

ABSTRACT
Polymyxin B (PB) has reemerged as a common treatment against multidrug-resistant Gram-negative pathogens. However, nephrotoxicity remains a significant dose-limiting side effect, and contemporary pharmacokinetic (PK) data are limited. This study sought to evaluate PB exposure differences in various loading and nonloading strategies according to total body weight (TBW) and adjusted body weight (ABW). Patients treated with PB had plasma samples obtained for clinical care and analyzed using liquid chromatography-tandem mass spectrometry. Compartmental PK models with linear and allometric scaling of TBW were explored. Semiparametric Monte Carlo simulation evaluated the total (i.e., protein bound plus unbound) area under the plasma concentration-time curve (AUCtotal) during the first 24 h of therapy and at 96 h posttherapy for each regimen at the 10th, 50th, and 90th percentiles of TBW and ABW in the derivation cohort. Literature-based values of the 24-h total AUC/MIC ratio (AUC/MICtotal) of ≥50 defined efficacy, and literature-based values of the 72- to 96-h AUCtotal of ≥100 μg · h/ml defined toxicity. Fifty-two patients contributed 156 PB plasma samples. A two-compartment model with allometric scaling of TBW produced a comparable fit (Akaike information criterion [AIC] = 376.7) to that achieved with linear scaling (AIC = 378). The regimen of a loading dose of 2.5 mg/kg of body weight plus a fixed dose of 100 mg every 12 h had the highest probability of achieving a 24-h AUC/MICtotal of ≥50 with the lowest probability of toxicity in all groups at 24 h, aside from those with the lowest 10th percentile of body weight. This is the first study to suggest that a weight-based loading and fixed maintenance (i.e., weight-independent) dosing strategy for polymyxin B may maximize efficacy while balancing toxicity concerns for most patients.
KEYWORDS: polymyxin B, pharmacokinetics, Gram-negative bacteria, Monte Carlo simulation
INTRODUCTION
The worldwide dissemination of multidrug resistance among important Gram-negative pathogens has led to a resurgence in the use of polymyxins as antibiotics of last resort. The two clinically used polymyxins, colistin and polymyxin B, both received marketing approval in the 1950s but fell out of favor following reports of severe nephrotoxicity and neurotoxicity (1–7). Increasing rates of resistance to beta-lactams, fluoroquinolones, and aminoglycosides among serious nosocomial pathogens, such as Pseudomonas aeruginosa, Acinetobacter baumannii, and Klebsiella pneumoniae, have prompted clinicians to revisit the clinical utility of these agents. Yet, uncertainty about the ideal dosing of polymyxins that avoids toxicity while maximizing efficacy for the treatment of infections caused by resistant Gram-negative pathogens has been a major barrier to the widespread use of these agents.
Despite the renewed interest in the polymyxins as a treatment for infections caused by resistant Gram-negative pathogens, optimal dosing strategies for these agents remain poorly defined. The polymyxins were approved prior to the requirement for rigorous pharmacokinetic (PK) data; thus, information in the package insert is sparse (8). Colistin, which is formulated as colistin methanesulfonate (CMS), was initially touted as the preferred polymyxin because there was more clinical experience with it (9, 10). However, CMS exhibits complex pharmacokinetics and variable bioconversion to colistin in vivo, which have led to difficulties in optimal dose selection (11, 12). On the other hand, polymyxin B appears to have more predictable pharmacokinetics (13, 14). In addition to inherent PK differences between the polymyxins, recent clinical data suggest that CMS may also be more nephrotoxic (15–18). Therefore, an improved understanding of the population PK of polymyxin B should yield important insights into effective and safer regimens for patients with serious Gram-negative bacterial infections. Adjustments to the polymyxin B dose for renal function and patient body weight are suggested in the package insert (8); however, it is not clear if total body weight (TBW) or an adjusted body weight (ABW) should be used, and it is unclear how a recommendation would change across the range of adult body weights. The objectives of the present study were to characterize the population PK of polymyxin B in acutely ill adults and compare the probabilities for safety and efficacy with alternate weight-based and fixed-dose polymyxin B dosing regimens using Monte Carlo simulation.
RESULTS
Patient characteristics.
A total of 52 patients contributed 156 plasma samples for polymyxin B concentration determination. Each patient contributed, on average, 3 clinical samples, of which 25% (n = 39) were samples with troughs, 15.4% (n = 24) were samples with peaks, and 59.6% (n = 93) were random samples. The 10th, 50th, and 90th percentiles were 50 kg, 75 kg, and 110 kg, respectively, for total body weight and 52 kg, 70 kg, and 85 kg, respectively, for adjusted body weight. Most patients were male (64%), and the median age was 47 years. The median weight of the population was 73 kg, with a range of 30 kg to 122 kg. The median calculated creatinine clearance (CLCR) was 68 ml/min, with a range of 16 to 389 ml/min. Thirty-one patients (60%) received a loading dose, and 21 patients (40%) did not. Among the patients who did receive a loading dose, the mean dose was 202.17 mg (range, 90 to 350 mg/day) or 2.79 mg/kg of body weight/day (range, 0.34 to 3.45 mg/kg/day). The mean maintenance dose was 180.88 mg/day (range, 20 to 360 mg/day) or 2.42 mg/kg/day (range, 0.34 to 3.45 mg/kg/day).
Pharmacokinetic models.
A two-compartment base model performed better than a one-compartment model (Akaike information criterion [AIC] and Bayes information criterion [BIC] of 376 and 391 versus 410 and 419, respectively); thus, a two-compartment model was used as the base model. Covariate analyses with the base model identified a weak relationship between (i) CLCR and apparent polymyxin B clearance and (ii) total body weight (TBW) and volume, with R2 values being 0.07 and 0.05, respectively (Fig. 1). In the models accounting for patient body weight, a two-compartment model with clearance allometrically scaled by a mean total body weight of 75 kg was found to be the most explanatory and parsimonious when it was compared to the linear scaled model (AIC, 376.7 versus 378; R2 values for observed versus predicted plot of the population, 0.507 versus 0.486; R2 values for Bayesian individual predictions, 0.889 versus 0.884). Predicted versus observed polymyxin B concentrations for the 52 patients are shown in Fig. 2. Bias and imprecision for the final allometrically scaled model were 0.232 μg/ml and 9.03 μg2/ml2, respectively, for the population model and −0.067 μg/ml and 0.774 μg2/ml2, respectively, for the individual Bayesian posterior model. Population and individual observed versus predicted polymyxin B concentrations for the model qualification cohort are shown in Fig. 3.
