

ABSTRACT
When bacterial cells are exposed to increasing concentrations of quinolone-class antibacterials, survival drops, reaches a minimum, and then recovers, sometimes to 100%. Despite decades of study, events underlying this paradoxical high-concentration survival remain obscure. Since reactive oxygen species (ROS) have been implicated in antimicrobial lethality, conditions generating paradoxical survival were examined for diminished ROS accumulation. Escherichia coli cultures were treated with various concentrations of nalidixic acid, followed by measurements of survival, rate of protein synthesis, and ROS accumulation. The last measurement used a dye (carboxy-H2DCFDA) that fluoresces in the presence of ROS; fluorescence was assessed by microscopy (individual cells) and flow cytometry (batch cultures). High, nonlethal concentrations of nalidixic acid induced lower levels of ROS than moderate, lethal concentrations. Sublethal doses of exogenous hydrogen peroxide became lethal and eliminated the nalidixic acid-associated paradoxical survival. Thus, quinolone-mediated lesions needed for ROS-executed killing persist at high, nonlethal quinolone concentrations, thereby implicating ROS as a key factor in cell death. Chloramphenicol suppressed nalidixic acid-induced ROS accumulation and blocked lethality, further supporting a role for ROS in killing. Nalidixic acid also inhibited protein synthesis, with extensive inhibition at high concentrations correlating with lower ROS accumulation and paradoxical survival. A catalase deficiency, which elevated ROS levels, overcame the inhibitory effect of chloramphenicol on nalidixic acid-mediated killing, emphasizing the importance of ROS. The data collectively indicate that ROS play a dominant role in the lethal action of narrow-spectrum quinolone-class compounds; a drop in ROS levels accounted for the quinolone tolerance observed at very high concentrations.
KEYWORDS: nalidixic acid, ROS, catalase, protein synthesis inhibition, paradoxical survival, Escherichia coli, tolerance, reactive oxygen species
INTRODUCTION
Antimicrobials recently emerged as important probes for studying bacterial responses to lethal stress, particularly for responses involving the accumulation of toxic reactive oxygen species (ROS) (1–6). While ROS-mediated killing raises the interesting possibility that bacteria self-destruct when experiencing severe stress (2), ROS involvement in the lethal action of multiple diverse antimicrobials has recently been challenged (7, 8). Consequently, we sought a new test for the ROS-lethal response hypothesis. Among the prominent probes for ROS effects are the quinolone-class inhibitors of DNA gyrase. One of the long-standing mysteries surrounding quinolone-mediated lethality is the loss of killing when concentrations of the compounds are very high (9). We reasoned that if ROS accumulation contributes to lethality, such accumulation might be reduced at the high drug concentrations that cause paradoxical survival (tolerance).
We are also interested in paradoxical high-concentration survival because new anti-topoisomerase compounds have emerged (10–13) that are likely to bypass existing quinolone resistance, a problem that is eroding the usefulness of fluoroquinolones (14). An issue with inhibitors of replication, such as the quinolones, is that they induce the mutagenic SOS response, which likely contributes to the emergence of resistance (for example, see reference 15). The effects of the SOS response are countered by lethal activity that rapidly reduces bacterial burden. Indeed, animal infection studies now show that several highly lethal antimicrobials severely restrict the emergence of resistance without needing to be maintained above the concentration threshold that blocks enrichment of resistant mutant subpopulations (16–19). Thus, understanding how the attack of DNA gyrase kills bacterial cells, including understanding the potential contribution of ROS, is important for finding ways to ensure the long-term utility of anti-topoisomerase agents.
Differences among quinolone-class derivatives can be important from an experimental perspective. Our current knowledge of lethality fits with two processes leading to bacterial death. In one, quinolone-mediated death requires protein synthesis and presumably the accumulation of toxic ROS (20, 21). In the second mechanism, quinolone-induced destabilization of quinolone-gyrase-DNA complexes (cleaved complexes) is proposed to directly fragment chromosomes and kill cells in the absence of ongoing protein synthesis and independent of putative ROS effects (22–24). Fluoroquinolones display features of both pathways (8, 25) and would, therefore, be less likely to produce unambiguous results than older quinolone derivatives that function largely through the pathway proposed to involve ROS.
In the present work, we examined the behavior of nalidixic acid, since almost all of its lethal activity appears to be ROS-dependent ([2, 24]; data obtained with the fluoroquinolone norfloxacin, which has dominated previous work concerning ROS [1, 7, 8, 26], would have been less readily interpreted due to multiple lethal mechanisms [25, 27]). Our findings indicate that at very high, quinolone-tolerant concentrations, ROS levels are lower than at moderate, lethal concentrations. Moreover, genetic and chemical perturbations that alter ROS levels also alter lethality, which may occur through attack of repairable DNA lesions generated by nalidixic acid. The data provide an explanation for tolerance to high concentrations of quinolone and solidify the hypothesis that ROS contribute to antimicrobial lethality.
RESULTS
Nalidixic acid-mediated killing correlates with ROS accumulation.
We began by examining the effect of nalidixic acid on survival of exponentially growing cultures of wild-type Escherichia coli (strain 3505). As expected from previous work (9), survival decreased as drug concentration increased over a low-to-moderate concentration range and reached a minimum at a nalidixic acid concentration of 400 μg/ml, which was about 50 times MIC (Fig. 1). Killing increased with incubation time, but the concentration that produced minimal survival remained the same (Fig. 1). When nalidixic acid concentration was raised to between 1,200 and 1,600 μg/ml, survival paradoxically rose to 100% (Fig. 1), regardless of incubation time.
FIG 1.
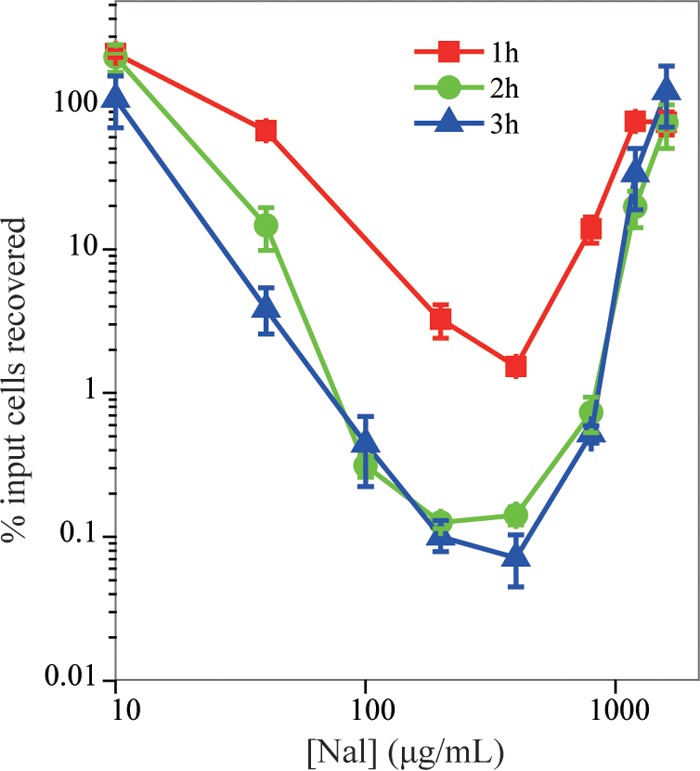
Paradoxical survival of wild-type E. coli is associated with high concentrations of nalidixic acid. Exponentially growing cultures of wild-type E. coli (strain 3505) were treated with nalidixic acid (Nal) at the indicated concentrations for 1 h (squares), 2 h (circles), or 3 h (triangles). Percentage of cells recovered was determined relative to CFU at the time of drug addition. Data are from at least three independent experiments; error bars represent standard errors of the mean.
