

ABSTRACT
Candida glabrata, the second most frequent cause of candidiasis after Candida albicans, is an emerging human fungal pathogen that is intrinsically drug tolerant. Currently, studies of C. glabrata genes involved in drug tolerance are limited. Ada2, a component serving as a transcription adaptor of the Spt-Ada-Gcn5 acetyltransferase (SAGA) complex, is required for antifungal drug tolerance and virulence in C. albicans. However, its roles in C. glabrata remain elusive. In this study, we found that ada2 mutants demonstrated severe growth defects at 40°C but only mild defects at 37°C or 25°C. In addition, C. glabrata ada2 mutants exhibited pleiotropic phenotypes, including susceptibility to three classes of antifungal drugs (i.e., azoles, echinocandins, and polyenes) and cell wall-perturbing agents but resistance to the endoplasmic reticulum stressor tunicamycin. According to RNA sequence analysis, the expression of 43 genes was downregulated and the expression of 442 genes was upregulated in the ada2 mutant compared to their expression in the wild type. C. glabrata ADA2, along with its downstream target ERG6, controls antifungal drug tolerance and cell wall integrity. Surprisingly, ada2 mutants were hypervirulent in a murine model of systemic infection, possibly due to the upregulation of multiple adhesin-like genes, increased agar invasion, and overstimulation of murine tumor necrosis factor alpha production.
KEYWORDS: Candida glabrata, antifungal drug susceptibility, cell wall integrity, hypervirulence
INTRODUCTION
The opportunistic human fungal pathogen Candida glabrata is the most common non-albicans Candida species that causes infections of mucosal surfaces or the bloodstream (i.e., systemic infection) in most geographic regions (1, 2). The rate of mortality from systemic infections caused by C. glabrata is relatively high, partly due to its rapidly evolving resistance to antifungal drugs, especially azoles (3, 4). The treatment of C. glabrata infections is challenging due to the limited knowledge of C. glabrata pathogenicity, the restricted antifungal drug susceptibility of C. glabrata, and the availability of few effective antifungal agents.
Infection with Candida species is initiated through adherence to host epithelial tissue and colonization within the host (5). In Candida albicans, the agglutinin-like sequence (Als) protein family and Hwp1 play crucial roles in adherence to host epithelial cells (6, 7). Similar to the Als family of C. albicans, the main adhesins in C. glabrata are those of the epithelial adhesin (EPA) family, which facilitate attachment to host epithelial cells and macrophages (8–10). After colonization on host cells, Candida cells destroy host tissues and undergo invasive growth, during which proteases, such as the secreted aspartyl proteases (Saps), and phospholipases act as key virulence factors (11–13). Additionally, the unique extracellular glycosylphosphatidylinositol (GPI)-linked aspartyl protease gene family (YPS genes) plays important roles in virulence and survival within macrophages (14).
Several studies have reported on the C. glabrata genes that are involved in virulence suppression. The most well characterized of these genes is C. glabrata ACE2 (CgACE2), a transcription factor that acts as a negative regulator of virulence in a model of systemic infection in immunocompromised mice (15). The evolved (Evo) strain, which has a single nucleotide mutation in the chitin synthase gene CHS2, is another hypervirulent C. glabrata strain (16). Both the ace2 mutant and the Evo strain have enhanced virulence; stimulate inflammatory response factors, such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6); and exhibit a clumpy pseudohypha-like structure. Other strains with enhanced virulence include a strain with the PDR1 gain-of-function mutation, a strain with mitochondrial dysfunction, and the anp1 and mnn2 glycosylation mutants (17–19).
The Spt-Ada-Gcn5 acetyltransferase (SAGA) complex is conserved in eukaryotes and influences the expression of approximately 10% of genes in the Saccharomyces cerevisiae genome (20, 21). Within the SAGA complex, Gcn5 is a histone acetyltransferase that mediates nucleosomal acetylation, and the site specificity of lysine acetylation on histones is potentiated by Ada2 and Ada3 (22). In S. cerevisiae, the loss of Ada2 results in slow growth under minimal glucose and low-phosphate conditions, temperature sensitivity, and sensitivity to the cell wall-perturbing agent Congo red (CR) (23–25). In C. albicans, the ada2/ada2 mutant is sensitive to Congo red, the endoplasmic reticulum (ER) stressor tunicamycin (TM), and antifungal drugs, such as caspofungin (CSF) and fluconazole (FLC) (26, 27). Meanwhile, the C. albicans ada2/ada2 mutant exhibits hyphal growth defects and attenuated virulence in murine systemic and Caenorhabditis elegans infection models (27, 28). In Cryptococcus neoformans, the ada2 mutant has severe growth defects at 39°C, and C. neoformans Ada2 (CnAda2) is required for capsule formation and virulence in a murine intranasal infection model (29). In the filamentous fungus Aspergillus nidulans, the adaB mutant has defects in growth, conidiation, and conidiophore formation (30). In the rice pathogen Fusarium fujikuroi, the ada2 mutant has impaired growth in solid media (31). However, the roles of C. glabrata Ada2 on growth, antifungal tolerance, and virulence have remained elusive.
In this study, we report that C. glabrata Ada2 has multiple roles in cellular functions, ranging from growth and cell wall integrity to antifungal drug tolerance and virulence suppression. Importantly, ERG6 also mediates cell wall integrity and antifungal drug tolerance. We found that C. glabrata mutants lacking ADA2 are hypervirulent in a murine model of systemic infection; this is distinct from findings in C. albicans and C. neoformans, indicating a divergent role of Ada2 in controlling virulence in human fungal pathogens. We further show evidence to support the suggestion that the hypervirulence of C. glabrata ada2 (Cgada2) mutants may be due to the upregulation of multiple adhesin-like genes, the enhanced invasive growth of C. glabrata, and increased murine TNF-α production.
RESULTS
Ada2 mediates acetylation of H3K9 but not H3K14 in C. glabrata.
To test the Ada2-mediated chromatin modification in C. glabrata, we analyzed the acetylation levels at lysine 9 and lysine 14, the main residues acetylated by the SAGA complex in S. cerevisiae, on the C. glabrata histone H3 protein (H3K9 and H3K14, respectively) (32). We extracted the total protein from the wild type, the ada2 mutants, and the complemented strain and performed a Western blot analysis with anti-acetylated H3K9 (anti-H3K9ac) or anti-acetylated H3K14 (anti-H3K14ac) antibody. The acetylation level of H3K9 was lower in ada2 mutants (Fig. 1A) than the wild-type strain; the P values for the significance of the difference between the wild-type strain and ada2 mutants YSJ4 and YSJ39 were <0.0001 and 0.0005, respectively, based on an unpaired t test (Fig. 1B). The H3K9 acetylation level of the complemented strain was similar to that of the wild type. The P values for the significance of the difference in the acetylation levels between the complemented strain and the wild type and ada2 mutants YSJ4 and YSJ39 were 0.1811, <0.0001, and 0.0012, respectively. Unlike the acetylation level of H3K14 in S. cerevisiae, the acetylation level of H3K14 in the C. glabrata ada2 mutants was similar to that in the wild-type and complemented strains (P > 0.05). These results indicate the convergent and divergent roles of Ada2 in the acetylation of H3K9 and H3K14, respectively, between C. glabrata and S. cerevisiae or, alternatively, that Ada2 regulates Gcn5 in C. glabrata differently than it does in S. cerevisiae, based on the finding that H3K14 was acetylated in S. cerevisiae but not C. glabrata.
FIG 1.
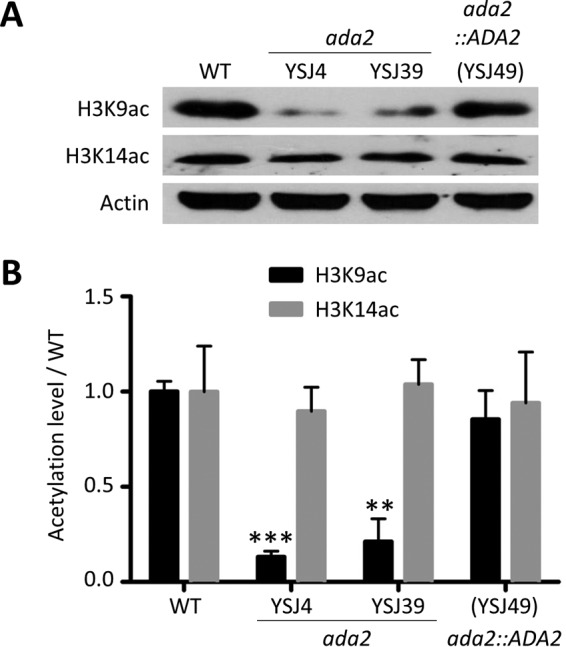
ADA2 deletion results in decreased acetylation of H3K9 but not H3K14. (A) C. glabrata strains were cultured, and the total proteins were extracted to determine the acetylation level of histone 3 via Western blotting. A total of 15 μg of protein was probed with anti-H3K9ac antibody and 45 μg was probed with anti-H3K14ac antibody. Actin was used as a loading control in all blots. (B) Acetylation levels of H3K9 and H3K14 in the C. glabrata wild type (WT), the ada2 mutants, and the complemented strain were determined from three independent Western blot experiments. The blots were analyzed using ImageJ software, and the signals were normalized to the actin signals. The results are represented as the mean ± standard deviation. Asterisks indicate statistically significant differences compared with the results for the wild type using an unpaired t test (**, P < 0.01; ***, P < 0.0001).
