

ABSTRACT
Polymicrobial intra-abdominal infections (IAI) involving Candida albicans and Staphylococcus aureus are associated with severe morbidity and mortality (∼80%). Our laboratory discovered that the immunomodulatory eicosanoid prostaglandin E2 (PGE2) plays a key role in the lethal inflammatory response during polymicrobial IAI using a mouse model of infection. In studies designed to uncover key PGE2 biosynthesis/signaling components involved in the response, selective eicosanoid enzyme inhibitors and receptor antagonists were selected and prescreened for antimicrobial activity against C. albicans or S. aureus. Unexpectedly, we found that the EP4 receptor antagonist L-161,982 had direct growth-inhibitory effects on S. aureus in vitro at the physiological concentration required to block the PGE2 interaction with EP4. This antimicrobial activity was observed with methicillin-sensitive S. aureus and methicillin-resistant S. aureus (MRSA) strains, with the MIC and minimum bactericidal concentration values for planktonic cells being 50 μg/ml and 100 μg/ml, respectively. In addition, L-161,982 inhibited S. aureus biofilm formation and had activity against preformed mature biofilms. More importantly, treatment of mice with L-161,982 following intraperitoneal inoculation with a lethal dose of MRSA significantly reduced the bioburden and enhanced survival. Furthermore, L-161,982 protected mice against the synergistic lethality induced by coinfection with C. albicans and S. aureus. The antimicrobial activity of L-161,982 is independent of EP4 receptor inhibitory activity; an alternative EP4 receptor antagonist exerted no antimicrobial or protective effects. Taken together, these findings demonstrate that L-161,982 has potent antimicrobial activity against MRSA and may represent a significant therapeutic alternative in improving the prognosis of mono- or polymicrobial infections involving MRSA.
KEYWORDS: antimicrobial activity; Staphylococcus aureus; MRSA; Candida albicans; polymicrobial infection; L-161,982; prostaglandin E2; EP4 receptor; antimicrobial activity
INTRODUCTION
Intra-abdominal infection (IAI) is a broad term used to describe infections that occur as a result of the perforation of the gastrointestinal tract. There is currently a 77% mortality rate associated with polymicrobial IAI involving fungal and bacterial species, and this rate far exceeds that of bacterial monomicrobial infections (20%) (1–4). A plethora of microorganisms can cause IAI; however, two microorganisms that are frequently coisolated are the fungal pathogen Candida albicans and the pathogenic bacterium Staphylococcus aureus (5). In patients with intra-abdominal perforations, isolation of C. albicans alone is indicative of a high mortality risk (6). Using animal models, it was demonstrated that coinfection with S. aureus raises the mortality rate even further, as a lethal synergistic association exists between these two pathogens (7, 8). Current research is aimed at understanding the mechanism underlying this lethal synergistic interaction as well as the host immune response to coinfection.
We recently developed a mouse model of IAI with C. albicans and/or S. aureus and demonstrated that the lethal outcome of coinfection, as opposed to monoinfection, was a result of an amplified host inflammatory response and not dependent on the microbial burden. In addition to proinflammatory cytokines, coinfected mice had significantly higher levels of the immunomodulatory prostanoid prostaglandin E2 (PGE2) than monoinfected mice (8, 9). Strikingly, treatment with the nonsteroidal anti-inflammatory drug (NSAID) indomethacin, which reduces PGE2 synthesis, prevented mortality by resolving the proinflammatory response, demonstrating that the excessive production of PGE2 is a key mediator of the lethal outcome (8). This finding was further supported by the observation that administration of exogenous PGE2 to indomethacin-treated mice restored PGE2 levels, production of proinflammatory cytokines, and mortality (8).
PGE2 is derived from polyunsaturated arachidonic acid by the enzymatic action of cyclooxygenases (COX; 2 isoforms, COX-1 and COX-2, exist in mammals) and can induce either pro- or anti-inflammatory responses (10, 11). PGE2 exerts its biological functions by interacting with one of four specific plasma membrane receptors (designated EP1 through EP4) coupled to GTP-binding regulatory proteins (G-proteins) (10). While using selective COX enzyme inhibitors and EP receptor antagonists to identify specific components of the prostanoid biosynthetic and signaling pathways involved in PGE2 production during C. albicans-S. aureus IAI, in vitro antimicrobial activity against C. albicans or S. aureus was assessed. Interestingly, the EP4 receptor antagonist L-161,982 exhibited growth-inhibitory activity toward S. aureus. The antimicrobial activity of L-161,982 has not been previously reported; therefore, the goal of these studies was to further investigate the antimicrobial activity of this compound using in vitro assays and in vivo infection models.
RESULTS
L-161,982 inhibits planktonic growth of S. aureus.
The antimicrobial efficacy of selective COX inhibitors and EP receptor antagonists (Table 1) against reference strains C. albicans DAY185 and S. aureus NRS383 was determined as a prerequisite analysis prior to their use in analyzing the role of the prostanoid biosynthetic and signaling pathway during C. albicans-S. aureus IAI. The pharmacological inhibitors have been used in vivo in animal models with no measurable mammalian cell cytotoxicity (12, 13) and thus were tested in vitro in the present study at the relevant physiological concentrations.
TABLE 1.
Selective and nonselective COX enzyme inhibitors and EP receptor antagonists
| Chemical name | Inhibitory target | Concn (mg/kg) |
|---|---|---|
| Indomethacin | COX1 and COX2 enzymes | 5 |
| SC-560 | COX1 enzyme | 20 |
| NS-398 | COX2 enzyme | 10 |
| SC 51322 | EP1 receptor | 10 |
| PF 04418948 | EP2 receptor | 10 |
| L-798,106 | EP3 receptor | 10 |
| L-161,982 | EP4 receptor | 10 |
| ONO AE3 208 | EP4 receptor | 10 |
The growth of C. albicans was not inhibited in the presence of COX inhibitors or EP receptor antagonists (Fig. 1A). Similarly, the growth of S. aureus was unaffected by the COX inhibitors as well as EP1 to EP3 receptor antagonists (Fig. 1B). Conversely, the EP4 receptor antagonist L-161,982 had a significant inhibitory effect on S. aureus growth (Fig. 1B, gray triangle). Based on this significant finding, we extended the antimicrobial susceptibility screen to include clinical methicillin-resistant S. aureus (MRSA) and methicillin-sensitive S. aureus (MSSA) strains isolated from a patient's catheter. In all cases, L-161,982 inhibited growth (Fig. 1C and D).