To determine whether ROS levels during nalidixic acid treatment are sensitive to quinolone concentration, cultured cells were treated with carboxy-H2DCFDA, a dye that becomes fluorescent in the presence of all forms of ROS (28, 29). The dye (10 μM) was added to cultures at the same time as nalidixic acid, and after a 2-h incubation, cells were examined by fluorescence microscopy. Fluorescent cells were readily distinguished (Fig. 2a). The intensity of fluorescence, which correlates with ROS level (28), was maximal at 400 μg/ml nalidixic acid (Fig. 2a), the concentration responsible for maximal killing (Fig. 1). High, nonlethal concentrations of the quinolone were associated with a drop in fluorescence to below detection levels (Fig. 2a).
FIG 2.
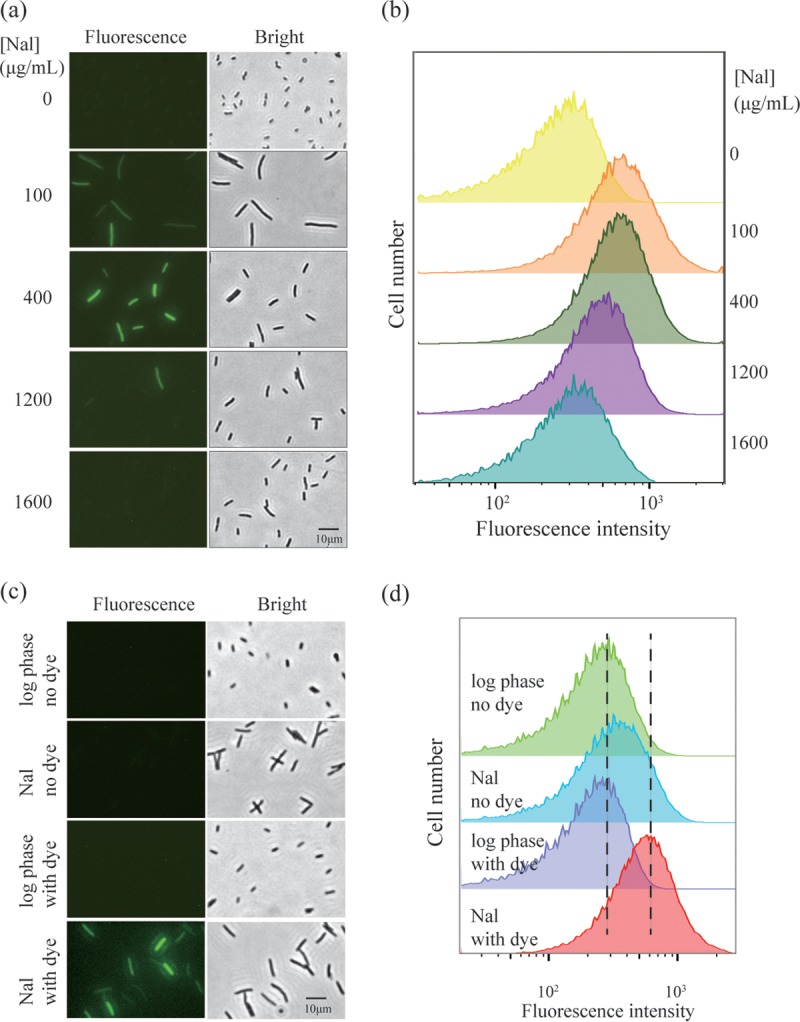
ROS accumulation correlates with nalidixic acid-mediated killing of E. coli. Exponentially growing cultures of wild-type E. coli (3505) were treated with carboxy-H2DCFDA (10 μM) and the indicated concentrations of nalidixic acid (Nal) for 2 h. Cells were then observed by fluorescence microscopy (a) or by flow cytometry (b). As an indicator of autofluorescence, samples containing only nalidixic acid (200 μg/ml) are shown for fluorescence microscopy (c) and flow cytometry (d). All panels show representative results from at least three independent experiments.
We also assessed ROS accumulation in bulk culture by flow cytometry of E. coli stained with carboxy-H2DCFDA. As shown in Fig. 2b, when nalidixic acid concentration increased, ROS levels increased and then decreased. This correlation between bacterial survival and ROS level is consistent with ROS participating in nalidixic acid-mediated lethality at low-to-moderate levels of quinolone; at very high drug concentration, ROS accumulation was suppressed, and cell death did not occur. Little background autofluorescence was observed (Fig. 2c and d). Since suppression of ROS parallels paradoxical survival and since the absence of ROS accumulation correlates with prevention of killing, the data support the idea that ROS are key factors in quinolone-mediated lethality.
Protein synthesis and killing by nalidixic acid.
Previous work showed that inhibition of protein synthesis by chloramphenicol or RNA synthesis by rifampin blocks killing by nalidixic acid (9). Chloramphenicol and rifampin, applied individually, also blocked the increase in ROS associated with lethal nalidixic acid treatment (Fig. 3).
FIG 3.
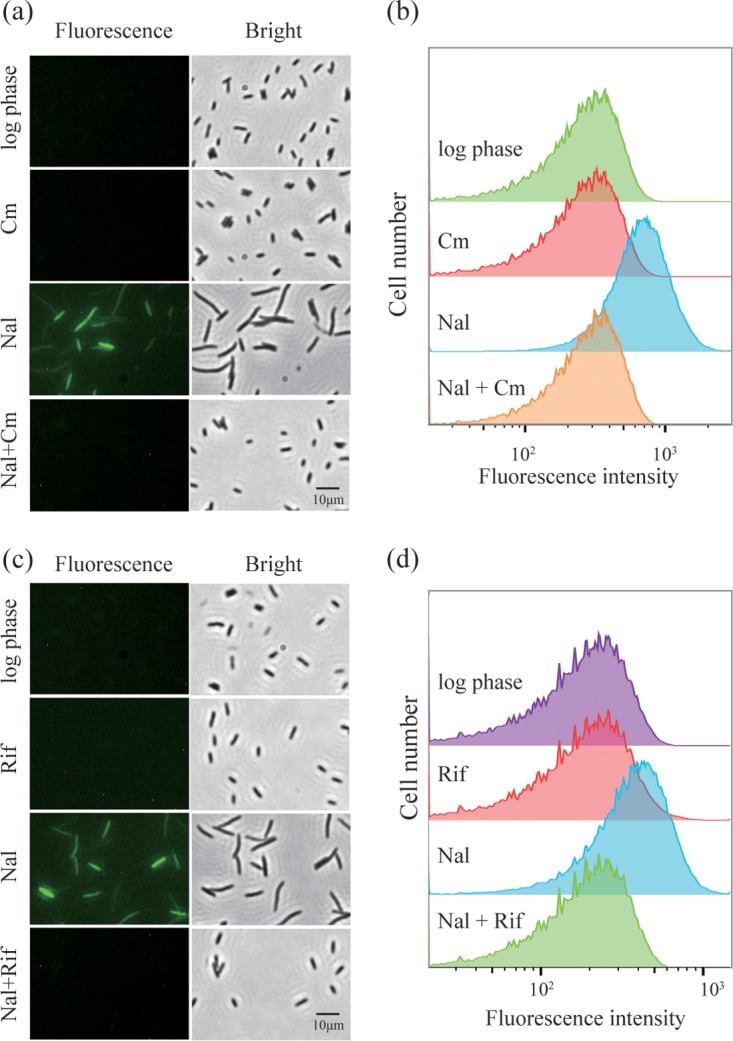
Chloramphenicol and rifampin block the increase in ROS associated with lethal nalidixic acid treatment. Exponentially growing cultures of wild-type E. coli (strain 3505) were treated with carboxy-H2DCFDA (10 μM) plus nalidixic acid (Nal, 200 μg/ml) for 2 h. Parallel samples were also treated with either chloramphenicol (Cm, 20 μg/ml, panels a and b) or rifampin (Rif, 50 μg/ml, panels c and d). Cells were then observed by fluorescence microscopy (a and c) or by flow cytometry (b and d). Representative results from three independent experiments are shown.