Ada2 is required for thermotolerance and growth in C. glabrata.
S. cerevisiae ada2 mutants grow slowly in minimal medium and exhibit temperature sensitivity (23), but the role of the ADA2 gene in C. glabrata remains unclear. In spotting assays, C. glabrata ada2 mutants grew slightly slower on solid YPD than the wild type at 25°C and 37°C but exhibited severe growth defects at 40°C that could be partially rescued with supplementation of the osmotic stabilizer sorbitol (Fig. 2A). We measured the growth kinetics of these strains for 72 h at 25°C, 37°C, and 40°C and found that the ada2 mutants grew slower at 40°C than at 25°C or 37°C. The complemented strain grew at a rate similar to that of the wild-type strain (Fig. 2B). We measured the growth rate (doubling time) of the wild type, the ada2 mutants, and the complemented strain in liquid yeast extract-peptone-dextrose (YPD) and found that the ada2 mutants exhibited a longer doubling time than the wild-type and complemented strains at 25°C and 40°C (Table 1). Interestingly, the ada2 mutants had a slightly shorter doubling time at 37°C than the wild-type and complemented strains (Table 1).
FIG 2.

Ada2 is required for thermotolerance and growth. (A) The growth of ada2 mutants was temperature sensitive, and the osmotic stabilizer sorbitol partially rescued the phenotype at 40°C. Cells were grown overnight in YPD at 37°C and washed twice with dH2O, 5-fold serially diluted, and spotted onto solid YPD with or without 1 M sorbitol. The cultures were incubated at the indicated temperature for 48 h. (B) Growth kinetics of C. glabrata strains at 25°C, 37°C, or 40°C. Cells were grown overnight in liquid YPD at 37°C, washed twice with dH2O, diluted to an OD600 of 0.1 unit/ml in fresh liquid YPD, and incubated at the indicated temperature with shaking at 150 rpm. The OD600 of the cultures was measured at 0, 3, 6, 9, 12, 15, 18, 24, 48, and 72 h. The results were plotted using Prism (v5.03) software, and the error bars represent standard deviations from triplicate technical experiments.
TABLE 1.
Doubling time of C. glabrata strains at various temperatures
| Strain | Doubling time (h) ata: |
||
|---|---|---|---|
| 25°C | 37°C | 40°C | |
| Wild type | 2.30 ± 0.23a,b,c | 2.04 ± 0.02g,h,i | 2.11 ± 0.08m,n,o |
| ada2 mutant (YSJ4) | 2.94 ± 0.04a,d,e | 1.86 ± 0.03g,j,k | 2.26 ± 0.07m,p,q |
| ada2 mutant (YSJ39) | 4.07 ± 0.37b,d,f | 1.89 ± 0.08h,j,l | 2.16 ± 0.02n,p,r |
| ada2::ADA2 mutant (YSJ49) | 2.38 ± 0.08c,e,f | 1.95 ± 0.06i,k,l | 1.98 ± 0.01o,q,r |
Data represent the mean ± standard error of the mean for three technical replicates. In the statistical analyses, P values were determined by an unpaired t test. The P values between two measurements with the same superscript letters were as follows: a, P = 0.049; b, P = 0.015; c, P = 0.739; d, P = 0.038; e, P = 0.003; f, P = 0.011; g, P = 0.016; h, P = 0.191; i, P = 0.228; j, P = 0.844; k, P = 0.345, l, P = 0.647; m, P = 0.258; n, P = 0.614; o, P = 0.189; p, P = 0.274; q, P = 0.021; r, P = 0.002.
Ada2 controls antifungal drug tolerance in C. glabrata.
To determine the role of Ada2 in antifungal drug tolerance, we performed serial dilution spotting, disk diffusion, and MIC assays to determine the role of Ada2 in antifungal drug tolerance. In the serial dilution spotting assay, ada2 mutants were susceptible to azoles (fluconazole [FLC], posaconazole [PSC], voriconazole [VRC]), echinocandins (micafungin [MCF], caspofungin [CSF], anidulafungin [ANF]), and a polyene (amphotericin B [AmB]) (Fig. 3A). In disk diffusion assays, the ada2 mutants demonstrated larger and/or clearer inhibition zones in the presence of fluconazole, micafungin, or amphotericin B than the wild-type and complemented strains (Fig. 3B; see also Fig. S2 in the supplemental material). The ada2 mutants showed 8-fold increased susceptibility to 2 μg/ml fluconazole and 2-fold increased susceptibility to 0.0625 μg/ml micafungin or 0.5 μg/ml amphotericin B compared with the susceptibilities of the wild-type and complemented strains (Table 2).
FIG 3.

Ada2 controls antifungal drug tolerance and cell wall integrity. (A) The ada2 mutants were susceptible to azoles, echinocandins, and amphotericin B. Cells were grown overnight in liquid YPD at 37°C, washed twice with dH2O, 5-fold serially diluted, and spotted onto solid YPD containing fluconazole (FLC), posaconazole (PSC), voriconazole (VRC), micafungin (MCF), caspofungin (CSF), or anidulafungin (ANF) at the indicated concentration. To test susceptibility to amphotericin B (AmB), strains were spotted onto synthetic complete (SC) medium with or without 50 ng/ml AmB. All plates were incubated at 37°C for 30 h. (B) A disk diffusion assay was used to test the susceptibility of the C. glabrata strains to FLC, MCF, or AmB. C. krusei ATCC 6258 and C. parapsilosis ATCC 20019 served as quality control strains. Cells were spread on the surface of Mueller-Hinton agar plus GMB medium plates. Disks containing 20 μg FLC, 40 ng MCF, or 2.5 μg AmB were placed on the agar surface, and the plates were incubated at 35°C for 24 h. (C) The ada2 mutants are sensitive to cell wall-perturbing agents but resistant to the ER stress chemical tunicamycin. Cells were grown overnight in liquid YPD at 37°C, 5-fold serially diluted, spotted onto solid YPD containing SDS, calcofluor white (CFW), Congo red (CR), dithiothreitol (DTT), or tunicamycin (TM), and incubated at 37°C for 30 h.
TABLE 2.
The MICs of C. glabrata strains and two quality control Candida isolates
| Strain | MIC (μg/ml) |
||
|---|---|---|---|
| Fluconazole | Micafungin | Amphotericin B | |
| Wild type | 16 | 0.125 | 1 |
| ada2 mutant (YSJ4) | 2 | 0.0625 | 0.5 |
| ada2 mutant (YSJ39) | 2 | 0.0625 | 0.5 |
| ada2::ADA2 mutant (YSJ49) | 16 | 0.125 | 1 |
| C. krusei ATCC 6258 | 32 | 0.125 | 2 |
| C. parapsilosis ATCC 22019 | 4 | 2 | 2 |
Ada2 is required for cell wall integrity but negatively regulates invasive growth in C. glabrata.
Most existing antifungal drugs target components or pathways involved in cell wall integrity (33). Since ada2 mutants are susceptible to three classes of antifungal drugs, we hypothesized that CgAda2 may play a role in controlling cell wall integrity. To test cell wall integrity in ada2 mutants, we exposed them to the cell wall-perturbing agents sodium dodecyl sulfate (SDS), calcofluor white (CFW), and Congo red (CR). The ada2 mutants were sensitive to these agents (Fig. 3C), indicating that Ada2 plays crucial roles in maintaining cell wall integrity. Furthermore, cell separation ability was reduced in ada2 mutants compared with that in the wild-type and complemented strains (Fig. S1). Because previous studies showed that cell wall integrity is linked with the unfolded protein response (UPR) (34), we investigated whether the UPR was affected in C. glabrata ada2 mutants when treated with the ER stress inducer tunicamycin (TM; which blocks the synthesis of N-linked glycoproteins in the ER). The tolerance of ada2 mutants to TM was increased compared with that of the wild-type and complemented strains (Fig. 3C). Interestingly, the C. glabrata ada2 mutants exhibited enhanced agar invasion compared with the wild-type and complemented strains (Fig. 4A); the ada2 mutants were observed microscopically to extensively invade agar from both the top and side views compared with the level of invasion by the wild-type and complemented strains (Fig. 4B).
FIG 4.

Inactivation of ADA2 enhances agar invasion. (A) C. glabrata strains were grown overnight in YPD at 37°C, washed twice with dH2O, and diluted to an OD600 of 1 unit/ml. Then, 3 μl of cells was spotted onto solid YPD and incubated at 37°C for 10 days. Colonies were photographed before and after washing with dH2O. (B) The colonies from the assay whose results are shown in panel A were washed, excised, and microscopically observed from the top and side. Arrows, the agar surface.
Inactivation of ADA2 leads to hypervirulence.
