

Abstract
A phase transfer catalyzed asymmetric alkylation of anthrones with cyclic allylic bromides using quinidine- or quinine-derived catalysts is described. Utilizing mild basic conditions and as low as 0.5 mol % catalyst loading, and achieving up to >99:1 dr selectivity, this asymmetric reaction was successfully applied to produce enantioselectively (−)- and (+)-viridicatumtoxins B, and thus allowed assignment of the absolute configuration of this naturally occurring antibiotic. While the developed asymmetric synthesis of C10 substituted anthrones is anticipated to find wider applications in organic synthesis, its immediate application to the construction of a variety of designed enantiopure analogues of viridicatumtoxin B led to the discovery of highly potent, yet simpler analogues of the molecule. These studies are expected to facilitate drug discovery and development efforts toward new antibacterial agents.
Graphical abstract
1. INTRODUCTION
The viridicatumtoxins [e.g., B (1)1 and A (2),2 Figure 1] and spirohexaline3 (3, originally reported structure, Figure 1) constitute a growing subgroup of the tetracycline class of naturally occurring antibiotics whose novel spirocyclic ring system (i.e., EF in 1a) and high potencies against drug-resistant bacterial strains offer challenges and opportunities for synthetic chemists and microbiologists alike. Our recently reported total synthesis of viridicatumtoxin B (1) provided the natural substance in its racemic form and resulted in revision of its originally assigned structure (from 1a to 1, Figure 1).4 It also rendered readily available a number of less complex racemic analogues of viridicatumtoxin B that proved equipotent to the natural substance against methicillin-resistant Staphylococcus aureus (MRSA) and Enterococcus faecium strains.4 In view of the importance and recent emphasis on new antibacterial agents to combat dangerous bacterial infections, we sought to develop a general asymmetric synthesis of members of this family of compounds that includes, in addition to viridicatumtoxins B (1) and A (2),2 spirohexaline (3, Figure 1).3 Interestingly, the latter compound was depicted in the original publication3 as shown in structure 3 despite its antipodal nature to that of its closest relative 2, whose absolute configuration was determined by X-ray crystallographic analysis.5 This puzzling difference heightened the intrigue over the absolute configuration of viridicatumtoxin B (1).
Figure 1.

Molecular structures of viridicatumtoxins (1 and 2) and spirohexaline (3).
2. RESULTS AND DISCUSSION
2.1. Asymmetric Alkylation of Anthrones
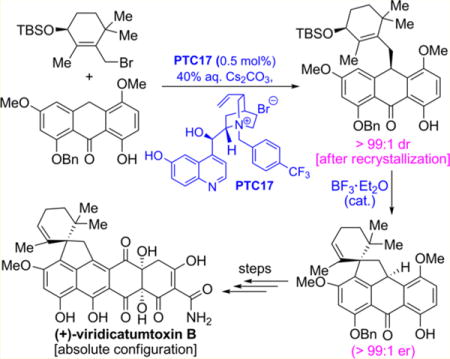
Figure 2 summarizes, in retrosynthetic format, the strategy employed in our first total synthesis of viridicatumtoxin B (1) in which the target molecule was traced back to key building blocks 5, 7, and 8 through advanced intermediates 4 and 6, with the latter being the first key chiral compound enroute to 1. The asymmetric generation of intermediate 6 through alkylation of anthrone 7 with cyclic allylic bromide 8, therefore, became the key challenge for the sought-after asymmetric synthesis of viridicatumtoxin B and its analogues. This challenge was expected to be a thorny problem not only due to the potential propensity of alkylation product 6 to C10 (anthrone numbering) racemization under the required basic conditions but also because of its perceived sensitivity to isomerize under the Lewis acid mediated spirocyclization conditions to form the next advanced intermediate, chiral pentacyclic compound 4 (viridicatumtoxin numbering). Although numerous asymmetric alkylations have been reported,6–11 a practical asymmetric alkylation of anthrones of type 7 was not available at the outset of this work.
Figure 2.
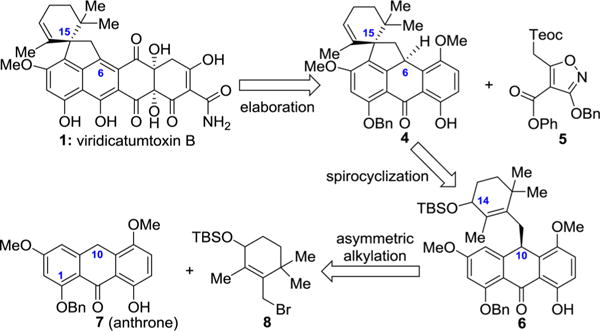
Proposed asymmetric total synthesis of viridicatumtoxin B through asymmetric alkylation of anthrone 7 with allylic bromide 8 in retrosynthetic format (labeling of 1 based on viridicatumtoxin B numbering; labeling of 6 and 7 based on anthrone numbering).
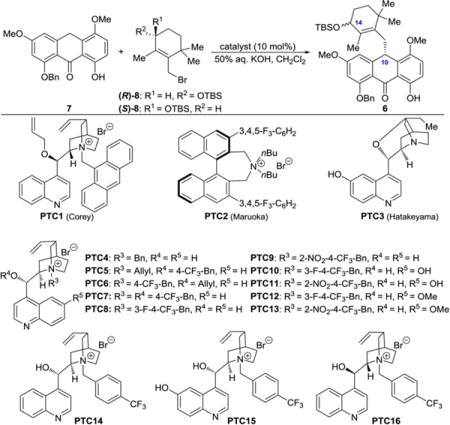
Focusing on known asymmetric alkylation reactions,6–11 our initial studies pointed to phase transfer catalysts as promising for further studies. Brief exploration of phase transfer catalysts PTC1 (Corey et al.)12 and PTC2 (Maruoka et al.)13 and chiral base catalyst PTC3 (Hatakeyama et al.)14 identified the Corey catalyst (PTC1) as the most practical and efficient to pursue as a lead scaffold for further optimization (see Table 1, entries 1–3). Thus, following this pathpointing observation, we tested a series of catalysts (i.e., newly synthesized: PTC5, PTC7− PTC13, and PTC15; previously reported: PTC4,15 PTC6,16 PTC14,17 and PTC16,18 Table 1, entries 4–18; for further catalysts synthesized and tested, see Supporting Information) for their efficiency in the anthrone alkylation reaction with racemic [(R,S)-] or enantiopure [(R)- or (S)-] allylic bromide 8 in a two-phase solvent system (50% aqueous KOH/CH2Cl2) at −78 to 0 °C. As seen in Table 1, the best results were obtained with PTC15 and the (R)-enantiomer of allylic bromide 8 [(R)-8, 99% substrate conversion, 75% yield, 89:11 dr, entry 17], although the reaction with (R,S)-8 also performed well (99% substrate conversion, 75% yield, and 86:14 dr, entry 15). The performance of PTC9 and PTC14 were also notable (entries 9 and 14, respectively, Table 1). The significant difference in diastereoselectivity observed with the (S)-enantiomer of 8 [(S)-8, entry 16, 83:17 dr] was noted with considerable interest. The ability to deliver the antipode of alkylation product 6 was demonstrated by using the (S)-enantiomer of allylic bromide 8 and catalyst PTC16 (pseudoenantiomer of PTC14, prepared from cinchonidine, see Supporting Information; 99% substrate conversion, 76% yield, 13:87 dr, entry 18, Table 1).
Table 1.
Catalyst Optimization of Alkylation of Anthrone 7 with Allylic Bromide 8 [(R) and/or (S)]a
| |||||
|---|---|---|---|---|---|
| Entry | Catalyst | 8 (R1, R2)b | Conv. (%) | Yield (%) | dr(%)c |
| 1 | PTC1 | (S) and (R) | >99 | 78 | 62:38 |
| 2 | PTC2 | (S) and (R) | 90 | 70 | 61:39 |
| 3 | PTC3 | (S) and (R) | >99 | 75 | 55:45 |
| 4 | PTC4 | (S) and (R) | >99 | 77 | 63:37 |
| 5 | PTC5 | (S) and (R) | >99 | 76 | 58:42 |
| 6 | PTC6 | (S) and (R) | >99 | 74 | 60:40 |
| 7 | PTC7 | (S) and (R) | >99 | 76 | 62:38 |
| 8 | PTC8 | (S) and (R) | >99 | 73 | 82:18 |
| 9 | PTC9 | (S) and (R) | 80 | 70 | 83:17 |
| 10 | PTC10 | (S) and (R) | >99 | 72 | 80:20 |
| 11 | PTC11 | (S) and (R) | >99 | 70 | 79:21 |
| 12 | PTC12 | (S) and (R) | >99 | 72 | 58:42 |
| 13 | PTC13 | (S) and (R) | >99 | 75 | 64:36 |
| 14 | PTC14 | (S) and (R) | >99 | 76 | 84:16 |
| 15 | PTC15 | (S) and (R) | >99 | 75 | 86:14 |
| 16 | PTC15 | (S) | >99 | 75 | 83:17 |
| 17 | PTC15 | (R) | >99 | 75 | 89:11 |
| 18 | PTC16e | (S) | >99 | 76 | 13:87 |
Reaction conditions: anthrone 7 (0.10 mmol), allylic bromide 8 (0.11 mmol), PTC cat. (10 mol %), CH2Cl2 (0.9 mL), 50% aqueous KOH (0.3 mL), −78 to 0 °C, 8 h.