Since nalidixic acid itself inhibits protein synthesis at lethal concentrations (9), it had been suggested that inhibition of protein synthesis by the quinolone accounted for the loss of lethal action observed at very high concentrations (9). To quantify the effects of nalidixic acid, we measured the rate of protein synthesis after a 1-h treatment with the drug. As nalidixic acid concentration increased, protein synthesis rate decreased; at the maximal lethal concentration of nalidixic acid, protein synthesis rate dropped by 80% (Fig. 4). At very high, nonlethal nalidixic acid concentrations, the protein synthesis rate dropped by about 95%, similar to that observed with chloramphenicol at the concentration used to block ROS accumulation (20 μg/ml) (Fig. 3). The loss of killing at high quinolone concentrations correlates with extensive inhibition of protein synthesis (greater than 80%).
FIG 4.
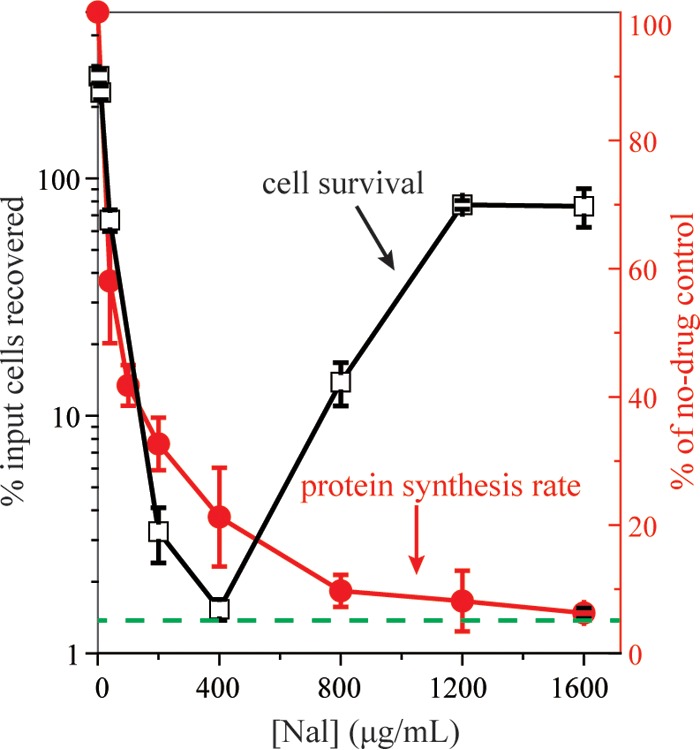
Effect of nalidixic acid concentration on survival and rate of protein synthesis. Exponentially growing cultures of wild-type cells (strain 3505) were treated with nalidixic acid (Nal) at the indicated concentrations for 1 h. Aliquots were diluted, plated, and incubated for determination of CFU and calculation of percent survival (empty squares). Rate of protein synthesis (filled circles) from a parallel culture was determined by pulse labeling with 35S-labeled methionine (1 μCi 35S-methionine per 200 μl culture) for 5 min followed by determination of acid-insoluble radioactivity. Inhibition of protein synthesis is expressed relative to the rate of a parallel untreated culture. Dashed line indicates inhibition of protein synthesis by 20 μg/ml chloramphenicol. Data are means from at least three independent experiments; error bars represent standard errors of the mean.
Suppression of ROS accumulation contributes to tolerance at high quinolone concentrations.
Since inhibition of protein synthesis suppresses respiration and ROS accumulation (30; Fig. 3), we next asked whether inhibition of protein synthesis contributes to high quinolone tolerance via suppression of ROS accumulation. To assess the interplay of ROS accumulation and inhibition of protein synthesis, we first examined the survival of a katG-deficient mutant following treatment with various concentrations of nalidixic acid. The product of katG, a catalase/peroxidase, detoxifies intracellular hydrogen peroxide; as expected, the katG mutation increased killing (reduced survival) during nalidixic acid treatment at low-to-moderate concentrations (Fig. 5a). Fluorescence microscopy revealed that treatment of wild-type cells produced an ROS signal in 73% of cells; this prevalence increased to 88% with the ΔkatG mutant (Fig. 5b shows typical microscopy fields). When ROS was measured by flow cytometry, the ΔkatG mutant exhibited higher levels of ROS (Fig. 5c). The elevated killing data, plus confirmation that ROS levels increase more in the ΔkatG mutant than in wild-type cells during nalidixic acid treatment, support the conclusion that ROS are involved in quinolone-mediated lethality. The katG-deficient mutant displayed paradoxical survival, with minimal survival occurring at a nalidixic acid concentration that was similar to that seen with wild-type cells (Fig. 5a). Thus, factors determining the maximal lethal concentration of nalidixic acid appear to be affected little by the catalase deficiency and elevated ROS levels.
FIG 5.

Paradoxical survival of a ΔkatG mutant following treatment with high concentrations of nalidixic acid. (a) Effect of a ΔkatG mutation on survival following nalidixic acid (Nal) treatment. Exponentially growing cultures of wild-type E. coli (strain 3505, squares) and a ΔkatG mutant (strain 3157, circles) were treated with nalidixic acid at the indicated concentrations for 2 h before samples were plated and incubated for determination of survival. (b) ROS determined by fluorescence microscopy. Exponentially growing cultures of wild-type E. coli (strain 3505) and a ΔkatG mutant (strain 3157) were treated with carboxy-H2DCFDA (10 μM) with or without nalidixic acid (Nal, 200 μg/ml) for 2 h, as indicated in the figure; they were then examined by fluorescence microscopy as described in Materials and Methods. Three independent experiments gave similar results. (c) ROS determined by flow cytometry. Samples were as in panel b. (d) Effect of chloramphenicol or a combination of 2,2′-bipyridyl plus thiourea on survival following nalidixic acid treatment. Exponentially growing cultures of a ΔkatG mutant (strain 3157) were treated with nalidixic acid alone (squares, Nal), nalidixic acid plus chloramphenicol (Cm) at 20 μg/ml (circles, Nal + Cm), or nalidixic acid plus 2,2′ bipyridyl and thiourea (BT) both at 1× MIC (triangles, Nal + BT at 1× MIC) or both at half MIC (diamonds, Nal + BT at 0.5× MIC) for 2 h at the indicated concentrations of nalidixic acid. For panels (a) and (d), percent survival was measured as described in Materials and Methods; data represent means from three independent experiments, and error bars represent standard errors of the mean. (e and f) ROS determined for cotreatment with chloramphenicol or thiourea plus bipyridyl. Exponentially growing cultures of a ΔkatG mutant (strain 3157) were treated with carboxy-H2DCFDA (10 μM) and nalidixic acid (Nal, 200 μg/ml), chloramphenicol (Cm, 20 μg/ml), 2,2′-bipyridyl plus thiourea (BT, both at 1× MIC), nalidixic acid plus chloramphenicol (Nal + Cm), or nalidixic acid plus 2,2′-bipyridyl and thiourea (Nal + BT, both at 1× MIC) for 2 h. Cells were observed by fluorescence microscopy (e) or flow cytometry (f), as described in Materials and Methods. Three independent experiments gave similar results.