FIG 1.
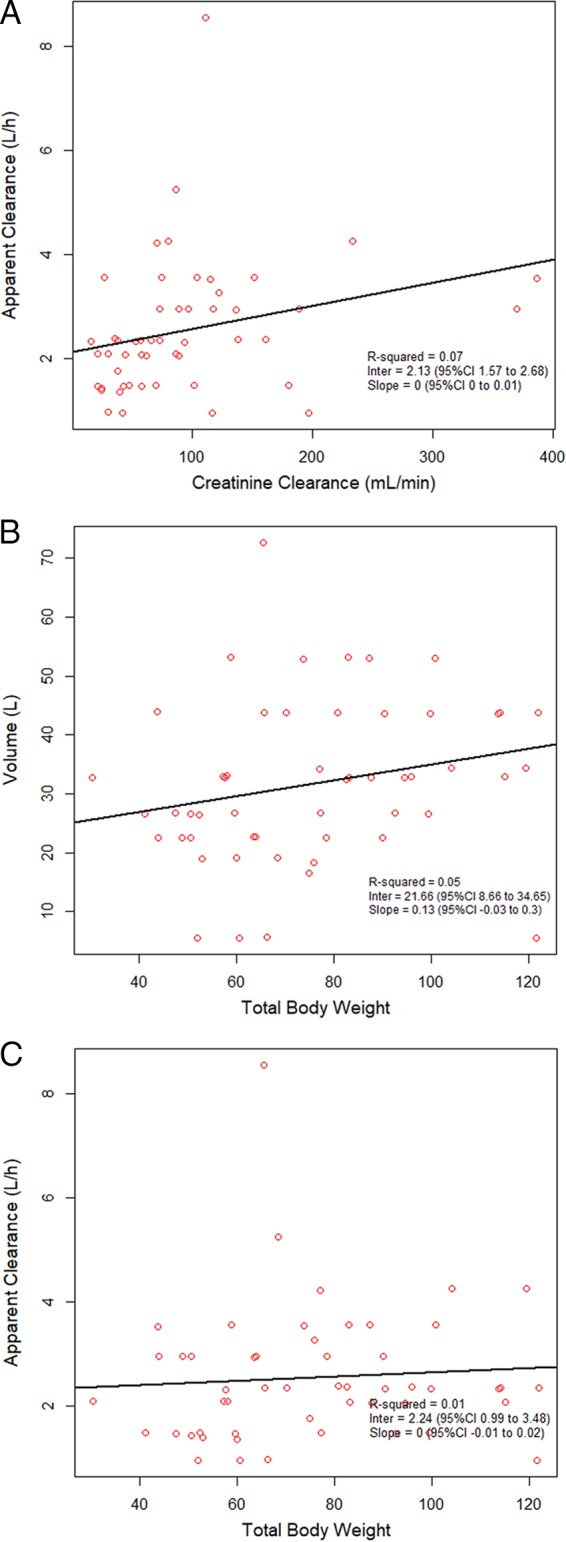
Covariate relationship using a two-compartment base model without scaling for weight of individual polymyxin B apparent clearance estimates versus creatinine clearance (A), individual total body weight versus the volume of distribution (B), and individual total body weight versus apparent clearance (C). CI, confidence interval.
FIG 2.

Individual and population fitted versus observed polymyxin B concentrations for a two-compartment allometrically scaled model in 52 acutely ill adult patients.
FIG 3.
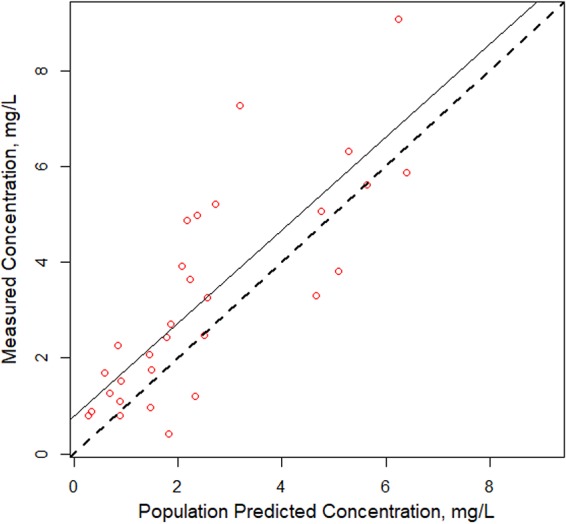
Individual and population fitted versus observed polymyxin B concentrations for a two-compartment allometrically scaled model in a 20% validation cohort.
The allometrically scaled two-compartment model identified 21 discrete support points. The final gamma value was 0.88. Parameter estimates and measures of central tendency are shown in Table 1. Patients had a mean estimated total polymyxin B total body clearance (CL) of 2.63 liters/h (standard deviation [SD], 1.41 liters/h) and a mean volume of distribution in the central compartment (Vc) of 33.77 liters (SD, 15.21 liters), according to the final population model (i.e., model 2).
TABLE 1.
Population PK parameter estimates, precisions of estimates (SD), and between-subject variability determined using the final two-compartment model with allometric scaling of TBWa
| PK parameter | Mean value | Median value | SD | Between-subject variability (% CV) |
|---|---|---|---|---|
| Vc (liters) | 33.77 | 31.49 | 15.21 | 45.03 |
| CL (liter/h) | 2.63 | 2.17 | 1.41 | 53.63 |
| Q (liter/h) | 2.32 | 2.30 | 1.33 | 57.41 |
| Vp (liters) | 78.20 | 100.00 | 37.46 | 47.90 |
Abbreviations: CV, coefficient of variation; Vc, volume of distribution in the central compartment; CL, total body clearance; Q, intercompartmental flow; Vp, volume of distribution in the peripheral compartment.