Allylic bromides (R)-8 and (S)-8 were prepared through a standard sequence4 involving CBS reduction19 of an intermediate enone as described in the Supporting Information.
The diastereoisomeric ratio (dr) was determined by HPLC using a chiralPak AD-H column; the dr ratios for entries 1–15 were determined from the HPLC peak areas corresponding to (10S)-6: (10R)-6 (see Supporting Information).
PTC16 is the pseudoenantiomer of PTC14.
The superiority of catalysts PTC8, PTC9, PTC15, and PTC16 (Table 1, entries 8, 9, and 15–18), all of which include an electron-withdrawing group on their benzyloxy residue, as opposed to relatively electron rich catalysts carrying a benzyloxy substituent (e.g., PTC4, entry 4, Table 1), is in line with Maruoka’s inspirational work.23 This trend is further corroborated with the observed superiority of catalysts PTC39, PTC40, and PTC41 (all of which contain fluorine atoms on their benzyloxy residue, see Supporting Information) over PTC38, whose benzyloxy moiety includes an electron-rich residue (i.e., Me; see Supporting Information).
Striving for higher asymmetric induction and efficiency and beyond catalyst and alkylating agent absolute configuration optimization, we then investigated the effect of reaction conditions and catalyst loading on the alkylation of anthrone 7 with TBS-protected alkylating agent (R)-8 and catalyst PTC15. Table 2 summarizes the results of this study that involved changes in the aqueous base, organic solvent, temperature, reaction time, and catalyst loading. Initial experiments (entries 1–6, Table 2) led to the identification of 40% aqueous Cs2CO3 and CH2Cl2 as the optimal base and solvent, respectively (entry 5, 0 °C, 10 mol % cat., 75% yield, 92:8 dr, Table 2). Changing the solvent from CH2Cl2 to (CH2)2Cl2 increased the dr slightly (93:7, entry 7, Table 2), while decreasing the temperature steadily from 0 to −30 °C led to further improvements in the dr with proportional increases in reaction time as expected (93:7; 94:6; 95:5; entries 12–14, respectively, Table 2). Stepwise decrease of catalyst loading from 10 to 0.1 mol % resulted in a slight increase of efficiency, albeit at the expense of reaction time (95:5–96:4 dr, entries 14–16, Table 2). The lower yield (due to decreased rate and conversion) reflected in entry 16 (Table 2, 15% yield) with 0.1 mol % catalyst loading provided an unaccepable limit, thereby leading us to adopt the 0.5 mol % catalyst loading as the most practical with regard to reaction rate, yield, and diastereoselectivity (72% yield, 95:5 dr, 40% aqueous Cs2CO3, (CH2)2Cl2, −30 °C, 180 h, entry 17, Table 2).
Table 2.
Optimization of Alkylation Conditions with PTC15a
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Base (aq. solution) |
Solvent | ϑ (°C) |
Time (h) |
Catb (mol%) |
Yield (%) |
drc |
| 1 | 50% KOH | CH2Cl2 | 0 | 8 | 10 | 77 | 89:11 |
| 2 | 50% NaOH | CH2Cl2 | 0 | 8 | 10 | 77 | 89:11 |
| 3 | 50% Cs2CO3 | CH2Cl2 | 0 | 8 | 10 | 77 | 91:9 |
| 4 | 50% K2CO3 | CH2Cl2 | 0 | 8 | 10 | 75 | 91:9 |
| 5 | 40% Cs2CO3 | CH2Cl2 | 0 | 8 | 10 | 75 | 92:8 |
| 6 | 30% Cs2CO3 | CH2Cl2 | 0 | 8 | 10 | 73 | 91:9 |
| 7 | 40% Cs2CO3 | (CH2)2C12 | 0 | 8 | 10 | 72 | 93:7 |
| 8 | 40% Cs2CO3 | CHCl3 | 0 | 8 | 10 | 60 | 85:15 |
| 9 | 40% Cs2CO3 | CCl4 | 0 | 8 | 10 | 50 | 81:19 |
| 10 | 40% Cs2CO3 | PhMe | 0 | 8 | 10 | 78 | 84:16 |
| 11 | 40% Cs2CO3 | EtOAc | 0 | 8 | 10 | 65 | 78:22 |
| 12 | 40% Cs2CO3 | (CH2)2C12 | −10 | 24 | 10 | 72 | 93:7 |
| 13 | 40% Cs2CO3 | (CH2)2Cl2 | −20 | 72 | 10 | 72 | 94:6 |
| 14 | 40% Cs2CO3 | (CH2)2Cl2 | −30 | 180 | 10 | 72 | 95:5 |
| 15 | 40% Cs2CO3 | (CH2)2Cl2 | −30 | 180 | 1 | 72 | 95:5 |
| 16d | 40% Cs2CO3 | (CH2)2Cl2 | −30 | 200 | 0.1 | 15 | 96:4 |
| 17d | 40% Cs2CO3 | (CH2)2Cl2 | −30 | 180 | 0.5 | 72 | 95:5 |
| 18 | 40% Cs2CO3 | (CH2)2C12 | −20 | 72 | 1 | 72 | 94:6 |
Reaction conditions: anthrone 7 (0.10 mmol), allylic bromide (R)-8 (0.11 mmol), PTC15, solvent (0.9 mL), aqueous base solution (0.3 mL).
Cat. = catalyst loading.
The dr [(10S)-6:(10R)-6] was determined by HPLC using a chiralPak AD-H column; see Supporting Information.
Reaction was run on 5 mmol scale (anthrone). For further studies on possible C10 racemization of the alkylation product under basic conditions, see Supporting Information.
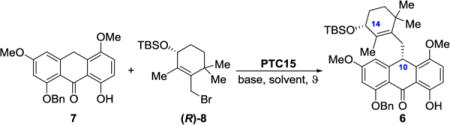
Inspired by the effect of the chirality of the alkylating agent on the diastereoselectivity of the anthrone alkylation reaction as described above (Table 1), we set out to investigate a series of allylic bromides (R)-8 varying in size of silyl protecting groups. For this investigation, we adopted catalyst PTC15 and the conditions of entry 18 rather than those of entry 17 (Table 2) due to the shorter reaction time (for convenience). As shown in Table 3, the results of this study indicated a correlation between the bulkiness of the silyl protecting group and asymmetric induction. Thus, the most effective groups inducing the highest diastereoselectivities were those leading to products 6d [tris(trimethylsilyl)silyl: 95:5 dr, 55% yield], 6b (thexyldimethylsilyl: 95:5 dr, 74% yield), and 6 (tert-butyldimethylsilyl: 94:6 dr, 72% yield). Because of the lower yield of the reaction with tris(trimethylsilyl)silyl-protected allylic bromide (R)-8d, alkylating agents (R)-8b (thexyldimethylsilyl), and (R)-8 (tert-butyldimethylsilyl) were chosen as the most attractive for further optimization.
Table 3.
Asymmetric Alkylation with Different Allylic Bromidesa

|
Reaction conditions: anthrone 7 (0.10 mmol), allylic bromides (R)-[8, 8a−8h] (0.11 mmol), PTC15 (1 mol %), (CH2)2Cl2 (0.9 mL), 40% aqueous Cs2CO3 (0.3 mL), −20 °C, 72 h.
Isolated yield.
The dr [(10S):(10R)] was determined by HPLC using a chiralPak AD-H column; see Supporting Information.
As shown in Table 2, this reaction was repeated in this study as a further confirmation of its diastereoselectivity. TBS = tert-butyldimethylsilyl; TBDPS = tert-butyldiphenylsilyl.
The absolute configuration of substituted anthrone 6a (mp 165–166 °C) obtained from the diastereoselective alkylation of anthrone 5, allylic bromide (R)-8a, and PTC15 as described in Table 3 was determined by X-ray crystallographic analysis (ORTEP, Figure 3, and CIF in the Supporting Information).20 Indeed, the crystallization of 6a that allowed its absolute configuration assignment at this point was a fortunate event, since all other products in Table 3 were foams in nature.
