

ABSTRACT
Optimized dosage regimens of aerosolized colistin (as colistin methanesulfonate [CMS]) are urgently required to maximize bacterial killing against multidrug-resistant Gram-negative bacteria while minimizing toxicity. This study aimed to develop a mechanism-based pharmacokinetic (PK)/pharmacodynamic (PD) model (MBM) for aerosolized colistin based upon PK/PD data in neutropenic infected mice and to perform a deterministic simulation with the PK of aerosolized colistin (as CMS) in critically ill patients. In vivo time-kill experiments were carried out with three different strains of Pseudomonas aeruginosa. An MBM was developed in S-ADAPT and evaluated by assessing its ability to predict the PK/PD index associated with efficacy in mice. A deterministic simulation with human PK data was undertaken to predict the efficacy of current dosage regimens of aerosolized colistin in critically ill patients. In the final MBM, the total bacterial population for each isolate consisted of colistin-susceptible and -resistant subpopulations. The antimicrobial efficacy of aerosolized colistin was best described by a sigmoidal Emax model whereby colistin enhanced the rate of bacterial death. Deterministic simulation with human PK data predicted that an inhalational dosage regimen of 60 mg colistin base activity (CBA) every 12 h is needed to achieve a ≥2-log10 bacterial reduction (as the number of CFU per lung) in critically ill patients at 24 h after commencement of inhaled therapy. In conclusion, the developed MBM is a useful tool for optimizing inhalational dosage regimens of colistin. Clinical studies are warranted to validate and refine our MBM for aerosolized colistin.
KEYWORDS: polymyxin, respiratory tract infections, pulmonary administration, Pseudomonas aeruginosa, multidrug-resistant Gram-negative bacteria, mechanism-based modeling, population pharmacokinetics and pharmacodynamics
INTRODUCTION
Respiratory tract infections caused by multidrug-resistant (MDR) Gram-negative bacteria, such as Pseudomonas aeruginosa, are increasingly worrisome and place a heavy burden on the global health care system (1, 2). Colistin is a cationic lipopeptide antibiotic administered as its inactive prodrug, colistin methanesulfonate (CMS) (3–5). It has been increasingly used as a last-line therapy for difficult-to-treat respiratory tract infections via intravenous administration or inhalation (2, 3, 5–12). Following intravenous administration, however, colistin fails to achieve effective exposure in the lungs owing to poor penetration into the epithelial lining fluid (ELF) (13–20). Pulmonary delivery of colistin (2.64 mg base/kg of body weight) in neutropenic infected mice can achieve high colistin exposure in the ELF over 24 h (area under the concentration-time curve for ELF [AUCELF] = 180 mg · h/liter), while following intravenous administration of the same dose, colistin is undetectable (i.e., it is present at levels below the limit of quantification [LOQ] of 2.21 mg/liter) in the ELF (14).
The current inhalation dosage regimens for CMS range from 0.5 million to 2 million international units (MIU; equivalent to 15 to 60 mg colistin base activity [CBA]) twice or thrice daily (21–24); however, these dosage regimens need to be reevaluated as they are based on empirical experience rather than contemporary pharmacokinetic (PK)/pharmacodynamic (PD) studies (15). Recently, Lin et al. identified the PK/PD index for aerosolized colistin against P. aeruginosa using a mouse lung infection model (14). The ratio of the AUC over the MIC (AUC/MIC) was the most predictive index that described the antimicrobial efficacy of aerosolized colistin against P. aeruginosa. The AUC/MIC targets to achieve bacteriostasis against MDR P. aeruginosa were 684 to 1,050 in ELF and 2.15 to 3.29 in plasma (14). However, there are several limitations associated with PK/PD indices, most notably, their determination based on efficacy at a fixed time point (i.e., 24 h), thereby neglecting the change in bacterial load over time (25, 26).
Mechanism-based PK/PD models (MBMs) that characterize the time course of PK and PD offer a greater potential for achieving optimal inhalational drug therapy (26–28). Recently, Khan et al. implemented an MBM that could predict the in vivo PK/PD indices of systemically administered colistin against P. aeruginosa in a murine thigh infection model (25). However, MBMs based on in vitro static time-kill data may not be adequately translated to aerosolized colistin due to the heterogeneous colistin distribution in the lungs following pulmonary administration (17). An MBM that characterizes the PK/PD relationship for aerosolized colistin in vivo would, therefore, be valuable in predicting the efficacy of different dosage regimens in humans.
The aims of this study were to develop an MBM that characterizes the time course of the colistin concentration in ELF and plasma as well as bacterial loads after the administration of different dosage regimens of colistin in neutropenic infected mice. Furthermore, we aimed to perform a deterministic simulation using the developed MBM and a previously developed population PK model of aerosolized CMS and formed colistin in critically ill patients (17) to predict the efficacy of inhalational dosage regimens of colistin (as CMS) in humans.
RESULTS
An MBM was developed to quantitatively describe the disposition of colistin in both ELF and plasma and the time course of bacterial killing in a mouse lung infection model.
Population PK model.
The population PK model offers a more quantitative understanding of the disposition of colistin following pulmonary and intravenous administration (Fig. 1). The final, fitted model-predicted PK parameters are provided in Table 1. A two-compartment model with linear elimination from the central plasma compartment (total body clearance [CLtotal] = 4.21 liters/h/kg; standard error [SE] = 10.3%; Table 1) was used to describe the disposition of colistin in plasma. Similarly, a two-compartment model was required to describe the disposition of colistin in ELF after inhalation of colistin. The rate of transfer of colistin across the lung epithelium was described by a first-order process (intercompartmental clearance from ELF to plasma [CLELF,plasma] = 7.01 × 10−3 liter/h/kg; SE = 6.49%; Table 1). The visual predictive check (VPC) showed that the developed PK model fit the data well, with the majority of the observed concentrations being contained within the 80% prediction interval (Fig. 2 and 3).
FIG 1.

Schematic diagram of the MBM based on in vivo time-kill data. Kdeath, natural death rate.
TABLE 1.
Estimated population PK parameters for unbound colistin following intravenous and pulmonary administration of colistin in neutropenic mice with lung infection caused by P. aeruginosa
| Parameter | Description | Units | Estimated value | SE (%) |
|---|---|---|---|---|
| V1 | ELF 1 vol of distribution (ELF compartment) | liters/kg | 1.07 × 10−3 | 22.1 |
| V2 | ELF 2 vol of distribution (peripheral lung compartment) | liters/kg | 8.11 × 10−3 | 15.9 |
| VC | Vol of distribution of central plasma compartment | liters/kg | 1.50 | 43.6 |
| VP | Vol of distribution of peripheral plasma compartment | liters/kg | 1.08 | 30.5 |
| CLELF,distribution | ELF intercompatmental clearance | liters/h/kg | 3.70 × 10−3 | 17 |
| CLplasma,distribution | Plasma intercompartmental clearance | liters/h/kg | 1.08 × 101 | 26.1 |
| CLtotal | Total body clearance | liters/h/kg | 4.21 | 10.3 |
| CLELF,plasma | Intercompartmental clearance, ELF to plasma | liters/h/kg | 7.01 × 10−3 | 6.49 |
| CLplasma,ELF | Intercompartmental clearance, plasma to ELF | liters/h/kg | 1.68 × 10−4 | 206 |
| fu | Fraction unbound in plasma | 8.40 × 10−2 (fixed)a | NAb | |
| F | Bioavailability after inhalation | % | 95 | 2.06 |
Data are from reference 29.