Model qualification.
Model qualification confirmed the robustness of the final population model and provided confidence that the original population estimates were not affected by subsets in the reported patient population. Parameter estimates and measures of central tendency were consistent within the full, 80% index, and 20% validation cohorts (Table 2). Estimates of the population pharmacokinetic parameters and the coefficient of variation (CV) in the index and validation cohorts were within ±0.25 SD of the full data set estimates for CL, intercompartmental flow (Q), and the volume of distribution in the peripheral compartment (Vp) and ±1.62 SD for the volume of distribution (V).
TABLE 2.
Population PK parameter estimates, precisions of estimates (SD), and between-subject variability for the full cohort, 80% index cohort, and 20% validation cohorta
| Population parameter estimate | Cohort | Mean value | SD | Between-subject variability (% CV) |
|---|---|---|---|---|
| Vc (liters) | Full | 33.71 | 15.21 | 45.03 |
| 80% index | 35.17 | 16.83 | 47.89 | |
| 20% validation | 35.12 | 16.83 | 47.86 | |
| CL (liters/h) | Full | 2.63 | 1.41 | 53.63 |
| 80% index | 2.62 | 1.48 | 54.37 | |
| 20% validation | 2.66 | 1.45 | 54.34 | |
| Q (liters/h) | Full | 2.32 | 1.33 | 57.41 |
| 80% index | 2.35 | 1.55 | 65.95 | |
| 20% validation | 2.35 | 1.55 | 65.95 | |
| Vp (liter) | Full | 78.21 | 37.46 | 47.90 |
| 80% index | 74.14 | 38.44 | 51.89 | |
| 20% validation | 74.13 | 38.44 | 51.86 |
Abbreviations: CV, coefficient of variation; Vc, volume of distribution in the central compartment; CL, total body clearance; Q, intercompartmental flow; Vp, volume of distribution in the peripheral compartment.
Simulations and PTA across MICs.
Results of analyses of the probability of target attainment (PTA) obtained using the final allometrically scaled model are shown in Table 3. When simulated concentration-time profiles were compared using a total goal of an area under the concentration-time curve (AUC) of 50 mg · h/liter for the first 24 h, all regimens aside from the regimen of 1.0 mg/kg every 12 h (q12h) achieved a PTA of ≥90% at an MIC of ≤0.5 mg/liter in all weight groups.
TABLE 3.
PTA for fixed and weight-based polymyxin B regimens according to the 10th, 50th, and 90th percentiles of TBW and ABWa
| Dosing regimen | Patient wt (kg) | PTA at the following MIC (mg/liter): |
||||||
|---|---|---|---|---|---|---|---|---|
| 0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | ||
| 100 mg q12h | 50 | 1 | 1 | 1 | 0.391 | 0.02 | 0.002 | 0 |
| 75 | 1 | 1 | 0.9992 | 0.247 | 0.011 | 0.001 | 0 | |
| 110 | 1 | 1 | 0.943 | 0.165 | 0.008 | 0 | 0 | |
| 1 mg/kg TBW q12h | 50 | 1 | 1 | 0.391 | 0.02 | 0.002 | 0 | 0 |
| 75 | 1 | 1 | 0.821 | 0.077 | 0.003 | 0 | 0 | |
| 110 | 1 | 1 | 0.976 | 0.244 | 0.0015 | 0.001 | 0 | |
| 1 mg/kg ABW q12h | 52 | 1 | 1 | 0.423 | 0.02 | 0.002 | 0 | 0 |
| 70 | 1 | 1 | 0.754 | 0.056 | 0.003 | 0 | 0 | |
| 85 | 1 | 1 | 0.901 | 0.112 | 0.003 | 0 | 0 | |
| 1.5 mg/kg TBW q12h | 50 | 1 | 1 | 0.935 | 0.107 | 0.003 | 0 | 0 |
| 75 | 1 | 1 | 0.997 | 0.402 | 0.021 | 0.001 | 0 | |
| 110 | 1 | 1 | 1 | 0.747 | 0.076 | 0.003 | 0 | |
| 1.5 mg/kg ABW q12h | 52 | 1 | 1 | 0.947 | 0.119 | 0.003 | 0 | 0 |
| 70 | 1 | 1 | 0.996 | 0.324 | 0.0017 | 0.001 | 0 | |
| 85 | 1 | 1 | 1 | 0.535 | 0.034 | 0.002 | 0 | |
| 2.5-mg/kg loading dose + 100 mg q12h | 50 | 1 | 1 | 1 | 0.613 | 0.031 | 0.002 | 0 |
| 75 | 1 | 1 | 1 | 0.813 | 0.082 | 0.003 | 0 | |
| 110 | 1 | 1 | 1 | 0.931 | 0.168 | 0.009 | 0 | |
| 2.5-mg/kg loading dose + 1.5 mg/kg TBW q12h | 50 | 1 | 1 | 1 | 0.45 | 0.02 | 0.002 | 0 |
| 75 | 1 | 1 | 1 | 0.855 | 0.09 | 0.003 | 0 | |
| 110 | 1 | 1 | 1 | 0.982 | 0.268 | 0.015 | 0.001 | |
| 2.5-mg/kg loading dose + 1.5 mg/kg ABW q12h | 52 | 1 | 1 | 1 | 0.502 | 0.025 | 0.002 | 0 |
| 70 | 1 | 1 | 1 | 0.804 | 0.075 | 0.003 | 0 | |
| 85 | 1 | 1 | 1 | 0.872 | 0.107 | 0.003 | 0 | |
The target was achievement of an AUC/MIC of ≥50 during the first 24 h of therapy.
A loading dose improved the probability of target attainment for all regimens in all weight strata. However, despite receipt of a loading dose, patients in the lowest body weight stratum were far from achieving a PTA of ≥90%. These patients achieved target attainment probabilities of only 0.613, 0.45, and 0.502 for the loading dose plus a fixed dose, a loading dose plus a TBW-based dose, and a loading dose plus an ABW-based dose, respectively, at an MIC of 1.0 mg/liter. None of the simulated regimens achieved adequate target attainment at MICs at or above the current CLSI and EUCAST breakpoint of 2 mg/liter.