Figure 3.
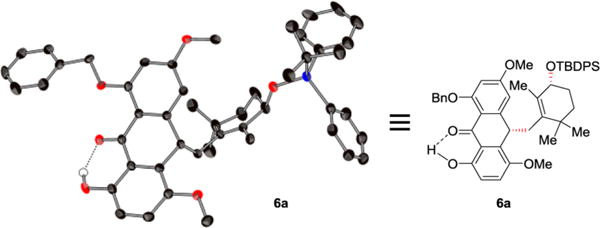
X-ray derived ORTEP representation of 6a.
Finally, we explored the C1 and C5 substituent effects on the diastereoselectivity of the anthrone alkylation using thexyldimethylsilyl-protected bromide (R)-8b, anthrones 7, 7i−7p, and PTC15 under the optimized conditions as shown in Table 4. These studies revealed that substituents at the C1 position of the anthrone substrates smaller than OBn (i.e., 6b, 95:5 dr) resulted in decreased diastereoselectivities (i.e., 6i: 90:10 dr; 6j: 93:7 dr), while larger substituents at this position led to increased diastereoselectivities (e.g., 6k: 92:8 dr; 6l: 98:2 dr). The significant increase in diastereoselectivity for product 6l carrying the p-trifluoromethyl benzyloxy group was notable, and a benzyloxy group at C5 (i.e., 6m) instead of methoxy increased the diastereoselectivity to 99:1 dr led to even more impressive improvements. Thus, the highest diastereoselectivities were obtained for the combinations of anthrone substrates (7n, 7o, and 7p) and alkylating agent (R)-8b, which led to highly enantioenriched products 6n (>99:1 dr), 6o (99:1 dr), and 6p (>99:1 dr) as shown in Table 4.
Table 4.
Asymmetric Alkylation with Different Anthronesa
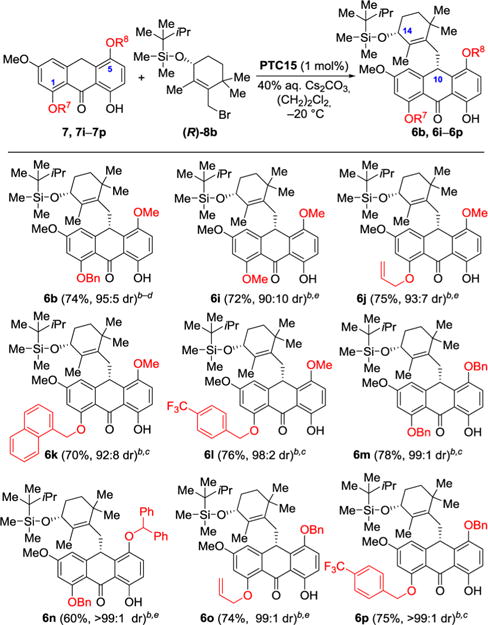
|
Reaction conditions: anthrones 7, 7i−7p (0.10 mmol), (R)-8b (0.11 mmol), PTC15 (1 mol %), (CH2)2Cl2 (0.9 mL), 40% aqueous Cs2CO3 (0.3 mL), −20 °C, 72 h.
Isolated yield.
The dr [(10S): (10R)] was determined by HPLC using chiralPak AD-H column; see Supporting Information.
As shown in Table 3, this reaction was repeated in this study as a further confirmation of its diastereoselectivity.
The dr was determined by HPLC using chiralPak AD-H column after benzylation of the product; see Supporting Information.
2.2. Enantioselective Total Synthesis and Absolute Configuration of (−)-Viridicatumtoxin B
Having developed an efficient and highly enantioselective synthesis of 10-substituted anthrones, we were in a position to undertake the total synthesis of enantiopure viridicatumtoxin B in an attempt to determine its absolute configuration. Given that the literature reports on the structures of viridicatumtoxin A (2, Figure 1; absolute configuration confirmed by X-ray crystallographic analysis)5 and spirohexaline3 (3, reported structure shown in Figure 1) suggested opposite absolute configurations for these two siblings, we were ambivalent as to which absolute configuration of viridicatumtoxin B to target first. Since our asymmetric synthesis of anthrones revealed a higher diastereoselectivity for PTC15 (leading to the corresponding C10-substituted anthrones, see Table 1) than that for PTC16 (leading to the antipodal enantiomer), we decided to employ the former as a means to reach one of the enantiomers of viridicatumtoxin B (1).
To this end, we adopted substrate anthrone 7, alkylating agent allylic bromide (R)-8, and catalyst PTC15 in our attept to construct the first chiral intermediate (6S,17R)-6 and convert it stereospecifically to the pending intermediate, spiropentacycle (6S,15R)-4, enroute to viridicatumtoxin B (Scheme 1). It was with considerable trepidation that we pondered the latter possibility, given the sensitivity of the substrate and the conditions involved in this spirocyclization. As shown in Scheme 1, the reaction of anthrone 7 (3.01 g, 8.00 mmol) with allylic bromide (R)-8 (3.06 g, 8.80 mmol) under the influence of PTC15 (0.5 mol %) was carried out on gram scale, delivering the expected substituted anthrone (6S,17R)-6 in 72% yield and 95:5 dr. The latter was subjected to optimized Lewis acid mediated spirocyclization conditions (BF3·Et2O, 5 mol %) to afford spiropentacycle (6S,15R)-4 in 74% yield and, much to our delight, with no loss of enantiopurity (95:5 er, see details in Supporting Information). Recrystallization of (6S,15R)-4 from hexanes/CH2Cl2 (50:1) led to further enantioenrichment of this intermediate (>99:1 er). Failing attempts to prepare a crystalline, heavy-atom-containing derivative of (6S,15R)-4 for absolute configuration determination purposes (including the newly formed spirocenter), we subjected the latter first to the action of diacetoxyiodo-benzene (PIDA) and then to camphorsulfonic acid (CSA) in MeOH/CH2Cl2, conditions that led to p-quinomethide (−)-9 (via the corresponding dimethoxyketal) in 81% overall yield. Treatment of (−)-9 with p-bromobenzoyl chloride under basic conditions furnished crystalline derivative (−)-11 (mp 220–221 °C), whose single crystal X-ray crystallographic analysis (ORTEP, Scheme 1, and CIF in the Supporting Information)21 revealed that indeed the single stereocenter (C15, viridicatumtoxin numbering) within intermediate (−)-9 (to be converted to virididatumtoxin B) was of the shown chirality. Chiral HPLC analysis confirmed its homogenuity as a single enantiomer (see details in Supporting Information). This absolute configuration corresponds to that suggested for spirohexaline (3),3 rather than that determined for viridicatumtoxin A (2).5
Scheme 1. Enantioselective Synthesis of the BCDEF Fragment (6S,15R)-4 of (−)-Viridicatumtoxin B [(−)-1] and Its Absolute Configurationa.
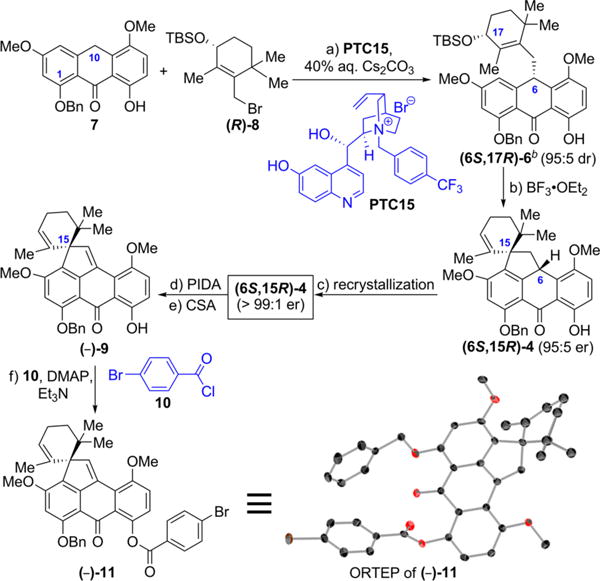
aReagents and conditions: (a) 7 (3.01 g, 8.00 mmol scale), PTC15 (0.5 mol %) 40% aqueous Cs2CO3, (CH2)2Cl2, −30 °C, 10 days, 72%, 95:5 dr; (b) BF3·Et2O (0.05 equiv), CH2Cl2, −78 to 0 °C, 30 min, 74%, 95:5 er; (c) hexanes/CH2Cl2 (50:1), 91%, > 99:1 er; (d) PIDA (1.2 equiv), MeOH/CH2Cl2 (1:1), 0 °C, 30 min, 25 °C, 30 min; (e) CSA (0.07 equiv), CH2Cl2, 0 °C, 5 min, 81% for two steps; (f) 10 (5.0 equiv), DMAP (10 equiv), Et3N (30 equiv), CH2Cl2, 25 °C, 6 h, 95%. bTo avoid confusion, the numbering on (6S,17R)-6, (6S,15R)-4, and (−)-9 in this scheme and Figure 4 is based on the viridicatumtoxin numbering, as opposed to the carbon numbering of compound (10S,14R)-6 (see Table 1), which is the same as (6S,17R)-6, the latter being designated using the anthrone numbering.