NA, Not applicable.
FIG 2.
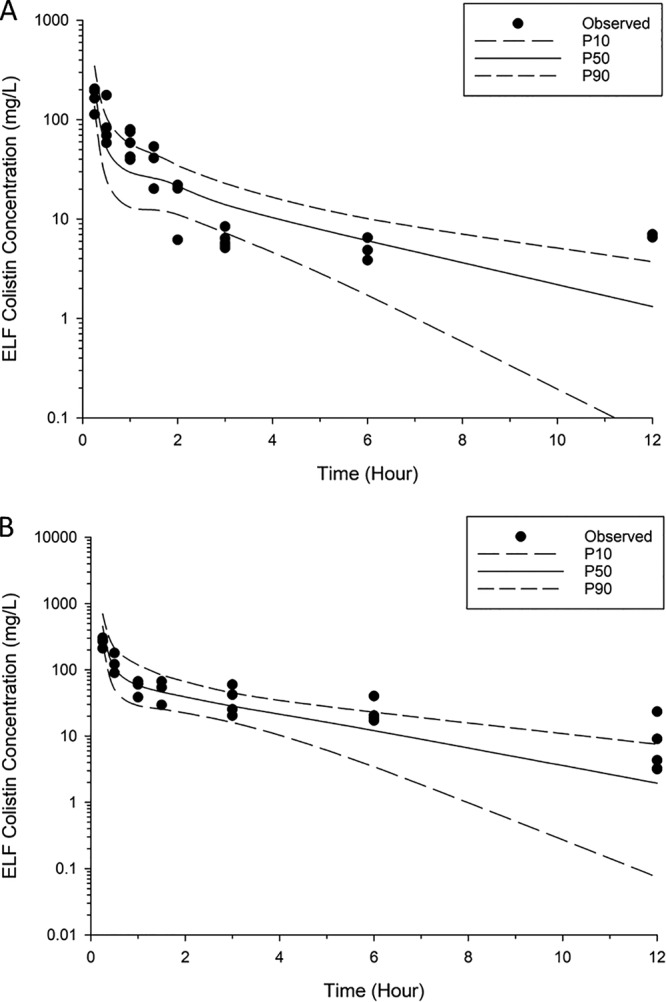
Visual predictive checks for colistin in ELF following pulmonary administration of 2.64 mg base/kg (A) and 5.28 mg base/kg (B). P50, median model-predicted bacterial load; P10, model-predicted 10th percentile bacterial load; P90, model-predicted 90th percentile bacterial load.
FIG 3.
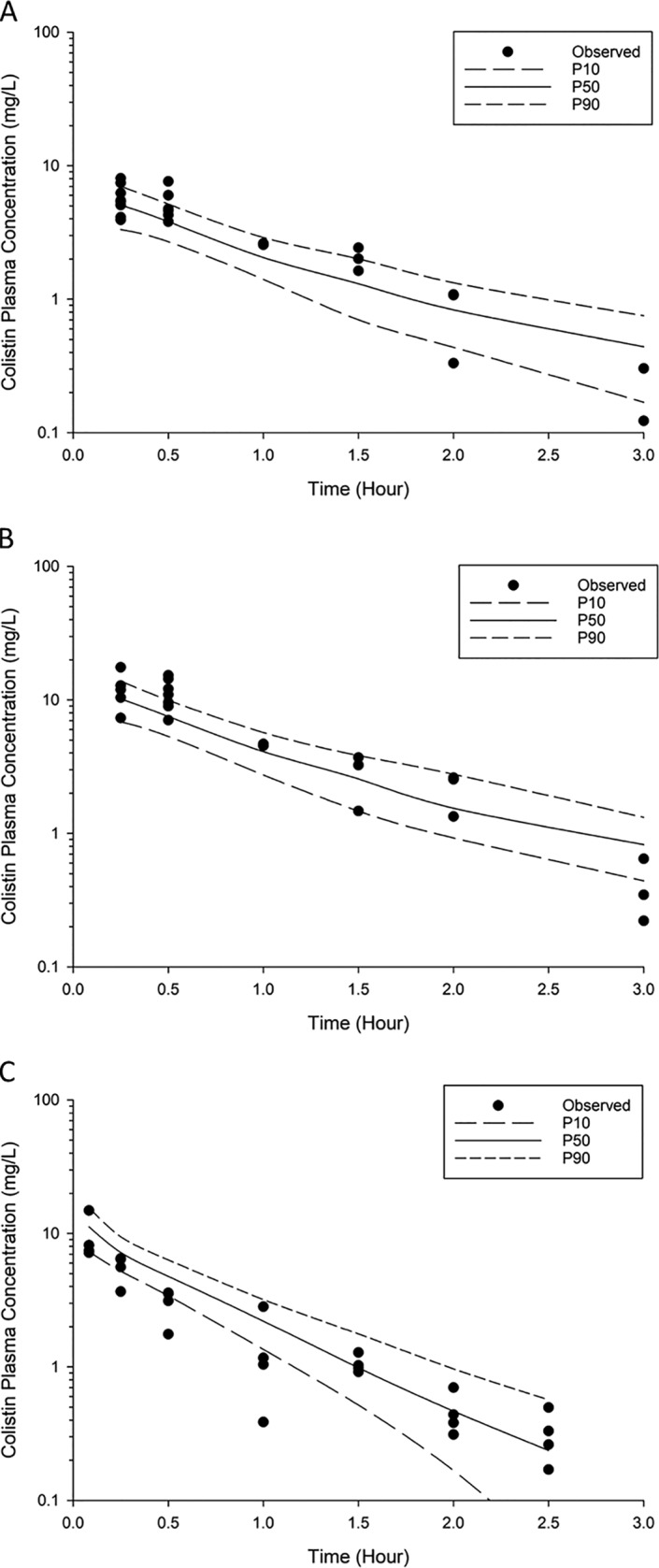
Visual predictive checks for colistin in plasma following pulmonary administration of 2.64 mg base/kg (A) and 5.28 mg base/kg (B) and intravenous administration of 2.64 mg base/kg (C). P50, median model-predicted bacterial load; P10, model-predicted 10th percentile bacterial load; P90, model-predicted 90th percentile bacterial load.
MBM for in vivo time-kill data.