FIG 1.

L-161,982 inhibits the growth of S. aureus strains. The antimicrobial activity of selective COX enzyme inhibitors or PGE2 EP receptor antagonists on the growth of C. albicans DAY185 (A), S. aureus NRS383 (B), and MRSA and MSSA clinical isolates (C) was determined. The growth of C. albicans and S. aureus in medium alone or in medium supplemented with DMSO, EP receptor antagonists, or COX enzyme inhibitors at physiologically relevant concentrations was monitored for up to 24 h. The data shown are representative of those from three independent experiments. TSB, tryptic soy broth.
Growth inhibition kinetics of L-161,982 against planktonic staphylococcal cells.
We next investigated the growth inhibition kinetics of L-161,982 against S. aureus and drug stability. The MIC of L-161,982 against planktonic S. aureus was 50 μg/ml, while the minimum bactericidal concentration (MBC) was 100 μg/ml. The growth inhibition kinetics of L-161,982 at the MIC of 50 μg/ml revealed that the inhibitory effect of L-161,982 on S. aureus was limited to 8 h (Fig. 2, black squares). To address whether the loss of inhibition was due to drug degradation over time (half-life) or to the adaptation of S. aureus cells to L-161,982, fresh L-161,982 (50 μg/ml) was added during the coincubation. The results showed that supplementation maintained inhibition over a 24-h period, indicating drug degradation or the drug's half-life (Fig. 2, gray circles).
FIG 2.
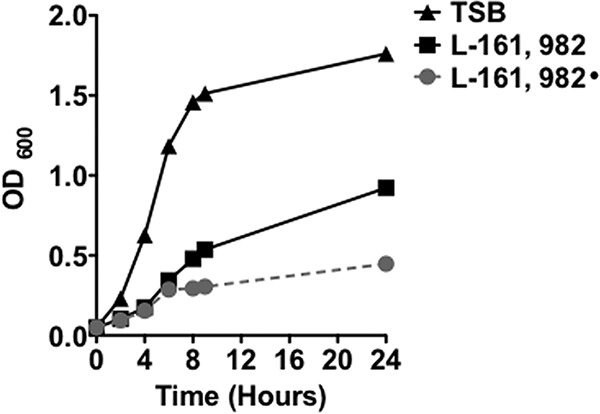
Growth inhibition kinetics of L-161,982. The growth of S. aureus NRS383 in medium alone or in medium supplemented with DMSO or 50 μg/ml L-161,982 was monitored for up to 24 h. After 6 h of coincubation, fresh L-161,982 was added to the growth medium (L-161,982•). The data shown are representative of those from three independent experiments.
L-161,982 has a narrow spectrum of activity.
The spectrum of activity of L-161,982 was investigated by testing several Gram-positive and Gram-negative bacteria. The activity of L-161,982 was restricted to the Gram-positive bacteria Staphylococcus epidermidis and Streptococcus mutans, as it had no inhibitory effect on another Gram-positive bacterium, Enterococcus faecium (Fig. 3). In addition, no inhibitory effects were observed against the Gram-negative bacteria tested (Escherichia coli and Klebsiella pneumoniae).
FIG 3.
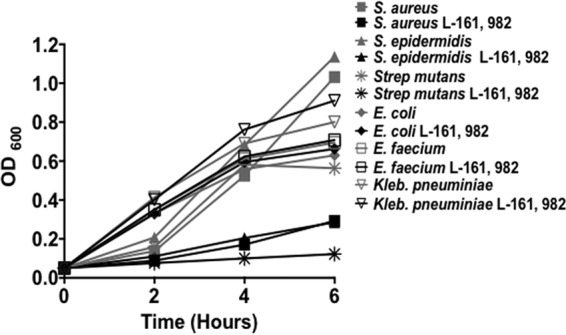
L-161,982 has a narrow spectrum of activity. Bacterial growth was monitored for 6 h in medium alone or in medium supplemented with 50 μg/ml of L-161,982. The data shown are representative of those from three independent experiments.
L-161,982 has no potentiating effect on oxacillin.
We next tested whether L-161,982 had synergistic or potentiating effects on the antibiotic activity of oxacillin against MRSA using the standard checkerboard broth microdilution assay. The fractional inhibitory concentration (FIC) index of 0.563 indicated indifference (data not shown) (a FIC index of ≤0.5 indicates synergy, a FIC index of 0.5 indicates indifference, and a FIC index of ≥4 indicates antagonism).
L-161,982 inhibits S. aureus biofilm formation.
As S. aureus and C. albicans form biofilms that are resistant to most antimicrobials, we examined the antibiofilm potential of L-161,982 against mono- and dual-species biofilms. For C. albicans biofilm formation, no significant inhibition of metabolic activity was observed between treated and untreated monospecies biofilms (Fig. 4A and B). In contrast, the metabolic activity of S. aureus was significantly inhibited compared to that of the untreated control at all concentrations tested (Fig. 4A). In addition, the number of CFU of S. aureus was significantly reduced by L-161,982 at concentrations ranging from 6.25 to 25 μg/ml, while no viable cells were recovered at concentrations of >50 μg/ml (Fig. 4B).
FIG 4.