The faithful transfer of absolute configuration from C6 (viridicatumtoxin numbering) in intermediate (6S,17R)-6 to C15 in (6S,15R)-4 is the consequence of the carbonium-mediated spirocyclization that requires attack of the C7 position on the aromatic ring onto C15 carbonium ion from the “top” side of a sterically controlled transition state (A: preferred; B: sterically congested), in which the gem-dimethyl groups play a non-negotiable steric control (see Figure 4).
Figure 4.
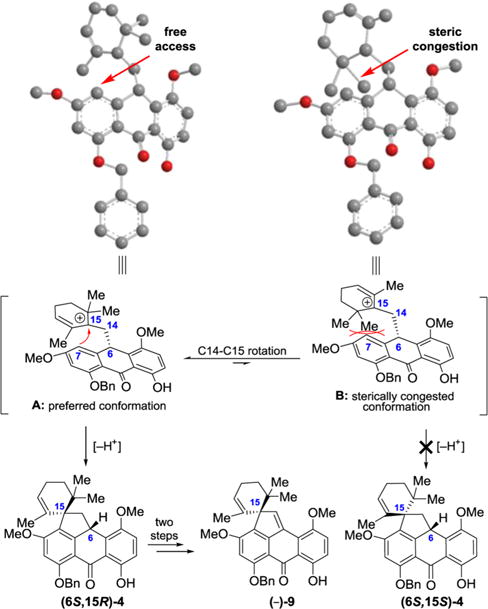
Proposed mechanism of stereoselective spirocyclization of substituted anthrone (6S,17R)-6 to pentacycle (6S,15R)-4 through carbonium species A (viridicatumtoxin numbering for all intermediates and compounds).
Our intention to obtain both enantiomeric forms of viridicatumtoxin B prompted us to push on with this enantiomer of key building block (−)-9, whose absolute configuration we just determined and whose single stereocenter (15R) was destined to control the stereochemistry of all others en route to the targeted molecule. We also took the opportunity along the way to optimize the process from the original one that delivered racemic viridicatumtoxin B (1).4 Scheme 2 summarizes the optimized synthesis of what turned out to be (−)-viridicatumtoxin B [(−)-1] starting with enantiopure precursor (−)-9. Thus, dearomatization of (−)-9 with PIDA in MeOH/CH2Cl2 furnished, in 90% yield, dimethoxy semiquinone (−)-12. In an effort to improve the previously low diastereoselectivity (ca. 2:1 dr)4 of the coupling of racemic 12 with oxazoline 5, we performed the reaction (t-BuOK, −50 °C, 48 h) in the presence of chiral catalyst PTC14 in the hope that the latter would exert a degree of stereocontrol. It was indeed with pleasure that we observed the formation of the expected heptacycle product (+)-13, in 88% yield and ca. 4:1 dr, as opposed to 91% yield and ca. 2:1 dr in the original prodedure, a welcome improvement since the major diastereoisomer was the desired one shown in Scheme 2. Complexation of ligand PTC14 with the initially formed anion of oxazoline 5 followed by preferred attack on substrate (−)-12 from the “bottom” side may explain the observed diastereoselectivity. For clarity, we note that the diastereoselectivity in this reaction refers to the C15 and C4/C4a stereocenters with the latter being of the syn configuration with respect to the H-residue on C4a. The 4:1 mixture of Teoc derivative (+)-13 was transformed to decarboxylated ketoenol (−)-14 as previously described.4 The hydroxylation of (−)-14 was improved in terms of yield and diastereoselectivity from the original (36%, 2:1 dr, 60% based on recovered starting material) through optimized conditions [THF, 0.2 equiv of Ni(acac)2, 3.0 equiv of DMDO, THF, −78 °C, 3 h, 52%, 4:1 dr, 72% based on recovered starting material]. This improvement may be attributed to increased reactivity of Ni(acac)2 in THF as solvent (as opposed to the originally used CH2Cl2), an effect that resulted in a cleaner and faster reaction. The reduction of (−)-15 (4:1 diastereomeric mixture) with NaCNBH3 allowed convenient chromatographic separation of resulting isomeric products (+)-16 and (−)-17, with the latter (and desired) now obtained from (−)-13 with improved overall diastereoselectivity (ca. 4:1) as opposed to ca. 2:1 in the original route.4 The ketal hydrolysis of major isomer (−)-17 with aqueous HCl proceeded well to afford hydroxy triketone (−)-18, whose regioselective reduction with NaBH(OAc)3 furnished dihydroxy diketone (−)-19 (46% yield). Optimization of conditions of the ensuing silylation of the latter with TBSOTf and 2,6-lutidine led to an improved yield of TBS-ether (+)-20 (76% vs 61%).4 The crucial hydroxylation of (+)-20 was also improved over that of the original procedure4 (32% yield, 55% based on 42% recovered starting material) by optimization of conditions that included freshly prepared Davis oxaziridine reagent. The recruiting steps proceeded as previously reported for the racemic series4 to provide enantiopure (−)-viridicatumtoxin B [(−)-1] through intermediates (−)-22 (HF·py, 72% yield; existing as an equilibrium mixture with its 1,5-lactol isomeric form (−)-22′) and (−)-23. The latter intermediate was obtained in 82% yield (as compared to 66% for the previous procedure)4 by adding the DMP as a solution in CH2Cl2 slowly and in portions, thereby resulting in further improvement in the process. Finally, exposure of precursor (−)-23 to hydrogenolysis conditions (H2, Pd black) furnished the coveted viridicatumtoxin B, whose levorotatory nature led to its absolute configuration assignment as enantiomer [(−)-1] of the natural product. Besides pointing to the absolute configuration of natural viridicatumtoxin B, this observation cast doubts over the depicted absolute configuration of spirohexaline in the isolation paper.3
Scheme 2. Total Synthesis of Enantiopure (−)-Viridicatumtoxin B [(−)-1]a.
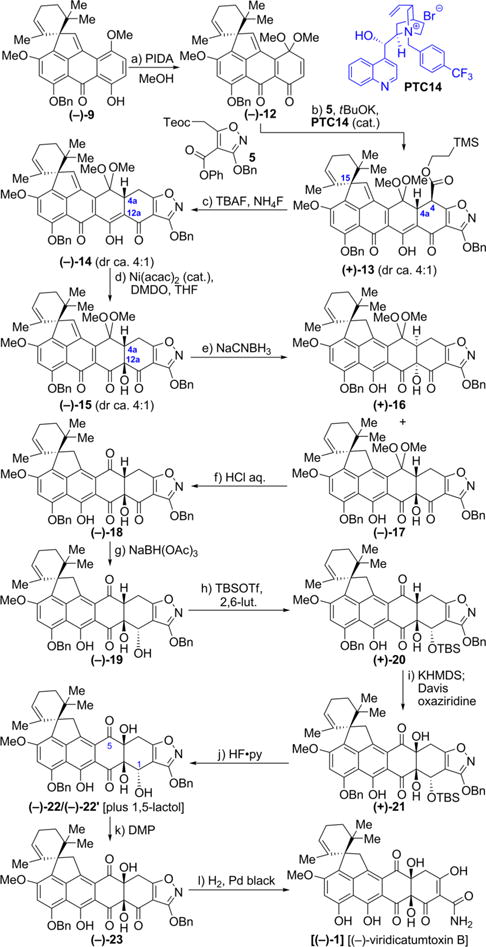
aReagents and conditions: (a) PIDA (1.2 equiv), MeOH/CH2Cl2 (10:1), 25 °C, 1.5 h, 86%; (b) 5 (1.1 equiv), t-BuOK (1.2 equiv), PTC14 (0.01 equiv), toluene, −50 °C, 48 h, 88%, 4:1 dr; (c) TBAF (10 equiv), NH4F (20 equiv), degassed THF, 25 °C, 5 min, 87%, 4:1 dr; (d) [Ni(acac)2] (0.2 equiv), DMDO (3.0 equiv), THF, −78 °C, 3 h, 52% (4:1 dr, 72% brsm), 28% recovered (−)-14; (e) NaCNBH3 (4 equiv), THF, −78 °C, 1.5 h, 48% for (−)-17, 12% for (+)-16, chromatographically separated; (f) 2 N aqueous HCl, THF, 25 °C, 5 h, quant.; (g) NaBH(OAc)3 (1.2 equiv), EtOAc/acetone (1:1), 40 °C, 2 h, 46%; (h) TBSOTf (4 × 10 equiv), 2,6-lutidine (4 × 15 equiv), CH2Cl2, 0 to 25 °C for four times, totaling 2 h, 76%; (i) KHMDS (3.4 equiv), THF, −78 °C, 1 h; then freshly prepared Davis oxaziridine (3.9 equiv), −78 °C, 2 h, 32% of (+)-21 (55% brsm) + 42% recovered (+)-20; (j) HF·pyridine (excess, added at 0 °C in four portions), MeCN, 0 to 55 °C for four times, totaling 20 h, 72% (product 22 exists in equilibrium with its 1,5-lactol isomeric form); (k) DMP solution in CH2Cl2 (0.3 M, 1.8 equiv, added in two portions at 0 °C), (CH2)2Cl2, 0 to 50 °C two times, totaling 2 h, 82%; (l) H2, Pd black (4.9 equiv), 1,4-dioxane/MeOH (1:1), 25 °C, 10 min, 96%.