The kinetics of bacterial killing and regrowth in neutropenic mice following aerosolized colistin were reasonably well described by the developed MBM (Fig. 4). The final model included two subpopulations: the colistin-susceptible and the colistin-resistant subpopulations (Fig. 1). The antimicrobial efficacy of aerosolized colistin was best described by the sigmoidal Emax and was assumed to enhance natural bacterial death. To improve numerical stability, the maximum bacterial load (the maximum number of CFU [CFUmax]) and the log10 initial inoculum (the initial number of CFU [CFU0]) were fixed at their experimental values (Table 2). The natural death rate (characterized by a first-order elimination rate constant [Kd]) of the isolate was fixed at 0.3. Furthermore, it was observed that the log10 Hill coefficient for bacterial killing (log10 Hill) was estimated to be a very large value (consistent with a very steep concentration-effect relationship), and hence, it was fixed at a value of 1.3 (equivalent to a Hill coefficient of 20 on a linear scale). The rates of natural bacterial growth (the maximal velocity of bacterial growth [VGmax] and the bacterial density at which growth rate is half maximal [CFUm]) and death (Kd) were assumed to be the same for both subpopulations of each isolate. Subpopulations of each isolate were assumed to have different initial inocula, and the colistin concentration causing 50% Emax (EC50) for each isolate were assumed to be different. The parameter estimates of the final model are listed in Table 2.
FIG 4.

Visual predictive checks for bacterial growth in the lungs for P. aeruginosa ATCC 27853 (A), PAO1 (B), and FADDI-PA022 (C). P50, median model-predicted bacterial load; P10, model-predicted 10th percentile bacterial load; P90, model-predicted 90th percentile bacterial load.
TABLE 2.
Estimated PD parameters from the 2-subpopulation model based on in vivo time-kill data
| Parameter | Abbreviation | ATCC 27853 |
PAO1 |
FADDI-PA022 |
|||
|---|---|---|---|---|---|---|---|
| Estimated value | SE (%) | Estimated value | SE (%) | Estimated value | SE (%) | ||
| Colistin concn causing 50% Emax for susceptible subpopulation (mg/liter) | EC50S | 1 | Fixed | 1 | Fixed | 1 | Fixed |
| Colistin concn causing 50% Emax for resistant-subpopulation (mg/liter) | EC50R | 51.6 | 8.99 | 187 | 4.57 | 229 | 11.2 |
| Maximum colistin-induced killing rate constant (1/h) | Emax,colistin | 5.56 | 43.2 | 23.2 | 3.72 | 38.9 | 18.5 |
| Natural bacterial death rate (1/h) | Kd | 0.3 | Fixed | 0.3 | Fixed | 0.3 | Fixed |
| Bacterial density at which growth rate is half maximal (CFU/lung) | Log10 CFUm | 8.98 | 0.386 | 9.38 | 0.278 | 9.30 | 0.539 |
| Maximum bacterial population (CFU/lung) | Log10 CFUmax | 9.33 | Fixed | 9.65 | Fixed | 9.56 | Fixed |
| Hill coefficient for bacterial killing | Log10 Hill | 1.3 | Fixed | 1.3 | Fixed | 1.3 | Fixed |
| Log10 initial inoculum for total bacterial population (CFU/lung) | Log10 CFU0 | 4.18 | Fixed | 6.29 | Fixed | 7.09 | Fixed |
| Log10 initial inoculum for CFURa | Log10 (initial inoculum for CFUR) | −1.16 | 45.1 | −1.75 | 16.9 | −1.07 | 28.3 |
CFUR, bacterial density for resistant subpopulation.
Deterministic simulation to predict the PK/PD index in neutropenic infected mice.
The developed MBM was qualified by assessing its ability to predict the PK/PD indices in neutropenic mice with lung infection caused by P. aeruginosa. The developed MBM was able to produce similar estimates of the PK/PD targets associated with bacteriostasis and 1-log10 kill, as previously observed for aerosolized colistin in a mouse lung infection model (14). The simulated AUC/MICs for the free drug (fAUC/MIC) in plasma required for bacteriostasis and 1-log10 kill in the lungs for strain PAO1 were 3.48 and 5.55, respectively, similar to previously reported values of 3.29 and 4.68, respectively (14) (Fig. 5 and Table 3).
FIG 5.
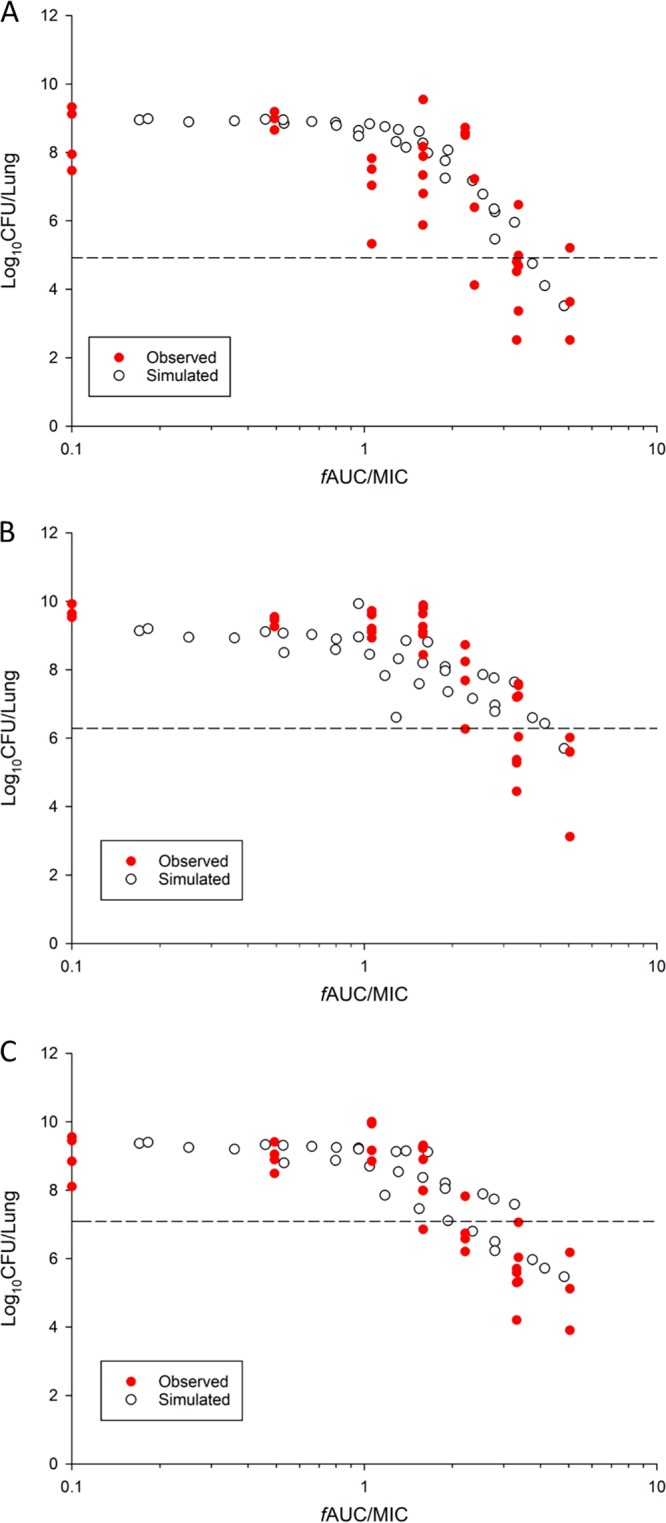
Log10 CFU per lung of P. aeruginosa ATCC 27853 (A), PAO1 (B), and FADDI-PA022 (C) at 24 h versus the colistin exposure in plasma (fAUC/MIC) in neutropenic infected mice. The observed bacterial load in neutropenic infected mice (14) and the simulated bacterial load obtained using the developed MBM are shown. Broken lines, the initial inoculum at the start of the experiment.