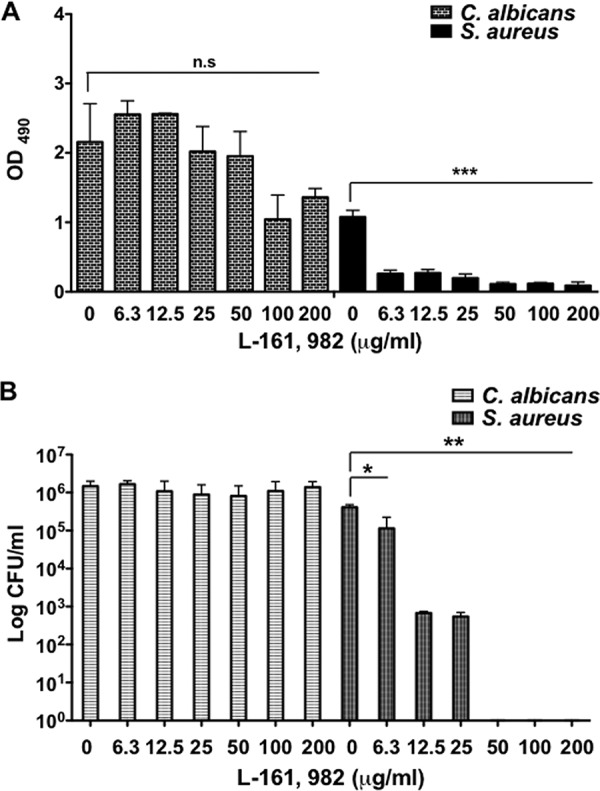
L-161,982 prevents MRSA biofilm formation. Mono- and dual-species C. albicans and/or S. aureus biofilms were grown on 96-well plates for 24 h with the indicated concentration of L-161,982. After incubation, the metabolic activity of the biofilms was quantified by the XTT assay (A), and then the biofilms were disrupted to determine the number of live CFU (B). The results are cumulative results from three independent experiments with duplicate samples. Statistical analysis was performed using one-way ANOVA with the Tukey post hoc test for the XTT assay. The Mann-Whitney U test was used to analyze the CFU data. Significance is indicated as follows: n.s, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. Error bars represent standard deviations.
Next we investigated the ability of L-161,982 to disrupt preformed mono- and dual-species biofilms and found no significant inhibitory effects on the metabolic activity of either C. albicans or S. aureus at any of the concentrations tested (Fig. 5A). Interestingly, though, at the higher concentrations of 100 and 200 μg/ml, no visible biofilm was observed following washing steps, and no CFU were recovered from S. aureus mono- and dual-species biofilms, suggesting that treatment with high concentrations of L-161,982 after the formation of the biofilms may have resulted in weak biofilm structures that were easily disrupted mechanically (Fig. 5B).
FIG 5.
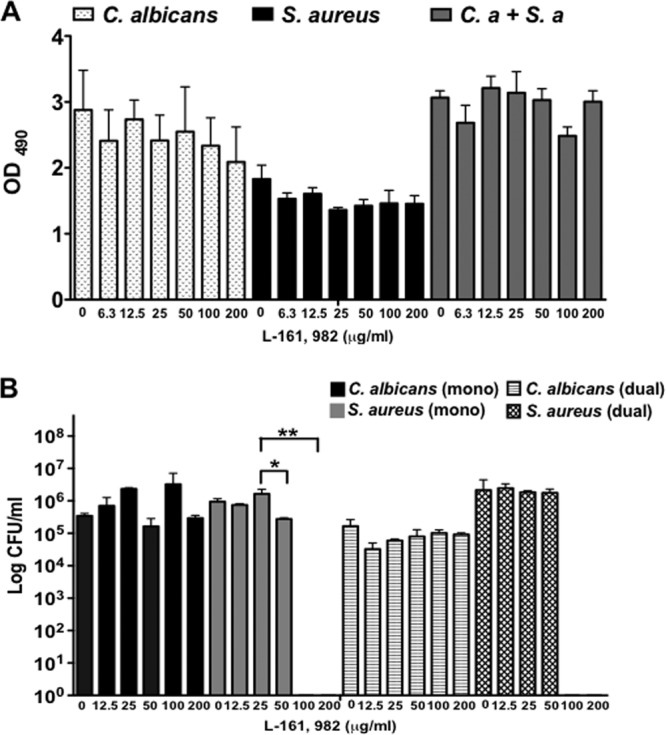
Effect of L-161,982 on preformed mono- and dual-species biofilms. The effect of L-161,982 on preformed mature biofilms was tested. Mono- and dual-species C. albicans (C.a) and/or S. aureus (S.a) biofilms were grown on 96-well plates for 24 h before being treated with the indicated concentration of L-161,982 for a further 24 h. After treatment, the metabolic activity of the biofilms was quantified by the XTT assay (A), and then the biofilms were disrupted to determine the number of live CFU (B). The results are cumulative results from three independent experiments with duplicate samples. Statistical analysis was performed using one-way ANOVA with the Tukey post hoc test for the XTT assay. The Mann-Whitney U test was used to analyze the CFU data. Significance is indicated as follows: ns, not significant; *, P < 0.05; **, P < 0.01. Error bars represent standard deviations.
L-161,982 protects against mono- and polymicrobial infections with MRSA.
We next evaluated the efficacy of L-161,982 in vivo using an established mouse model of peritonitis/sepsis. Mice were inoculated with three lethal inocula of S. aureus and treated with L-161,982 (10 mg/kg of body weight) intraperitoneally (i.p.) at 2 h postinoculation and once daily afterwards through day 4. The control group received the vehicle at the same dose interval.
Results showed that at the intermediate inoculum of 5 × 106 CFU, L-161,982 increased the rate of survival from 25 to 55% by day 2 postinoculation (Fig. 6A). At the lowest inoculum of 4 × 106 CFU, survival increased from 40 to ∼85% during the same 2-day period. Continued dosing of L-161,982 sustained survival in the intermediate- and lowest-inoculum groups through day 10, whereas the rate of survival was 20% in the control group. At the high inoculum of 2 × 107 CFU, the 80% mortality by day 2 could not be reversed by L-161,982 treatment. We also investigated the ability of L-161,982 to aid in bacterial clearance in the host by examining changes in the bioburden of S. aureus in the peritoneal cavity and spleen after two or three doses of L-161,982 (24 h or 48 h postinoculation, respectively). Compared to no treatment, treatment with L-161,982 significantly reduced the S. aureus burden in the peritoneal cavity and spleen by 3 logs at 24 h, and an additional 3-log reduction was achieved after a second dose of L-161,982 (48 h postinfection) (Fig. 6B).
