ΔKU80 pyrG1 ΔAfssn3::Afssn3 | This study |
| ΔAfssn8 | ΔKU80, pyrG1 ΔAfssn8::hph | This study |
| ΔAfcyp51A | ΔKU80 pyrG1 ΔAfcyp51A::pyr4 | This study |
| ΔAfssn3ΔAfcyp51A | ΔKU80 pyrG1 ΔAfssn3::pyr4 ΔAfcyp51A::hph | This study |
| ΔAfssn3ΔAfssn8 | ΔKU80, pyrG1 ΔAfssn3::pyr4 ΔAfssn8::hph | This study |
| ΔAfssn3OE::Afssn8 | ΔKU80 pyrG1 ΔAfssn3::pyr4 gpdA(p)-Afssn8 hph | This study |
| WTOE::Afssn3-gfp | ΔKU80 pyrG1 gpdA(p)-Afssn3-gfp hph | This study |
| WTOE::Afssn3 | ΔKU80 pyrG1 gpdA(p)-Afssn3 hph | This study |
| WTOE::Afssn8 | ΔKU80 pyrG1 gpdA(p)-Afssn8 hph | This study |
| WTOE::aurA | ΔKU80 pyrG1 gpdA(p)-aurA pyr4 | This study |
| WTOE::ksrA | ΔKU80 pyrG1 gpdA(p)-ksrA pyr4 | This study |
FGSC, Fungal Genetics Stock Center.
ATMT transformation and screening of mutants.
Agrobacterium-mediated transformation was carried out according to a previously described protocol (22). Basically, A. tumefaciens strain EHA105 harboring plasmid pPK2, which contains a hygromycin resistance marker, was cocultured with A. fumigatus on induction medium supplemented with 200 mM acetosyringone, which stimulates Agrobacterium to transfer T-DNA into the fungal genome. After two days of coincubation, A. fumigatus transformants were selected on YAG medium supplemented with hygromycin (300 μg/ml) and cefotaxime (200 μg/ml). After 48 h at 37°C, plates containing putative transformed colonies were checked for their susceptibility to azoles and caspofungin by inoculating spores onto YAG plates with azoles and caspofungin for 48 h. Mutants with azoles and caspofungin susceptibilities clearly different from the parental isolate were selected for further analysis.
Molecular analysis of T-DNA insertion mutants.
PCR detection of the hph gene in putative mutants was performed using primers HPH-1 and HPH-2. The fungal genomic sequence flanking the T-DNA insertions of the putative mutants were cloned and sequenced following TAIL-PCR as described previously (45) with the primer sequences shown in Table S2 in the supplemental material. Flanking sequences recovered by TAIL-PCR were analyzed by using the BLAST tool hosted by the NCBI against the GenBank database.
Deletion and complementation of Afssn3 and/or Afssn8.
All primers used in this study are shown in Table S1 in the supplemental material. To generate the Afssn3 deletion cassette, we used fusion PCR. Briefly, upstream and downstream flanking sequences ∼1 kb in length of the Afssn3 gene were amplified using the primers Afssn3 P1 and P3 and the primers Afssn3 P4 and P6, respectively. The pyr4 fragment used as a selection marker was amplified from plasmid pAL5 with the primers pyr4 F and R. These three fragments were then mixed and used as a template to construct the Afssn3 deletion cassette using the primers Afssn3 P2 and P5. The Afssn3 deletion cassette was then transformed into A1160, which belonged to a ku80-null mutant using the method described to generate the Afssn3-null mutant. To generate the Afssn8 deletion cassette, a similar strategy was used except that the selection marker used for Afssn8 was the hygromycin B resistance gene hph, which was amplified from the plasmid pAN7-1 using the primers hph F and R. For double deletions of Afssn3 and Afssn8, Afssn8 was deleted in the background of an Afssn3-null mutant by the same method. To complement the Afssn3-null mutant, the full-length Afssn3 gene was amplified using the primer pair Afssn3 P1 and P6, which includes the native promoter, the 5′ untranslated region (UTR), the Afssn3 gene coding sequences, and the 3′ UTR. The complementation cassette was subsequently transformed into ΔAfssn3 and screened using 5-fluoroorotic acid as previously described (44).
Construction of the Afssn3, Afssn8, ksrA, and aurA overexpression strains.