TABLE 3.
Observed and model-simulated fAUC/MIC targets in plasma associated with bacteriostasis and 1-log10 kill against three strains of P. aeruginosa in neutropenic infected micea
| Kill level |
fAUC/MIC |
|||||
|---|---|---|---|---|---|---|
| ATCC 27853 |
PAO1 |
FADDI-PA022 |
||||
| Observed value | Simulated value | Observed value | Simulated value | Observed value | Simulated value | |
| Stasis | 2.99 (2.47–4.17) | 3.62 (2.00–4.10) | 3.29 (2.90–3.91) | 3.48 (1.52–10.4) | 2.15 (1.90–2.45) | 2.85 (0.80–4.12) |
| 1-log10 kill | ND | ND | 4.68 (3.48–5.23) | 5.55 (1.99–14.82) | 2.60 (2.25–3.50) | 4.36 (0.88–4.55) |
Data for observed values are from reference 14. Values in parentheses represent the 10th to 90th percentile range generated from the Monte Carlo simulation. ND, not determined.
Deterministic simulation with human PK data for aerosolized colistin.
The proposed MBM was combined with a human population PK model of aerosolized CMS to predict the clinical efficacy of inhaled CMS in humans. Deterministic simulation with human PK data for aerosolized colistin (as CMS) predicted that an inhalational dosage regimen of 60 mg CBA every 12 h is required to achieve a ≥2-log10 bacterial reduction in critically ill patients infected with P. aeruginosa at 24 h after the commencement of inhaled therapy (Fig. 6).
FIG 6.

(A) Deterministic simulation of population average ELF concentration-versus-time profile of formed colistin following pulmonary nebulization (I.T.) of CMS at 30 and 60 mg CBA twice daily in critically ill patients. Deterministic simulation of bacterial growth in the lungs following exposure to aerosolized CMS at 30 and 60 mg CBA twice daily in critically ill patients for P. aeruginosa ATCC 27853 (B), PAO1 (C), and FADDI-PA022 (D).
DISCUSSION
The current inhalational dosage regimens of CMS are empirical and lack scientific evaluation with well-designed PK/PD studies. Using a mouse lung infection model, our recent study revealed that AUC/MIC best describes the antimicrobial efficacy of aerosolized colistin against MDR P. aeruginosa (14). The calculated AUC/MIC targets provided a good prediction of aerosolized colistin efficacy and the required magnitude of bacterial kill in treating respiratory tract infections. Regrowth was observed in the in vitro and in vivo time course studies (Fig. 4; see also Fig. S2 in the supplemental material), indicating that bacterial susceptibility may rapidly change over time following exposure to aerosolized colistin. It was thus necessary to utilize an MBM to evaluate the efficacy of inhalational dosage regimens of colistin. In this study, an MBM was developed to describe the disposition of colistin following intravenous and pulmonary administration, the growth of bacterial subpopulations, and the mechanism of colistin-induced killing.
In general, the observed ELF and plasma colistin concentration-time profiles were adequately described by the developed population PK model (Fig. 1 to 3), even though model misspecification existed in the ELF profile (for times of <15 min). The values of the parameters estimated by the model (Table 1) are close to those previously reported from studies performed using noncompartmental analysis (14), with the majority of the observed concentrations being within the 80% prediction interval (Fig. 2 and 3).
Previously, Khan et al. developed an MBM using in vitro time-kill curves to predict the PK/PD indices of colistin observed in a mouse thigh infection model (25). In the current study, a similar approach was utilized to evaluate whether an MBM based on in vitro static time-kill data could be used to predict aerosolized colistin activity against respiratory tract infections in a mouse lung infection model. The proposed static time-kill MBM consisted of two to three preexisting bacterial subpopulations (Fig. S1 and Table S1). On the basis of in vitro static time-kill data, the proposed MBM predicts that a single inhalational dose (2.64 mg base/kg) of colistin would result in the complete eradication of bacteria (data not shown). In clinical practice, a single dose is unlikely to clear all bacteria without regrowth. One proposed reason for the discrepancies between the in vivo and in vitro results is that bacteria are exposed to a constant colistin concentration over time in static time-kill experiments, while the in vivo drug distribution is not constant and the concentrations achieved at the respiratory tract and infection site vary (17).
To develop an in silico model that can be translated into the clinical setting, we built an MBM based on in vivo time-kill data in neutropenic infected mice. The proposed MBM shares a structure similar to that of the static time-kill model. Furthermore, only two subpopulations were fitted for in vivo time-kill data, instead of the three subpopulations in the model used for the in vitro time-kill data for isolates PAO1 and FADDI-PA022. This was most likely due to the inherent nature of our data set, i.e., destructive sampling and live animal data with an inherent variability higher than that in the in vitro data; the available data could support only the simpler parameterization. Further investigation enabling direct quantification of the different bacterial subpopulations is needed to determine their relative contribution to the total inoculum. Since a narrow dose range (2.64 and 5.28 mg base/kg) was tested due to toxicity concerns, it was important to qualify our model by assessing its ability to predict the PK/PD indices in neutropenic mice with lung infection caused by P. aeruginosa (14). The predicted values closely resembled the observed data, with some minor differences (Fig. 5). Through simulation, we observed that the PK/PD targets for bacteriostasis and 1-log10 kill are similar to those reported previously in a lung infection model in neutropenic mice (Table 3) (14). Here, for the first time, we have shown that an MBM based on in vivo time-kill data can be utilized to calculate the PK/PD indices for dose optimization of aerosolized colistin.