2.3. Enantioselective Total Synthesis and Absolute Configuration of (+)-Viridicatumtoxin B
With an enantioselective and improved synthetic route at hand, the road to what was sure to be the correct enantiomer of viridicatumtoxin B was now open. For optimum results we chose to use, in addition to anthrone 7, the (S)-enantiomer of allylic bromide 8 [(S)-8], and the new catalyst PTC17 (derived from quinine), the latter being the pseudoenantiomer of PTC15 (derived from quinidine) rather than PTC16 that was used in the methodology development study described above (see Table 1). Equipped with the phenolic moiety on its quinoline domain, PTC15 proved its superiority over PTC16, which lacks this phenolic group, as seen in Table 1 (entries 17 and 18). This crucial observation prompted us to synthesize PTC17 (Scheme 3; for details of the synthesis, see Supporting Information), as we expected it to perform better than PTC16 in the anthrone alkylation step.
Scheme 3. Enantioselective Synthesis of the BCDEF Fragment (6R,15S)-4 of (+)-Viridicatumtoxin B and Its Absolute Configurationa.
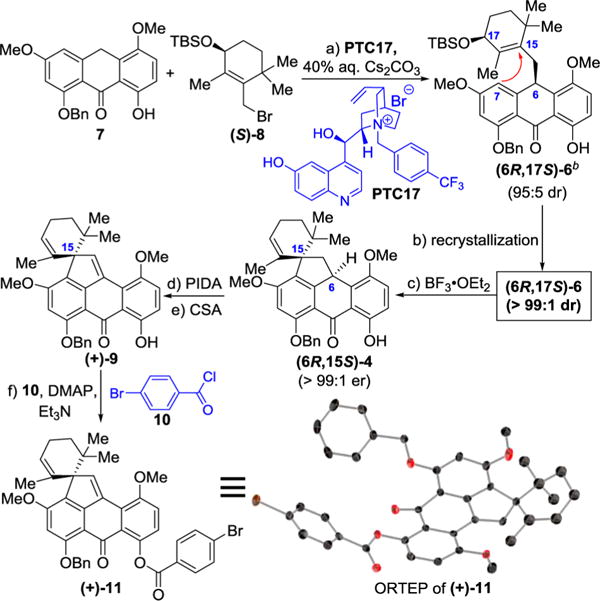
aReagents and conditions: (a) 7 (6.02 g, 16.0 mmol scale), PTC17 (0.5 mol %), 40% aqueous Cs2CO3, (CH2)2Cl2, −30 °C, 10 days, 73%, 95:5 dr; (b) hexanes, 91%, >99:1 dr; (c) BF3·Et2O (0.05 equiv), CH2Cl2, −78 to 0 °C, 30 min, 74%, >99:1 er; (d) PIDA (1.2 equiv), MeOH/CH2Cl2 (1:1), 0 °C, 30 min, 25 °C, 30 min; (e) CSA (0.07 equiv), CH2Cl2, 0 °C, 5 min, 80% for two steps; (f) 10 (5.0 equiv), DMAP (10 equiv), Et3N (30 equiv), CH2Cl2, 25 °C, 6 h, 95%. bTo avoid confusion, the numbering on (6R,17S)-6, (6R,15S)-4 and (+)-9 in this scheme and Figure 4 is based on the viridicatumtoxin numbering, as opposed to the carbon numbering of compound (10R,14S)-6 (see Supporting Information), which is the same as (6R,17S)-6, the latter being designated using the anthrone numbering.
Indeed, as shown in Scheme 3, catalyst PTC17 performed well in ensuring high diastereoselectivity in the alkylation of anthrone 7 (6.02 g scale, 16.0 mmol) with allylic bromide (S)-8, affording alkylated anthrone (6R,17S)-6 in 72% yield and 95:5 dr (as opposed to 87:13 dr obtained with PTC16, see Table 1, entry 18). The obtained product was purified by recrystallization from hexanes, the racemate crystallizing out of the solution and the enriched material [i.e., (6R,17S)-6] being recovered from the mother liquor. The latter was then subjected to the developed spirocyclization reaction conditions [BF3·Et2O (cat.)] furnishing pentacycle (6R,15S)-4 as expected. The absolute configuration of this intermediate was determined by conversion to its p-bromobenzoate derivative (+)-11, obtained via intermediate (+)-9, as summarized in Scheme 3. The X-ray crystallographic analysis (ORTEP representation, Scheme 3, and CIF in the Supporting Information)22 of (+)-11 (mp 220–221 °C) confirmed its (S) absolute configuration as expected from the results shown in Scheme 3 for its enantiomer.
The total synthesis of (+)-viridicatumtoxin B [(+)-1] from key building block spiropentacycle (+)-9 was successfully carried out through the same route and conditions (Figure 5), and in similar yields, as those employed for its enantiomeric form [(−)-1] (see Scheme 3 and the Supporting Information for more details). Synthetic viridicatumtoxin B exhibited the same sign of optical rotation as the one reported in the literature for the natural substrate { , c = 0.1, EtOH for synthetic (+)-1; , c = 0.2, EtOH for natural (+)-1}.1 The higher value for the synthetic material may reflect its higher purity, while the lower value reported for the natural substance may be due to purification and measurement difficulties due to its low natural abundance.1 However, the possibility of the latter occurring in nature in its scalemic form cannot be excluded at this time.
Figure 5.
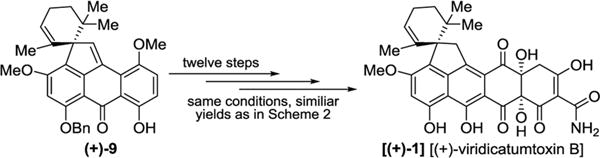
Total synthesis of enantiopure (+)-viridicatumtoxin B [(+)-1].
2.4. Enantioselective Synthesis of Viridicatumtoxin B Analogues
With a practical and enantioselective route to viridicatumtoxin B [(+)-1] and its antipode [(−)-1] and their precursors available to us, we decided to apply it to the construction of a number of their enantiopure analogues (see Figure 6).
Figure 6.
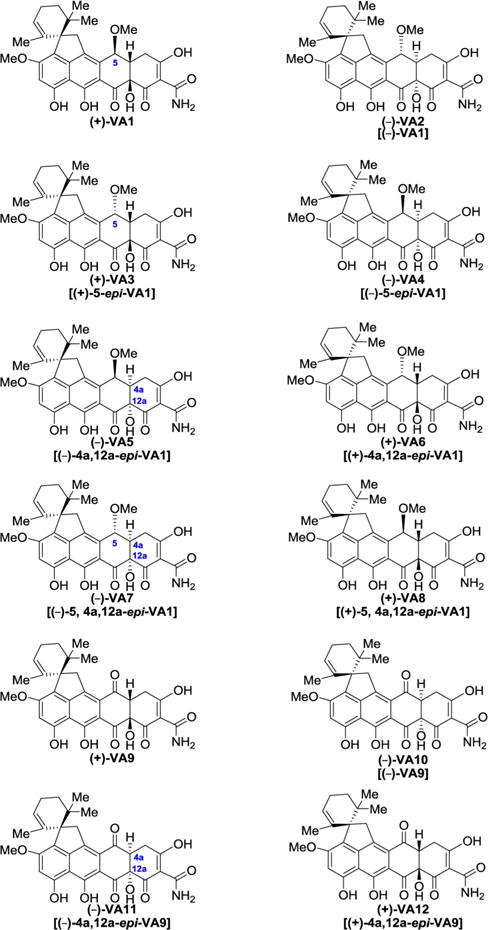
Synthesized viridicatumtoxin B analogues (+)-VA1, (−)-VA2, (+)-VA3, (−)-VA4, (−)-VA5, (+)-VA6, (−)-VA7, (+)-VA8, (+)-VA9, (−)-VA10, (−)-VA11, and (+)-VA12.