To overexpress Afssn3 under the control of gpdA, we used the following strategy. Briefly, the ORF of Afssn3 was amplified from the genomic DNA using the primers OE-Afssn3 F and R. A purified fragment was then subcloned into the ClaI site of pBARGPE-1 (46) to generate the overexpression plasmid and then transformed into A1160. A similar strategy was used to construct the Afssn8, ksrA, and aurA overexpression strains.
Construction of AfSsn3 GFP-tagged strains.
To label AfSsn3 with a GFP tag, we first amplified the Afssn3 ORF (without a termination codon) and gfp (including a termination codon) fragments using the primers Afssn3-gfp P1 and P2 and the primers Afssn3-gfp P3 and P4, respectively. Then, the two fragments were combined and used as a template to generate the Afssn3-gfp cassette using the primers Afssn3-gfp P1 and P4. The fused fragment was then subcloned into the ClaI site of pBARGPE-1 as described above to construct a Afssn3-gfp vector in which Afssn3-gfp was under the control of gpdA and then transformed into A1160.
Protein isolation and Western blotting.
For protein isolation and Western blotting, fresh mycelia were harvested, ground in liquid nitrogen, and suspended in pre-cold protein extraction buffer (50 mM HEPES [pH 7.4], 137 mM KCl, and 10% glycerol containing 1 mM EDTA, 1 μg/ml pepstatin A, 1 μg/ml leupeptin, and 1 mM phenylmethylsulfonyl fluoride). The samples were incubated on ice for 15 min and vortex mixed every 5 min for 30 s. Cell pellets were removed using centrifugation at 16,000 × g and 4°C for 10 min. The protein concentration of the supernatant was measured by using a Bio-Rad protein assay kit. The GFP antibody used in this study was purchased from Roche and diluted 1:3,000. Anti-actin antibody was purchased from ICN Biomedicals, Inc., and used at 1:20,000. Peroxidase activity was detected by using an enhanced chemiluminescence kit (Roche).
Microscopic observations.
To visualize the subcellular localization of AfSsn3, the GFP-labeled strain was incubated in 2 ml of liquid YUU medium on coverslips for 16 h with or without ITC stimulation. For nucleus staining, DAPI (4′,6-diamidino-2-phenylindole) was used at a final concentration of 0.8 μg/ml. Images were obtained by using an Axio Imager A1 microscope (Zeiss, Jena, Germany).
Accumulated rhodamine-123 detection.
We measure the cellular accumulation of azole-mimicking reagent Rh123 as described previously (47). Briefly, 107 spores of parental wild-type and mutant strains were incubated in 100 ml of liquid YAG medium at 37°C for 4 h in a rotary shaker at 220 rpm. A final concentration of 10 μM Rh123 was added to the conidial suspension, followed by incubation at 37°C for another 1 h. Spores were obtained by centrifuge at 5,000 × g for 5 min and then washed three times with phosphate-buffered saline (PBS) to remove extracellular Rh123. The fluorescent signal was quantified by using flow cytometry (BD FACSCalibur) with 488 nm as the excitation wavelength and 546 nm as the emission wavelength. Data acquisition and manipulation were performed as described in the manufacturer's instruction manuals for CellQuest and FCS Express v3.
Cellular drug detection.
We performed cellular drug detection as previously described (20). Briefly, 107 spores of the indicated strain were cultured in 100 ml of liquid YAG medium at 37°C and 220 rpm for 18 h. A final ITC concentration of 1 mg/ml was added to the medium, followed by incubation for 1 h. Mycelia were harvested and washed with distilled water to remove the extracellular ITC and then lyophilized. Approximately 50 mg of lyophilized mycelia was incubated in 1 ml of methanol and homogenized using ceramic beads. The cell debris and ceramic beads were then removed by centrifugation at 16,000 × g for 10 min. The supernatant harboring the ITC was analyzed using HPLC on an AQ-C18 column (250 mm by 4.6 mm, 5 μm) with a methanol flow rate of 1 ml/min at 265 nm.
Detection of cell wall composition and medium residual glucose.