In addition to analysis of the PTA, the probability of efficacious and toxic polymyxin B exposures associated with each of the simulated regimens was obtained from the Monte Carlo simulations (Table 4). The highest efficacy was seen in the regimens with loading doses, with comparable probabilities being achieved in all three groups among patients with middle to high weights. Among these regimens, the strategy of a loading dose plus a fixed maintenance dose yielded the best balance between efficacy and toxicity in patients with average to above-average weights. However, the regimens with loading doses still failed to produce probabilities of efficacious exposures in patients with body weights in the lowest 10th percentile, with rates of only 43%, 47.7%, and 58.2% in the regimens with a loading dose plus a TBW-based dose, a loading dose plus an ABW-based dose, and a loading dose plus a fixed dose, respectively.
TABLE 4.
Probability of efficacious and toxic exposures predicted by Monte Carlo simulation for each simulated regimen at the 10th, 50th, and 90th percentiles of TBW and ABWa
| Dosing regimen | Patient wt | Probability of efficacyb (%) | Probability of toxicityc (%) |
|---|---|---|---|
| 100 mg q12h | 50 kg TBW | 37.1 | 36.4 |
| 75 kg TBW | 23.6 | 22.6 | |
| 110 kg TBW | 15.7 | 14.6 | |
| 1.0 mg/kg q12h | 50 kg TBW | 1.8 | 5.4 |
| 75 kg TBW | 7.4 | 11.1 | |
| 110 kg TBW | 22.9 | 18.7 | |
| 52 kg ABW | 1.8 | 5.9 | |
| 70 kg ABW | 5.3 | 10 | |
| 85 kg ABW | 10.9 | 13.5 | |
| 1.5 mg/kg q12h | 50 kg TBW | 10.4 | 18.2 |
| 75 kg TBW | 38.1 | 29.6 | |
| 110 kg TBW | 67.1 | 42.3 | |
| 52 kg ABW | 11.6 | 16.9 | |
| 70 kg ABW | 30.7 | 27.5 | |
| 85 kg ABW | 50.1 | 33.4 | |
| 2.5 mg/kg + 100 mg q12h | 50 kg TBW | 58.2 | 37.2 |
| 75 kg TBW | 73.1 | 24.8 | |
| 110 kg TBW | 76.3 | 17.6 | |
| 2.5 mg/kg + 1.5 mg/kg q12h | 50 kg TBW | 43 | 20.0 |
| 75 kg TBW | 76.5 | 31.0 | |
| 110 kg TBW | 71.4 | 43.4 | |
| 52 kg ABW | 47.7 | 20.9 | |
| 70 kg ABW | 72.9 | 29.4 | |
| 85 kg ABW | 76.5 | 23.8 |
Abbreviations: TBW, total body weight; ABW, adjusted body weight.
Efficacy was a total AUC0–24 of ≥50 and <100 μg · h/ml with the assumption that the MIC was 1 mg/liter.
Toxicity was a total AUC72–96 of ≥100 mg · h/liter.
DISCUSSION
The increase in the rates of antimicrobial resistance among Gram-negative bacilli has led to the clinical need for polymyxin B; however, population pharmacokinetic data for this agent remain scarce. Polymyxin B has a narrow therapeutic window and is known for its propensity to cause dose-limiting nephrotoxicity. Despite this renewed interest, the plasma concentrations associated with nephrotoxicity remain ill defined, and optimal exposures that balance efficacy and toxicity have yet to be elucidated.
To date, there have been limited population pharmacokinetic studies of polymyxin B (13, 19, 20). Contemporary dosing strategies are largely based on population pharmacokinetic data presented by Sandri et al. suggesting that a weight-based strategy of 2.5 to 3 mg/kg/day, using total body weight, is needed to achieve efficacious exposures. That study also found that creatinine clearance did not significantly influence the clearance of polymyxin B and definitively stated that dosing should not be adjusted in the setting of renal impairment (13). While that study is seminal to contemporary dosing strategies, it did not evaluate exposures associated with fixed-dose regimens or with weight classifications other than total body weight. Further, one could argue that the analysis was constrained, as patients at the extremes of body weight were not evaluated, as only two patients included in the analysis were at the large extremes of weight (41 kg and 250 kg) (21).
The present study utilized nonparametric, compartment pharmacokinetic modeling and simulation strategies to estimate population pharmacokinetic parameters and quantify the range of possible exposures associated with various fixed and weight-based doses of polymyxin B. To allow increased adoption for clinical application, we also completed a one-compartment parametric analysis using Monolix 2016R1 software (Lixoft, Orsay, France). The full analysis of this model, along with the Simulx code that allows clinicians to use patient-specific data to determine the population-derived estimates for exposure, can be found in our companion paper (22). A likely explanation for the difference in model selection between the analysis with Monolix software and the analysis with the Pmetrics package is that the latter captured the observed bimode in peripheral volume to a better extent (data not shown). Thus, it was felt that the nonparametric analysis (i.e., that with the Pmetrics package) was the better analysis for simulation, and the parametric analysis remains useful for the creation of clinical population dosing parameters.
This analysis, which used a two-compartment allometrically scaled pharmacokinetic model, adequately described the serum concentrations of polymyxin B in 52 patients across a broad weight distribution. We identified one potential outlier in the clinical cohort during evaluation of each patient's individual pharmacokinetic profile. This particular profile suggested that the patient may have received a dose without subsequent documentation, though we opted to keep this patient in the analysis. Model fits were better without this patient (data not shown); however, we were unable to verify the error in order to warrant exclusion in the final model.