FIG 6.
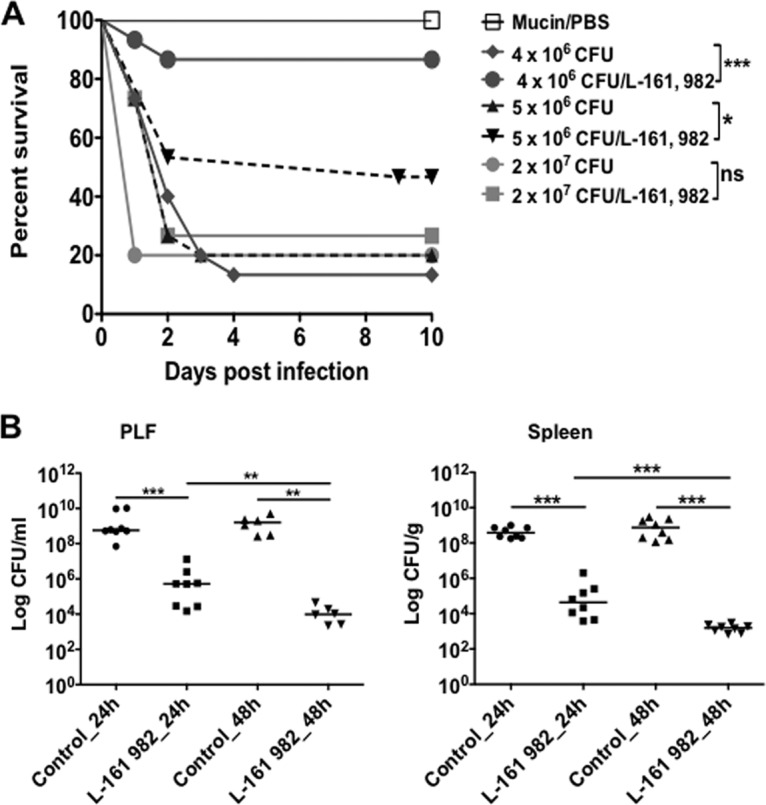
Effect of L-161,982 on survival and microbial burden. Mice were infected intraperitoneally with 0.2 ml of the indicated inoculum of S. aureus NRS383 in 3% hog gastric mucin–PBS. One group of mice per inoculum received either the vehicle or L-161,982 at a dose of 10 mg/kg by i.p. injection, as indicated in Materials and Methods. (A) Mortality was assessed using the Kaplan-Meier test and was compared to that of the control groups (*, P < 0.05; ***, P < 0.001; ns, not significant; n = 15/group). (B) The microbial burden in peritoneal lavage fluid (PLF) and spleen between the untreated (control) and treated (L-161,982) groups was assessed at 24 and 48 h postinoculation (n = 6 to 8/group). Values are plotted as the median number of CFU and were compared using the Mann-Whitney U test (**, P < 0.01; ***, P < 0.001). Data shown are representative of those from three replicate experiments.
In a final set of experiments, we evaluated the ability of L-161,982 to reduce lethality in coinfected mice. Mice coinoculated with the standard inocula of 7 × 106 C. albicans CFU and 8 × 107 S. aureus CFU (8.7 × 107 CFU total) were treated with L-161,982 (10 mg/kg) at 2 h postinfection and once daily afterwards until day 5. To ensure that the beneficial effect of L-161,982 was not a result of blocking of PGE2-EP4 receptor signaling, we included an alternative antagonist, ONO AE3 208, which was confirmed to have no antimicrobial effect on S. aureus in vitro and in vivo (Fig. 1 and 7). Compared to the vehicle control group, the rate of survival in mice given L-161,982 improved from 10 to 50%, while ONO AE3 208 had no effect (Fig. 7).
FIG 7.
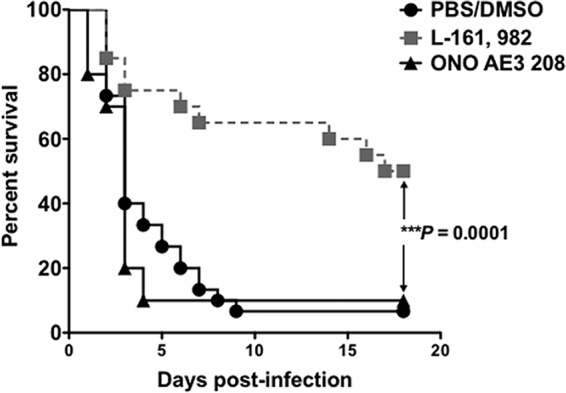
Antibacterial effect of L-161,982 on MRSA enhances survival in a mouse model of C. albicans-S. aureus polymicrobial intra-abdominal infection. The effect of inhibition of EP4 receptor signaling on survival is shown. Mice received either vehicle or an EP4 receptor antagonist at a dose of 10 mg/kg by i.p. injection, as indicated in Materials and Methods. Mortality compared to that in the control groups was assessed using the Kaplan-Meier test (***, P < 0.001; n = 20/group). The data shown are representative of those from three replicate experiments.
DISCUSSION
In studies designed to uncover the specific pathways involved in PGE2 synthesis and downstream signaling via EP receptors, we discovered that the EP4 receptor antagonist L-161,982 had direct antimicrobial activity against S. aureus. This is the first report of a PGE2 receptor antagonist having antimicrobial activity, although some NSAIDs have been known to have antimicrobial activity, in addition to anti-inflammatory activity (14–17). Therefore, this novel activity represents a major finding that could be exploited as a therapeutic alternative. It is noteworthy that the S. aureus reference strain used in this study (NRS383) is methicillin resistant, which makes this finding more clinically significant, given that MRSA is currently among the most common multidrug-resistant pathogens worldwide and the need to identify novel antibiotics to combat infections with these pathogens is urgent. The equivalent activity of L-161,982 against other clinical MRSA and MSSA isolates obtained from contaminated catheters enhances the potential that L-161,982 could be a formidable alternative therapeutic against any S. aureus strains.