To study the role of C. glabrata Ada2 in virulence, we used a murine model of systemic infection. Immunocompromised mice were infected with a wild-type strain, an ada2 mutant (YSJ4 or YSJ39), or the complemented strain (YSJ49) in order to determine the survival curve, organ fungal burden, and histopathology. The ada2 mutants (YSJ4 and YSJ39) showed hypervirulence and produced 100% and 90% mortality, respectively, while the wild-type and complemented strains produced only 40% and 20% mortality, respectively, within 3 weeks postinfection (Fig. 5A). The P values for the difference in the rates of mortality between mice infected with the wild-type strain and those infected with ada2 mutant YSJ4 or YSJ39 were 0.001 and 0.012, respectively, as determined by the log-rank test (Fig. 5A). To determine the ability of C. glabrata mutant strains to infect tissues, we performed fungal burden analyses of the spleen, kidneys, and brain. At 48 h postinfection, organs were removed and halved. The organ halves were homogenized, and the homogenates were serially diluted and plated onto solid YPD containing 100 μg/ml chloramphenicol to determine the number of CFU per gram of organ tissue. The ada2 mutants showed increased fungal burdens in the spleen (YSJ4, P < 0.0001; YSJ39, P = 0.0011), kidney (YSJ4, P = 0.0002; YSJ39, P = 0.0002), and brain (YSJ4, P = 0.0003; YSJ39, P = 0.0007) (P values were determined by the Mann-Whitney test), whereas the burden of the complemented strain in the spleen or brain exhibited no difference from that of the wild type, but the burden of the complemented strain in the kidney was slightly lower than that of the wild type (P = 0.017) (Fig. 5B). We performed histopathology analysis using the other halves of the organs, which were sliced and periodic acid-Schiff (PAS) stained to visualize colonization of the spleen, kidney, and brain tissues. The ada2 mutants exhibited a greater ability than the wild-type or complemented strain to colonize the spleen and kidneys (Fig. 5C). However, we did not observe obvious fungal cells in the brain.
FIG 5.

C. glabrata ada2 mutants are hypervirulent in a model of systemic infection in immunosuppressed mice. (A) Survival curves for mice infected with the wild type, the ada2 mutants, or the complemented strain. A total of 10 5- to 6-week-old male ICR mice per strain were immunocompromised with cyclophosphamide (200 mg/kg at days −3, 0, and +1). Each mouse was intravenously challenged with 7 × 107 yeast cells in 200 μl of PBS and monitored for 21 days. (B) The fungal burdens of the spleens, kidneys, and brains of the immunocompromised mice were determined at 48 h after challenge with 7 × 107 cells (in 200 μl) via lateral tail vein injection. The P values for the significance of the differences between the wild type and the mutants or the complemented strain, based on the log-rank test, are shown. (C) Histopathological sections of spleens and kidneys were dissected from immunocompromised mice infected with 7 × 107 cells of the wild type, the ada2 mutants, or the complemented strain. The mice were sacrificed at 48 h postinfection. PAS staining was used to observe C. glabrata colonization. Arrows, C. glabrata cells.
C. glabrata ada2 mutants overstimulate TNF-α production in mice.
Because the C. glabrata ada2 mutants showed hypervirulence in a murine model of systemic infection, we determined murine TNF-α and IL-6 levels. Mice were infected with the wild-type strain, an ada2 mutant, or the complemented strain, and the controls received phosphate-buffered saline (PBS). We collected serum at 18 h postinfection. The levels of TNF-α and IL-6 in PBS-treated mice were undetermined or barely detected, respectively (Table 3). The levels of TNF-α in mice infected with ada2 mutant YSJ4 or YSJ39 were significantly increased compared with those in mice infected with the wild type (P = 0.0008 and P = 0.0185, respectively) or the complemented strain (P < 0.0001 and P = 0.0007, respectively) (P values are based on an unpaired t test). We observed no difference in the levels of IL-6 production between the wild type and the ada2 mutants (Table 3).
TABLE 3.
TNF-α and IL-6 levels in the blood of mice infected with C. glabrata strains
| Strain | Cytokine concna (pg/ml) |
|
|---|---|---|
| TNF-α | IL-6 | |
| Control (PBS) | UD | 0.3 ± 0.2 |
| Wild type | 39.8 ± 7.5a,b,c | 411.0 ± 48.1g,h,i |
| ada2 mutant (YSJ4) | 130.3 ± 12.9a,d,e | 410.3 ± 85.8g,j,k |
| ada2 mutant (YSJ39) | 101.8 ± 16.9b,d,f | 336.6 ± 100.6h,j,l |
| ada2::ADA2 mutant (YSJ49) | 10.2 ± 1.6c,e,f | 216.6 ± 45.5i,k,l |
Data represent the mean ± standard deviation. In the statistical analyses, P values were determined by an unpaired t test. The P values between two measurements with the same superscript letter were as follows: a, P = 0.0008; b, P = 0.018; c, P = 0.0037; d, P = 0.218; e, P = <0.0001; f, P = 0.0007; g, P = 0.994; h, P = 0.560; i, P = 0.022; j, P = 0.592; k, P = 0.081; l, P = 0.308. UD, undetected.
Genome-wide analysis of Ada2-mediated genes in C. glabrata.
To further understand the pleiotropic phenotypes of ada2 mutants and identify Ada2-mediated genes in C. glabrata, we performed RNA sequencing to compare the genome-wide transcriptome profiles of the wild type and ada2 mutant YSJ4. Real-time quantitative reverse-transcription PCR (qRT-PCR) analysis confirmed that the transcription of ADA2 was abolished in the ada2 mutant (Fig. 6A). A total of 485 genes were differentially regulated (i.e., a >2-fold change in expression was detected) between the ada2 mutant and the wild-type strain when grown in liquid YPD (Fig. 6B). Among these Ada2-mediated genes, 442 genes were upregulated and 43 were downregulated in the ada2 mutant. C. glabrata contains more adhesin-like genes than other human fungal pathogens; approximately 67 adhesin-like GPI proteins have been found in the C. glabrata genome (35). Among the identified upregulated genes, 26 were adhesin-like genes (representing 39% of C. glabrata adhesin-like genes); in contrast, only 2 adhesin-like genes (∼3% of C. glabrata adhesin-like genes) were downregulated (Table 4). Among the 26 upregulated adhesin-like genes were 9 EPA genes (EPA1, EPA6, EPA8, EPA10, EPA11, EPA12, EPA15, EPA20, EPA23) and 3 PWP (PA14 domain-containing wall protein) genes (PWP2, PWP6, PWP7). The two downregulated adhesin-like genes in the ada2 mutant were EPA7 and AWP2. In addition to the adhesin-like genes, three genes (YPS4, YPS8, YPS10) belonging to the YPS gene cluster (14) were also upregulated in the ada2 mutant. To confirm the reliability of RNA sequencing analysis, we used real-time qRT-PCR to determine the transcription levels of four upregulated genes (EPA23, EPA20, YPS4, YPS10) and four downregulated genes (GAS3, CAGL0L01771g, CAGL0G05269g, ERG6). The real-time qRT-PCR results were consistent with the RNA sequencing results (Fig. 6C and D).
FIG 6.

Genes regulated by Ada2 in C. glabrata. Strains were grown overnight in liquid YPD at 24°C, washed twice with dH2O, diluted to an OD600 of 0.2 unit/ml with fresh YPD, and incubated at 24°C for 3 h. Log-phase cells were collected for total RNA extraction and further RNA sequencing analyses. (A) The expression of ADA2, measured by real-time qRT-PCR, was undetectable in the ada2 mutant. (B) A total of 485 genes underwent changes in their levels of expression (442 were upregulated and 43 were downregulated) in the ada2 mutant compared to their levels of expression in the wild type. (C) Real-time qRT-PCR was used to confirm the expression of four downregulated genes (GAS3 [CAGL0G1056g], CAGL0L01771g, CAGL0G05269g, and ERG6 [CAGL0H04653g]). (D) Real-time qRT-PCR confirmed the expression of EPA23, YPS4, EPA20, and YPS10, which were upregulated in the ada2 mutant. The results are presented as the mean ± standard deviation. Asterisks indicate statistically significant differences compared with the results for the wild type using an unpaired t test (*, P < 0.01; **, P < 0.001).
TABLE 4.