When we examined the effect of chloramphenicol at high, growth-inhibiting concentration (20 μg/ml, 5× MIC) with the ΔkatG mutant, we found that this inhibitor of protein synthesis failed to block either the lethal action of nalidixic acid at moderate concentrations (Fig. 5d) or ROS accumulation (indicated by chloramphenicol having no effect on fluorescence intensity; Fig. 5e and f). Chloramphenicol inhibited protein synthesis by more than 90% with the ΔkatG mutant, indicating that the drug was active in this mutant. These data suggest that chloramphenicol, through its inhibition of protein synthesis, can keep nalidixic acid-induced ROS below a lethal threshold in wild-type but not in ΔkatG mutant cells. However, the effect of the ΔkatG mutation on ROS accumulation is inadequate to eliminate the tolerance seen at very high concentrations of nalidixic acid (see Discussion).
Treatment with a combination of 2,2′-bipyridyl plus thiourea (BT) at inhibitory concentration (1× MIC) blocked killing by nalidixic acid, in contrast to the killing seen with chloramphenicol plus nalidixic acid treatment of the ΔkatG mutant (Fig. 5d). Interference of nalidixic acid-mediated killing was also seen at a subinhibitory concentration (0.5× MIC) of the bipyridyl-thiourea combination, as the survival curve was shifted upward relative to that seen when chloramphenicol was present with nalidixic acid. 2,2′-Bipyridyl is an iron chelator that blocks the formation of hydroxyl radicals from hydrogen peroxide and also likely reduces respiration; thiourea is a scavenger of hydroxyl radicals. The distinct effects of bipyridyl plus thiourea (Fig. 5e and f) are likely due to direct suppression of ROS accumulation. Thus, a catalase deficiency, which is associated with elevated ROS levels, overcomes the inhibitory effects of protein synthesis inhibition by chloramphenicol, but not the inhibitory effects of bipyridyl plus thiourea. In conclusion, inhibition of protein synthesis per se is not sufficient to block nalidixic acid-mediated killing, whereas inhibition of ROS accumulation is.
In all but one experiment described above, the combination of bipyridyl plus thiourea was applied at sub-MIC levels, since in the absence of growth (stationary phase or resuspension of cells in saline), nalidixic acid is known to lack the ability to kill E. coli (27). But at these sub-MIC levels, the bipyridyl-thiourea combination does lower the growth rate (to about 1/3 the untreated level; see Fig. S1a in the supplemental material). To assess the effect of this growth rate reduction on killing, we reduced the growth rate by the same amount using chloramphenicol. The bipyridyl-thiourea combination exhibited a more striking inhibition of killing than did chloramphenicol (Fig. S1b). Thus, the bipyridyl-thiourea combination has inhibitory effects on nalidixic acid-mediated lethality that cannot be easily accounted for by reduction in the growth rate. As expected, measurement of ROS showed that the bipyridyl-thiourea combination restricted the accumulation of ROS more than did chloramphenicol (Fig. S1c).
We also assessed the effect of the bipyridyl-thiourea combination on protein synthesis. The bipyridyl-thiourea combination interfered with protein synthesis to an extent similar to that observed with nalidixic acid at 100 μg/ml (Fig. 6a), a concentration that was submaximal with respect to killing. The bipyridyl-thiourea combination failed to add to the inhibition of protein synthesis elicited by nalidixic acid alone (Fig. 6a); however, the bipyridyl-thiourea combination elevated survival during nalidixic acid treatment by 100-fold (Fig. 6b). These data support the idea that bipyridyl plus thiourea, at a sub-MIC level, protects from quinolone-mediated lethality by a means other than inhibiting protein synthesis.
FIG 6.
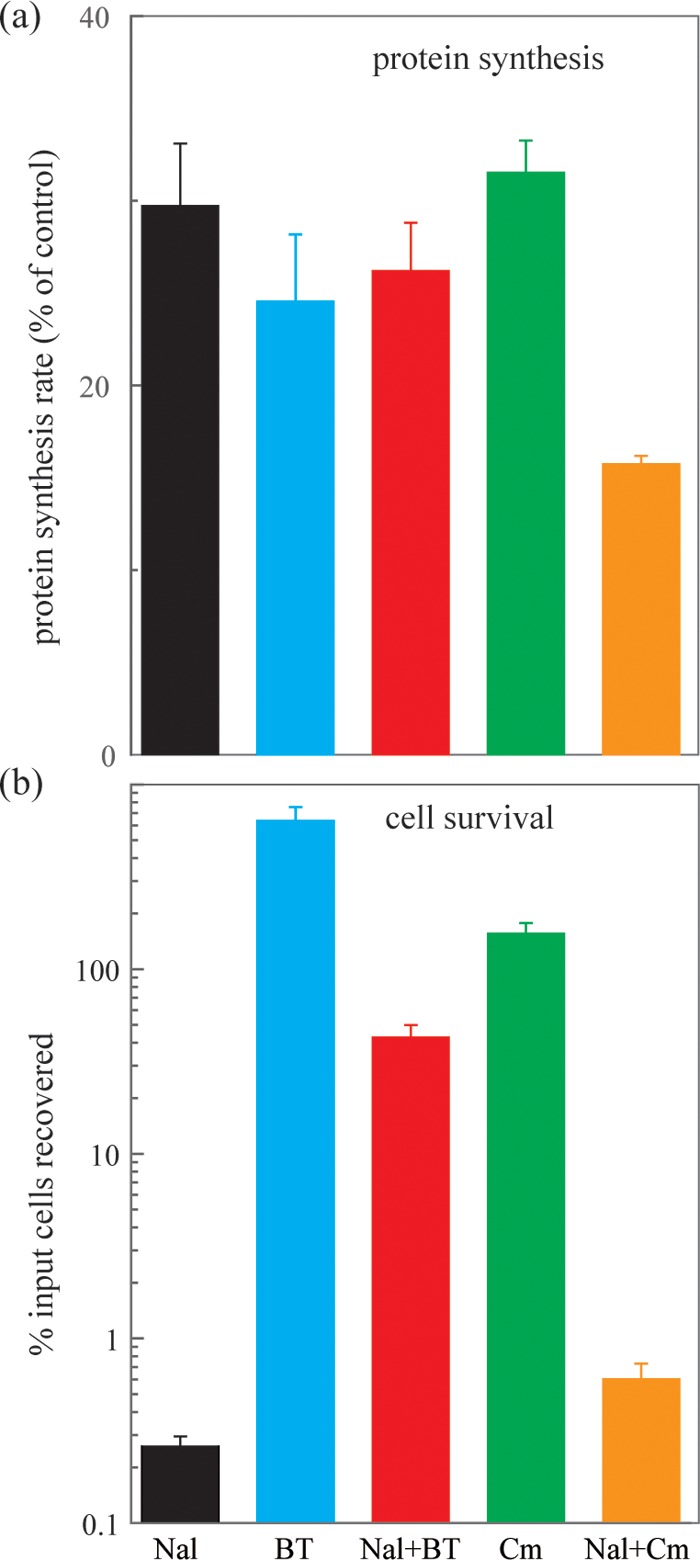
Nalidixic acid-mediated inhibition of protein synthesis and bacterial survival. (a) Inhibition of protein synthesis. Exponentially growing cultures of wild-type E. coli (strain 3505) were treated with the indicated inhibitors for 1 h, and rate of protein synthesis was measured as described in Materials and Methods. (b) Bacterial survival. Exponentially growing cultures of wild-type E. coli were treated with the indicated inhibitors for 3 h, and then survival was measured relative to an untreated control at the time of inhibitor addition. Nal, nalidixic acid, 100 μg/ml; BT, bipyridyl plus thiourea, both at half MIC; Cm, chloramphenicol, 2 μg/ml; Nal + BT, combination of nalidixic acid, 100 μg/ml, with BT, both at half MIC; Nal + Cm, combination of nalidixic acid 100, μg/ml, with chloramphenicol, 2 μg/ml. Data represent means from three independent experiments; error bars represent standard errors of the mean.