The MIC for the reference strain was 50 μg/ml, while the MBC was 100 μg/ml. Continued dosing of L-161,982 was required to maintain the growth-inhibitory effect, and the need for continued dosing could have been due to either drug degradation (half-life) or depletion. Interestingly, the spectrum of activity of L-161,982 extended to other Gram-positive bacteria, including S. epidermidis and S. mutans, but it had no activity against E. faecium or several Gram-negative bacterial species. While E. faecium is classified within the phylum Firmicutes, order Bacilli, similar to staphylococci and streptococci, it belongs to a different bacterial family (Enterococcaceae). This suggests that it has a spectrum of activity even narrower than that of other drug classes targeting Gram-positive bacteria, such as glycopeptides (18). However, further testing is needed to confirm whether this range of activity holds true for all Gram-positive versus Gram-negative species. We also explored the possible synergistic activity of L-161,982 with oxacillin and the potential for L-161,982 to lower the MIC of oxacillin against MRSA; however, we found the activity of L-161,982 to be mutually exclusive of that of oxacillin against MRSA strains.
Both C. albicans and S. aureus are notorious for forming biofilms on medical devices that are extremely resistant to antimicrobial agents and host defense mechanisms, and this becomes more pronounced with mixed-species biofilms. We show here that concentrations of L-161,982 below the MIC (50 μg/ml) for planktonic growth inhibited S. aureus monospecies growth during biofilm formation, whereas the MBC of 100 μg/ml was required to weaken preformed mature biofilms. Unfortunately, L-161,982 showed no inhibitory effect in a dual-species biofilm with C. albicans, suggesting that the presence of C. albicans might have prevented L-161,982 accessibility. This finding was not surprising, as the resulting extracellular matrix (ECM) produced by C. albicans, which is composed of secreted β-1,3-glucan molecules, has been shown to coat bacterial cells within the mixed-species biofilm, thereby preventing drug penetration (19–21). Nevertheless, the antimicrobial activity of L-161,982 against S. aureus or other biofilm-forming bacteria in a mixed-species biofilm could enhance the efficacy of other antimicrobials.
Our in vivo studies also highlight the potential use of L-161,982 against mono- and polymicrobial IAI with MRSA. In a mouse model of MRSA peritonitis, L-161,982 significantly decreased the bioburden in the peritoneal cavity and spleen with just one dose administered at 2 h postinoculation. We also found that, depending on the infectious dose of S. aureus, one treatment with L-161,982 raised the survival rate by at least 50%. Consistent with the results of the in vitro study, sustained survival required continued dosing. There was an upper limit to the activity, as L-161,982 was ineffective when the inoculum was very high (2 × 107 CFU), despite continued dosing. The protective role of L-161,982 was further demonstrated in a mouse model of polymicrobial intra-abdominal infection with C. albicans, where survival was enhanced by 40% (50% survival), which was likely due to the antimicrobial effect of L-161,982 on S. aureus.
The antimicrobial activity of L-161,982 was independent of effects on PGE2 signaling, as treatment of mice with a different EP4 receptor antagonist (ONO AE3 208) had no effect on enhancing survival. If it is presumed that the antagonist effectively blocked PGE2 signaling, it is unlikely that PGE2 promotes lethal inflammation via EP4 in our model. Further studies will be aimed at evaluating the pharmacodynamics and pharmacokinetics of L-161,982 as an antimicrobial agent, as well higher dose-responses to determine its upper limits of activity. Although extensive toxicological studies have not been carried out, the toxic dose low (TDLO) values reported for L-161,982 subcutaneous administrations in mice are 100 μg/kg to 333.33 μg/kg (Tocris). In our studies using peritoneal administration, L-161,982 was given at 10 mg/kg of body weight on the basis of previous reports with no apparent toxic effects (22, 23). Further studies are required to establish the full range of toxicological properties for L-161,982.
In summary, this study provides the first evidence of L-161,982 antibacterial activity via several in vitro study designs and in vivo protection against S. aureus mono- and polymicrobial IAI. In addition, the results of the preliminary studies presented here indicate that it has in vitro activity against two other clinically relevant Gram-positive bacteria. Further studies are required to uncover the growth-inhibitory mechanism of L-161,982 and determine its safety as a therapeutic agent in clinical trials. If successful, L-161,982 could be used as an alternative preventive or therapeutic agent with activity against mono- and polymicrobial infections with S. aureus, including MRSA.
MATERIALS AND METHODS
Strains and growth conditions.
The methicillin-resistant S. aureus strain NRS383 used in all experiments was obtained from the Network on Antimicrobial Resistance in S. aureus (NARSA) data bank. NRS383 is positive for the toxic shock syndrome toxin (tst) and δ-toxin genes. SJ-MRSA 6 and SJ-SA5 are clinical methicillin-resistant S. aureus (MRSA) and methicillin-sensitive S. aureus (MSSA) strains, respectively, isolated from a patient's catheter. Other bacterial species used in this study included Escherichia coli (ATCC 25922, clinical isolate), S. epidermidis (ATCC 12228), Klebsiella pneumoniae (ATCC 43816), Enterococcus faecalis (ATCC 51299, clinical isolate), and Streptococcus mutans (UA159) (24). The S. mutans strain was maintained in brain heart infusion (BHI) broth, while the other bacterial species were maintained in Luria-Bertani broth. Frozen stocks were obtained at −80°C and streaked onto agar plates at 37°C prior to use. A single colony was transferred to 10 ml of culture broth and grown at 37°C overnight with aeration at 200 rpm. Overnight cultures were diluted 1:100 in fresh growth medium and shaken at 37°C for 3 h to obtain cells in mid-log-growth phase, with the exception of S. mutans cultures, which were grown without agitation in the presence of 5% CO2.