Genes regulated by Ada2 in C. glabrata
| Gene regulation, function, and ORF | Gene | Fold change in expressiona | S. cerevisiae ortholog |
|---|---|---|---|
| Upregulated adhesin/adhesin-like genes (selected, 26/442) | |||
| CAGL0F09273g | 8.8 | ||
| CAGL0I11011g | 8.3 | ||
| CAGL0G10219g | AWP12 | 5.2 | |
| CAGL0L00157g | 5.1 | ||
| CAGL0I10098g | PWP7 | 3.2 | FLO5 |
| CAGL0I00220g | EPA23 | 2.7 | FLO1 |
| CAGL0I00110g | 2.6 | ||
| CAGL0E00110g | 2.3 | ||
| CAGL0E00275g | EPA20 | 2.2 | FLO5 |
| CAGL0J00110g | 1.8 | ||
| CAGL0E01661g | 1.8 | ||
| CAGL0A01284g | EPA10 | 1.7 | FLO1 |
| CAGL0C00847g | EPA8 | 1.6 | FLO10 |
| CAGL0J01774g | 1.6 | ||
| CAGL0L00227g | 1.5 | ||
| CAGL0G00110g | 1.4 | ||
| CAGL0L10092g | 1.4 | ||
| CAGL0I10246g | PWP2 | 1.3 | FLO1 |
| CAGL0C00110g | EPA6 | 1.3 | FLO1 |
| CAGL0E06644g | EPA1 | 1.3 | FLO1 |
| CAGL0M14069g | PWP6 | 1.2 | FLO9 |
| CAGL0L13299g | EPA11 | 1.2 | FLO9 |
| CAGL0M00132g | EPA12 | 1.2 | FLO1 |
| CAGL0J11968g | EPA15 | 1.2 | FLO5 |
| CAGL0C00968g | 1.2 | ||
| CAGL0I07293g | 1.1 | ||
| Down-regulated genes (43/43) | |||
| Enzyme/enzyme regulator activity | |||
| CAGL0G01056g | GAS3 | −5.9 | GAS2 |
| CAGL0A03102g | ARO10 | −2.4 | ARO10 |
| CAGL0M00176g | BAT2 | −1.7 | BAT2 |
| CAGL0K04235g | −1.5 | NQM1 | |
| CAGL0H04653g | ERG6 | −1.3 | ERG6 |
| CAGL0I01166g | TRR1 | −1.3 | TRR2 |
| CAGL0J11242g | −1.3 | RHO5 | |
| CAGL0F00363g | OPI3 | −1.2 | OPI3 |
| CAGL0F02563g | HPT1 | −1.2 | HPT1 |
| CAGL0B01122g | SAM1 | −1.1 | SAM1 |
| CAGL0M08602g | −1.1 | CCC2 | |
| CAGL0B00990g | HBN1 | −1.1 | FRM2 |
| CAGL0H04081g | ERG13 | −1.1 | HCS1 |
| CAGL0K06259g | TSA1 | −1.1 | TSA2 |
| Transcription factor/DNA, RNA, protein, or lipid binding activity | |||
| CAGL0L01771g | −4.6 | PHD1 | |
| CAGL0B04213g | −1.6 | RGC1 | |
| CAGL0H08712g | −1.6 | ||
| CAGL0H08195g | −1.3 | STI1 | |
| CAGL0B00792g | −1.3 | SRO9 | |
| CAGL0K08756g | AP5 | −1.2 | YAP5 |
| CAGL0H05511g | −1.2 | RPS9A | |
| CAGL0B04675g | −1.2 | DOM34 | |
| CAGL0D05368g | −1.1 | SRB6 | |
| CAGL0K04741g | SSB2 | −1.0 | SSB2 |
| Molecular function | |||
| CAGL0G05269g | −2.5 | FMP16 | |
| CAGL0G03531g | −2.2 | SPR6 | |
| CAGL0H07469g | −1.9 | ICS2 | |
| CAGL0G06798g | −1.7 | LSO1 | |
| CAGL0F00187g | FET4 | −1.2 | FET4 |
| CAGL0A01474g | −1.2 | SCW11 | |
| CAGL0M00902g | −1.2 | DIF1 | |
| CAGL0L04510g | −1.1 | RPS28A | |
| CAGL0J11462g | −1.1 | YNL190W | |
| Transporter | |||
| CAGL0M06281g | −2.4 | DTR1 | |
| CAGL0A02321g | HXT3 | −1.4 | HXT5 |
| CAGL0H08393g | −1.2 | BAP2 | |
| CAGL0H10076g | −1.1 | YRO2 | |
| Adhesin/adhesin-like genes | |||
| CAGL0K00110g | AWP2 | −1.5 | |
| CAGL0C05643g | EPA7 | −1.1 | FLO9 |
| Unknown function | |||
| CAGL0F00715g | −1.5 | MAK16 | |
| CAGL0K03069g | −1.3 | IRC21 | |
| CAGL0G06050g | −1.1 |
Log2 ratio, ada2 mutant/wild type.
C. glabrata ADA2 mediates antifungal drug tolerance and cell wall integrity via the downstream target ERG6.
To identify potential downstream targets of ADA2, we disrupted several genes that we found were regulated by ADA2, including GAS3, CAGL0L01771g, and ERG6. The gas3 and CAGL0L01771g mutants did not exhibit phenotypes similar to those of the ada2 mutants (data not shown). Deletion of ERG6, a C24 sterol methyltransferase involved in the ergosterol biosynthesis pathway, demonstrated echinocandin susceptibility similar to that of the ada2 mutants, while fluconazole susceptibility was intermediate between that of the wild type and the ada2 mutants (Fig. 7). In addition, the erg6 mutants phenocopied ada2 mutants in terms of sensitivity to the cell wall-perturbing agents SDS, CFW, CR, and dithiothreitol (DTT) (Fig. 7). However, while the ada2 mutants were tolerant to tunicamycin, the erg6 mutants showed sensitivity to tunicamycin compared with the wild type (Fig. 7).
FIG 7.

ADA2 control of antifungal drug tolerance and cell wall integrity is mediated by ERG6. The erg6 mutants were susceptible to echinocandins, fluconazole, cell wall-perturbing agents, and the ER stress chemical tunicamycin but not amphotericin B. Cells were grown overnight in YPD at 37°C and washed twice with dH2O, 5-fold serially diluted, and spotted onto YPD medium containing MCF, CSF, ANF, FLC, PSC, VRC, SDS, CFW, CR, DTT, or TM at the indicated concentration. Strains were spotted onto SC medium with or without 50 ng/ml AmB to test tolerance to amphotericin B. All plates were incubated at 37°C for 30 h.
DISCUSSION
Roles of Ada2 in thermotolerance and cell wall integrity.
Thermotolerance and adaptation to elevated temperatures are important traits for human fungal pathogens to successfully establish infection. The transcription adaptor Ada2 is involved in thermotolerance in several fungi. For example, the S. cerevisiae ada2 mutant exhibits temperature sensitivity at 37°C (36), while C. neoformans strains without ADA2 have growth defects at 37°C and 39°C (29). In contrast, C. albicans Ada2 does not mediate thermotolerance (27). In this study, we showed that C. glabrata ada2 mutants exhibit severe growth defects at 40°C that can be partially rescued with supplementation of the osmotic stabilizer sorbitol, suggesting that CgAda2 contributes to thermotolerance via maintenance of cell wall integrity. Recent research showed that S. cerevisiae SAGA complex subunits, such as Gcn5, are recruited to the promoters of the cell wall integrity response genes in an Slt2-, Rlm1-, and SWI/SNF-dependent manner, and deletion mutants of GCN5 are sensitive to CR, thus establishing a potential link between the SAGA complex and cell wall integrity (37). However, whether this phenomenon is conserved in C. glabrata requires further study.
In this study, we showed that C. glabrata ada2 mutants are sensitive to elevated temperatures but resistant to the ER stress chemical tunicamycin. Accumulation of unfolded proteins in the ER triggers the unfolded protein response (UPR) pathway, and the ER stress response is related to thermotolerance. For example, in C. glabrata the UPR pathway is mainly mediated by the calcineurin pathway and is partially mediated by the Slt2 pathway (38). Inactivation of calcineurin or SLT2 in C. glabrata results in increased sensitivity to tunicamycin and elevated temperatures (38–40). Unlike the tunicamycin sensitivity of cell wall-defective strains, such as calcineurin mutants (40), the C. glabrata ada2 mutants showed resistance to tunicamycin, indicating that C. glabrata Ada2 and calcineurin might control tunicamycin tolerance in distinct pathways.
Antifungal drug tolerance.
Ada2 controls not only cell wall integrity but also antifungal drug tolerance in C. glabrata as well as C. albicans (41). It is known that C. albicans Ada2 binds to the promoter of the multidrug transporter CDR1 or the multidrug efflux pump MDR1, and the gene expression levels of CDR1 and MDR1 in the C. albicans ada2/ada2 mutant are lower than those in the wild type when exposed to fluconazole (27). However, in C. glabrata we did not find that Ada2 regulates ABC transporter-encoding genes, such as CDR1, CDR2 (PDH1), or SNQ2, based on RNA sequencing results. Possible explanations for this difference may involve divergent roles of Ada2 between C. glabrata and C. albicans in controlling the expression of the multidrug transporter or efflux pump genes or simply different incubation conditions. In contrast to the C. glabrata and C. albicans Ada2 proteins, C. neoformans Ada2 does not influence fluconazole tolerance (29), suggesting divergent roles of Ada2 in fluconazole tolerance between Candida and Cryptococcus species. Meanwhile, this study showed that Cgada2 mutants are susceptible to amphotericin B, which is distinct from the findings for the C. neoformans ada2 mutant, which does not exhibit amphotericin B susceptibility (29). However, it is unknown whether the C. albicans ada2/ada2 mutant is susceptible to amphotericin B.
Hypervirulence.
Despite the defects of the C. glabrata ada2 mutants in cell wall integrity and growth at elevated temperatures (except for a shorter doubling time in liquid medium at 37°C), we found, surprisingly, that the ada2 mutants have enhanced virulence in a model of systemic infection in immunosuppressed mice. Previous studies showed that C. albicans ada2/ada2 and C. neoformans ada2 mutants exhibit attenuated virulence in murine models (27, 29), which is distinct from the increased virulence of the C. glabrata ada2 mutant, indicating divergent roles of Ada2 in governing virulence between C. glabrata and C. albicans or C. neoformans. The slightly shorter doubling time of the C. glabrata ada2 mutants in the exponential phase at 37°C might partially explain the observed hypervirulence.
The proinflammatory cytokines TNF-α and IL-6 are crucial for host defense, and their levels reflect the severity of Candida infection in humans (42). The ace2 mutant is the first reported hypervirulent strain in C. glabrata, with hypervirulence being attributed to overstimulation of the host cytokines IL-6 and TNF-α (15). For example, the Evo isolate is a hypervirulent C. glabrata strain, and infection with this strain results in increased production of IL-6 and TNF-α in the host brain tissue instead of the serum during early murine infection (16). Based on our results, we suggest that the hypervirulence of the Cgada2 mutants is due at least in part to the overstimulation of TNF-α during murine infection (Table 3 and Fig. 8).
FIG 8.