Our recent PK/PD study showed that pulmonary administration of colistin is superior to systemic administration in treating respiratory tract infections (14). The fAUC/MIC in plasma required to achieve bacteriostasis in the lungs following pulmonary administration is approximately 11-fold lower than that required following subcutaneous administration (2.99 versus 34.1, respectively) (14, 29). The MBM developed in the current study provided an explanation for the superiority of pulmonary delivery in treating respiratory tract infections. The more resistant subpopulation was estimated to have a relatively large EC50 across the three strains (Table 2, EC50R). These high EC50 values were most likely due to the binding of colistin to mucin in the mucus (30) and surfactants in ELF (31), as well as the heterogeneous distribution of colistin aerosol in the lung (19). Such high values imply that the so-called resistant subpopulation is unlikely to respond to the range of ELF concentrations obtained following systemic delivery. To confirm this finding, the antimicrobial efficacy of systemic colistin was assessed following the intraperitoneal delivery of colistin in treating respiratory tract infections. Following treatment with intraperitoneal colistin (2.64 and 5.28 mg base/kg), no statistically significant reduction in the bacterial load was observed over 24 h (Fig. S3). Notably, the colistin ELF concentration achieved following the intraperitoneal delivery of colistin was below the limit of quantitation (<2.21 mg/liter) over the 12-h sampling period (data not shown). Taken together, our results have clearly demonstrated the superiority of aerosolized colistin over systemically delivered colistin to treat respiratory tract infections caused by P. aeruginosa in a mouse lung infection model.
The developed MBM serves as an important pharmacological tool that can be used in conjunction with Monte Carlo simulation to evaluate the efficacy of clinically utilized aerosolized colistin dosing regimens. These simulations are performed using a small population PK model of nebulized colistin (as CMS) in critically ill patients (17). Given the large degree of variability (i.e., the high interindividual variability [IIV], random unexplained variability [RUV], and large standard errors [SE] associated with IIV) in the population PK model of nebulized colistin (as CMS) (17), a deterministic simulation was performed using the model-predicted median value of formed colistin ELF concentrations without the incorporation of IIV (32, 33). Deterministic simulation with human PK data predicted that an inhalational dosage regimen of 30 mg CBA every 12 h (equivalent to 1 MIU twice daily) was unable to achieve a ≥2-log10 bacterial reduction in critically ill patients infected with P. aeruginosa at 24 h after the commencement of inhaled therapy (Fig. 6). However, 60 mg CBA every 12 h (equivalent to 2 MIU twice daily) was able to achieve a ≥2-log10 bacterial reduction. In line with the previous recommendation (33), the 60-mg CBA dose should be preferred over the lower doses (e.g., 15 to 30 mg CBA) for the treatment of respiratory tract infections caused by P. aeruginosa. However, translation of these findings to critically ill patients remains challenging, as the lung anatomy and transport mechanisms of mice differ from those of humans. Importantly, since a naive pooled approach and deterministic simulation were used, the MBM did not take the variability in PK/PD responses into consideration. Well-designed clinical studies are needed to characterize the extent of potential patient- and device-specific differences in the PK of aerosolized colistin for bacterial killing. As a detailed and robust human population PK model of aerosolized colistin becomes available, our proposed MBM can be used to evaluate the clinical efficacy of other commonly used inhalational dosage regimens of colistin (as CMS) in humans (e.g., 75 to 150 mg CBA every 12 h). Although optimization of inhalational dosage regimens of colistin remains an important task, the presence of regrowth for some dosage regimens (e.g., 30 CBA twice daily) suggests that inhalational polymyxin monotherapy may be inadequate to treat respiratory tract infections. Future clinical studies are needed to evaluate nebulized colistin as mono- and combination therapy for the treatment of life-threatening respiratory tract infections caused by MDR P. aeruginosa.
To the best of our knowledge, this is the first in vivo MBM that describes the disposition of colistin in both ELF and plasma following pulmonary administration and the antibacterial effect on MDR P. aeruginosa. Deterministic simulations suggest that an inhalational dose of 60 mg CBA every 12 h may be required to achieve ≥2-log10 killing at 24 h after the commencement of inhaled therapy. The MBM, in combination with a validated human population PK model, may have significant potential in facilitating the development of optimal inhalational dosage regimens of colistin in patients to treat life-threatening respiratory tract infections caused by Gram-negative superbugs.
MATERIALS AND METHODS
Chemicals and bacterial strains.
Colistin solution (sulfate; ≥15,000 U/mg; lot number 08M1526V; Sigma-Aldrich, St. Louis, MO, USA) was freshly prepared in sterile 0.9% sodium chloride. The solution was filtered through a 0.22-μm-pore-size filter and stored at 4°C. Three strains of MDR P. aeruginosa (two reference strains [ATCC 27853 and PAO1] and one MDR clinical isolate [FADDI-PA022]; MIC = 1 mg/liter for all strains) were examined. All strains were stored in tryptone soy broth with 20% glycerol at −80°C and subcultured onto a nutrient agar plate prior to each experiment (34).
Population PK modeling.
The PK data for intravenous and pulmonary administration of colistin have been published previously (14). Single-dose PK studies were performed with neutropenic mice after intravenous (2.64 mg base/kg) and pulmonary (2.64 and 5.28 mg base/kg) administration of colistin. Animal experiments were approved by the Animal Ethics Committee of the Monash Institute of Pharmaceutical Sciences, Monash University, and conducted in accordance with the guideline of the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. Mice received either intravenous or pulmonary administration of colistin. For pulmonary administration, mice were briefly anesthetized and placed on a Perspex support in a vertical upright position. A Penn-Century MicroSprayer (Penn-Century, Philadelphia, PA, USA) was used to administer colistin solution (2.64 and 5.28 mg base/kg in 25 μl) to the mouse lungs (14). Blood samples were collected at a total of 6 time points over a 6-h period. In addition, for PK analysis bronchoalveolar lavage fluid (BALF) was also collected using 0.5 ml of sterile saline (13) at a total of 7 unique time points (n = 3 or more) over a 12-h period.
Population PK modeling of colistin in ELF and plasma following intravenous and pulmonary administration was performed using the Monte Carlo parametric expectation maximization algorithm (pmethod = 4) in the S-ADAPT (version 1.57) program, facilitated by the S-ADAPT TRAN program (35). PK structural models with one or two compartments were evaluated to fit the plasma concentrations following intravenous administration. Subsequently, colistin plasma concentrations following administration by both routes were simultaneously modeled. Similarly, the concentration of colistin in ELF following pulmonary administration was comodeled with the plasma concentration. Since a naive pooled approach was utilized, the IIV of the PK parameters was not estimated. A combined additive and proportional error model was utilized to describe the RUV. The most predictive PK structural model was selected on the basis of the corrected Akaike information criterion (AIC) value, the objective function value (reported as −1 × log likelihood in S-ADAPT), the precision of the fitted parameters, the goodness of fit, and visual inspection of standard visual diagnostic plots (36–38). The final model was evaluated using a VPC. A decrease in the objective function of 1.92 units (chi-square test with 1 degree of freedom) was considered statistically significant.
Mouse lung infection model.
Female Swiss mice (age, 9 to 10 weeks) were obtained from the Monash Animal Center (Clayton, Australia). Animals were housed in the animal facility (Monash University, Parkville Campus, Australia) and acclimatized for 3 days prior to each experiment with free access to water and food.