The C. albicans strain used in these experiments was DAY185, a prototrophic control strain of strain BWP17 (an auxotrophic derivative of strain SC5314) into which the HIS1, URA3, and ARG4 genes were reinserted (25). Frozen stocks were obtained at −80°C and streaked onto yeast extract-peptone-dextrose (YPD) agar prior to use. A single colony was transferred to 20 ml of YPD broth and grown at 30°C for 18 h with aeration at 200 rpm. Prior to inoculation into mice, both C. albicans and S. aureus were rinsed 3 times by centrifugation in sterile phosphate-buffered saline (PBS; pH 7.4), counted on a hemocytometer, and diluted in sterile PBS to prepare standardized inocula.
Planktonic antimicrobial assay.
C. albicans and S. aureus were grown as described above. On the following day, cultures were diluted 1:100 in fresh medium containing the following chemical inhibitors at the indicated concentrations: indomethacin (50 μg/ml; a nonselective COX inhibitor; Cayman Chemicals), SC-560 (200 μg/ml; a selective COX1 inhibitor; Cayman Chemicals), NS-398 (100 μg/ml; a selective COX2 inhibitor; Cayman Chemicals), SC 51322 (100 μg/ml; an EP1 receptor antagonist; Tocris), PF 04418948 (100 μg/ml; an EP2 receptor antagonist; Tocris), L-798,106 (100 μg/ml; an EP3 receptor antagonist; Tocris), L-161,982 (100 μg/ml; an EP4 receptor antagonist; Tocris), and ONO AE3 208 (100 μg/ml; an EP4 receptor antagonist; Tocris) in 0.28% dimethyl sulfoxide (DMSO) or 0.28% DMSO alone to serve as a control (Table 1). Because the average peritoneal volume of a Swiss Webster mouse is approximately 2 ml, the concentration of each inhibitor and DMSO was chosen to reflect the physiologically delivered final concentrations. C. albicans and S. aureus cells were grown with shaking as described above, aliquots were removed each hour in duplicate and diluted 1:100, and the optical density at 600 nm (OD600) and OD660 were measured with a spectrophotometer (Biomate 3S; Thermo Scientific).
MIC/MBC assay.
The CLSI reference method for antimicrobial susceptibility testing of bacteria was used to determine the MIC of L-161,982 (26). Cells were diluted in sterile Mueller-Hinton broth (Sigma) to a final concentration of 5.0 × 105 CFU/ml, and 200-μl aliquots (1 × 105 CFU/well) were added to the wells of sterile round-bottom 96-well plates. A stock solution of L-161,982 was diluted in sterile Mueller-Hinton broth prior to addition to the cell suspension. The drug concentrations tested ranged from 1 to 100 μg/ml and were tested in triplicate. The plates were incubated at 37°C for 24 h, and the MIC was determined to be the lowest drug concentration that inhibited visible growth. Bacteriostatic or bactericidal activity was assessed by plating 20 μl of broth from all wells with no visible growth on Mueller-Hinton agar plates, and the plates were incubated at 37°C for 24 to 48 h. The MBC was the lowest drug concentration that prevented growth. MIC and MBC determinations were done in triplicate, and oxacillin was used as a positive control.
Test for synergy between L-161,982 and oxacillin.
Due to the newfound antimicrobial activity of L-161,982 against S. aureus, the checkerboard method (27) with the broth microdilution method was used to test for synergy between L-161,982 and oxacillin, a drug similar to methicillin used more commonly in the clinical setting. Synergy was determined using the fractional inhibitory concentration (FIC) index method (27, 28). With this method, the MIC of the antibiotic compound in the combination is divided by the MIC of the compound alone, giving the fractional contribution of each drug component in the combination. Quotients for all compounds in combination are summed, and drug interactions are scored using the formula FIC index = [(MIC AcombA + B)/MICA] + [(MIC BcombA + B)/MICB)], where MIC AcombA + B is the MIC of compound A in the combination of compounds A and B, MICA is the MIC of compound A, MIC BcombA + B is the MIC of compound B in the combination of compounds A and B, and MICB is the MIC of compound B.
The stock solutions and serial 2-fold dilutions of oxacillin ranged from 8× the MIC to 32× below the MIC, and those of L-161,982 ranged from 8× the MIC to 2,000× below the MIC. Oxacillin was serially diluted along the ordinate, while L-161,982 was diluted along the abscissa. The resulting checkerboard contained each combination of two drugs, with the wells that contained the highest concentration of each antibiotic being at opposite corners. Each well of the microtiter plate was inoculated with 1 × 105 CFU. The plates were incubated at 37°C for 24 h, and the OD600 was measured using a microplate spectrophotometer. A FIC index of ≤0.5 indicates synergy, a FIC index of between 0.5 and 4 indicates indifference, and a FIC index of >4 indicates antagonism (28, 29).
Biofilm formation on polystyrene.
Following growth as described above, the microbes were washed in phosphate-buffered saline (PBS) by centrifugation, counted on a hemocytometer, and adjusted to 2 × 107 CFU/ml in RPMI buffered with MOPS (morpholinepropanesulfonic acid; 165 mM). For monomicrobial biofilms, 50 μl of adjusted C. albicans or S. aureus culture was added to each well of a sterile 96-well cell culture polystyrene microtiter plate (1 × 106 CFU per well); to this, 50 μl of sterile RPMI plus MOPS was added. For polymicrobial biofilms, 50 μl of each organism was added per well (1 × 106 CFU of each organism per well). The plates were incubated for 24 h at 37°C to induce biofilm formation. Following confirmation of mature biofilm growth by macro- and microscopic examination, the plates were washed once with 200 μl sterile saline, and then 200 μl RPMI plus MOPS containing L-161,982 at various concentrations (6.25 to 200 μg/ml) was added to each well. L-161,982-free RPMI plus MOPS was added to wells with and without microbes to serve as positive and negative controls, respectively. The plates were incubated at 37°C for 24 h. After incubation, the plates were washed once with PBS and processed for the sodium 2,3-bis (2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino) carbonyl]-2H-tetrazolium hydroxide inner salt (XTT) metabolic activity assay, followed by CFU enumeration. To dislodge the biofilms, the plates were sonicated for 5 to 10 s and suspensions were pipetted up and down prior to serial dilutions, which were done with constant vortexing (5 to 10 s) in between dilutions and prior to plating out.