Proposed role of Ada2 in C. glabrata. C. glabrata Ada2 functions as a positive or a negative regulator governing pleiotropic phenotypes. CgAda2 positively regulates growth, cell wall integrity, and echinocandin and fluconazole (FLC) tolerance, which is mediated at least in part by Erg6. Additionally, Ada2 contributes to tolerance to posaconazole (PSC), voriconazole (VRC), amphotericin B, and thermal stress. Meanwhile, Ada2 negatively regulates, possibly with mediation by Erg6, ER stress tolerance, which potentially inhibits genes such as ADA2 or other undiscovered genes that negatively regulate ER stress tolerance. Interestingly, Ada2 also negatively regulates virulence in a murine model of systemic infection. The hypervirulence of ada2 mutants might be caused by the upregulation of multiple adhesin-like genes, increased agar invasion, and overstimulation of murine TNF-α production. Dashed lines, potential relationships.
The enhanced agar invasion observed in the Cgada2 mutants may be linked to the hypervirulent phenotype, although the relationship between agar invasion and virulence in C. glabrata is unclear. However, C. glabrata mutants, such as the tpo1_1 and tpo1_2 mutants, which have a reduced agar invasion ability compared with that of the wild type, show reduced virulence in a Galleria mellonella infection model (43). Further, the C. albicans fluconazole-resistant strain CO23RFLC and the micafungin-resistant strain CO23RFK, both of which are derived from the fluconazole- and micafungin-susceptible strain CO23S, showed increased virulence in a murine model of systemic infection and formed branching hyphae that invaded the surface of the agar, unlike the hyphae formed by CO23S, and that were longer than the hyphae formed by CO23S (44), suggesting a possible relationship between increased virulence and agar invasion in both C. glabrata and C. albicans. Thus, it is possible that the enhanced agar invasion of Cgada2 mutants might be linked with hypervirulence (Fig. 4 and 8).
Among a total of 67 C. glabrata adhesion genes, 26 genes were upregulated in the Cgada2 mutant, while only 2 genes were downregulated in the Cgada2 mutant. Thus, the loss of ADA2, resulting in the activation of 26 adhesin-like genes, might potentially contribute to the hypervirulence of Cgada2 mutants (Table 4 and Fig. 8). Previous research showed that silencing mutations in C. glabrata cells can result in the derepression of three EPA genes (EPA1, EPA6, and EPA7) and hyperadhesion to several epithelial cell lines (45). For instance, a hyperadherent C. glabrata sir3 mutant exhibited increased colonization in murine kidneys compared to the wild type (46), suggesting that adherence might be correlated with virulence in C. glabrata.
In this study, we demonstrated that C. glabrata Ada2 is required for growth, thermotolerance, cell wall integrity, and tolerance to three classes of antifungal drugs. Importantly, Ada2 control of cell wall integrity and antifungal drug tolerance is partially mediated by Erg6. The loss of Ada2 in C. glabrata results in hypervirulence in a murine model of systemic infection, which is in contrast to the attenuated virulence seen in C. albicans and C. neoformans ada2 mutants, suggesting a divergent role of Ada2 in governing virulence between different human fungal pathogens. The upregulation of 26 adhesin-like genes, increased agar invasion, and increased TNF-α production in host mice may contribute to the observed hypervirulence (Fig. 8). Future investigations on the signaling pathways and proteins or DNA regions that physically interact with C. glabrata Ada2 or other SAGA complex components, such as Ada3 or Gcn5, will allow us to further decipher the mechanisms governing antifungal drug tolerance and virulence in C. glabrata.
MATERIALS AND METHODS
Yeast strains, media, and chemicals.
The C. glabrata strains used in this study are listed in Table 5. Yeast extract-peptone-dextrose (YPD; 1% yeast extract, 2% peptone, 2% glucose) liquid and agar (2%), synthetic complete (SC) medium [0.17% yeast nitrogen base without amino acids, 0.5% (NH4)2SO4, 2% glucose, and 2% agar], Mueller-Hinton agar plus GMB medium (Mueller-Hinton agar, 2% glucose, 0.5 μg/ml methylene blue), and RPMI 1640 medium (Sigma, St. Louis, MO, USA) were used in this study. YPD containing 100 μg/ml nourseothricin (Werner BioAgents, Jena, Germany) was used to select transformants. Sodium dodecyl sulfate (SDS; Bioman, New Taipei City, Taiwan), calcofluor white (CFW; Fluorescent Brighter 28; Sigma), Congo red (CR; Genzyme, Cambridge, MA, USA), fluconazole (FLC; Selleckchem, Houston, TX, USA), posaconazole (PSC; Merck, Rahway, NJ, USA), voriconazole (VRC; Sigma), micafungin (MCF; Astellas Pharma Inc., Deerfield, IL, USA), caspofungin (CSF; Merck), anidulafungin (ANF; Pfizer Inc., Groton, CT, USA), dithiothreitol (DTT; BioShop, Burlington, ON, Canada), and tunicamycin (TM, Sigma) were added to the media at the concentrations indicated above and below.
TABLE 5.
Candida strains used in this study
| Species | Strain | Genotype | Parent strain | Reference or source |
|---|---|---|---|---|
| C. glabrata | CBS138/ATCC 2001 | Prototrophic wild type | Clinical isolate | 47 |
| C. glabrata | YSJ4 | ada2Δ::SAT1 | CBS138 | This study |
| C. glabrata | YSJ39 | ada2Δ::SAT1-FLP | CBS138 | This study |
| C. glabrata | YSJ43 | ada2Δ::FRT | YSJ39 | This study |
| C. glabrata | YSJ49 | ada2Δ::FRT::ADA2-SAT1 | YSJ43 | This study |
| C. glabrata | YSJ93 | erg6Δ::SAT1-FLP | CBS138 | This study |
| C. glabrata | YSJ95 | erg6Δ::SAT1-FLP | CBS138 | This study |
| C. krusei | CBS573/ATCC 6258 | Prototrophic wild type | Clinical isolate | 52 |
| C. parapsilosis | CBS604/ATCC 20019 | Prototrophic wild type | Clinical isolate | 52 |
Gene disruption and complementation in C. glabrata.
All deletion mutants were generated from the prototrophic wild-type strain CBS138 (47) using the SAT1 marker or SAT1 flipper (48). To disrupt the ADA2 (CAGL0K06193g) gene, approximately 1 kb of the 5′ and 3′ noncoding region (NCR) of the ADA2 open reading frame (ORF) (for homologous recombination) was PCR amplified with primers JC641/JC764 (for the 5′ NCR) and JC765/JC644 (for the 3′ NCR) (see Table S1 in the supplemental material) from the genomic DNA of the CBS138 strain. The 1.8 kb of the SAT1 marker was amplified from the plasmid pSFS2A (48) with primers JC723/JC724. Three PCR products, including the 5′ NCR of ADA2 (5′ ADA2NCR), the 5′ NCR of the SAT1 marker, and the 3′ NCR of ADA2 (3′ ADA2NCR), were treated with the ExoSAP-IT reagent (USB Corp., Cleveland, OH, USA) to remove the remaining primers and deoxynucleoside triphosphates (dNTPs), and we measured the DNA concentration using a SpectraMax 190 microplate reader (Molecular Devices, CA, USA). The products were combined at a 1:3:1 molar ratio (5′ ADA2NCR/SAT1 marker/3′ ADA2NCR) to generate an ∼3.8-kb disruption cassette by overlap PCR with primers JC641/JC644. Wild-type strain CBS138 was then transformed with 1 to 5 μg of the 5′ ADA2NCR-SAT1 marker-3′ ADA2NCR disruption cassette using a modified lithium acetate transformation method (49), and the first nourseothricin-resistant ada2 mutant (YSJ4) was obtained. To make a second independent nourseothricin-resistant ada2 mutant, approximately 1 kb of the 5′ and 3′ NCRs of the ADA2 gene were PCR amplified from the genomic DNA of the CBS138 strain with primers JC885/JC886 (for the 5′ NCR) and JC887/JC888 (for the 3′ NCR). The 3′ and 5′ NCR PCR products of the ADA2 gene were each digested with two restriction enzymes: SacII/SacI and KpnI/ApaI, respectively. The SacII/SacI-digested PCR products of 3′ ADA2NCR were gel purified and cloned into the plasmid pSFS2A, resulting in plasmid pYSJ32 (Table S2). The KpnI/ApaI-digested 5′ ADA2NCR PCR products were gel purified and cloned into pYSJ32 to make the ADA2 disruption plasmid pYSJ36. The pYSJ36 plasmids were digested with KpnI and SacI to release the 5′ ADA2NCR–SAT1-FLP–3′ ADA2NCR disruption cassette and transform the CBS138 strain to obtain YSJ39, the second independent ada2 mutant with SAT1-FLP.
To obtain an ADA2 complementation strain, we restored the ADA2 gene in the ada2 mutant background (YSJ43). SAT1-FLP was removed by culturing YSJ39 in YPD medium and replica plating onto nourseothricin-containing medium to confirm the loss of SAT1-FLP. Approximately 200 bp of the 5′ NCR and the ADA2 ORF was PCR amplified from the CBS138 strain with primers JC893/JC894 and digested with the restriction enzymes KpnI and HindIII. The digested PCR products were gel purified and cloned into pYSJ32 to generate the reconstitution plasmid pYSJ46 and then digested with KpnI and SacI to release the complementation cassette 5′ ADA2NCR-ADA2 ORF-SAT1-3′ ADA2NCR. We transformed this cassette into an ada2 mutant background (YSJ43) to obtain the ADA2-complemented strain YSJ49 (Table 5).