This analysis did not identify a significant relationship between TBW and clearance, suggesting that weight may not modify pharmacokinetic parameters, as previously reported (13). Additionally, models that allowed the allometric exponent for weight adjustment to vary did not produce better fits to the data (data not shown); thus, they were not used. Despite the independence of weight from volume, we forced weight into our model to allow simulation strategies to be used to better understand the variability in exposures associated with higher doses (as polymyxin B is presently dosed on the basis of body weight). Ultimately, an allometric weight-scaled model was used for all patients in our cohort. Notably, weight adjustment is the most common contemporary dosing strategy used in practice and the FDA-approved dosing strategy (8). The inclusion of weight in our model allowed us to simulate and understand the variability in exposures with increasing doses; however, it was unable to convincingly answer the question about whether any one weight-based dosing scheme should be used. To this end, our results indicate that dosing on the basis of total body weight is not likely a good strategy. For efficacy, the use of higher doses should be considered up front, but maintenance doses should be independent of weight (i.e., fixed dosing should be used). Future studies will need to be designed specifically to address the exact strategies that should be utilized.
To facilitate a comparison between our final parameter estimates and those previously published by Sandri et al. (13), our data were also similarly fit to a weight-based, linearly scaled, two-compartment model. Notably, our estimate for clearance was similar to that of Sandri et al. (13) (2.45 liters/h and 1.87 liters/h, respectively) but volume and flow parameters were not (Vc = 34.13 liters and 6.35 liters, respectively; Vp = 99.99 liters and 22.3 liters, respectively; Q = 1.60 liters/h and 9.86 liters/h, respectively). This highlights the need for further investigation to better define the peripheral distribution of polymyxin B.
We also assessed the relationship between CLCR and apparent polymyxin B clearance. A general lack of a significant linear relationship between CLCR and polymyxin B clearance was found in this patient population (R2 = 0.07), similar to the findings of other studies (13). Future work should quantify the full relationship between measured creatinine clearance and polymyxin B clearance.
We believe that this study is the first to simulate fixed and weight-based regimens using total and adjusted body weight. In this analysis, none of the simulated regimens could achieve a ≥90% PTA at the current CLSI or EUCAST breakpoint of ≤2 mg/liter, and a loading dose was needed to achieve this goal with an MIC of ≤1 mg/liter in most patients, with the exceptions being those in the lowest weight stratum. While the commonly used weight-based regimen of 1.5 mg/kg q12h was able to achieve efficacious AUCs at MICs of ≤0.5 mg/liter, a loading dose is likely warranted in serious infections, as MICs are often not known prior to initiation of the first dose. All values of AUC during the first 24 h of therapy (AUC24) for strategies of a loading dose plus a fixed dose and a loading dose plus a weight-based dose were above the proposed therapeutic target of ≥50 mg · h/liter. Thus, determination of optimal dosing strategies should be based on the balance between the comparative propensity of each regimen to cause toxicity at steady state and the need for early efficacious exposures (i.e., exposures efficacious in the first 24 h). A regimen of a loading dose plus a weight-based dose produced a total AUC (AUCtotal) above the proposed toxicity threshold of 100 mg · h/liter in patients with total body weights in the 50th and 90th percentiles. When adjusted body weights were used in the simulations for this regimen, the AUCs for each weight stratum indicated that the efficacies were similar to those based on total body weight and were associated with a probability of toxicity lower than that obtained in the simulations based on total body weight.
To our knowledge, this is the first study to suggest that total body weight-based dosing strategies may not be the most optimal in producing polymyxin B exposures that best balance efficacy and toxicity. Our results suggest that the use of higher doses should be considered up front but maintenance doses should be weight independent (i.e., fixed dosing should be used); however, we recognize that our results are unable to convincingly answer the question about whether any one weight-based dosing scheme should be used. Future studies will need to be designed specifically to address the exact strategies that should be utilized.
Several data suggest the need to cap doses on the basis of excess patient body weight. Weight-based dosing strategies have been associated with increased toxicity in higher-weight patients (23, 24). This study is the first to simulate a fixed-dose regimen of 100 mg q12h with and without a weight-based loading dose. The regimen of a loading dose plus a fixed dose achieved an efficacious AUC24 in most weight groups simulated, but it was associated with toxicity in patients with total body weights in the lower 10th percentile at steady state (within the 75th percentile of the simulated AUCs). When PTA values were compared, the regimen of a loading dose plus a fixed dose produced values higher than those achieved with the regimens of a loading dose plus a weight-based dose for both total and adjusted body weight in the low-weight stratum, with values of 0.613, 0.45, and 0.502, respectively.
Our study has several limitations. First, we studied acutely ill adult patients with all the known potentials for study variation. Plasma samples were obtained as part of clinical care, and both rich and sparse sampling schedules that differed between patients were included. However, the patient population used in this study most closely matches patient populations that need polymyxin B as a therapeutic agent. Second, the efficacy and toxicity thresholds have not been rigorously analyzed and were not confirmed with clinical outcomes in this study. Clinical outcome studies are warranted. Further, the exposures presented in this study are based on categorical weight distributions and cannot be definitively translated to all patients.
Conclusion.
To our knowledge, this is the first study to suggest that a strategy involving a combination of a weight-based loading dose with a fixed maintenance dose for polymyxin B may produce efficacious results while balancing toxicity concerns. Notably in the first 24 h, efficacious exposures are generally not achievable without risking toxic exposures when MICs are ≥1 mg/liter. Our data also suggest that if weight-based strategies are used, calculations should be based on the adjusted body weight rather than the total body weight. Ultimately, clinical care requires the balancing of efficacious exposures that also result in the lowest realized toxicity. The severity and the urgency of the infection need to dictate the relative importance for targeting any given exposure. Real-time drug assays are needed to help clinicians understand and achieve this balance.
MATERIALS AND METHODS
Polymyxin B administration and sample collection.
Patients admitted to New York-Presbyterian Hospital between January 2009 and December 2015 were considered for inclusion. Polymyxin B was administered in the setting of a suspected or documented Gram-negative bacterial infection. The dose, duration of use, and use of polymyxin B and other antimicrobials were determined by the treating primary medical team on the basis of institutional dosing guidelines. Methodological details and the institutional dosing protocol can be found in our companion paper (22). Patients received therapeutic drug monitoring of intravenous polymyxin B as part of their clinical care. The study was approved by the Columbia University Irving Medical Center Institutional Review Board with a waiver for informed consent and by Midwestern University for deidentified data analysis.