The XTT reduction assay was used to determine the metabolic activity of biofilms following treatment with L-161,982 (30). Briefly, the wells were washed once in PBS, the plates were incubated at 37°C for 2 h with 200 μl XTT working reagent (0.5 mg/ml XTT, 1 μM menadione), and the resulting absorbance at 490 nm was measured on a microplate spectrophotometer. After L-161,982 treatment and metabolic activity measurement, enumeration of CFU was carried out as previously described (31).
Mouse model of infection.
All animals were housed and handled according to institutionally recommended guidelines. All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the LSU Health Sciences Center, New Orleans, LA. Mice were given access to food and water ad libitum. In all experiments, 5- to 7-week-old female outbred NIH Swiss mice, purchased from Charles Rivers, Frederick, MD, were used.
The mouse peritonitis/intra-abdominal infection model was performed as previously described (8). In brief, for MRSA peritonitis, mice were injected intraperitoneally (i.p.) with 4 × 106 CFU, 5 × 106 CFU, or 2 × 107 CFU of S. aureus in 0.2 ml of PBS containing 3% hog gastric mucin type III (Sigma-Aldrich). Mice injected i.p. with 0.2 ml of mucin-PBS served as a negative infection control. For intra-abdominal infection with C. albicans and S. aureus, mice were injected i.p. with 7 × 106 CFU of C. albicans and 8 × 107 CFU of S. aureus (8.7 × 107 CFU total) in 0.2 ml of PBS. After inoculation, the mice were observed over 10 days for signs of morbidity (hunched posture, inactivity, lethargy, and ruffled fur) and mortality. Mice that were significantly moribund were euthanized according to institutionally recommended guidelines. In the MRSA peritonitis experiments, mice were sacrificed at 24 or 48 h postinoculation to assess the microbial burden. The peritoneal cavities were lavaged by injection of 2 ml of sterile PBS, followed by gentle massaging of the peritoneal cavity. Peritoneal lavage fluid was then removed using a pipette inserted into a small incision in the abdominal cavity. The spleens were removed, weighed, and mechanically homogenized prior to analysis of the number of CFU. The microbial burdens in the peritoneal lavage fluid and spleen were enumerated as described above.
Inhibitor treatment.
Each inhibitor was prepared fresh as a concentrated stock (20 to 25 mg/ml) in DMSO and diluted to a working concentration of 10 mg/ml in sterile PBS (final DMSO concentration, 2.3%). A sham treatment containing only 2.3% DMSO in PBS was also prepared. Groups of 5 to 10 mice were intraperitoneally administered 0.1 ml of inhibitor (as indicated in Table 1). For the MRSA peritonitis study, the EP4 receptor antagonist was administered 2 h after S. aureus inoculation and once daily thereafter until day 4 postinoculation. For the IAI coinfection study, a COX inhibitor or vehicle control was administered 4 h prior to and 4 and 8 h after inoculation with C. albicans and S. aureus, while the EP receptor antagonists were administered daily starting 1 day prior to and 5 days after infection. All other procedures were performed as described above. Control groups received the vehicle at the same dose interval.
Statistical analysis.
All experiments used groups of 5 to 10 mice and were repeated in duplicate, except where noted. All assays were repeated in triplicate, and the results were averaged. Survival data were analyzed using the Kaplan-Meier test. The metabolic activities of the treated biofilms were compared to those of the controls using one-way analysis of variance (ANOVA) with the Tukey post hoc test. The Mann-Whitney U test was used to analyze all CFU data. In all tests, differences were considered significant at a P value of <0.05. All statistical analyses were performed with GraphPad Prism software.
ACKNOWLEDGMENT
Funding from the National Institutes of Health supported this work (NIAID grant R01-AI116025 to M.C.N.).
REFERENCES
- 1.Blot SI, Vandewoude KH, De Waele JJ. 2007. Candida peritonitis. Curr Opin Crit Care 13:195–199. doi: 10.1097/MCC.0b013e328028fd92. [DOI] [PubMed] [Google Scholar]
- 2.Dupont H, Paugam-Burtz C, Muller-Serieys C, Fierobe L, Chosidow D, Marmuse JP, Mantz J, Desmonts JM. 2002. Predictive factors of mortality due to polymicrobial peritonitis with Candida isolation in peritoneal fluid in critically ill patients. Arch Surg 137:1341–1346. doi: 10.1001/archsurg.137.12.1341. [DOI] [PubMed] [Google Scholar]
- 3.Montravers P, Gauzit R, Muller C, Marmuse JP, Fichelle A, Desmonts JM. 1996. Emergence of antibiotic-resistant bacteria in cases of peritonitis after intra-abdominal surgery affects the efficacy of empirical antimicrobial therapy. Clin Infect Dis 23:486–494. doi: 10.1093/clinids/23.3.486. [DOI] [PubMed] [Google Scholar]
- 4.Calandra T, Bille J, Schneider R, Mosimann F, Francioli P. 1989. Clinical significance of Candida isolated from peritoneum in surgical patients. Lancet ii:1437–1440. [DOI] [PubMed] [Google Scholar]