We determined the cell wall composition of the mycelia as described previously (48). For cell wall extraction, 107 spores of each strain were grown in 100 ml of liquid YAG medium for 24 h at 37°C at 220 rpm. Mycelia were harvested and washed three times using double-distilled H2O (ddH2O) to remove the extracellular glucose. Mycelia were ground in liquid nitrogen and suspended in 1 ml of protein extraction buffer (2% sodium dodecyl sulfate, 40 mM β-mercaptoethanol, 50 mM Tris-HCl, 5 mM EDTA [pH 7.4]) with 0.5 ml of acid-washed glass beads. Mycelial cell walls were then broken using a homogenizer. After 10 cycles of 30 s at full speed, with 30-s intervals, the supernatant was collected, the glass beads were washed five times with 1 ml of protein extraction buffer, and then the aliquots were pooled. The cell wall debris were collected by centrifugation at 16,000 × g at 4°C for 10 min, washed with ddH2O, and then lyophilized. Acid hydrolysis of the cell walls was performed. To determine the monosaccharide composition, the supernatants of the acid-hydrolyzed cell wall were analyzed by high-performance anion-exchange chromatography with pulsed amperometric detection as described previously. A similar strategy was used to detect the residual glucose in the media.
SEM and TEM.
For ultrathin sections, germlings grown on YAG plates for 48 h were fixed for 5 h in 5% buffered glutaraldehyde (pH 7.0) with 0.1 M phosphate buffer and then postfixed for 2 h in buffered 1% osmium tetroxide. The whole fixation procedure was carried out at 4°C. After dehydration with a graded ethanol series, specimens were embedded in epoxy resin. The sections, cut on an ultramicrotome using a glass knife, were stained with uranyl acetate and lead citrate and observed in a JEOL transmission electron microscope (JEM-1010). A. fumigatus biofilms for SEM were developed in six-well polystyrene plates at 37°C in YAG liquid culture medium for 48 h. The biofilms were washed with PBS and fixed with 2% glutaraldehyde for 2 h. The biofilms were then postfixed with 1% osmium tetroxide for 2 h. The bottoms of the polystyrene plates were cut with a hot punch, and the intact biofilm was obtained. The samples were dehydrated with a graded ethanol series. The biofilms were then lyophilized and coated with ionized gold for 400 s at 15,000 kV and 10 μA. The samples were observed in a scanning electron microscope (JEOL, Tokyo, Japan).
RNA isolation and qRT-PCR analysis.
Total RNA of the overexpression strains was isolated from fresh mycelia by using TRIzol as described in the manufacturer's instructions. qRT-PCR analysis was performed as described previously (49).
Ergosterol extraction and analysis.
For total ergosterol extraction from A. fumigatus strains, 108 spores were incubated in 100 ml of liquid YAG medium at 37°C and 220 rpm for 24 h. Mycelia were obtained via filtration with gauze, washed three times with distilled water, and lyophilized. About 200 mg of dry mycelia was extracted and analyzed using HPLC as described previously (20).
Antifungal susceptibility testing.
The conidia of tested Aspergillus samples were harvested, adjusted to 107 spores per ml in YAG broth, and then inoculated into liquid or solid media in the presence of different concentrations of the azole antifungals ITC, VRC, and caspofungin. ITC and VRC were prepared in the stock solution with dimethyl sulfoxide. Broth microdilution was performed according to CLSI-M38-A2 (20). Briefly, 2-fold serial drug dilutions were prepared in flat-bottom 96-well microtiter plates (100 μl per well), Drug-free wells were used as controls. Each well was inoculated with 100 μl of freshly isolated spores (2 × 104 CFU/ml) suspended in RPMI 1640. After 48 h of incubation at 35°C, the MIC was recorded as the lowest drug concentration at which no growth was observed.
Metabolomics.