The demographic information collected at the baseline included the following: age, sex, weight, the serum creatinine concentration prior to polymyxin B therapy, and details of intravenous polymyxin B therapy, including the timing of every dose and sampling of blood for plasma assay. Creatinine clearance was estimated using the Cockcroft-Gault equation and the actual or adjusted body weight. Adjusted body weight was calculated as ideal body weight + 0.4(actual body weight − ideal body weight) (25). Patients receiving any form of renal replacement therapy or treatment via any other extracorporeal device (e.g., extracorporeal membrane oxygenation) at the start of polymyxin B treatment or at the time of sampling as well as any patient with cystic fibrosis were excluded from the analysis. Polymyxin B concentrations (i.e., the polymyxin B1 concentration plus the polymyxin B2 concentration) were analyzed using a validated high-performance liquid chromatography-mass spectrometry method. As previously reported (26), the assay was linear between 100 and 2,500 ng/ml. For polymyxin B1 and polymyxin B2, the interday precisions were 5.9% and 3.4%, respectively, at 100 ng/ml and 5.3% and 4.0%, respectively, at 2,000 ng/ml. For polymyxin B1 and polymyxin B2, accuracies were 80.2% and 82.2%, respectively, at 100 ng/ml and 99.9% and 109.6%, respectively, at 2,000 ng/ml (26).
Pharmacokinetic modeling procedures.
Model fitting was performed using the nonparametric adaptive grid (NPAG) algorithm within the Pmetrics (version 1.4.0) package (Laboratory of Applied Pharmacokinetics and Bioinformatics, Los Angeles, CA) for R (R Foundation for Statistical Computing, Vienna, Austria) (27). Pmetrics was used to create nonparametric mixed-effects models with model parameters described as a set of discrete support points and the corresponding probability for each vector of values. One- and two-compartment models were considered; the Akaike information criterion (AIC) and Bayesian information criterion (BIC) were used to set the base model. The covariates collected during the initial chart review included total body weight (TBW), height, sex, age, and serum creatinine and albumin concentrations. Ideal body weight and creatinine clearance were calculated for each patient. The relationship between relevant covariates (TBW, ideal body weight, and CLCR) and PK model parameters were visualized through scatter plots and evaluated using stepwise linear regressions. Relationships with R2 values of >0.4 were further explored. Covariate relationships between polymyxin B clearance and (i) TBW and (ii) creatinine clearance (CLCR) were assessed a priori. Because polymyxin B dosing is currently weight based according to the package insert (8), the covariate of TBW was forced into all models. Additionally, the effect of CLCR on polymyxin B CL was explored since adjustments to the dose according to renal function are suggested by the package insert (8).
The goodness of fit for the covariate-adjusted models was evaluated by visual inspection of the population predicted versus observed-predicted plots, visual inspection of the individual weighted residuals, and the interindividual variability for each population pharmacokinetic parameter. Predictive performance evaluation was based on bias and imprecision for the population. The best-fit model was determined by the rule of parsimony and AIC. Stepwise changes in objective function and interindividual variability terms can be found in Table 5. The individual fitted pharmacokinetic profiles obtained using the final model were visually assessed for each patient in the cohort.
TABLE 5.
Changes in objective function values and interindividual variability resulting from stepwise covariate model buildinga
| Compartment | Parameters | −2LL | AIC | BIC | Population bias | Population imprecision | Bayesian bias | Bayesian imprecision |
|---|---|---|---|---|---|---|---|---|
| 1 | Vc, CL | 404 | 410.1 | 419.1 | 0.32909 | 7.34 | −0.14382 | 0.7396 |
| 2 | Vc, CL, Q, Vp | 365.3 | 375.7 | 390.6 | 0.07646 | 7.56 | −0.1559 | 0.7747 |
| 2 | Vc, CL · (TBW/75)0.75, Q, Vp | 366.3 | 376.7 | 391.6 | 0.23222 | 9.033 | −0.06711 | 0.7742 |
| 2 | Vc, CL · (TBW/75), Q, Vp | 367.6 | 378 | 392.8 | 0.21602 | 9.929 | −0.1415 | 0.8196 |
Abbreviations: −2LL, −2 log likelihood; AIC, Akaike information criterion; BIC, Bayes information criterion; Vc, volume of distribution in the central compartment; CL, total body clearance; Q, intercompartmental flow; Vp, volume of distribution in the peripheral compartment; TBW, total body weight.
Two covariate-adjusted structural PK models were assessed wherein polymyxin B clearance was modified by TBW. For each two-compartment PK model, model parameters included total clearance (CLT), the volumes of distribution in the central and peripheral compartments (Vc and Vp, respectively), and intercompartmental clearance (Q). Mass transit for compartment 1 (X1) and compartment 2 (X2) [(dX1/dt) and (dX1/dt), respectively, where t is time] for the two-compartment clearance model was described using the following differential equations (where Rate IV is the model input, or rate of the intravenous administration):
| (1) |
| (2) |
Model 1 standardized clearance to TBW relative to a 75-kg referent weight as follows:
| (3) |
where CL0 is the baseline clearance.
Model 2 also standardized clearance to TBW but added an allometric scaling factor of 0.75 as follows:
| (4) |
Assay error (standard deviation [SD]) was accounted for using an error polynomial as a function of the measured concentration, Y (i.e., SD = C0 + C1Y). Coefficient values (C0 and C1) were empirically set to 0.1 and 0.15, respectively. The inverse of the estimated assay variance (SD2) was used as the first estimate for weighting in the PK modeling. Final weighting was accomplished using the multiplicative gamma value [i.e., error = (SD × gamma value)] to capture both the experimental observation variance and process noise, with the initial gamma value being set at 5 and the final value being estimated by the NPAG algorithm.
The final PK model was internally qualified using a data-splitting procedure consistent with FDA guidance (28). Model predictions were tested against a randomly withheld subset (random subject selection was done using Stata software [version 14; Stata Corp., College Station, TX]). First, 80% of the full study cohort was randomly selected as the index data set to be used for PK model development and selection. After the final PK model fitting was completed using the index data set, the Bayesian priors for the PK parameters (from the 80% cohort) were utilized to generate Bayesian posterior predictions (for the 20% validation cohort). The accuracy and precision of the Bayesian posterior predictions were calculated.