Chloramphenicol behaved differently from the bipyridyl-thiourea combination. When chloramphenicol was administered at 2 μg/ml, it inhibited protein synthesis by roughly the same amount as the bipyridyl-thiourea combination (Fig. 6a). But when chloramphenicol was combined with nalidixic acid, the protein synthesis rate dropped further (to about half), suggesting an additive effect of the two drugs (Fig. 6a). Moreover, the protective effect of chloramphenicol against killing by nalidixic acid was 100-fold less than that seen with bipyridyl plus thiourea (Fig. 6b), even though more inhibition of protein synthesis was seen with the former than the latter treatment. These data support the conclusion that the protective effect of the bipyridyl-thiourea combination differs from that of chloramphenicol, an inhibitor of protein synthesis. As pointed out above, that difference was expected to be due to direct suppression of ROS accumulation by the bipyridyl-thiourea combination rather than indirect suppression by chloramphenicol.
Collectively, the data indicate that protein synthesis is required for the accumulation of enough ROS for nalidixic acid-mediated lethality in wild-type cells. However, when a restriction on ROS accumulation is relaxed, as with a katG deficiency, an exogenous inhibitor of protein synthesis loses its ability to block killing and ROS accumulation. Agents that block ROS accumulation, such as a combination of bipyridyl and thiourea, inhibit killing by nalidixic acid beyond the effects they may have on growth rate or inhibition of protein synthesis.
Effect of exogenous hydrogen peroxide on quinolone lethality.
To further test ROS as contributors to quinolone-mediated killing, we treated wild-type E. coli cultures with various concentrations of nalidixic acid for 2 h and then added hydrogen peroxide for an additional 20 min at 2 mM. At this concentration, hydrogen peroxide by itself caused little killing during a 60-min incubation (data not shown; see also Fig. S2 in the supplemental material). Hydrogen peroxide (2 mM) modestly increased the killing observed with nalidixic acid for concentrations up to 200 μg/ml; however, at the elevated quinolone concentrations that exhibited tolerance, peroxide showed a striking lethal effect, as it eliminated paradoxical survival (Fig. 7). These data indicate that a quinolone-mediated substrate for ROS (peroxide)-mediated killing is present at very high, nonlethal concentrations of nalidixic acid. Chloramphenicol did not alter the ability of normally sublethal concentrations of exogenous peroxide to kill E. coli treated with nalidixic acid (Fig. S2 in the supplemental material). Thus, inhibition of protein synthesis is unlikely to perturb lesions created by nalidixic acid, but inhibition of protein synthesis reduces ROS to below a lethal threshold level at quinolone concentrations that are responsible for paradoxical survival.
FIG 7.
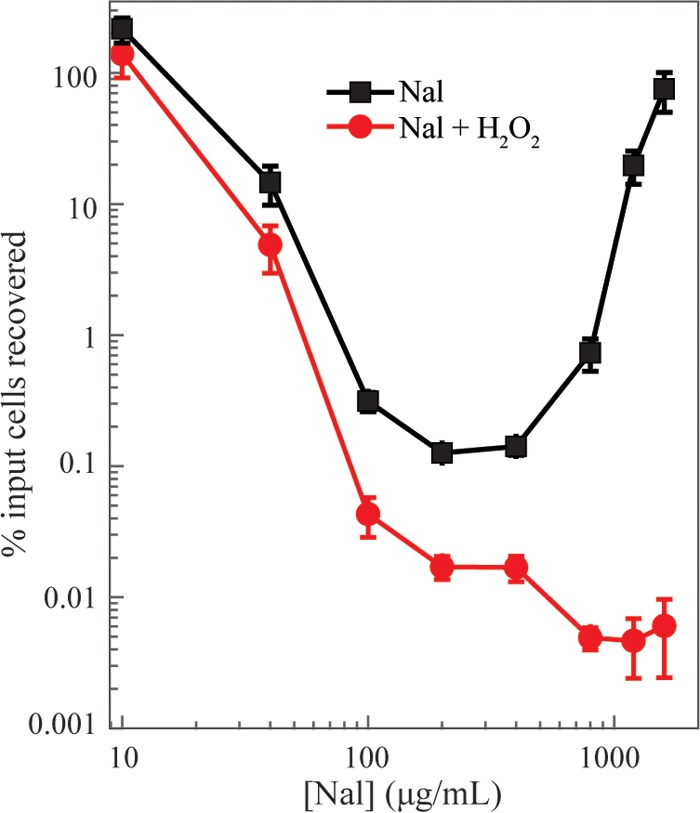
Exogenous ROS can eliminate nalidixic acid-mediated paradoxical survival. Exponentially growing cultures of wild-type E. coli (strain 3505) were treated for 2 h with the indicated concentrations of nalidixic acid (Nal), and survival was measured as described in Materials and Methods (squares). In a parallel culture, hydrogen peroxide (H2O2), at a sublethal final concentration of 2 mM, was added to cultures for an additional 20 min (circles). Data represent the means from three independent experiments; error bars represent standard errors of the mean.
Overall, restoring killing at high concentrations of nalidixic acid using exogenous peroxide supports the idea that high quinolone concentrations prevent ROS from exceeding a lethal threshold and that ROS are directly involved in killing by nalidixic acid.
DISCUSSION
The present work addressed two related issues: the underlying cause of paradoxical bacterial survival at very high concentrations of quinolone-class antibacterials and the contribution of reactive oxygen species to antibacterial lethality. When nalidixic acid concentration was increased, bacterial survival decreased, reached a minimum, and then increased. A reciprocal increase and decrease occurred with intracellular ROS, as detected by a fluorescent probe with individual cells and with bulk bacterial cultures (Fig. 1 and 2). Additional ROS-survival correlations were seen during cotreatment with chloramphenicol (Fig. 3), associated with a ΔkatG mutation (Fig. 5c), and during cotreatment with a combination of bipyridyl and thiourea (Fig. 5e and f). The katG deficiency overcame the protective effect of chloramphenicol (Fig. 5d), emphasizing the importance of ROS (at moderate, lethal concentrations of nalidixic acid, the katG-deficiency-mediated elevation of ROS must be greater than suppression of ROS associated with nalidixic acid-mediated inhibition of protein synthesis). Since exogenous peroxide eliminated paradoxical survival, it is likely that at high concentrations of nalidixic acid, peroxide-sensitive lesions persist (Fig. 7). Collectively, these data provide novel support for the idea that ROS contribute to the lethal action of quinolone-like compounds, extend that conclusion from studies focused on norfloxacin (1, 8, 26, 31), and offer a solution to the quinolone paradox (9, 23, 32–43).