To create an erg6 mutant, approximately 1 kb of the 5′ and 3′ NCRs of the ERG6 (CAGL0H04653g) gene was PCR amplified from the genomic DNA of the CBS138 strain with primers JC974/JC975 (for the 3′ NCR) and JC972/JC973 (for the 5′ NCR). The 3′ and 5′ NCR PCR products of the ERG6 gene were each digested with two restriction enzymes: SacII/SacI and KpnI/ApaI, respectively. The SacII/SacI-digested PCR products of 3′ ERG6NCR were gel purified and cloned into the plasmid pSFS2A, resulting in the plasmid pYSJ89 (Table S2). We then gel purified the KpnI/ApaI-digested 5′ ERG6NCR PCR products and cloned them into pYSJ89 to make the ERG6 disruption plasmid pYSJ91. The pYSJ91 plasmid was digested with KpnI and SacI to release the 5′ ERG6NCR–SAT1-FLP–3′ ERG6NCR disruption cassette and transform it into the CBS138 strain. YSJ93 and YSJ95, two independent erg6 mutants derived from two separate transformations, were obtained. All strains were confirmed by amplification of the ORF and 5′ and 3′ NCR integrations of the disruption cassettes.
Determination of acetylation level of H3K9 and H3K14.
For Western blot analyses (50), cells were grown overnight at 37°C, diluted to an optical density at 600 nm (OD600) of 0.5 unit/ml with 50 ml fresh YPD broth, and incubated for 4 h at 37°C with shaking at 200 rpm. Cells were then centrifuged, washed once with distilled H2O (dH2O), and resuspended in 800 μl extraction buffer (25 mM Tris-HCl, pH 7.4, 0.5 mM EDTA, 0.5 mM EGTA, 10 mM β-mercaptoethanol, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride, 1:50 protease inhibitor cocktail [Roche Diagnostics, Mannheim, Germany]). Total protein was isolated using the glass bead method with 0.5-mm-diameter glass beads; samples were homogenized five times for 1 min each time at 1,750 rpm with a 2010 Geno/Grinder homogenizer (Spex SamplePrep, Metuchen, NJ, USA). Samples were placed on ice between each homogenization. Protein concentrations were determined by the Bradford method (Bio-Rad, Hercules, CA, USA). Then, 15 μg (for H3K9ac) or 45 μg (for H3K14ac) of total protein was separated on SDS-PAGE gels and transferred to nitrocellulose membranes. The membranes were blocked with 5% Silk milk (Fonterra, Auckland City, New Zealand) in Tris-buffered saline with Tween 20 (TBST; 20 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Tween 20) and incubated with primary antiactin antibody (catalog number MAB1501; Merck Millipore), anti-H3K9ac antibody (catalog number ab4441; Abcam, Cambridge, MA, USA), or anti-H3K14ac antibody (catalog number ab52946; Abcam, Cambridge, MA, USA) overnight at 4°C with shaking. The membranes were washed five times with TBST buffer and then incubated with secondary horseradish peroxidase-conjugated goat anti-mouse IgG antibody (catalog number AP124P; Merck Millipore, Billerica, MA, USA) or goat anti-rabbit IgG antibody (catalog number ab205718; Abcam, Cambridge, MA, USA). Chemiluminescence signals were detected using an enhanced chemiluminescence system (T-Pro Biotechnology, New Taipei City, Taiwan). The Western blot analyses were repeated three times independently, and signals were analyzed using ImageJ software (https://imagej.nih.gov). The statistical analysis of P values was performed using an unpaired t test. Bar graphs of the quantity normalized to that of the wild type were created with Prism (v5.03) software.
Serial dilution spotting assays.
Cells were grown overnight at 37°C and washed twice with dH2O. We measured the absorbance of the cells at 600 nm (OD600) with an Ultrospec 10 cell density meter (Amersham Biosciences, Uppsala, Sweden). Cells were resuspended to an OD600 of 1 unit/ml with dH2O, and 3 μl of a 5-fold serial dilution of each strain was spotted onto solid agar plates and incubated at the temperatures and for the times indicated above.
Growth kinetics and doubling time measurement.
For growth kinetics assays, cells were grown overnight at 37°C, washed twice with dH2O, and diluted to an OD600 of 0.1 with fresh YPD broth. Then, 5 ml of diluted cells was cultured at 25°C, 37°C, and 40°C with shaking at 150 rpm. We measured the cell concentrations after 0, 3, 6, 9, 12, 15, 18, 24, 48, and 72 h using a SpectraMax 190 microplate reader. The experiments were performed in triplicate, and the data were plotted using Prism (v5.03) software. Measurement of the doubling time of each strain was calculated with the following formula: Tln 2/[ln(ODT/ODT0)], where ODT and ODT0 represent the OD600 at time T and time zero, respectively (51). The exponential-phase time points from 3 to 9 h were chosen to calculate the doubling time. The P values were determined using a nonparametric t test.
Observation of cell separation ability.
C. glabrata strains were grown overnight in YPD broth at 37°C, washed twice with dH2O, stained with 1 mg/ml calcofluor white (Fluorescent Brighter 28; Sigma) for 5 min at room temperature, and then washed once with dH2O. Stained cell suspensions were spotted onto slides and visualized at a ×1,000 magnification under a bright field and UV light.
Disk diffusion susceptibility assays.
Disk diffusion susceptibility assays with fluconazole, micafungin, and amphotericin B were performed following Clinical and Laboratory Standards Institute (CLSI) guideline M44-A2 (52). In brief, C. glabrata strains were plated onto Mueller-Hinton agar plus GMB medium plates, and sterile disks were applied onto the agar surface. Then, 20 μg of fluconazole, 40 ng of micafungin, or 2.5 μg of amphotericin B was dropped into each disk. The plates were incubated for 24 h at 35°C and photographed.
Determination of MICs.
To determine the MICs, we followed CLSI guideline M27-A3 (53). In brief, we added 100 μl of serially diluted drugs (2-fold the final concentration) into 100-μl cell suspensions in a 96-well polystyrene plate. The final cell concentrations were between 0.5 × 103 and 2.5 × 103 CFU/ml. The plates were incubated for 24 h at 35°C without shaking. The quality control strains Candida krusei ATCC 6258 and Candida parapsilosis ATCC 22019 were used to ensure that the drugs, medium, and procedure were reliable.
Agar invasion assay.
C. glabrata strains were grown overnight in YPD broth at 37°C and washed twice with dH2O. The cell suspensions were diluted to an OD600 of 1 unit/ml, and 3 μl was spotted onto solid YPD medium and incubated at 37°C for 10 days. Cells were removed from the surface by rubbing with gloved fingers, and then the plates were washed with 200 ml dH2O. Colonies were photographed before and after removal from the agar surface. The agar was excised after the cells were removed from the agar surface. We microscopically observed and photographed the invading cells from the top and side.
Murine model of systemic infection.
Animal studies were conducted in the Department of Plant Pathology and Microbiology at National Taiwan University (NTU). The murine systemic infection models were reviewed and approved by the NTU Institutional Animal Care and Use Committee (IACUC) under approval number NTU-102-EL-36.
Five- to 6-week-old male ICR mice (BioLasco Taiwan Co., Ltd.) were used in the study. For the survival test, groups of 10 mice were immunosuppressed with cyclophosphamide (200 mg/kg of body weight; Sigma) at day −3, day 0, and day +1. C. glabrata strains were grown in liquid YPD overnight at 37°C with shaking at 200 rpm, washed twice with phosphate-buffered saline (PBS), and resuspended in PBS. The cells were vigorously vortexed for 2 min in order to reduce the clumping of ada2 mutant cells and then counted with a hemocytometer and diluted with an appropriate amount of PBS to obtain an infection inoculum concentration of 3.5 × 108 cells/ml. Then, 200 μl (7 × 107 cells) was used to infect mice through the lateral tail vein. To assess cell viability, appropriate dilutions of the cells were plated on solid YPD and incubated at 37°C for 48 h. The course of infection was monitored for 21 days. Statistical analyses by the Mantel-Cox log-rank test were used to determine the P values for the significance of the difference between the survival curves of each strain; statistical significance was set at a P value of <0.05. For fungal burden analyses, 10 mice were immunosuppressed and infected with C. glabrata strains as described above. The mice were sacrificed and dissected at 48 h after inoculation. The spleens, kidneys, and brains of C. glabrata-infected mice were removed, weighed, and transferred to 2-ml sample tubes containing 1 ml ice-cold PBS. Samples were homogenized with steel beads for 2 min at 1,750 rpm. Tissue homogenates were serially diluted, and 100 μl of each dilution was plated on solid YPD containing 100 μg/ml chloramphenicol and incubated at 37°C for 48 h in order to determine the number of CFU per gram of organ tissue. The identity of the colonies recovered from the organs was confirmed by colony PCR. The P values for the significance of the difference between the fungal burdens of each strain were determined via the Mann-Whitney test. For histopathological analyses, half of the spleen and kidney was fixed with 10% formamide (Sigma). Periodic acid-Schiff (PAS) staining was performed at the Department of Veterinary Medicine at National Taiwan University.
Analyses of immune modulators in mice.