- 5.Burke M, Hawley CM, Badve SV, McDonald SP, Brown FG, Boudville N, Wiggins KJ, Bannister KM, Johnson DW. 2011. Relapsing and recurrent peritoneal dialysis-associated peritonitis: a multicenter registry study. Am J Kidney Dis 58:429–436. doi: 10.1053/j.ajkd.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 6.Sandven P, Qvist H, Skovlund E, Giercksky KE. 2002. Significance of Candida recovered from intraoperative specimens in patients with intra-abdominal perforations. Crit Care Med 30:541–547. doi: 10.1097/00003246-200203000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Carlson E. 1982. Synergistic effect of Candida albicans and Staphylococcus aureus on mouse mortality. Infect Immun 38:921–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters BM, Noverr MC. 2013. Candida albicans-Staphylococcus aureus polymicrobial peritonitis modulates host innate immunity. Infect Immun 81:2178–2189. doi: 10.1128/IAI.00265-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nash EE, Peters BM, Palmer GE, Fidel PL, Noverr MC. 2014. Morphogenesis is not required for Candida albicans-Staphylococcus aureus intra-abdominal infection-mediated dissemination and lethal sepsis. Infect Immun 82:3426–3435. doi: 10.1128/IAI.01746-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller SB. 2006. Prostaglandins in health and disease: an overview. Semin Arthritis Rheum 36:37–49. doi: 10.1016/j.semarthrit.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 11.Ricciotti E, FitzGerald GA. 2011. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dey I, Lejeune M, Chadee K. 2006. Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br J Pharmacol 149:611–623. doi: 10.1038/sj.bjp.0706923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones VC, Birrell MA, Maher SA, Griffiths M, Grace M, O'Donnell VB, Clark SR, Belvisi MG. 2016. Role of EP2 and EP4 receptors in airway micro vascular leak induced by prostaglandin E2. Br J Pharmacol 173:992–1004. doi: 10.1111/bph.13400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Matos RF, Mendonca LC, da Silva Souza KG, Fonseca AA, Costa EM, de Lima MV, Vieira JM, de Brito MT, Monteiro MC. 2017. Nimesulide inhibits pathogenic fungi: PGE2-dependent mechanisms. Folia Microbiol (Praha) 62:169–174. doi: 10.1007/s12223-016-0483-6. [DOI] [PubMed] [Google Scholar]
- 15.Guzman-Beltran S, Rubio-Badillo MA, Juarez E, Hernandez-Sanchez F, Torres M. 2016. Nordihydroguaiaretic acid (NDGA) and alpha-mangostin inhibit the growth of Mycobacterium tuberculosis by inducing autophagy. Int Immunopharmacol 31:149–157. doi: 10.1016/j.intimp.2015.12.027. [DOI] [PubMed] [Google Scholar]
- 16.El-Mowafy SA, Abd El Galil KH, El-Messery SM, Shaaban MI. 2014. Aspirin is an efficient inhibitor of quorum sensing, virulence and toxins in Pseudomonas aeruginosa. Microb Pathog 74:25–32. doi: 10.1016/j.micpath.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Abdelmegeed E, Shaaban MI. 2013. Cyclooxygenase inhibitors reduce biofilm formation and yeast-hypha conversion of fluconazole resistant Candida albicans. J Microbiol 51:598–604. doi: 10.1007/s12275-013-3052-6. [DOI] [PubMed] [Google Scholar]
- 18.Nailor MD, Sobel JD. 2011. Antibiotics for Gram-positive bacterial infections: vancomycin, teicoplanin, quinupristin/dalfopristin, oxazolidinones, daptomycin, telavancin, and ceftaroline. Med Clin North Am 95:723–742. doi: 10.1016/j.mcna.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 19.Kong EF, Tsui C, Kucharikova S, Andes D, Van Dijck P, Jabra-Rizk MA. 2016. Commensal protection of Staphylococcus aureus against antimicrobials by Candida albicans biofilm matrix. mBio 7:e01365-16. doi: 10.1128/mBio.01365-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harriott MM, Noverr MC. 2009. Candida albicans and Staphylococcus aureus form polymicrobial biofilms: effects on antimicrobial resistance. Antimicrob Agents Chemother 53:3914–3922. doi: 10.1128/AAC.00657-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harriott MM, Noverr MC. 2010. Ability of Candida albicans mutants to induce Staphylococcus aureus vancomycin resistance during polymicrobial biofilm formation. Antimicrob Agents Chemother 54:3746–3755. doi: 10.1128/AAC.00573-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greaves E, Horne AW, Jerina H, Mikolajczak M, Hilferty L, Mitchell R, Fleetwood-Walker SM, Saunders PT. 2017. EP2 receptor antagonism reduces peripheral and central hyperalgesia in a preclinical mouse model of endometriosis. Sci Rep 7:44169. doi: 10.1038/srep44169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Machwate M, Harada S, Leu CT, Seedor G, Labelle M, Gallant M, Hutchins S, Lachance N, Sawyer N, Slipetz D, Metters KM, Rodan SB, Young R, Rodan GA. 2001. Prostaglandin receptor EP4 mediates the bone anabolic effects of PGE2. Mol Pharmacol 60:36–41. [DOI] [PubMed] [Google Scholar]
- 24.Ajdic D, McShan WM, McLaughlin RE, Savic G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S, Jia H, Lin S, Qian Y, Li S, Zhu H, Najar F, Lai H, White J, Roe BA, Ferretti JJ. 2002. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc Natl Acad Sci U S A 99:14434–14439. doi: 10.1073/pnas.172501299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis D, Edwards JE Jr, Mitchell AP, Ibrahim AS. 2000. Candida albicans RIM101 pH response pathway is required for host-pathogen interactions. Infect Immun 68:5953–5959. doi: 10.1128/IAI.68.10.5953-5959.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clinical and Laboratory Standards Institute. 2015. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 10th ed CLSI document M07-A10. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 27.Berenbaum MC. 1978. A method for testing for synergy with any number of agents. J Infect Dis 137:122–130. doi: 10.1093/infdis/137.2.122. [DOI] [PubMed] [Google Scholar]
- 28.Berenbaum MC. 1989. What is synergy? Pharmacol Rev 41:93–141. [PubMed] [Google Scholar]
- 29.Saiman L. 2007. Clinical utility of synergy testing for multidrug-resistant Pseudomonas aeruginosa isolated from patients with cystic fibrosis: ‘the motion for.’ Paediatr Respir Rev 8:249–255. doi: 10.1016/j.prrv.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 30.Pierce CG, Uppuluri P, Tummala S, Lopez-Ribot JL. 2010. A 96 well microtiter plate-based method for monitoring formation and antifungal susceptibility testing of Candida albicans biofilms. J Vis Exp 2010:2287. doi: 10.3791/2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peters BM, Ward RM, Rane HS, Lee SA, Noverr MC. 2013. Efficacy of ethanol against Candida albicans and Staphylococcus aureus polymicrobial biofilms. Antimicrob Agents Chemother 57:74–82. doi: 10.1128/AAC.01599-12. [DOI] [PMC free article] [PubMed] [Google Scholar]