For LC-MS analysis, 300-ml cultures of liquid YAG were inoculated with 3 × 107 spores, followed by incubation at 220 rpm and 37°C for 36 h. Mycelia were removed from the culture supernatant using Miracloth and quenched immediately in liquid nitrogen for 30 min. All strains were grown in duplicates. A 60-mg accurately weighed sample was transferred to a 1.5-ml Eppendorf tube. Then, 20 μl of 2-chloro-l-phenylalanine (0.3 mg/ml) dissolved in methanol as an internal standard and 2 ml of methanol-water (4:1 [vol/vol]) were added to each sample. The mixture was blended by using a TissueLyser II (Qiagen, Dusseldorf, Germany) at a frequency of 50 Hz for 3 min and then transferred to a 4-ml glass vial. Next, 400 μl of chloroform was added to each aliquot, dispersing the samples by pipette. We used an ultrasonic homogenizer to break up the cells for 6 min at 500 W. All of the mixtures of each sample were then extracted by ultrasonication for 20 min in an ice-water bath. The extract was centrifuged at 16,000 × g and 4°C for 10 min. Quality control samples were prepared by mixing aliquots of all samples to serve as a pooled sample. Then, 500-μl portions of the supernatants of each tube were transferred to LC vials. Six biological replicates were used for metabolite profiling with a 1290 Infinity UHPLC system coupled with a 6538 UHD QTOF mass spectrometer (LC-MS) (Agilent Technologies, Inc., Los Angeles, CA). Differential metabolites were identified by the Human Metabolome Database (HMDB) and the METLIN metabolite database. For GC-MS analysis, 300-ml cultures of liquid YAG were inoculated with 3 × 107 spores, followed by incubation at 220 rpm and 37°C for 18 h. A similar strategy was used for pretreatment of the samples. Derivatization of the samples before GC-MS analysis was performed as described previously (50). A total of six to eight biological replicates was used for metabolite profiling by a TSQ-8000 triple quadrupole GC-MS approach (Thermo Fisher Scientific, Bremen, Germany). The P value was obtained by a Student t test in SPSS 17.0, the significance level was set as 0.05, and differential metabolites were identified by the National Institute of Standards and Technology (NIST) metabolite database.
Supplementary Material
ACKNOWLEDGMENTS
We are very grateful for the suggestions and advice of Xinyu Yu during the analysis of our metabolomics data. We also thank Ling Lu for the donation of strains (Af293 and A1160) and plasmids.
This study was financially supported by the National Natural Science Foundation young investigator grant program of China (grant NSFC31500055), the Natural Science Foundation young investigator grant program of Jiangsu (grant BK20140898), the Natural Science Foundation (grant 14KJB330004), and a project funded by the Priority Academic Program Development of Jiangsu Education Department to G.Z. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01978-17.
REFERENCES
- 1.Tekaia F, Latge JP. 2005. Aspergillus fumigatus: saprophyte or pathogen? Curr Opin Microbiol 8:385–392. doi: 10.1016/j.mib.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 2.Wingard JR. 2005. The changing face of invasive fungal infections in hematopoietic cell transplant recipients. Curr Opin Oncol 17:89–92. doi: 10.1097/01.cco.0000152975.65477.7c. [DOI] [PubMed] [Google Scholar]
- 3.Marr KA, Boeckh M, Carter RA, Kim HW, Corey L. 2004. Combination antifungal therapy for invasive aspergillosis. Clin Infect Dis 39:797–802. doi: 10.1086/423380. [DOI] [PubMed] [Google Scholar]
- 4.Snelders E, van der Lee HA, Kuijpers J, Rijs AJ, Varga J, Samson RA, Mellado E, Donders AR, Melchers WJ, Verweij PE. 2008. Emergence of azole resistance in Aspergillus fumigatus and spread of a single resistance mechanism. PLoS Med 5:e219. doi: 10.1371/journal.pmed.0050219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arendrup MC. 2014. Update on antifungal resistance in Aspergillus and Candida. Clin Microbiol Infect 20(Suppl 6):S42–S48. doi: 10.1111/1469-0691.12360. [DOI] [PubMed] [Google Scholar]
- 6.Chowdhary A, Sharma C, Hagen F, Meis JF. 2014. Exploring azole antifungal drug resistance in Aspergillus fumigatus with special reference to resistance mechanisms. Future Microbiol 9:697–711. doi: 10.2217/fmb.14.27. [DOI] [PubMed] [Google Scholar]