Five- to 6-week-old male ICR mice (BioLasco Taiwan Co., Ltd.) were used in the study. Mice were immunosuppressed with cyclophosphamide (200 mg/kg; Sigma) at day −3 and day 0. The preparation of the C. glabrata inoculum was as described above. Four mice were infected with 7 × 107 cells of the C. glabrata wild type (200 μl) by lateral tail vein injection, while five mice were infected with PBS as controls; one mouse from the group infected with the wild type expired during the lateral tail vein injection. Blood was collected via the submandibular blood collection method at 18 h postinfection. The tube of blood was allowed to stand for 1 h and then was centrifuged for 20 min at 1,750 rpm at room temperature to collect serum, which was stored at −20°C. The interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) serum levels were determined by the use of mouse IL-6 and TNF-α enzyme-linked immunosorbent assay Ready-Set-Go kits (eBioscience, San Diego, CA, USA), respectively. Statistical analysis to determine P values for the significance of differences between the groups was performed with an unpaired t test.
RNA sequencing experiments.
Strains were grown overnight in liquid YPD at 24°C, washed twice with dH2O, diluted to an OD600 of 0.2 unit/ml with fresh YPD, and incubated for 3 h at 24°C with shaking at 250 rpm. Then, 20 ml of the culture was immediately added to 30 ml of methanol precooled to −80°C in order to stop cellular processes and RNase activity. Cells were then centrifuged at −4°C for 10 min at 3,250 rpm. Total RNA was extracted using a RiboPure-Yeast kit (Ambion) and treated with DNase I (Ambion) to degrade contaminating DNA. mRNA was enriched with oligo(dT) magnetic beads. Fragmentation buffer allowed the mRNA to be shortened into ∼200-base fragments. Then, the first strand of cDNA was synthesized by the use of a random hexamer, buffer, dNTPs, and RNase H, and the second strand of cDNA was synthesized by the use of DNA polymerase I. The double-stranded cDNAs were purified with magnetic beads. End preparation and 3′ end single nucleotide adenine addition were performed, and sequencing adaptors were ligated to the fragments, which were enriched by PCR amplification. During the quality control step, an Agilent 2100 bioanalyzer and ABI StepOnePlus real-time PCR system were used to qualify and quantify the sample library. The library products were sequenced via an Illumina HiSeq 2000 instrument.
Real-time qRT-PCR.
For real-time qRT-PCR, total RNA extracted as described above was treated with the reagents from a Turbo DNA-free kit (Invitrogen, Carlsbad, CA, USA) to eliminate genomic DNA contamination. Then, 2 μg of DNA-free total RNAs was reverse transcribed to cDNA by a high-capacity cDNA reverse transcription kit (Applied Biosystems). The 20-μl reaction volume of the real-time PCR mixtures included 6 μl cDNA (6 ng), 10 μl of 2× quantitative PCR master mix (Fast SYBR green master mix; Applied Biosystems), 2 μl of 2.5 μM forward primer, and 2 μl of 2.5 μM reverse primer. The primers were designed using Primer Express (v3.0) software (Applied Biosystems) (Table S1). Quantitative PCR conditions were as follows: 95°C for 10 min for denaturation, 95°C for 3 s and 60°C for 30 s (40 cycles), 95°C for 15 s, 60°C for 60 s, and 95°C for 15 s (melting curve). A StepOnePlus system and a StepOne (v2.2) system (Applied Biosystems) were used to determine the cycle threshold (CT) values, and the relative expression levels were calculated on the basis of the 2−ΔΔCT value. Bar graphs of the ACT1-normalized relative quantity compared with that for the wild type (strain CBS138) were created with Prism (v5.03) software. P values for the significance of the difference in the RT-quantitative PCR results between the wild type and the ada2 mutant (YSJ4) were determined by an unpaired t test.
Accession number(s).
We deposited the RNA sequences in the NCBI Gene Expression Omnibus (GEO) database under accession number GSE76338 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM1981429).
Supplementary Material
ACKNOWLEDGMENTS
We thank Joachim Morschhäuser for providing the pSFS2A plasmid and Cecelia Wall for language editing.
This work was financially supported by grants 102-2320-B-002-041-MY2, 104-2320-B-002-063-MY3, and 106-2923-B-002-001-MY3 from the Ministry of Science & Technology and grant 104AS-10.7.3-BQ-B1(5) from the Bureau of Animal and Plant Health Inspection and Quarantine in Taiwan.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01924-17.
REFERENCES
- 1.Azie N, Neofytos D, Pfaller M, Meier-Kriesche H-U, Quan S-P, Horn D. 2012. The PATH (Prospective Antifungal Therapy) Alliance® registry and invasive fungal infections: update 2012. Diagn Microbiol Infect Dis 73:293–300. doi: 10.1016/j.diagmicrobio.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 2.Guinea J. 2014. Global trends in the distribution of Candida species causing candidemia. Clin Microbiol Infect 20:5–10. doi: 10.1111/1469-0691.12539. [DOI] [PubMed] [Google Scholar]
- 3.Pfaller M, Diekema D. 2007. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klevay MJ, Horn DL, Neofytos D, Pfaller MA, Diekema DJ. 2009. Initial treatment and outcome of Candida glabrata versus Candida albicans bloodstream infection. Diagn Microbiol Infect Dis 64:152–157. doi: 10.1016/j.diagmicrobio.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Calderone RA, Fonzi WA. 2001. Virulence factors of Candida albicans. Trends Microbiol 9:327–335. doi: 10.1016/S0966-842X(01)02094-7. [DOI] [PubMed] [Google Scholar]
- 6.Hoyer LL, Green CB, Oh S-H, Zhao X. 2008. Discovering the secrets of the Candida albicans agglutinin-like sequence (ALS) gene family—a sticky pursuit. Med Mycol 46:1–15. doi: 10.1080/13693780701435317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nobile CJ, Schneider HA, Nett JE, Sheppard DC, Filler SG, Andes DR, Mitchell AP. 2008. Complementary adhesin function in C. albicans biofilm formation. Curr Biol 18:1017–1024. doi: 10.1016/j.cub.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cormack BP, Ghori N, Falkow S. 1999. An adhesin of the yeast pathogen Candida glabrata mediating adherence to human epithelial cells. Science 285:578–582. doi: 10.1126/science.285.5427.578. [DOI] [PubMed] [Google Scholar]
- 9.Kuhn DM, Vyas VK. 2012. The Candida glabrata adhesin Epa1p causes adhesion, phagocytosis, and cytokine secretion by innate immune cells. FEMS Yeast Res 12:398–414. doi: 10.1111/j.1567-1364.2011.00785.x. [DOI] [PubMed] [Google Scholar]
- 10.Brunke S, Hube B. 2013. Two unlike cousins: Candida albicans and C. glabrata infection strategies. Cell Microbiol 15:701–708. doi: 10.1111/cmi.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hube B, Sanglard D, Odds FC, Hess D, Monod M, Schäfer W, Brown A, Gow N. 1997. Disruption of each of the secreted aspartyl proteinase genes SAP1, SAP2, and SAP3 of Candida albicans attenuates virulence. Infect Immun 65:3529–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Bernardis F, Arancia S, Morelli L, Hube B, Sanglard D, Schäfer W, Cassone A. 1999. Evidence that members of the secretory aspartyl proteinase gene family, in particular SAP2, are virulence factors for Candida vaginitis. J Infect Dis 179:201–208. doi: 10.1086/314546. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y-L, de Bernardis F, Yu S-J, Sandini S, Kauffman S, Tams RN, Bethea E, Reynolds TB. 2015. Candida albicans OPI1 regulates filamentous growth and virulence in vaginal infections, but not inositol biosynthesis. PLoS One 10:e0116974. doi: 10.1371/journal.pone.0116974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaur R, Ma B, Cormack BP. 2007. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc Natl Acad Sci U S A 104:7628–7633. doi: 10.1073/pnas.0611195104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamran M, Calcagno A-M, Findon H, Bignell E, Jones MD, Warn P, Hopkins P, Denning DW, Butler G, Rogers T. 2004. Inactivation of transcription factor gene ACE2 in the fungal pathogen Candida glabrata results in hypervirulence. Eukaryot Cell 3:546–552. doi: 10.1128/EC.3.2.546-552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brunke S, Seider K, Fischer D, Jacobsen ID, Kasper L, Jablonowski N, Wartenberg A, Bader O, Enache-Angoulvant A, Schaller M. 2014. One small step for a yeast-microevolution within macrophages renders Candida glabrata hypervirulent due to a single point mutation. PLoS Pathog 10:e1004478. doi: 10.1371/journal.ppat.1004478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, Rohde B, Bauser C, Bader O, Sanglard D. 2009. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog 5:e1000268. doi: 10.1371/journal.ppat.1000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrari S, Sanguinetti M, De Bernardis F, Torelli R, Posteraro B, Vandeputte P, Sanglard D. 2011. Loss of mitochondrial functions associated with azole resistance in Candida glabrata results in enhanced virulence in mice. Antimicrob Agents Chemother 55:1852–1860. doi: 10.1128/AAC.01271-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.West L, Lowman DW, Mora-Montes HM, Grubb S, Murdoch C, Thornhill MH, Gow NA, Williams D, Haynes K. 2013. Differential virulence of Candida glabrata glycosylation mutants. J Biol Chem 288:22006–22018. doi: 10.1074/jbc.M113.478743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Mizzen C, Ying C, Candau R, Barlev N, Brownell J, Allis CD, Berger SL. 1997. Histone acetyltransferase activity is conserved between yeast and human GCN5 and is required for complementation of growth and transcriptional activation. Mol Cell Biol 17:519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee TI, Causton HC, Holstege FC, Shen W-C, Hannett N, Jennings EG, Winston F, Green MR, Young RA. 2000. Redundant roles for the TFIID and SAGA complexes in global transcription. Nature 405:701–704. doi: 10.1038/35015104. [DOI] [PubMed] [Google Scholar]