Mice were rendered neutropenic by intraperitoneal injection of two doses of cyclophosphamide on day −4 and day −1 before the experiment (14, 29). To establish an infection, a Penn-Century MicroSprayer (model IA-1C; Penn-Century, Philadelphia, PA, USA) was employed to deliver a 25-μl bacterial suspension (approximately 106 CFU/ml in early logarithmic phase) directly into the lungs (39).
Colistin treatment was initiated at 2 h postinoculation. The bacterial burden in the lungs of the saline-treated control animals and the colistin-treated animals was determined at 0, 1, 3, 6, 12, and 24 h posttreatment. At least 3 animals were included per time point. To minimize any potential drug carryover, lung tissue samples were washed once with 1 ml sterile 0.9% saline solution before harvesting in 8-ml sterile saline. The lung tissue samples were then homogenized, filtered, diluted accordingly, and spiral plated on nutrient agar plates to determine the bacterial load (34). The limit of detection was 164 CFU/lung (equivalent to one colony per plate). The bacterial load in the infected lungs was comodeled with the different PK profiles using mechanistic modeling, as described below.
Mechanism-based pharmacodynamic modeling.
A general mechanistic model developed by Bulitta et al. (40), Cheah et al. (41), and Meagher et al. (42) was used as a starting point for MBM development in the current study. In our model, bacterial cells were partitioned into two to three preexisting subpopulations. The number of subpopulations needed to describe the data was tested using the corrected Akaike information criterion. The subpopulations consisted of colistin-susceptible (S), intermediately susceptible (I), and mostly resistant (R) populations with capacity-limited (saturable) growth and first-order rate constants for natural death (see Fig. 1 and S1 in the supplemental material for in vivo and in vitro data, respectively). The initial inoculum of all subpopulations was estimated. The subpopulations differed at least in colistin susceptibility (EC50) and the initial individual inoculum, which were fitted for all subpopulations. The initial inoculum of the resistant subpopulations was estimated as a fraction of the total initial inoculum (CFU0). The total bacterial load is represented by equation 1:
| (1) |
where CFUS, CFUI, and CFUR are the numbers of CFU for the colistin-susceptible, intermediately susceptible, and mostly resistant populations, respectively. The rate of bacterial replication (Kreplication) (equation 2) was modeled as capacity limited, dependent on the maximal velocity of bacterial growth (VGmax) and the bacterial density at which the rate of replication is half maximal (CFUm) (43). Models with the same growth constant and different growth constants (i.e., different VGmax and CFUm) were tested.
| (2) |
where CFUtotal is the total number of CFU. VGmax may be computed as Kd × (CFUm + CFUmax). The rate of natural bacterial death was characterized by a first-order elimination rate constant (Kd). In the current model, colistin was assumed to enhance the rate of bacterial killing (natural death) and was described with Emax models based on the bacterial subpopulation (41–45), as represented in equation 3:
| (3) |
where Emax,colistin is the maximum colistin killing rate constant, Kcolistin is the colistin-induced killing rate constant, and EC50i is the colistin concentration causing 50% Emax for the ith subpopulation, Ccolistin,effective is the effective colistin concentration, and γ is the Hill coefficient.
The differential equations that describe each bacterial subpopulation are outlined below.
| (4) |
| (5) |
| (6) |
where t is time.
For the static time-kill data, to account for the competitive displacement of bound divalent cations from their bacterial target, the lipid A moiety of lipopolysaccharide (LPS), the following equations were utilized (43):
| (7) |
| (8) |
where Fcations is the fractional occupancy of the receptor for the cations Mg2+ and Ca2+, Kd,cations is the dissociation constant for the cations Mg2+ and Ca2+, Kd,colistin is the dissociation constant for colistin, Ccation is the sum of the molar concentrations of the two cations (Mg2+ and Ca2+), Ccolistin is the colistin concentration, and Mmolecular is the average molecular weight of the two predominant components of colistin (i.e., colistin A and B).
The time course of the antimicrobial effect on bacterial killing and regrowth was comodeled in S-ADAPT (version 1.57) (46), facilitated by S-ADAPT TRAN (35, 40), using a Monte Carlo parametric expectation maximization algorithm (pmethod = 4). All bacterial counts were transformed to the log10 scale. RUV was described by an additive error model on the log10 scale, and the interexperiment variability was not estimated. The final model was assessed by the precision of the fitted parameters, the goodness of fit, visual inspection of diagnostic plots, and VPC (36–38, 42).
Deterministic simulation to derive the PK/PD index in neutropenic infected mice.
The final parameters estimated from the final in vivo MBM for P. aeruginosa were used to simulate the modified dose fractionation studies outlined in the work of Lin et al. (14) for neutropenic infected mice. Subsequently, the inhibitory sigmoid dose-effect model was applied to these simulated data to calculate the PK/PD index for different killing effects (i.e., fAUC/MIC).
Deterministic simulation with human PK data.
The final parameters estimated from the current in vivo MBM for all three isolates of P. aeruginosa (ATCC 27853, PAO1, and FADDI-PA022) and a previously reported PK model for aerosolized CMS in critically ill patients (17) were employed to simulate the time course of bacterial killing and colistin concentration-time profiles, respectively. Deterministic simulations were performed using the model-predicted median value of formed colistin ELF concentrations and PD parameters for each isolate in the Berkeley Madonna (version 8.3.18) program without interindividual or residual variabilities. The following clinically available inhalational doses were evaluated against each isolate: (i) 30 mg CBA every 12 h and (ii) 60 mg CBA every 12 h. The inhalational dosage regimens were chosen to represent those that are commonly utilized in humans (47, 48).
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the financial support from the National Health and Medical Research Council (NHMRC, APP1065046). Q.T.Z., A.F., and J.L. are supported by research grants from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965 [J.L. and A.F.] and R01 AI132681 [Q.T.Z., J.L., and A.F.]). Y.-W.L. is a recipient of an Australian postgraduate award. J.L. is an Australian NHMRC senior research fellow. Q.T.Z. is a recipient of the Ralph W. and Grace M. Showalter Research Trust Award.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01965-17.