We were particularly interested in comparing the antibacterial properties of the antipodal simpler viridicatumtoxin analogues shown in Figure 6 [(+)-VA1, (−)-VA2, (+)-VA3, (−)-VA4, (−)-VA5, (+)-VA6, (−)-VA7, (+)-VA8, (+)-VA9, (−)-VA10, (−)-VA11, and (+)-VA12]. Their synthesis proceeded smoothly from their required precursors, all of which were encountered in our synthetic studies toward (+)-and (−)-viridicatumtoxin B as described above.
Scheme 4 summarizes the synthesis of (+)-VA1 and (+)-VA3 from precursor (−)-17. Thus, reduction of precursor (−)-17 with NaCNBH3 in AcOH at ambient temperature resulted in the formation of a mixture of 5β-methyl ether (+)-24 (48% yield) and its epimer 5α-methyl ether (−)-5-epi-24 (32% yield), which were chromatographically separated. Hydrogenolysis of (+)-24 with Pd black as a catalyst then furnished viridicatumtoxin B analogue (+)-VA1 in 95% yield. Similar treatment of (−)-5-epi-24 led to analogue (+)-VA3 in comparable yield as for (+)-VA1 (see Scheme 4). Analogues (−)-VA5 and (−)-VA7 were prepared from precursor (+)-16 in two steps, while the NaCNBH3 reduction gave (−)-4a,12a-epi-24 in 17% yield and (+)-5,4a,12a-epi-24 in 51% yield, and the following hydrogenolysis of each produced analogues (−)-VA5 and (−)-VA7 in similar yields (see Scheme 5). Analogues (−)-VA2 and (−)-VA4 were similarly prepared from their precursor (+)-17 in yields comparable to those for (+)-VA1 and (+)-VA3 as summarized in Scheme 6. Analogues (+)-VA6 and (+)-VA8 were similarly prepared from their precursor (−)-16 in yields comparable to those for (−)-VA5 and (−)-VA7 as summarized in Scheme 7. Analogue (+)-VA9 was obtained by hydrogenolysis of triketone (−)-18 in 95% yield (Scheme 8, A). Analogue (−)-VA11 was synthesized from hydrolysis of precursor (+)-16 (quant. yield), followed by hydrogenolysis with Pd black (Scheme 8, B). Analogues (−)-VA10 and (+)-VA12 were similarly prepared from their respective precursors as summarized in Scheme 8 C,D.
Scheme 4. Enantioselective Synthesis of Viridicatumtoxin B Analogues (+)-VA1 and (+)-VA3a.

aReagents and conditions: (a) NaCNBH3 (4.0 equiv), AcOH, 25 °C, 30 min, 48% for (+)-24, 32% for (−)-5-epi-24; (b) H2, Pd black (4.1 equiv), THF/MeOH 1:1, 25 °C, 10 min, 95%.
Scheme 5. Enantioselective Synthesis of Viridicatumtoxin B Analogues {(−)-VA5 [(−)-4a,12a-epi-VA1], (−)-VA7 [(−)-5,4a,12a-epi-VA1a.
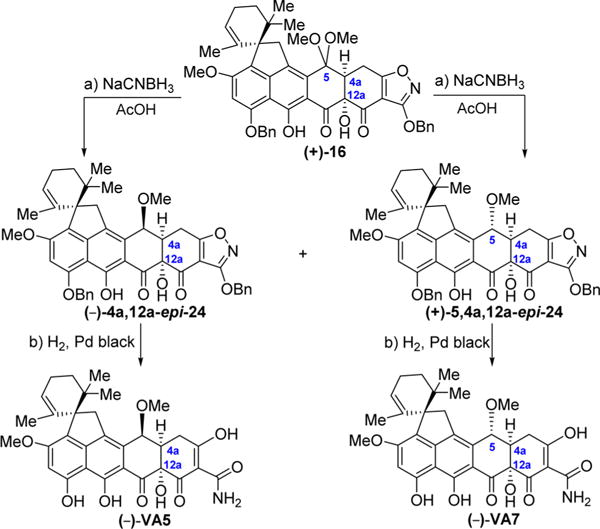
aReagents and conditions: (a) NaCNBH3 (4.0 equiv), AcOH, 25 °C, 30 min, 17% for (−)-4a,12a-epi-24, 51% for (+)-5,4a,12a-epi-24; (b) H2, Pd black (4.1 equiv), THF/MeOH 1:1, 25 °C, 10 min, 95%.
Scheme 6. Enantioselective Synthesis of Viridicatumtoxin B Analogues (−)-VA2 and (−)-VA4a.
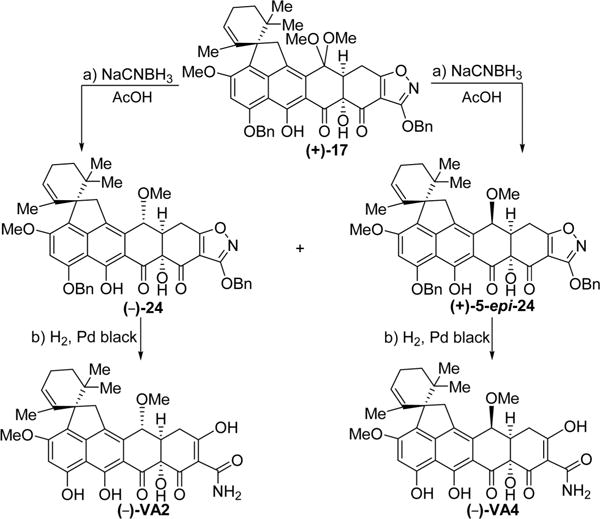
aReagents and conditions: (a) NaCNBH3 (4.0 equiv), AcOH, 25 °C, 30 min, 45% for (−)-24, 30% for (+)-5-epi-24; (b) H2, Pd black (4.1 equiv), THF/MeOH 1:1, 25 °C, 10 min, 95%.
Scheme 7. Enantioselective Synthesis of Viridicatumtoxin B Analogues (+)-VA6 and (+)-VA8a.
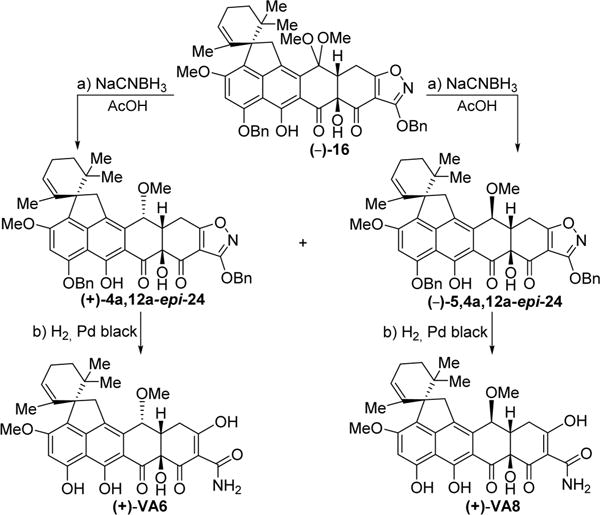
aReagents and conditions: (a) NaCNBH3 (4.0 equiv), AcOH, 25 °C, 30 min, 16% for (+)-4a,12a-epi-24, 48% for (−)-5,4a,12a-epi-24; (b) H2, Pd black (4.1 equiv), THF/MeOH 1:1, 25 °C, 10 min, 95%.
Scheme 8. Enantioselective Synthesis of Viridicatumtoxin B Analogues (+)-VA9, (−)-VA10, (−)-VA11, and (+)-VA12a.
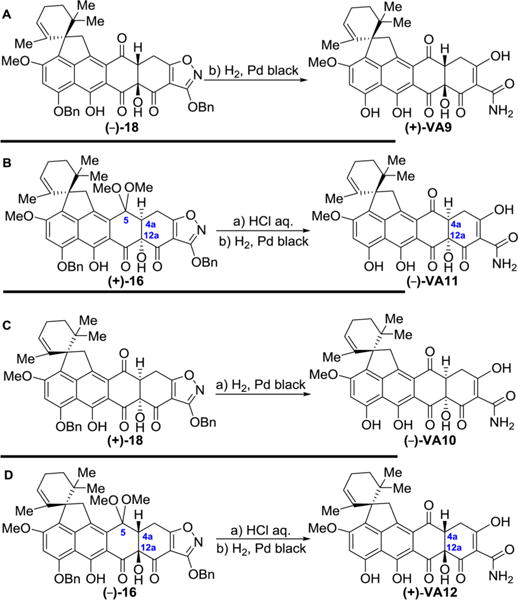
aReagents and conditions: Panel A: (a) H2, Pd black (4.1 equiv), THF/MeOH 1:1, 25 °C, 10 min, 95%. Panel B: (a) 2 N aqueous HCl, THF, 25 °C, 5 h, quant.; (b) H2, Pd black (4.1 equiv), THF/MeOH 1:1, 25 °C, 10 min, 95%. Panel C: (a) H2, Pd black (4.1 equiv), THF/MeOH 1:1, 25 °C, 10 min, 95%. Panel D: (a) 2 N aqueous HCl, THF, 25 °C, 5 h, quant.; (b) H2, Pd black (4.1 equiv), THF/MeOH 1:1, 25 °C, 10 min, 95%.