Simulations and probabilities of target attainment (PTA).
For covariate simulations, a multimodal, multivariate sampling method was conducted in the Pmetrics package. This semiparametric sampling method was employed to best capture nonnormally distributed data (27). Simulations were constrained to a lower limit of zero times the final model parameter estimates and an upper limit of three times the final model parameter estimates. Monte Carlo sampling from this weighted, multivariate, multimodal distribution generated a simulated patient population with 1,000 sets of unique PK parameters. From each of the 1,000 sets of simulated PK parameters, concentration-time profiles were created by applying fixed and weight-based polymyxin B dosing regimens to the simulated patient population. Serum concentrations were generated every 30 min for the first 24 h and over the range of 72 to 96 h. Covariate simulations utilizing TBW as a covariate of interest were conducted using the 10th, 50th, and 90th percentiles of the total and adjusted body weights observed within the full patient sample. The following fixed and weight-based regimens were evaluated: 100 mg every 12 h, 1 mg/kg every 12 h, and 1.5 mg/kg every 12 h. Additionally, regimens consisting of a single loading dose of 2.5 mg/kg followed by either 100 mg every 12 h or 1.5 mg/kg every 12 h were also evaluated.
AUCs were calculated for each simulated dosing regimen and each TBW stratum after 24 or 96 h of therapy to evaluate the probabilities of efficacy and toxicity, respectively. An AUC from time zero to 24 h (AUC0–24) of 50 mg · h/liter was taken to be the efficacy exposure of interest for the first 24 h. This exposure threshold was selected because previous studies with colistin found that total AUC0–24 values (i.e., AUC0–24 values of the protein-bound plus protein-unbound drug) between 50 and 96 mg · h/liter are necessary to produce a 2-log10 kill in murine thigh and lung infection models, and an AUC/MIC of ∼50 has been established as the PK/pharmacodynamic target for polymyxins (29, 30). Likewise, a therapeutic window for polymyxin B of 50 to 100 mg · h/liter has also been recently proposed (31). A total AUC threshold value of 100 mg · h/liter for nephrotoxicity was also taken from the proposed therapeutic window and applied to the time period between 72 and 96 h (31). The percentages of the 1,000 simulated concentration-time profiles wherein the exposures exceeded the AUC thresholds for efficacy or toxicity were calculated for each dosing regimen and body weight stratum. Additionally, the probability of achieving the target AUC/MIC for efficacy was also calculated for doubling MICs of between 0.125 and 8 mg/liter over the first 24 h of therapy.
Footnotes
For a companion article on this topic, see https://doi.org/10.1128/AAC.01493-17.
REFERENCES
- 1.Falagas ME, Kasiakou SK. 2005. Colistin: the revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin Infect Dis 40:1333–1341. doi: 10.1086/429323. [DOI] [PubMed] [Google Scholar]
- 2.Storm DR, Rosenthal KS, Swanson PE. 1977. Polymyxin and related peptide antibiotics. Annu Rev Biochem 46:723–763. doi: 10.1146/annurev.bi.46.070177.003451. [DOI] [PubMed] [Google Scholar]
- 3.Koch-Weser J, Sidel VW, Federman EB, Kanarek P, Finer DC, Eaton AE. 1970. Adverse effects of sodium colistimethate. Manifestations and specific reaction rates during 317 courses of therapy. Ann Intern Med 72:857–868. [DOI] [PubMed] [Google Scholar]
- 4.Tallgren LG, Liewendahl K, Kuhlbaeck B. 1965. The therapeutic success and nephrotoxicity of colistin in acute and chronic nephropathies with impaired renal function. Acta Med Scand 177:717–728. doi: 10.1111/j.0954-6820.1965.tb01882.x. [DOI] [PubMed] [Google Scholar]
- 5.Baines RD Jr, Rifkind D. 1964. Intravenous administration of sodium colistimethate. JAMA 190:278–281. doi: 10.1001/jama.1964.03070170019004. [DOI] [PubMed] [Google Scholar]
- 6.Fekety FR Jr, Norman PS, Cluff LE. 1962. The treatment of gram-negative bacillary infections with colistin. The toxicity and efficacy of large doses in forty-eight patients. Ann Intern Med 57:214–229. [DOI] [PubMed] [Google Scholar]
- 7.Olesen S, Madsen PO. 1967. Intravenous administration of sodium colistimethate in urinary tract infections. Curr Ther Res Clin Exp 9:283–287. [PubMed] [Google Scholar]
- 8.Bedford Laboratories 2011. Polymyxin B package insert. U.S. Food and Drug Administration, Rockville, MD: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/060716Orig1s020Lbl.pdf. [Google Scholar]
- 9.Lim LM, Ly N, Anderson D, Yang JC, Macander L, Jarkowski A III, Forrest A, Bulitta JB, Tsuji BT. 2010. Resurgence of colistin: a review of resistance, toxicity, pharmacodynamics, and dosing. Pharmacotherapy 30:1279–1291. doi: 10.1592/phco.30.12.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nation RL, Li J. 2009. Colistin in the 21st century. Curr Opin Infect Dis 22:535–543. doi: 10.1097/QCO.0b013e328332e672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nation RL, Garonzik SM, Thamlikitkul V, Giamarellos-Bourboulis EJ, Forrest A, Paterson DL, Li J, Silveira FP. 2016. Dosing guidance for intravenous colistin in critically-ill patients. Clin Infect Dis 64:565–571. doi: 10.1093/cid/ciw839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plachouras D, Karvanen M, Friberg LE, Papadomichelakis E, Antoniadou A, Tsangaris I, Karaiskos I, Poulakou G, Kontopidou F, Armaganidis A, Cars O, Giamarellou H. 2009. Population pharmacokinetic analysis of colistin methanesulfonate and colistin after intravenous administration in critically ill patients with infections caused by Gram-negative bacteria. Antimicrob Agents Chemother 53:3430–3436. doi: 10.1128/AAC.01361-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandri AM, Landersdorfer CB, Jacob J, Boniatti MM, Dalarosa MG, Falci DR, Behle TF, Bordinhao RC, Wang J, Forrest A, Nation RL, Li J, Zavascki AP. 2013. Population pharmacokinetics of intravenous polymyxin B in critically ill patients: implications for selection of dosage regimens. Clin Infect Dis 57:524–531. doi: 10.1093/cid/cit334. [DOI] [PubMed] [Google Scholar]