REFERENCES
- 1.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Nation RL, Li J, Cars O, Couet W, Dudley MN, Kaye KS, Mouton JW, Paterson DL, Tam VH, Theuretzbacher U. 2015. Framework for optimisation of the clinical use of colistin and polymyxin B: the Prato polymyxin consensus. Lancet Infect Dis 15:225–234. doi: 10.1016/S1473-3099(14)70850-3. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Nation RL, Turnidge JD, Milne RW, Coulthard K, Rayner CR, Paterson DL. 2006. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis 6:589–601. doi: 10.1016/S1473-3099(06)70580-1. [DOI] [PubMed] [Google Scholar]
- 4.Cheah S-E, Li J, Bergen PJ, Nation RL. 2016. Polymyxin pharmacokinetics and pharmacodynamics, p 221–260. In Rotschafer JC, Andes DR, Rodvold KA (ed), Antibiotic pharmacodynamics. Springer, New York, NY. [Google Scholar]
- 5.Couet W, Gregoire N, Marchand S, Mimoz O. 2012. Colistin pharmacokinetics: the fog is lifting. Clin Microbiol Infect 18:30–39. doi: 10.1111/j.1469-0691.2011.03667.x. [DOI] [PubMed] [Google Scholar]
- 6.Langton Hewer SC, Smyth AR. 2009. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Library 11:CD004197. doi: 10.1002/14651858.CD004197.pub3. [DOI] [PubMed] [Google Scholar]
- 7.Quon BS, Goss CH, Ramsey BW. 2014. Inhaled antibiotics for lower airway infections. Ann Am Thorac Soc 11:425–434. doi: 10.1513/AnnalsATS.201311-395FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boisson M, Gregoire N, Couet W, Mimoz O. 2013. Colistin in critically ill patients. Minerva Anestesiol 79:200–208. [PubMed] [Google Scholar]
- 9.Falagas ME, Kasiakou SK, Saravolatz LD. 2005. Colistin: the revival of polymyxins for the management of multidrug-resistant Gram-negative bacterial infections. Clin Infect Dis 40:1333–1341. doi: 10.1086/429323. [DOI] [PubMed] [Google Scholar]
- 10.Landman D, Georgescu C, Martin DA, Quale J. 2008. Polymyxins revisited. Clin Microbiol Rev 21:449–465. doi: 10.1128/CMR.00006-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zavascki AP, Goldani LZ, Li J, Nation RL. 2007. Polymyxin B for the treatment of multidrug-resistant pathogens: a critical review. J Antimicrob Chemother 60:1206–1215. doi: 10.1093/jac/dkm357. [DOI] [PubMed] [Google Scholar]
- 12.Ratjen F, Rietschel E, Kasel D, Schwiertz R, Starke K, Beier H, Van Koningsbruggen S, Grasemann H. 2006. Pharmacokinetics of inhaled colistin in patients with cystic fibrosis. J Antimicrob Chemother 57:306–311. doi: 10.1093/jac/dki461. [DOI] [PubMed] [Google Scholar]
- 13.Landersdorfer CB, Nguyen T-H, Lieu LT, Nguyen G, Bischof RJ, Meeusen EN, Li J, Nation RL, McIntosh MP. 2017. Substantial targeting advantage achieved by pulmonary administration of colistin methanesulfonate in a large-animal model. Antimicrob Agents Chemother 61:e01934-16. doi: 10.1128/AAC.01934-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin Y-W, Zhou Q, Cheah S-E, Zhao J, Chen K, Wang J, Chan H-K, Li J. 2017. Pharmacokinetics/pharmacodynamics of pulmonary delivery of colistin against Pseudomonas aeruginosa in a mouse lung infection model. Antimicrob Agents Chemother 61:e02025-16. doi: 10.1128/AAC.02025-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yapa SW, Li J, Patel K, Wilson JW, Dooley MJ, George J, Clark D, Poole S, Williams E, Porter CJ. 2014. Pulmonary and systemic pharmacokinetics of inhaled and intravenous colistin methanesulfonate in cystic fibrosis patients: targeting advantage of inhalational administration. Antimicrob Agents Chemother 58:2570–2579. doi: 10.1128/AAC.01705-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yapa SW, Li J, Porter CJ, Nation RL, Patel K, McIntosh MP. 2013. Population pharmacokinetics of colistin methanesulfonate in rats: achieving sustained lung concentrations of colistin for targeting respiratory infections. Antimicrob Agents Chemother 57:5087–5095. doi: 10.1128/AAC.01127-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boisson M, Jacobs M, Grégoire N, Gobin P, Marchand S, Couet W, Mimoz O. 2014. Comparison of intrapulmonary and systemic pharmacokinetics of colistin methanesulfonate (CMS) and colistin after aerosol delivery and intravenous administration of CMS in critically ill patients. Antimicrob Agents Chemother 58:7331–7339. doi: 10.1128/AAC.03510-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchand S, Bouchene S, de Monte M, Guilleminault L, Montharu J, Cabrera M, Grégoire N, Gobin P, Diot P, Couet W. 2015. Pharmacokinetics of colistin methansulphonate (CMS) and colistin after CMS nebulisation in baboon monkeys. Pharm Res 32:3403–3414. doi: 10.1007/s11095-015-1716-0. [DOI] [PubMed] [Google Scholar]
- 19.Lu Q, Girardi C, Zhang M, Bouhemad B, Louchahi K, Petitjean O, Wallet F, Becquemin M-H, Le Naour G, Marquette C-H. 2010. Nebulized and intravenous colistin in experimental pneumonia caused by Pseudomonas aeruginosa. Intensive Care Med 36:1147–1155. doi: 10.1007/s00134-010-1879-4. [DOI] [PubMed] [Google Scholar]
- 20.Gontijo AVL, Grégoire N, Lamarche I, Gobin P, Couet W, Marchand S. 2014. Biopharmaceutical characterization of nebulized antimicrobial agents in rats: 2. Colistin. Antimicrob Agents Chemother 58:3950–3956. doi: 10.1128/AAC.02819-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen C, Pressler T, Høiby N. 2008. Early aggressive eradication therapy for intermittent Pseudomonas aeruginosa airway colonization in cystic fibrosis patients: 15 years experience. J Cyst Fibros 7:523–530. doi: 10.1016/j.jcf.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 22.Döring G, Conway S, Heijerman H, Hodson M, Høiby N, Smyth A, Touw D, Consensus Committee . 2000. Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis: a European consensus. Eur Respir J 16:749–767. doi: 10.1034/j.1399-3003.2000.16d30.x. [DOI] [PubMed] [Google Scholar]
- 23.Döring G, Hoiby N, Consensus Study Group . 2004. Early intervention and prevention of lung disease in cystic fibrosis: a European consensus. J Cyst Fibros 3:67–91. doi: 10.1016/j.jcf.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 24.Banerjee D, Stableforth D. 2000. The treatment of respiratory Pseudomonas infection in cystic fibrosis. Drugs 60:1053–1064. doi: 10.2165/00003495-200060050-00006. [DOI] [PubMed] [Google Scholar]
- 25.Khan DD, Friberg LE, Nielsen EI. 2016. A pharmacokinetic-pharmacodynamic (PKPD) model based on in vitro time-kill data predicts the in vivo PK/PD index of colistin. J Antimicrob Chemother 71:1881–1884. doi: 10.1093/jac/dkw057. [DOI] [PubMed] [Google Scholar]