2.5. Biological Evaluation of Synthetic (+)- and (−)-Viridicatumtoxin B and Analogues
The synthesized simpler enantiopure analogues (+)-VA1, (−)-VA2, (+)-VA3, (−)-VA4, (−)-VA5, (+)-VA6, (−)-VA7, (+)-VA8, (+)-VA9, (−)-VA10, (−)-VA11, and (+)-VA12 (see Figure 6 for structures), all lacking the C4a-hydroxyl group so cumbersome to install, together with (+)- and (−)-viridicatumtoxin B [(+)-1 and (−)-1] were tested against a number of bacterial strains and compared to natural viridicatumtoxin B [(+)-1, taking reported values from ref1 for Enterococcus faecalis KCTC5191, Enterococcus faecium KCTC3122, methicillin-resistant Staphylococcus aureus CCARM3167 (MRSA CCARM3167), Acinetobacter calcoaceticus KCTC2357, and Escherichia coli CCARM1356], minocycline (Minocin, CK1), and tigecycline (Tygacil, CK2) (see Figure 7 for structures).
Figure 7.
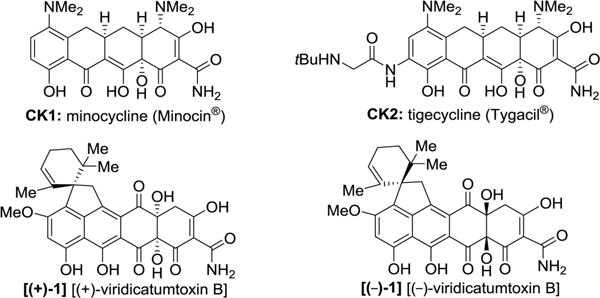
Molecular structure of tetracycline drugs monocycline (CK1), tigecycline (CK2), and (+)- and (−)-viridicatumtoxin B [(+)-1 and (−)-1].
As shown in Table 5, most of the viridicatumtoxins and analogues tested exhibited antibacterial efficacy against Gram-positive bacteria [(E. faecalis S613, E. faecium 105, and methicillin-resistant S. aureus 371 (MRSA 371)] but were less active against Gram-negative bacteria (i.e., Acinetobacter baumannii AB210). Thus, synthetic viridicatumtoxins B [(+)-1 and (−)-1] exhibited comparable antibacterial properties against these strains [E. faecalis S613, E. faecium 105, and MRSA 371: MIC = 2, 2, and 4 μg/mL, respectively, for (+)-1; MIC = 4, 4, and 8 μg/mL, respectively, for (−)-1] to those reported1 for natural viridicatumtoxin B [(+)-1] against similar strains (E. faecalis KCTC5191, E. faecium KCTC3122, MRSA CCARM3167: MIC = 2, 0.5, and 0.5 μg/mL, respectively). Based on these data, it seems that synthetic (+)-viridicatumtoxin B [(+)-1] is twice as potent as its antipode (−)-viridicatumtoxin B [(−)-1]. The Clinical and Laboratory Standards Institute (CLSI) states that MIC testing has a 2-fold error, which makes the values for (+)-viridicatumtoxin B and (−)-viridicatumtoxin B within the error range for their MIC values. Nonetheless, (+)-viridicatumtoxin B displayed high activity against the Gram-positive strains across the three independent replicates.
Table 5.
Minimum Inhibitory Concentration (MIC) Data of Compounds against Gram-Positive and Gram-Negative Bacteria and Comparison with Selected Literature Data
| Gram-(+) | Gram-(−) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| this studya
|
ref1
|
this studya
|
ref1
|
||||||
| Entry |
E. faecalis S613 |
E. faecium 105 |
MRSA 371 |
E. faecalis KCTC5191b |
E. faecium KCTC3122b |
MRSA CCARM3167b |
A. baumannii AB210 |
A. calcoaceticus KCTC2357b |
E. coli CCARM1356b |
| CK1 | 8 | 8 | 1 | 4 | |||||
| CK2 | 0.5 | 0.25 | 2 | 0.5 | |||||
| (+)-1 | 2 | 2 | 4 | 2c | 0.5c | 0.5c | 64 | 1c | >64c |
| (−)-1 | 4 | 4 | 8 | 64 | |||||
| (+)-VA1 | 8 | 8 | 8 | 64 | |||||
| (−)-VA2 | 16 | 16 | 32 | 64 | |||||
| (+)-VA3 | 16 | 16 | 8 | 64 | |||||
| (−)-VA4 | 8 | 16 | 64 | 64 | |||||
| (−)-VA5 | 128 | 64 | >128 | 64 | |||||
| (+)-VA6 | 1 | 2 | 64 | 64 | |||||
| (−)-VA7 | 4 | 8 | 8 | 64 | |||||
| (+)-VA8 | 2 | 8 | 8 | 64 | |||||
| (+)-VA9 | 0.5 | 2 | 2 | 64 | |||||
| (−)-VA10 | 1 | 1 | 2 | 64 | |||||
| (−)-VA11 | 1 | 0.5 | 2 | 64 | |||||
| (+)-VA12 | 1 | 1 | 2 | 64 | |||||
For the 5-methoxy analogues (+)-VA1, (−)-VA2, (+)-VA3, (−)-VA4, (−)-VA5, (+)-VA6, (−)-VA7, and (+)-VA8, potencies against the tested strains were generally lower than those of synthetic viridicatumtoxin B [(+)-1], except for analogue (+)-VA6, which exhibited comparable potencies against E. faecalis S613 (MIC = 1 μg/mL) and E. faecium 105 (MIC = 2 μg/mL). However, all four 5,12-diketo analogues [(+)-VA9, (−)-VA10, (−)-VA11, and (+)-VA12] demonstrated antibacterial properties stronger than those of synthetic viridicatumtoxin B [(+)-1] against E. faecalis S613 [(+)-VA9: MIC = 0.5 μg/mL; (−)-VA10: MIC = 1 μg/mL; (−)-VA11: MIC = 1 μg/mL; (+)-VA12: MIC = 1 μg/mL], E. faecium 105 [(+)-VA9: MIC = 2 μg/mL; (−)-VA10: MIC = 1 μg/mL; (−)-VA11: MIC = 0.5 μg/mL; (+)-VA12: MIC = 1 μg/mL], and MRSA 371 [(+)-VA9: MIC = 2 μg/mL; (−)-VA10: MIC = 2 μg/mL; (−)-VA11: MIC = 2 μg/mL; (+)-VA12: MIC = 2 μg/mL] (see Table 4). These results are significant in that they show that (a) the C-4a hydroxyl group of the viridicatumtoxin B molecule is not necessary for antibacterial activity and (b) the chirality of the molecule is not important for the tested bioactivity.