Previous work suggests that lethal stress initiates a pathway involving several proteins (MazF, EF-4, Cpx, and Arc) (2, 3, 6, 44–46) that are protective when stress is moderate, but contribute to ROS accumulation and bacterial self-destruction when stress is severe (2, 3, 45, 46). Lesions from the primary stressor perturb respiration and lead to elevated levels of peroxide and superoxide (2, 3, 26), which can dismutate to peroxide. Peroxide can convert to highly toxic hydroxyl radicals via the Fenton reaction (47); these radicals, which can break DNA (20), are the likely cause of death (48). Variations probably occur among stressors, since thymineless death is unaffected by deficiencies in the protein factors that affect quinolone-mediated killing (48).
The ability of nalidixic acid to render low concentrations of peroxide lethal indicates that nalidixic acid creates peroxide-sensitive lesions in DNA. A comparable phenomenon occurs during thymineless death (48). In the latter case, the RecQ helicase increases the number of single-stranded DNA lesions, which then serve as the substrates for ROS attack. RecQ also increases the lethal action of nalidixic acid (more than 10-fold; data not shown). Thus, it is likely that an increase in single-stranded DNA accompanies nalidixic acid treatment.
Challenges to the ROS-lethal stress hypothesis (7, 8) raised a variety of issues, many of which were subsequently addressed experimentally (5). Previous work had shown that studies of norfloxacin require attention to concentration to distinguish ROS-dependent and ROS-independent modes of killing (25). When norfloxacin concentration was considered, the report by Keren et al., (8) confirmed that the behavior of norfloxacin at low concentration fits with an ROS contribution. Failure to observe anaerobic effects (7) likely resulted from use of a single concentration (25) and from unspecified differences between laboratories.
It has also been suggested that endogenous ROS levels are unlikely to be high enough to kill bacterial cells (31). However, we see that nalidixic acid creates primary lesions that serve as vulnerable substrates for ROS attack and thereby amplify the lethal action of ROS (Fig. 7). Examples of sensitization of DNA to ROS attack are also observed during thymineless death (48) and ultraviolet (UV) irradiation (Y. Hong and X. Zhao, unpublished observations). Stress-mediated ROS accumulation may also be self-amplifying, that is, the DNA damage caused by ROS could serve as an additional stress that leads to even higher levels of ROS once the systems that protect from oxidative damage are saturated. It is likely that the combination of stress-induced endogenous ROS and lesions derived from primary stress are adequate to kill bacteria. Overall, challenges to the ROS-lethality hypothesis have been largely answered.
An important question is whether the accumulation of ROS causes death or whether the two are simply associated. Causality is suggested by the following observation. When cells are treated with quinolone under lethal conditions and then plated on drug-free agar to measure survival, addition of an antioxidant (thiourea) to the agar revives cells thought to have been killed by quinolone treatment (2; unpublished observations). Thus, ROS are the cause rather than consequence of cell death. ROS persist even after removal of the primary stressor and kill cells in what appears to be a form of poststress bacterial programmed cell death (2).
Previous work leads to a scenario for how nalidixic acid limits its own lethality. At moderate concentrations, quinolone-containing cleaved complexes block RNA polymerase movement and interfere with RNA synthesis (9, 49, 50), perhaps contributing to the bacteriostatic action of the drugs. At high, lethal quinolone concentrations, chromosome fragmentation occurs (9, 22); DNA supercoiling, which is important for initiation of transcription (51), is lost (22). That would further reduce transcription, protein synthesis, and ROS production. Consequently, at very high quinolone concentrations, ROS production would be low. Since chromosome fragmentation associated with narrow-spectrum quinolone treatment is repaired during the long incubation required to score survival (X. Wang and X. Zhao, unpublished observations), low ROS levels allow repair and survival. Still unknown is how the Lon protease facilitates paradoxical survival (37).
The ROS-lethal stress hypothesis has at least two potential applications. One is that antioxidant consumption, in the form of food or nutritional supplements, may be counterproductive during antimicrobial therapy (52, 53). Another is the extension of the present work to potent fluoroquinolones. Nalidixic acid was chosen for the present work because it exhibits the most readily interpretable ROS-mediated effects of the gyrase poisons. However, high-concentration, paradoxical tolerance has been observed with a variety of bacterial species (Enterococcus faecalis, Escherichia coli, Mycobacterium bovis strain BCG, Mycobacterium smegmatis, Staphylococcus aureus, and Streptococcus pneumoniae) and for many quinolone and naphthyridone derivatives (9, 23, 32–43, 54). Thus, it appears to be a general feature of this antimicrobial class. Understanding and avoiding high-concentration tolerance with future derivatives may be important for obtaining the highly lethal agents needed to counter the induction of resistance. A next step is to find ways to enhance ROS accumulation during drug treatment.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and reagents.
E. coli K-12 strain BW25113 (3505) was used as the parental, wild-type strain. A ΔkatG mutant (3157) was constructed by antibiotic marker excision from strain 3137 (ΔkatG 729::kan, CGSC 10827) (26). E. coli cultures were grown aerobically at 37°C in LB liquid medium and as colonies on LB agar (55). 5(6)-Carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) was purchased from Thermo Fisher Scientific Corp. (Waltham, MA). Other reagents were obtained from Sigma-Aldrich Corp. (St. Louis, MO).
Fluorescence microscopy.
ROS levels in E. coli cells were assessed by fluorescence microscopy following treatment with carboxy-H2DCFDA. Exponentially growing cultures of E. coli (optical density at 600 nm [OD600] = 0.15) were treated with 10 μM carboxy-H2DCFDA and various concentrations of nalidixic acid for 2 h, avoiding ambient light by wrapping culture tubes with aluminum foil. Samples were removed, washed with 0.9% NaCl by centrifugation (9,000 × g for 40 s), and resuspended at the original volume in 0.9% NaCl. They were then concentrated 10-fold, also by centrifugation, and resuspended in 0.9% NaCl, followed by mixing at 40°C with one-third volume 1.5% low-melting-point agarose (FMC Corp., Philadelphia, PA). Samples were immediately spread on a microscope slide using a coverslip. Microscopy was performed with a Nikon Eclipse TS100 (Melville, NY) inverted microscope equipped with filters for excitation and emission of green fluorescent protein. Automated, unbiased image acquisition was carried out with NIS Elements BR imaging software (Nikon Corp., Melville, NY). A no-dye sample was included to control for autofluorescence.
Flow cytometry.
Exponentially growing cultures of E. coli were treated with various concentrations of nalidixic acid and 10 μM carboxy-H2DCFDA for 2 h. Samples were removed and washed with the same volume of 0.9% NaCl by centrifugation (9,000 × g for 40 s), and cells were resuspended in 0.9% NaCl. A no-dye sample was included to control for autofluorescence. A total of 100,000 ungated events for each sample was determined with a BD Accuri C6 Plus flow cytometer (BD Biosciences, San Jose, CA) using 20-mV laser power and a 533/30-nm bandpass filter (FL1-channel). Data were analyzed using BD Accuri C6 software.
Measurement of protein synthesis rate.
Exponentially growing cultures of E. coli were treated with various concentrations of nalidixic acid as indicated in the figures. Then aliquots (200-μl) were transferred to prewarmed tubes containing 1 μCi 35S-methionine (product no. NEG709A500UC; PerkinElmer) and incubated at 37°C for 5 min. Incorporation of the isotope was stopped by adding 1 ml ice-cold 10% (wt/vol) trichloroacetic acid to radioactively labeled samples on ice. Radioactive protein samples were collected on Whatman GF/A filters (24-mm diameter), and unincorporated radioactivity was removed by washing successively with 1N HCl, ice-cold water, and 95% ethanol. After drying, the filters were wetted with scintillation cocktail (Opti-Fluor O, product no. 6013339; PerkinElmer), and scintillation counting was performed with a Beckman LS6500 Multi-Purpose Scintillation counter (Beckman-Coulter, Fullerton, CA).