- 22.Balasubramanian R, Pray-Grant MG, Selleck W, Grant PA, Tan S. 2002. Role of the Ada2 and Ada3 transcriptional coactivators in histone acetylation. J Biol Chem 277:7989–7995. doi: 10.1074/jbc.M110849200. [DOI] [PubMed] [Google Scholar]
- 23.Berger SL, Piña B, Silverman N, Marcus GA, Agapite J, Regier JL, Triezenberg SJ, Guarente L. 1992. Genetic isolation of ADA2: a potential transcriptional adaptor required for function of certain acidic activation domains. Cell 70:251–265. doi: 10.1016/0092-8674(92)90100-Q. [DOI] [PubMed] [Google Scholar]
- 24.Hoke SM, Genereaux J, Liang G, Brandl CJ. 2008. A conserved central region of yeast Ada2 regulates the histone acetyltransferase activity of Gcn5 and interacts with phospholipids. J Mol Biol 384:743–755. doi: 10.1016/j.jmb.2008.09.088. [DOI] [PubMed] [Google Scholar]
- 25.Sinha H, David L, Pascon RC, Clauder-Münster S, Krishnakumar S, Nguyen M, Shi G, Dean J, Davis RW, Oefner PJ. 2008. Sequential elimination of major-effect contributors identifies additional quantitative trait loci conditioning high-temperature growth in yeast. Genetics 180:1661–1670. doi: 10.1534/genetics.108.092932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruno VM, Kalachikov S, Subaran R, Nobile CJ, Kyratsous C, Mitchell AP. 2006. Control of the C. albicans cell wall damage response by transcriptional regulator Cas5. PLoS Pathog 2:e21. doi: 10.1371/journal.ppat.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sellam A, Askew C, Epp E, Lavoie H, Whiteway M, Nantel A. 2009. Genome-wide mapping of the coactivator Ada2p yields insight into the functional roles of SAGA/ADA complex in Candida albicans. Mol Biol Cell 20:2389–2400. doi: 10.1091/mbc.E08-11-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pukkila-Worley R, Peleg AY, Tampakakis E, Mylonakis E. 2009. Candida albicans hyphal formation and virulence assessed using a Caenorhabditis elegans infection model. Eukaryot Cell 8:1750–1758. doi: 10.1128/EC.00163-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haynes BC, Skowyra ML, Spencer SJ, Gish SR, Williams M, Held EP, Brent MR, Doering TL. 2011. Toward an integrated model of capsule regulation in Cryptococcus neoformans. PLoS Pathog 7:e1002411. doi: 10.1371/journal.ppat.1002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reyes-Dominguez Y, Narendja F, Berger H, Gallmetzer A, Fernandez-Martin R, Garcia I, Scazzocchio C, Strauss J. 2008. Nucleosome positioning and histone H3 acetylation are independent processes in the Aspergillus nidulans prnD-prnB bidirectional promoter. Eukaryot Cell 7:656–663. doi: 10.1128/EC.00184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rösler SM, Kramer K, Finkemeier I, Humpf HU, Tudzynski B. 2016. The SAGA complex in the rice pathogen Fusarium fujikuroi: structure and functional characterization. Mol Microbiol 102:951–974. doi: 10.1111/mmi.13528. [DOI] [PubMed] [Google Scholar]
- 32.Church M, Smith KC, Alhussain MM, Pennings S, Fleming AB. 2017. Sas3 and Ada2 (Gcn5)-dependent histone H3 acetylation is required for transcription elongation at the de-repressed FLO1 gene. Nucleic Acids Res 45:4413–4430. doi: 10.1093/nar/gkx028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cowen LE, Sanglard D, Howard SJ, Rogers PD, Perlin DS. 2014. Mechanisms of antifungal drug resistance. Cold Spring Harb Perspect Med 5:a019752. doi: 10.1101/cshperspect.a019752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krishnan K, Askew DS. 2014. Endoplasmic reticulum stress and fungal pathogenesis. Fungal Biol Rev 28:29–35. doi: 10.1016/j.fbr.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Groot PW, Kraneveld EA, Yin QY, Dekker HL, Groß U, Crielaard W, de Koster CG, Bader O, Klis FM, Weig M. 2008. The cell wall of the human pathogen Candida glabrata: differential incorporation of novel adhesin-like wall proteins. Eukaryot Cell 7:1951–1964. doi: 10.1128/EC.00284-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pina B, Berger S, Marcus GA, Silverman N, Agapite J, Guarente L. 1993. ADA3: a gene, identified by resistance to GAL4-VP16, with properties similar to and different from those of ADA2. Mol Cell Biol 13:5981–5989. doi: 10.1128/MCB.13.10.5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanz AB, García R, Rodríguez-Peña JM, Nombela C, Arroyo J. 2016. Cooperation between SAGA and SWI/SNF complexes is required for efficient transcriptional responses regulated by the yeast MAPK Slt2. Nucleic Acids Res 44:7159–7172. doi: 10.1093/nar/gkw324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyazaki T, Nakayama H, Nagayoshi Y, Kakeya H, Kohno S. 2013. Dissection of Ire1 functions reveals stress response mechanisms uniquely evolved in Candida glabrata. PLoS Pathog 9:e1003160. doi: 10.1371/journal.ppat.1003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyazaki T, Inamine T, Yamauchi S, Nagayoshi Y, Saijo T, Izumikawa K, Seki M, Kakeya H, Yamamoto Y, Yanagihara K. 2010. Role of the Slt2 mitogen-activated protein kinase pathway in cell wall integrity and virulence in Candida glabrata. FEMS Yeast Res 10:343–352. doi: 10.1111/j.1567-1364.2010.00611.x. [DOI] [PubMed] [Google Scholar]
- 40.Chen Y-L, Konieczka JH, Springer DJ, Bowen SE, Zhang J, Silao FGS, Bungay AAC, Bigol UG, Nicolas MG, Abraham SN. 2012. Convergent evolution of calcineurin pathway roles in thermotolerance and virulence in Candida glabrata. G3 (Bethesda) 2:675–691. doi: 10.1534/g3.112.002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramírez-Zavala B, Mogavero S, Schöller E, Sasse C, Rogers PD, Morschhäuser J. 2014. SAGA/ADA complex subunit Ada2 is required for Cap1- but not Mrr1-mediated upregulation of the Candida albicans multidrug efflux pump Mdr1. Antimicrob Agents Chemother 58:5102–5110. doi: 10.1128/AAC.03065-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Netea MG, Van der Graaf CA, Vonk AG, Verschueren I, Van der Meer JW, Kullberg BJ. 2002. The role of Toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J Infect Dis 185:1483–1489. doi: 10.1086/340511. [DOI] [PubMed] [Google Scholar]
- 43.Santos R, Costa C, Mil-Homens D, Romão D, Carvalho CC, Pais P, Mira NP, Fialho AM, Teixeira MC. 2017. The multidrug resistance transporters CgTpo1_1 and CgTpo1_2 play a role in virulence and biofilm formation in the human pathogen Candida glabrata. Cell Microbiol 19:12686. doi: 10.1111/cmi.12686. [DOI] [PubMed] [Google Scholar]
- 44.Angiolella L, Stringaro AR, De Bernardis F, Posteraro B, Bonito M, Toccacieli L, Torosantucci A, Colone M, Sanguinetti M, Cassone A. 2008. Increase of virulence and its phenotypic traits in drug-resistant strains of Candida albicans. Antimicrob Agents Chemother 52:927–936. doi: 10.1128/AAC.01223-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaur R, Domergue R, Zupancic ML, Cormack BP. 2005. A yeast by any other name: Candida glabrata and its interaction with the host. Curr Opin Microbiol 8:378–384. doi: 10.1016/j.mib.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 46.Castaño I, Pan SJ, Zupancic M, Hennequin C, Dujon B, Cormack BP. 2005. Telomere length control and transcriptional regulation of subtelomeric adhesins in Candida glabrata. Mol Microbiol 55:1246–1258. [DOI] [PubMed] [Google Scholar]
- 47.Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, De Montigny J, Marck C, Neuvéglise C, Talla E. 2004. Genome evolution in yeasts. Nature 430:35–44. doi: 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- 48.Reuß O, Vik Å, Kolter R, Morschhäuser J. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119–127. doi: 10.1016/j.gene.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 49.Gietz D, St Jean A, Woods RA, Schiestl RH. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res 20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burnette WN. 1981. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem 112:195–203. doi: 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- 51.Chen Y-L, Yu S-J, Huang H-Y, Chang Y-L, Lehman VN, Silao FGS, Bigol UG, Bungay AAC, Averette A, Heitman J. 2014. Calcineurin controls hyphal growth, virulence, and drug tolerance of Candida tropicalis. Eukaryot Cell 13:844–854. doi: 10.1128/EC.00302-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clinical and Laboratory Standards Institute. 2009. Method for antifungal disk diffusion susceptibility testing of yeasts; approved guideline M44-A2, 2nd ed Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 53.Clinical and Laboratory Standards Institute. 2008. Reference methods for broth dilution antifungal susceptibility testing of yeasts: approved standard, 2nd ed Document M27-A3. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.