The modified synthetic route delivers enantiopure viridicatumtoxin B in 0.985% overall yield from key building block 7 as compared to 0.267% yield for the original route to racemic viridicatumtoxin B from the same prochiral intermediate (7), thus resulting in a 3.7-fold improved efficiency. Furthermore, these studies revealed a potential drug candidate [i.e., (−)-VA10] that is biologically superior and molecularly simpler than viridicatumtoxin B. This analogue can be derived from precursor 32 in one step and 95% yield as opposed to the natural product that requires six steps for its generation from the same intermediate in only 11% overall yield. These improvements bode well for compound (−)-VA10 and its siblings [such as (+)-VA9, (−)-VA11, and (+)-VA12] as potential drug candidates for further development. Despite the previously reported activity of viridicatumtoxin B against several Gram-negative bacterial strains,1 our tested compounds proved inactive against A. baumannii AB210. Further improvements of the antibacterial and pharmacological profiles of these compounds may be achieved by incorporating beneficial structural motifs such as the C4-dimethylamino group, which proved important for imparting the broad-spectrum activity observed for both minocycline (CK1) and tigecycline (CK2).23
3. CONCLUSION
An efficient and practical catalytic asymmetric alkylation of anthrones with cyclic allylic bromides and new phase transfer catalysts derived from quinidine and quinine has been developed. Achieving up to 99.6:0.4 selectivities and employing as low as 0.5 mol % catalyst, this method was applied to the total synthesis of both enantiomeric forms of viridicatumtoxin B. Guided by X-ray crystallographic analysis of precursor intermediates that revealed their absolute configurations and the signs of optical rotations of synthetic and natural viridicatumtoxin B (1),1 the absolute configuration of this antibiotic was deduced. This configuration is in agreement with that of viridicatumtoxin A (2) determined by X-ray crytallographic analysis and reported in 1982.20 However, it differs from that depicted for spirohexaline (3),3,23 a closely related member of the viridicatumtoxin family of natural products. Since no experimental data were reported for the given structure to spirohexaline (3),3 we suggest that its absolute configuration is most likely the same as those of viridicatumtoxins B (1) and A (2). The developed catalytic asymmetric alkylation of anthrones may find applications in organic synthesis in general, while its application to the synthesis of enantiopure natural or designed members of the viridicatumtoxin class and related compounds may lead to attractive and urgently needed drug candidates to be developed as new antibacterial agents to combat menacing infections by drug-resistant bacteria. Simpler viridicatumtoxin B analogues (+)-VA9, (−)-VA10, (−)-VA11, and (+)-VA12 are attractive candidates for further investigation along these lines.
Supplementary Material
Acknowledgments
Financial support for this work was provided by the National Institutes of Health (USA) (grant AI055475), the Cancer Prevention & Research Institute of Texas (CPRIT), and The Welch Foundation (grant C1819). A fellowship from the Postdoctoral International Exchange Program of China Postdoctoral Science Foundation (CPSF) to G.L. is gratefully acknowledged. We thank Prof. W. G. Kim for graciously providing us with copies of 1H and 13C NMR spectra of natural viridicatumtoxin B. We also thank Dr. L. B. Alemany and Dr. Q. Kleerekoper from Rice University and Mrs. Tamara Meinel, and Dr. Lothar Hennig from Leipzig University for NMR spectroscopic assistance, Dr. J. D. Korp (University of Houston) for X-ray crystallographic analysis assistance, and Dr. I. Riddington (University of Texas at Austin) and Dr. C. Pennington (Rice University) for mass spectrometric assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b12654.
Full experimental details and characterization data (PDF)
Crystallographic information file for compound 6a (CIF)
Crystallographic information file for compound (−)-11 (CIF)
Crystallographic information file for compound (+)-11 (CIF)
ORCID
K. C. Nicolaou: 0000-0001-5332-2511
Yousif Shamoo: 0000-0001-9241-8962
Notes
The authors declare no competing financial interest.
References
- 1.Zheng CJ, Yu HE, Kim EH, Kim WG. J Antibiot. 2008;61:633–637. doi: 10.1038/ja.2008.84. [DOI] [PubMed] [Google Scholar]
- 2.Hutchison RD, Steyn PS, van Rensburg SJ. Toxicol Appl Pharmacol. 1973;24:507–509. doi: 10.1016/0041-008x(73)90057-4. [DOI] [PubMed] [Google Scholar]
- 3.Inokoshi J, Nakamura Y, Hongbin Z, Uchida R, Nonaka K, Masuma R, Tomoda H. J Antibiot. 2013;66:37–41. doi: 10.1038/ja.2012.83. [DOI] [PubMed] [Google Scholar]
- 4.(a) Nicolaou KC, Nilewski C, Hale CRH, Ioannidou HA, ElMarrouni A, Koch LG. Angew Chem, Int Ed. 2013;52:8736–8741. doi: 10.1002/anie.201304691. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nicolaou KC, Hale CRH, Nilewski C, Ioannidou HA, ElMarrouni A, Nilewski LG, Beabout K, Wang TT, Shamoo Y. J Am Chem Soc. 2014;136:12137–12160. doi: 10.1021/ja506472u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silverton JV, Kabuto C, Akiyama T. Acta Crystallogr, Sect B: Struct Crystallogr Cryst Chem. 1982;38:3032–3037. [Google Scholar]
- 6.For reviews on Pd-catalyzed asymmetric alkylations, see; (a) Trost BM, Machacek MR, Aponick A. Acc Chem Res. 2006;39:747–760. doi: 10.1021/ar040063c. [DOI] [PubMed] [Google Scholar]; (b) Trost BM. J Org Chem. 2004;69:5813–5837. doi: 10.1021/jo0491004. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Crawley ML. Chem Rev. 2003;103:2921–2944. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Van Vranken DL. Chem Rev. 1996;96:395–422. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; (e) Trost BM. Acc Chem Res. 1996;29:355–364. [Google Scholar]
- 7.For selected reviews on asymmetric phase-transfer catalysis, see; (a) Maruoka K, Ooi T. Chem Rev. 2003;103:3013–3028. doi: 10.1021/cr020020e. [DOI] [PubMed] [Google Scholar]; (b) Ooi T, Maruoka K. Angew Chem, Int Ed. 2007;46:4222–4266. doi: 10.1002/anie.200601737. [DOI] [PubMed] [Google Scholar]
- 8.Trost BM, Bunt RC. J Am Chem Soc. 1994;116:4089–4090. [Google Scholar]
- 9.Shih HW, Vander Wal MN, Grange RL, MacMillan DWC. J Am Chem Soc. 2010;132:13600–13603. doi: 10.1021/ja106593m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Liu Y, Provencher BA, Bartelson KJ, Deng L. Chem Sci. 2011;2:1301–1304. doi: 10.1039/c1sc00137j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Provencher BA, Bartelson KJ, Liu Y, Foxman BM, Deng L. Angew Chem, Int Ed. 2011;50:10565–10569. doi: 10.1002/anie.201105536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ceban V, Tauchman J, Meazza M, Gallagher G, Light ME, Gergelitsová I, Veselý J, Rios R. Sci Rep. 2015;5:16886. doi: 10.1038/srep16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Corey EJ, Xu F, Noe MC. J Am Chem Soc. 1997;119:12414–12415. [Google Scholar]; (b) Corey EJ, Bo Y, Busch-Petersen J. J Am Chem Soc. 1998;120:13000–13001. [Google Scholar]
- 13.Kitamura M, Shirakawa S, Maruoka K. Angew Chem, Int Ed. 2005;44:1549–1551. doi: 10.1002/anie.200462257. [DOI] [PubMed] [Google Scholar]
- 14.Iwabuchi Y, Nakatani M, Yokoyama N, Hatakeyama S. J Am Chem Soc. 1999;121:10219–10220. [Google Scholar]
- 15.(a) Lee TBK, Wong GSK. J Org Chem. 1991;56:872–875. [Google Scholar]; (b) Lian M, Li Z, Du J, Meng Q, Gao Z. Eur J Org Chem. 2010;34:6525–6530. [Google Scholar]
- 16.(a) Kobbelgaard S, Bella M, Jørgensen KA. J Org Chem. 2006;71:4980–4987. doi: 10.1021/jo060627l. [DOI] [PubMed] [Google Scholar]; (b) Liang J, Pan J, Xu D, Xie J. Tetrahedron Lett. 2014;55:6335–6338. [Google Scholar]
- 17.(a) Zeng X, Miao C, Wang S, Xia C, Sun W. Chem Commun. 2013;49:2418–2420. doi: 10.1039/c2cc38436a. [DOI] [PubMed] [Google Scholar]; (b) Adam W, Rao PB, Degen HG, Levai A, Patonay T, Saha-Möller CR. J Org Chem. 2002;67:259–264. doi: 10.1021/jo0162078. [DOI] [PubMed] [Google Scholar]; (c) Xie JW, Xu ML, Zhang RZ, Pan JY, Zhu WD. Adv Synth Catal. 2014;356:395–400. [Google Scholar]
- 18.(a) Johnston CP, Kothari A, Sergeieva T, Okovytyy SI, Jackson KE, Paton RS, Smith MD. Nat Chem. 2015;7:171–177. doi: 10.1038/nchem.2150. [DOI] [PubMed] [Google Scholar]; (b) Li M, Woods PA, Smith MD. Chem Sci. 2013;4:2907–2911. [Google Scholar]
- 19.Corey EJ, Bakshi RK, Shibata S. J Am Chem Soc. 1987;109:5551–5553. [Google Scholar]
- 20.CCDC 1447911 contains the supplementary crystallographic data of 6a for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 21.CCDC 1503479 contains the supplementary crystallographic data of (−)-11 for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 22.CCDC 1503480 contains the supplementary crystallographic data of (+)-11 for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 23.Nelson M, Hillen W, Greenwald RA, editors. Tetracyclines in Biology, Chemistry, and Medicine. Birkhauser Verlag; Basel: 2001. p. 26. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.