Statistical analysis.
All experiments were performed independently at least three times. Each value was presented as the mean value plus/minus standard error.
Supplementary Material
ACKNOWLEDGMENTS
We thank Marila Gennaro for critical comments on the manuscript.
Author contributions: G.L. conducted most of the experiments, Y.H. and G.L. jointly conducted flow cytometry and microscopy assays, and G.L., Y.H., K.D., and X.Z. designed the study, analyzed data, and wrote the manuscript.
This work was supported by the National Institutes of Health (grants AI073491 and DP2OD007423) and by the National Natural Science Foundation of China (grant 81473251). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We have no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01622-17.
REFERENCES
- 1.Kohanski M, Dwyer D, Hayete B, Lawrence C, Collins J. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 2.Dorsey-Oresto A, Lu T, Mosel M, Wang X, Salz T, Drlica K, Zhao X. 2013. YihE kinase is a novel regulator of programmed cell death in bacteria. Cell Reports 3:528–537. doi: 10.1016/j.celrep.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao X, Drlica K. 2014. Reactive oxygen species and the bacterial response to lethal stress. Current Opin Microbiol 21:1–6. doi: 10.1016/j.mib.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao X, Hong Y, Drlica K. 2015. Moving forward with ROS involvement in antimicrobial lethality. J Antimicrob Chemother 70:639–642. doi: 10.1093/jac/dku463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dwyer D, Belenky P, Yang J, MacDonald I, Martell J, Takahashi N, Chan C, Lobritz M, Braff D, Schwarz E, Ye J, Pati M, Vercruysse M, Ralifo P, Allison K, Khalil A, Ting A, Walker G, Collins J. 2014. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci U S A 111:E2100–E2109. doi: 10.1073/pnas.1401876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dwyer D, Collins J, Walker G. 2015. Unraveling the physiological complexities of antibiotic lethality. Annu Rev Pharmacol Toxicol 55:9.1-9.20. doi: 10.1146/annurev-pharmtox-010814-124712. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Imlay J. 2013. Cell death from antibiotics without the involvement of reactive oxygen species. Science 339:1210–1213. doi: 10.1126/science.1232751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keren I, Wu Y, Inocencio J, Mulcahy L, Lewis K. 2013. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339:1213–1216. doi: 10.1126/science.1232688. [DOI] [PubMed] [Google Scholar]
- 9.Crumplin GC, Smith JT. 1975. Nalidixic acid: an antibacterial paradox. Antimicrob Agents Chemother 8:251–261. doi: 10.1128/AAC.8.3.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drlica K, Mustaev A, Towle T, Luan G, Kerns R, Berger J. 2014. Bypassing fluoroquinolone resistance with quinazolinediones: studies of drug-gyrase-DNA complexes having implications for drug design. ACS Chem Biol 9:2895–2904. doi: 10.1021/cb500629k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malik M, Mustaev A, Schwanz H, Luan G, Shah N, Oppegard L, deSouza E, Hiasa H, Zhao X, Kerns R, Drlica K. 2016. Suppression of gyrase-mediated resistance by C7 aryl fluoroquinolones. Nucleic Acids Res 44:3304–3316. doi: 10.1093/nar/gkw161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bax B, Chan P, Eggleston D, Fosberry A, Gentry D, Gorrec F, Giordano I, Hann M, Hennessy A, Hibbs M, Huang J, Jones E, Jones J, Brown K, Lewis C, May E, Saunders M, Singh O, Spitzfaden C, Shen C, Shillings A, Theobald A, Wohlkonig A, Pearson N, Gwynn M. 2010. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466:935–940. doi: 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- 13.Chan P, Germe T, Bax B, Huang J, Thalji R, Bacqué E, Checchia A, Chen D, Cui H, Ding X, Ingraham K, McCloskey L, Raha K, Srikannathasan V, Maxwell A, Stavenger R. 2017. Thiophene antibacterials that allosterically stabilize DNA-cleavage complexes with DNA gyrase. Proc Natl Acad Sci U S A 114:E4492–E4500. doi: 10.1073/pnas.1700721114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dalhoff A. 2012. Resistance surveillance studies: a multifaceted problem—the fluoroquinolone example. Infection 40:239–262. doi: 10.1007/s15010-012-0257-2. [DOI] [PubMed] [Google Scholar]
- 15.Malik M, Hoatam G, Chavda K, Kerns R, Drlica K. 2010. Novel approach for comparing quinolones for emergence of resistant mutants during quinolone exposure. Antimicrob Agents Chemother 54:149–156. doi: 10.1128/AAC.01035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui J, Liu Y, Wang R, Tong W, Drlica K, Zhao X. 2006. The mutant selection window demonstrated in rabbits infected with Staphylococcus aureus. J Infect Dis 194:1601–1608. doi: 10.1086/508752. [DOI] [PubMed] [Google Scholar]
- 17.Xiong M, Wu X, Ye X, Zhang L, Zeng S, Huang Z, Wu Y, Sun J, Ding H. 2016. Relationship between cefquinome PK/PD parameters and emergence of resistance of Staphylococcus aureus in rabbit tissue-cage infection model. Front Microbiol 7:874. doi: 10.3389/fmicb.2016.00874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ni W, Song X, Cui J. 2014. Testing the mutant selection window hypothesis with Escherichia coli exposed to levofloxacin in a rabbit tissue cage infection model. Eur J Clin Microbiol Infect Dis 33:385–389. doi: 10.1007/s10096-013-1968-8. [DOI] [PubMed] [Google Scholar]
- 19.Zhang B, Gu X, Li Y, Li X, Gu M, Zhang N, Shen X, Ding H. 2014. In vivo evaluation of mutant selection window of cefquinome against Escherichia coli in piglet tissue-cage model. BMC Vet Res 10:297. doi: 10.1186/s12917-014-0297-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi S, Ueda K, Komano T. 1990. The effects of metal ions on the DNA damage induced by hydrogen peroxide. Agric Biol Chem 54:69–76. [PubMed] [Google Scholar]
- 21.Foti J, Devadoss B, Winkler J, Collins J, Walker G. 2012. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen C-R, Malik M, Snyder M, Drlica K. 1996. DNA gyrase and topoisomerase IV on the bacterial chromosome: quinolone-induced DNA cleavage. J Mol Biol 258:627–637. doi: 10.1006/jmbi.1996.0274. [DOI] [PubMed] [Google Scholar]
- 23.Malik M, Zhao X, Drlica K. 2006. Lethal fragmentation of bacterial chromosomes mediated by DNA gyrase and quinolones. Mol Microbiol 61:810–825. doi: 10.1111/j.1365-2958.2006.05275.x. [DOI] [PubMed] [Google Scholar]
- 24.Wang X, Zhao X, Malik M, Drlica K. 2010. Contribution of reactive oxygen species to pathways of quinolone-mediated bacterial cell death. J Antimicrob Chemother 65:520–524. doi: 10.1093/jac/dkp486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malik M, Hussain S, Drlica K. 2007. Effect of anaerobic growth on quinolone lethality with Escherichia coli. Antimicrob Agents Chemother 51:28–34. doi: 10.1128/AAC.00739-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Zhao X. 2009. Contribution of oxidative damage to antimicrobial lethality. Antimicrob Agents Chemother 53:1395–1402. doi: 10.1128/AAC.01087-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Howard BM, Pinney RJ, Smith JT. 1993. 4-Quinolone bactericidal mechanisms. Arzneimittelforschung/Drug Res 43:1125–1129. [PubMed] [Google Scholar]