- 7.Al-Abdely HM, Alothman AF, Salman JA, Al-Musawi T, Almaslamani M, Butt AA, Al Thaqafi AO, Raghubir N, Morsi WE, Yared NA. 2014. Clinical practice guidelines for the treatment of invasive Aspergillus infections in adults in the Middle East region: expert panel recommendations. J Infect Public Health 7:20–31. doi: 10.1016/j.jiph.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Warrilow AG, Melo N, Martel CM, Parker JE, Nes WD, Kelly SL, Kelly DE. 2010. Expression, purification, and characterization of Aspergillus fumigatus sterol 14-alpha demethylase (CYP51) isoenzymes A and B. Antimicrob Agents Chemother 54:4225–4234. doi: 10.1128/AAC.00316-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cowen LE, Sanglard D, Howard SJ, Rogers PD, Perlin DS. 2014. Mechanisms of antifungal drug resistance. Cold Spring Harb Perspect Med 5:a019752. doi: 10.1101/cshperspect.a019752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.da Silva Ferreira ME, Capellaro JL, dos Reis Marques E, Malavazi I, Perlin D, Park S, Anderson JB, Colombo AL, Arthington-Skaggs BA, Goldman MH, Goldman GH. 2004. In vitro evolution of itraconazole resistance in Aspergillus fumigatus involves multiple mechanisms of resistance. Antimicrob Agents Chemother 48:4405–4413. doi: 10.1128/AAC.48.11.4405-4413.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meneau I, Coste AT, Sanglard D. 2016. Identification of Aspergillus fumigatus multidrug transporter genes and their potential involvement in antifungal resistance. Med Mycol 54:616–627. doi: 10.1093/mmy/myw005. [DOI] [PubMed] [Google Scholar]
- 12.Ben-Ami R, Zimmerman O, Finn T, Amit S, Novikov A, Wertheimer N, Lurie-Weinberger M, Berman J. 2016. Heteroresistance to fluconazole is a continuously distributed phenotype among Candida glabrata clinical strains associated with in vivo persistence. mBio 7:e00655-. doi: 10.1128/mBio.00655-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajendran R, Mowat E, McCulloch E, Lappin DF, Jones B, Lang S, Majithiya JB, Warn P, Williams C, Ramage G. 2011. Azole resistance of Aspergillus fumigatus biofilms is partly associated with efflux pump activity. Antimicrob Agents Chemother 55:2092–2097. doi: 10.1128/AAC.01189-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei XL, Zhang YW, Lu L. 2015. The molecular mechanism of azole resistance in Aspergillus fumigatus: from bedside to bench and back. J Microbiol 53:91–99. doi: 10.1007/s12275-015-5014-7. [DOI] [PubMed] [Google Scholar]
- 15.Kumamoto CA. 2002. Candida biofilms. Curr Opin Microbiol 5:608–611. doi: 10.1016/S1369-5274(02)00371-5. [DOI] [PubMed] [Google Scholar]
- 16.Fanning S, Mitchell AP. 2012. Fungal biofilms. PLoS Pathog 8:e1002585. doi: 10.1371/journal.ppat.1002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seidler MJ, Salvenmoser S, Muller FM. 2008. Aspergillus fumigatus forms biofilms with reduced antifungal drug susceptibility on bronchial epithelial cells. Antimicrob Agents Chemother 52:4130–4136. doi: 10.1128/AAC.00234-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bueid A, Howard SJ, Moore CB, Richardson MD, Harrison E, Bowyer P, Denning DW. 2010. Azole antifungal resistance in Aspergillus fumigatus: 2008 and 2009. J Antimicrob Chemother 65:2116–2118. doi: 10.1093/jac/dkq279. [DOI] [PubMed] [Google Scholar]
- 19.Bowyer P, Mosquera J, Anderson M, Birch M, Bromley M, Denning DW. 2012. Identification of novel genes conferring altered azole susceptibility in Aspergillus fumigatus. FEMS Microbiol Lett 332:10–19. doi: 10.1111/j.1574-6968.2012.02575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei X, Chen P, Gao R, Li Y, Zhang A, Liu F, Lu L. 2017. Screening and characterization of a non-cyp51A mutation in an Aspergillus fumigatus cox10 strain conferring azole resistance. Antimicrob Agents Chemother 61:e02101-. doi: 10.1128/AAC.02101-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mullins ED, Kang S. 2001. Transformation: a tool for studying fungal pathogens of plants. Cell Mol Life Sci 58:2043–2052. doi: 10.1007/PL00000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugui JA, Chang YC, Kwon-Chung KJ. 2005. Agrobacterium tumefaciens-mediated transformation of Aspergillus fumigatus: an efficient