- 26.Nielsen EI, Friberg LE. 2013. Pharmacokinetic-pharmacodynamic modeling of antibacterial drugs. Pharmacol Rev 65:1053–1090. doi: 10.1124/pr.111.005769. [DOI] [PubMed] [Google Scholar]
- 27.Nielsen EI, Cars O, Friberg LE. 2011. Pharmacokinetic/pharmacodynamic (PK/PD) indices of antibiotics predicted by a semimechanistic PKPD model: a step toward model-based dose optimization. Antimicrob Agents Chemother 55:4619–4630. doi: 10.1128/AAC.00182-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nielsen EI, Viberg A, Lowdin E, Cars O, Karlsson MO, Sandstrom M. 2007. Semimechanistic pharmacokinetic/pharmacodynamic model for assessment of activity of antibacterial agents from time-kill curve experiments. Antimicrob Agents Chemother 51:128–136. doi: 10.1128/AAC.00604-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheah S-E, Wang J, Turnidge JD, Li J, Nation RL. 2015. New pharmacokinetic/pharmacodynamic studies of systemically administered colistin against Pseudomonas aeruginosa and Acinetobacter baumannii in mouse thigh and lung infection models: smaller response in lung infection. J Antimicrob Chemother 70:3291–3297. doi: 10.1093/jac/dkv267. [DOI] [PubMed] [Google Scholar]
- 30.Huang JX, Blaskovich MA, Pelingon R, Ramu S, Kavanagh A, Elliott AG, Butler MS, Montgomery AB, Cooper MA. 2015. Mucin binding reduces colistin antimicrobial activity. Antimicrob Agents Chemother 59:5925–5931. doi: 10.1128/AAC.00808-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwameis R, Erdogan-Yildirim Z, Manafi M, Zeitlinger M, Strommer S, Sauermann R. 2013. Effect of pulmonary surfactant on antimicrobial activity in vitro. Antimicrob Agents Chemother 57:5151–5154. doi: 10.1128/AAC.00778-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Llanos-Paez CC, Hennig S, Staatz CE. 2017. Population pharmacokinetic modelling, Monte Carlo simulation and semi-mechanistic pharmacodynamic modelling as tools to personalize gentamicin therapy. J Antimicrob Chemother 72:639–667. doi: 10.1093/jac/dkw461. [DOI] [PubMed] [Google Scholar]
- 33.Boisson M, Grégoire N, Cormier M, Gobin P, Marchand S, Couet W, Mimoz O. 2017. Pharmacokinetics of nebulized colistin methanesulfonate in critically ill patients. J Antimicrob Chemother 72:2607–2612. doi: 10.1093/jac/dkx167. [DOI] [PubMed] [Google Scholar]
- 34.Dudhani RV, Turnidge JD, Nation RL, Li J. 2010. fAUC/MIC is the most predictive pharmacokinetic/pharmacodynamic index of colistin against Acinetobacter baumannii in murine thigh and lung infection models. J Antimicrob Chemother 65:1984–1990. doi: 10.1093/jac/dkq226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bulitta JB, Landersdorfer CB. 2011. Performance and robustness of the Monte Carlo importance sampling algorithm using parallelized S-ADAPT for basic and complex mechanistic models. AAPS J 13:212–226. doi: 10.1208/s12248-011-9258-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Landersdorfer CB, Kirkpatrick CM, Kinzig-Schippers M, Bulitta JB, Holzgrabe U, Drusano GL, Sörgel F. 2007. Population pharmacokinetics at two dose levels and pharmacodynamic profiling of flucloxacillin. Antimicrob Agents Chemother 51:3290–3297. doi: 10.1128/AAC.01410-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bulitta JB, Zhao P, Arnold RD, Kessler DR, Daifuku R, Pratt J, Luciano G, Hanauske A-R, Gelderblom H, Awada A. 2009. Mechanistic population pharmacokinetics of total and unbound paclitaxel for a new nanodroplet formulation versus Taxol in cancer patients. Cancer Chemother Pharmacol 63:1049–1063. doi: 10.1007/s00280-008-0827-2. [DOI] [PubMed] [Google Scholar]
- 38.Tsuji BT, Okusanya OO, Bulitta JB, Forrest A, Bhavnani SM, Fernandez PB, Ambrose PG. 2011. Application of pharmacokinetic-pharmacodynamic modeling and the justification of a novel fusidic acid dosing regimen: raising Lazarus from the dead. Clin Infect Dis 52:S513–S519. doi: 10.1093/cid/cir166. [DOI] [PubMed] [Google Scholar]
- 39.Bivas-Benita M, Zwier R, Junginger HE, Borchard G. 2005. Non-invasive pulmonary aerosol delivery in mice by the endotracheal route. Eur J Pharm Biopharm 61:214–218. doi: 10.1016/j.ejpb.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 40.Bulitta JB, Ly NS, Yang JC, Forrest A, Jusko WJ, Tsuji BT. 2009. Development and qualification of a pharmacodynamic model for the pronounced inoculum effect of ceftazidime against Pseudomonas aeruginosa. Antimicrob Agents Chemother 53:46–56. doi: 10.1128/AAC.00489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheah S-E, Li J, Tsuji BT, Forrest A, Bulitta JB, Nation RL. 2016. Colistin and polymyxin B dosage regimens against Acinetobacter baumannii: differences in activity and the emergence of resistance. Antimicrob Agents Chemother 60:3921–3933. doi: 10.1128/AAC.02927-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meagher AK, Forrest A, Dalhoff A, Stass H, Schentag JJ. 2004. Novel pharmacokinetic-pharmacodynamic model for prediction of outcomes with an extended-release formulation of ciprofloxacin. Antimicrob Agents Chemother 48:2061–2068. doi: 10.1128/AAC.48.6.2061-2068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bulitta JB, Yang JC, Yohonn L, Ly NS, Brown SV, D'Hondt RE, Jusko WJ, Forrest A, Tsuji BT. 2010. Attenuation of colistin bactericidal activity by high inoculum of Pseudomonas aeruginosa characterized by a new mechanism-based population pharmacodynamic model. Antimicrob Agents Chemother 54:2051–2062. doi: 10.1128/AAC.00881-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tam VH, Kabbara S, Vo G, Schilling AN, Coyle EA. 2006. Comparative pharmacodynamics of gentamicin against Staphylococcus aureus and Pseudomonas aeruginosa. Antimicrob Agents Chemother 50:2626–2631. doi: 10.1128/AAC.01165-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuck EL, Dalhoff A, Stass H, Derendorf H. 2005. Pharmacokinetic/pharmacodynamic (PK/PD) evaluation of a once-daily treatment using ciprofloxacin in an extended-release dosage form. Infection 2:22–28. doi: 10.1007/s15010-005-8204-0. [DOI] [PubMed] [Google Scholar]
- 46.Bauer RJ, Guzy S, Ng C. 2007. A survey of population analysis methods and software for complex pharmacokinetic and pharmacodynamic models with examples. AAPS J 9:E60–E83. doi: 10.1208/aapsj0901007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Profile Pharma Ltd. 2010. Promixin package insert. Profile Pharma Ltd, West Sussex, United Kingdom. [Google Scholar]
- 48.Phebra Pty Ltd. 2011. Tadam package insert. Phebra Pty Ltd, Lane Cove, NSW, Australia. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.