- 28.Vowells SJ, Sekhsaria S, Malech HL, Shalit M, Fleisher TA. 1995. Flow cytometric analysis of the granulocyte respiratory burst: a comparison study of fluorescent probes. J Immunol Methods 178:89–97. doi: 10.1016/0022-1759(94)00247-T. [DOI] [PubMed] [Google Scholar]
- 29.Eruslanov E, Kusmartsev S. 2010. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol 594:57–72. doi: 10.1007/978-1-60761-411-1_4. [DOI] [PubMed] [Google Scholar]
- 30.Lobritz MA, Belenky P, Porter CBM, Gutierrez A, Yang JH, Schwarz EG, Dwyer DJ, Khalil AS, Collins JJ. 2015. Antibiotic efficacy is linked to bacterial cellular respiration. Proc Natl Acad Sci U S A 112:8173–8180. doi: 10.1073/pnas.1509743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imlay J. 2015. Diagnosing oxidative stress in bacteria: not as easy as you might think. Curr Opin Microbiol 24:124–131. doi: 10.1016/j.mib.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong Y, Xu C, Zhao X, Domagala J, Drlica K. 1998. Fluoroquinolone action against mycobacteria: effects of C8 substituents on bacterial growth, survival, and resistance. Antimicrob Agents Chemother 42:2978–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.George J, Morrissey I. 1997. The bactericidal activity of levofloxacin compared with ofloxacin, D-ofloxacin, ciprofloxacin, sparfloxacin and cefotaxime against Streptococcus pneumoniae. J Antimicrob Chemother 39:719–723. doi: 10.1093/jac/39.6.719. [DOI] [PubMed] [Google Scholar]
- 34.Lewin C, Morrissey I, Smith J. 1991. The mode of action of quinolones: the paradox in activity of low and high concentrations and activity in the anaerobic environment. Eur J Clin Microbiol Infect Dis 10:240–248. doi: 10.1007/BF01966996. [DOI] [PubMed] [Google Scholar]
- 35.Lu T, Zhao X, Li X, Drlica-Wagner A, Wang J-Y, Domagala J, Drlica K. 2001. Enhancement of fluoroquinolone activity by C-8 halogen and methoxy moieties: action against a gyrase resistance mutant of Mycobacterium smegmatis and a gyrase-topoisomerase IV double mutant of Staphylococcus aureus. Antimicrob Agents Chemother 45:2703–2709. doi: 10.1128/AAC.45.10.2703-2709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu T, Zhao X, Li X, Hansen G, Blondeau J, Drlica K. 2003. Effect of chloramphenicol, erythromycin, moxifloxacin, penicillin, and tetracycline concentration on the recovery of resistant mutants of Mycobacterium smegmatis and Staphylococcus aureus. J Antimicrob Chemother 52:61–64. doi: 10.1093/jac/dkg268. [DOI] [PubMed] [Google Scholar]
- 37.Malik M, Capecci J, Drlica K. 2009. Lon protease is essential for paradoxical survival of Escherichia coli when exposed to high concentrations of quinolone. Antimicrob Agents Chemother 53:3103–3105. doi: 10.1128/AAC.00019-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morrissey I, Clark S, Mathias I. 2000. The bactericidal activity of gemifloxacin (SB-265805). J Med Microbiol 49:841–844. doi: 10.1099/0022-1317-49-9-841. [DOI] [PubMed] [Google Scholar]
- 39.Morrissey I, George J. 2000. Bactericial activity of gemifloxacin and other quinolones against Streptococcus pneumoniae. J Antimicrob Chemother 45 Suppl S 1:107–110. doi: 10.1093/jac/45.suppl_3.107. [DOI] [PubMed] [Google Scholar]
- 40.Morrissey I, Smith J. 1996. Bactericidal activity of trovafloxacin (CP-99,219). J Antimicrob Chemother 38:1061–1066. doi: 10.1093/jac/38.6.1061. [DOI] [PubMed] [Google Scholar]
- 41.Morrissey I, Smith JT. 1995. Bactericidal activity of the new 4-quinolones DU-6859a and DV-7751a. J Med Microbiol 43:4–8. doi: 10.1099/00222615-43-1-4. [DOI] [PubMed] [Google Scholar]
- 42.Zhao X, Wang J-Y, Xu C, Dong Y, Zhou J, Domagala J, Drlica K. 1998. Killing of Staphylococcus aureus by C-8-methoxy fluoroquinolones. Antimicrob Agents Chemother 42:956–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao X, Xu C, Domagala J, Drlica K. 1997. DNA topoisomerase targets of the fluoroquinolones: a strategy for avoiding bacterial resistance. Proc Natl Acad Sci U S A 94:13991–13996. doi: 10.1073/pnas.94.25.13991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. 2008. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135:679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li L, Hong Y, Luan G, Mosel M, Malik M, Drlica K, Zhao X. 2014. Ribosomal elongation factor 4 promotes cell death associated with lethal stress. mBio 5:e01708. doi: 10.1128/mBio.01708-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu X, Wang X, Drlica K, Zhao X. 2011. A toxin-antitoxin module in Bacillus subtilis can both mitigate and amplify effects of lethal stress. PLoS One 6:e23909. doi: 10.1371/journal.pone.0023909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Repine JE, Fox RB, Berger EM. 1981. Hydrogen peroxide kills Staphylococcus aureus by reacting with staphylococcal iron to form hydroxyl radical. J Biol Chem 256:7094–7096. [PubMed] [Google Scholar]
- 48.Hong Y, Li L, Luan G, Drlica K, Zhao X. 2017. Contribution of reactive oxygen species to thymineless death in Escherichia coli. Nat Microbiol 2:1667–1675. doi: 10.1038/s41564-017-0037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manes SH, Pruss GJ, Drlica K. 1983. Inhibition of RNA synthesis by oxolinic acid is unrelated to average DNA supercoiling. J Bacteriol 155:420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willmott CJR, Critchlow SE, Eperon IC, Maxwell A. 1994. The complex of DNA gyrase and quinolone drugs with DNA forms a barrier to transcription by RNA polymerase. J Mol Biol 242:351–363. doi: 10.1006/jmbi.1994.1586. [DOI] [PubMed] [Google Scholar]
- 51.Gamper H, Hearst J. 1982. A topological model for transcription based on unwinding angle analysis of E. coli RNA polymerase binary, initiation, and ternary complexes. Cell 29:81–90. doi: 10.1016/0092-8674(82)90092-7. [DOI] [PubMed] [Google Scholar]
- 52.Marathe S, Kumar R, Ajitkumar P, Nagaraja V, Chakravortty D. 2013. Curcumin reduces the antimicrobial activity of ciprofloxacin against Salmonella Typhimurium and Salmonella Typhi. J Antimicrob Chemother 68:139–152. doi: 10.1093/jac/dks375. [DOI] [PubMed] [Google Scholar]
- 53.Liu Y, Zhou J, Qu Y, Yang X, Shi G, Wang X, Hong Y, Drlica K, Zhao X. 2016. Resveratrol antagonizes antimicrobial lethality and stimulates ecovery of bacterial mutants. PLoS One 11:e0153023. doi: 10.1371/journal.pone.0153023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lewin CS, Amyes SGB, Smith JT. 1989. Bactericidal activity of enoxacin and lomefloxacin against Escherichia coli KL16. Eur J Clin Microbiol Infect Dis 8:731–733. doi: 10.1007/BF01963763. [DOI] [PubMed] [Google Scholar]
- 55.Miller J. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.