tool for insertional mutagenesis and targeted gene disruption. Appl Environ Microbiol 71:1798–1802. doi: 10.1128/AEM.71.4.1798-1802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuchin S, Yeghiayan P, Carlson M. 1995. Cyclin-dependent protein kinase and cyclin homologs SSN3 and SSN8 contribute to transcriptional control in yeast. Proc Natl Acad Sci U S A 92:4006–4010. doi: 10.1073/pnas.92.9.4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elmlund H, Baraznenok V, Lindahl M, Samuelsen CO, Koeck PJB, Holmberg S, Hebert H, Gustafsson CM. 2006. The cyclin-dependent kinase 8 module sterically blocks Mediator interactions with RNA polymerase II. Proc Natl Acad Sci U S A 103:15788–15793. doi: 10.1073/pnas.0607483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindsay AK, Morales DK, Liu Z, Grahl N, Zhang A, Willger SD, Myers LC, Hogan DA. 2014. Analysis of Candida albicans mutants defective in the Cdk8 module of mediator reveal links between metabolism and biofilm formation. PLoS Genet 10:e1004567. doi: 10.1371/journal.pgen.1004567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Firestein R, Hahn WC. 2009. Revving the throttle on an oncogene: CDK8 takes the driver seat. Cancer Res 69:7899–7901. doi: 10.1158/0008-5472.CAN-09-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu W, Ji JY. 2011. Dysregulation of CDK8 and cyclin C in tumorigenesis. J Genet Genomics 38:439–452. doi: 10.1016/j.jgg.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dickson RC, Lester RL. 2002. Sphingolipid functions in Saccharomyces cerevisiae. Biochem Biophys Acta 1583:13–25. doi: 10.1016/S1388-1981(02)00210-X. [DOI] [PubMed] [Google Scholar]
- 29.Thakur JK, Arthanari H, Yang F, Pan SJ, Fan X, Breger J, Frueh DP, Gulshan K, Li DK, Mylonakis E, Struhl K, Moye-Rowley WS, Cormack BP, Wagner G, Naar AM. 2008. A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 452:604–609. doi: 10.1038/nature06836. [DOI] [PubMed] [Google Scholar]
- 30.Shao PL, Huang LM, Hsueh PR. 2007. Recent advances and challenges in the treatment of invasive fungal infections. Int J Antimicrob Agents 30:487–495. doi: 10.1016/j.ijantimicag.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 31.van der Linden JW, Snelders E, Kampinga GA, Rijnders BJ, Mattsson E, Debets-Ossenkopp YJ, Kuijper EJ, Van Tiel FH, Melchers WJ, Verweij PE. 2011. Clinical implications of azole resistance in Aspergillus fumigatus, The Netherlands, 2007-2009. Emerg Infect Dis 17:1846–1854. doi: 10.3201/eid1710.110226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mowat E, Butcher J, Lang S, Williams C, Ramage G. 2007. Development of a simple model for studying the effects of antifungal agents on multicellular communities of Aspergillus fumigatus. J Med Microbiol 56:1205–1212. doi: 10.1099/jmm.0.47247-0. [DOI] [PubMed] [Google Scholar]
- 33.Gonzalez-Ramirez AI, Ramirez-Granillo A, Medina-Canales MG, Rodriguez-Tovar AV, Martinez-Rivera MA. 2016. Analysis and description of the stages of Aspergillus fumigatus biofilm formation using scanning electron microscopy. BMC Microbiol 16:243. doi: 10.1186/s12866-016-0859-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kathiravan MK, Salake AB, Chothe AS, Dudhe PB, Watode RP, Mukta MS, Gadhwe S. 2012. The biology and chemistry of antifungal agents: a review. Bioorg Med Chem 20:5678–5698. doi: 10.1016/j.bmc.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 35.Bachmann SP, VandeWalle K, Ramage G, Patterson TF, Wickes BL, Graybill JR, Lopez-Ribot JL. 2002. In vitro activity of caspofungin against Candida albicans biofilms. Antimicrob Agents Chemother 46:3591–3596. doi: 10.1128/AAC.46.11.3591-3596.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cateau E, Rodier MH, Imbert C. 2008. In vitro efficacies of caspofungin or micafungin catheter lock solutions on Candida albicans biofilm growth. J Antimicrob Chemother 62:153–155. doi: 10.1093/jac/dkn160. [DOI] [PubMed] [Google Scholar]
- 37.Chen X, Wu H, Cao Y, Yao X, Zhao L, Wang T, Yang Y, Lv D, Chai Y, Cao Y, Zhu Z. 2014. Ion-pairing chromatography on a porous graphitic carbon column coupled with time-of-flight mass spectrometry for targeted and untargeted profiling of amino acid biomarkers involved in Candida albicans biofilm formation. Mol Biosyst 10:74–85. doi: 10.1039/C3MB70240E. [DOI] [PubMed] [Google Scholar]