- 14.Tran TB, Velkov T, Nation RL, Forrest A, Tsuji BT, Bergen PJ, Li J. 2016. Pharmacokinetics/pharmacodynamics of colistin and polymyxin B: are we there yet? Int J Antimicrob Agents 48:592–597. doi: 10.1016/j.ijantimicag.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akajagbor DS, Wilson SL, Shere-Wolfe KD, Dakum P, Charurat ME, Gilliam BL. 2013. Higher incidence of acute kidney injury with intravenous colistimethate sodium compared with polymyxin B in critically ill patients at a tertiary care medical center. Clin Infect Dis 57:1300–1303. doi: 10.1093/cid/cit453. [DOI] [PubMed] [Google Scholar]
- 16.Phe K, Lee Y, McDaneld PM, Prasad N, Yin T, Figueroa DA, Musick WL, Cottreau JM, Hu M, Tam VH. 2014. In vitro assessment and multicenter cohort study of comparative nephrotoxicity rates associated with colistimethate versus polymyxin B therapy. Antimicrob Agents Chemother 58:2740–2746. doi: 10.1128/AAC.02476-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rigatto MH, Oliveira MS, Perdigao-Neto LV, Levin AS, Carrilho CM, Tanita MT, Tuon FF, Cardoso DE, Lopes NT, Falci DR, Zavascki AP. 2016. Multicenter prospective cohort study of renal failure in patients treated with colistin versus polymyxin B. Antimicrob Agents Chemother 60:2443–2449. doi: 10.1128/AAC.02634-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuon FF, Rigatto MH, Lopes CK, Kamei LK, Rocha JL, Zavascki AP. 2014. Risk factors for acute kidney injury in patients treated with polymyxin B or colistin methanesulfonate sodium. Int J Antimicrob Agents 43:349–352. doi: 10.1016/j.ijantimicag.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Kwa AL, Lim TP, Low JG, Hou J, Kurup A, Prince RA, Tam VH. 2008. Pharmacokinetics of polymyxin B1 in patients with multidrug-resistant Gram-negative bacterial infections. Diagn Microbiol Infect Dis 60:163–167. doi: 10.1016/j.diagmicrobio.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 20.Onufrak NJ, Rao GG, Forrest A, Pogue JM, Scheetz MH, Nation RL, Li J, Kaye KS. 2017. Critical need for clarity in polymyxin B dosing. Antimicrob Agents Chemother 61:e00208-17. doi: 10.1128/AAC.00208-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pai MP. 2013. Polymyxin B dosing in obese and underweight adults. Clin Infect Dis 57:1785. doi: 10.1093/cid/cit604. [DOI] [PubMed] [Google Scholar]
- 22.Kubin CJ, Nelson BC, Miglis C, Scheetz MH, Rhodes N, Avedissian S, Cremers S, Yin MT. 2018. Population pharmacokinetics of intravenous polymyxin B from clinical samples. Antimicrob Agents Chemother 62:e01493-17. doi: 10.1128/AAC.01493-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rigatto MH, Behle TF, Falci DR, Freitas T, Lopes NT, Nunes M, Costa LW, Zavascki AP. 2015. Risk factors for acute kidney injury (AKI) in patients treated with polymyxin B and influence of AKI on mortality: a multicentre prospective cohort study. J Antimicrob Chemother 70:1552–1557. doi: 10.1093/jac/dku561. [DOI] [PubMed] [Google Scholar]
- 24.Kubin CJ, Ellman TM, Phadke V, Haynes LJ, Calfee DP, Yin MT. 2012. Incidence and predictors of acute kidney injury associated with intravenous polymyxin B therapy. J Infect 65:80–87. doi: 10.1016/j.jinf.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 25.Winter M. 2010. Basic clinical pharmacokinetics. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 26.Thomas TA, Broun EC, Abildskov KM, Kubin CJ, Horan J, Yin MT, Cremers S. 2012. High performance liquid chromatography-mass spectrometry assay for polymyxin B1 and B2 in human plasma. Ther Drug Monit 34:398–405. doi: 10.1097/FTD.0b013e31825c827a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. 2012. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit 34:467–476. doi: 10.1097/FTD.0b013e31825c4ba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. 1999. Guidance for industry: population pharmacokinetics. Center for Biologics Evaluation and Research, Rockville, MD: https://www.fda.gov/downloads/drugs/guidances/UCM072137.pdf. [Google Scholar]
- 29.Dudhani RV, Turnidge JD, Nation RL, Li J. 2010. fAUC/MIC is the most predictive pharmacokinetic/pharmacodynamic index of colistin against Acinetobacter baumannii in murine thigh and lung infection models. J Antimicrob Chemother 65:1984–1990. doi: 10.1093/jac/dkq226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheah SE, Wang J, Nguyen VT, Turnidge JD, Li J, Nation RL. 2015. New pharmacokinetic/pharmacodynamic studies of systemically administered colistin against Pseudomonas aeruginosa and Acinetobacter baumannii in mouse thigh and lung infection models: smaller response in lung infection. J Antimicrob Chemother 70:3291–3297. doi: 10.1093/jac/dkv267. [DOI] [PubMed] [Google Scholar]
- 31.Lakota EA, Rao GG, Landersdorfer CB, Nation RL, Li J, Kaye KS, Forrest A. 2015. Improving clinical utility of polymyxin B through a meta-analysis of toxicodynamics and development of an adaptive feedback control algorithm, abstr O-8. Abstr Internatl Soc Anti-infective Pharmacol 2nd Internatl Conf Polymyxins, San Diego, CA. [Google Scholar]