- 38.Jia X, Wang Y, Jia Y, Gao P, Xu Y, Wang L, Cao Y, Cao Y, Zhang L, Jiang Y. 2009. RTA2 is involved in calcineurin-mediated azole resistance and sphingoid long-chain base release in Candida albicans. Cell Mol Life Sci 66:122–134. doi: 10.1007/s00018-008-8409-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Healey KR, Katiyar SK, Raj S, Edlind TD. 2012. CRS-MIS in Candida glabrata: sphingolipids modulate echinocandin-Fks interaction. Mol Microbiol 86:303–313. doi: 10.1111/j.1365-2958.2012.08194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Healey KR, Challa KK, Edlind TD, Katiyar SK. 2015. Sphingolipids mediate differential echinocandin susceptibility in Candida albicans and Aspergillus nidulans. Antimicrob Agents Chemother 59:3377–3384. doi: 10.1128/AAC.04667-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernandes CM, de Castro PA, Singh A, Fonseca FL, Pereira MD, Vila TV, Atella GC, Rozental S, Savoldi M, Del Poeta M, Goldman GH, Kurtenbach E. 2016. Functional characterization of the Aspergillus nidulans glucosylceramide pathway reveals that LCB Δ8-desaturation and C9-methylation are relevant to filamentous growth, lipid raft localization and Psd1 defensin activity. Mol Microbiol 102:488–505. doi: 10.1111/mmi.13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Albarrag AM, Anderson MJ, Howard SJ, Robson GD, Warn PA, Sanglard D, Denning DW. 2011. Interrogation of related clinical pan-azole-resistant Aspergillus fumigatus strains: G138C, Y431C, and G434C single nucleotide polymorphisms in cyp51A, upregulation of cyp51A, and integration and activation of transposon Atf1 in the cyp51A promoter. Antimicrob Agents Chemother 55:5113–5121. doi: 10.1128/AAC.00517-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mellado E, Garcia-Effron G, Alcazar-Fuoli L, Melchers WJ, Verweij PE, Cuenca-Estrella M, Rodriguez-Tudela JL. 2007. A new Aspergillus fumigatus resistance mechanism conferring in vitro cross-resistance to azole antifungals involves a combination of cyp51A alterations. Antimicrob Agents Chemother 51:1897–1904. doi: 10.1128/AAC.01092-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang H, Shen Y, Liu W, Lu L. 2014. Deletion of the putative stretch-activated ion channel Mid1 is hypervirulent in Aspergillus fumigatus. Fungal Genet Biol 62:62–70. doi: 10.1016/j.fgb.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Combier JP, Melayah D, Raffier C, Gay G, Marmeisse R. 2003. Agrobacterium tumefaciens-mediated transformation as a tool for insertional mutagenesis in the symbiotic ectomycorrhizal fungus Hebeloma cylindrosporum. FEMS Microbiol Lett 220:141–148. doi: 10.1016/S0378-1097(03)00089-2. [DOI] [PubMed] [Google Scholar]
- 46.Song J, Zhai P, Zhang Y, Zhang C, Sang H, Han G, Keller NP, Lu L. 2016. The Aspergillus fumigatus damage resistance protein family coordinately regulates ergosterol biosynthesis and azole susceptibility. mBio 7:e01919-15. doi: 10.1128/mBio.01919-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clark FS, Parkinson T, Hitchcock CA, Gow NA. 1996. Correlation between rhodamine 123 accumulation and azole sensitivity in Candida species: possible role for drug efflux in drug resistance. Antimicrob Agents Chemother 40:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Munster JM, Nitsche BM, Krijgsheld P, van Wijk A, Dijkhuizen L, Wosten HA, Ram AF, van der Maarel MJ. 2013. Chitinases CtcB and CfcI modify the cell wall in sporulating aerial mycelium of Aspergillus niger. Microbiology 159:1853–1867. doi: 10.1099/mic.0.067967-0. [DOI] [PubMed] [Google Scholar]
- 49.Zhong G, Wei W, Guan Q, Ma Z, Wei H, Xu X, Zhang S, Lu L. 2012. Phosphoribosyl pyrophosphate synthetase, as a suppressor of the sepH mutation in Aspergillus nidulans, is required for the proper timing of septation. Mol Microbiol 86:894–907. doi: 10.1111/mmi.12026. [DOI] [PubMed] [Google Scholar]
- 50.Gao X, Pujos-Guillot E, Sebedio JL. 2010. Development of a quantitative metabolomic approach to study clinical human fecal water metabolome based on trimethylsilylation derivatization and GC/MS analysis. Anal Chem 82:6447–6456. doi: 10.1021/ac1006552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.