

Abstract
Increasing evidence shows that conventional cardiovascular risk factors are incompletely predictive of cardiovascular disease, particularly in elderly individuals, suggesting that there may still be unidentified causal risk factors. While the accumulation of somatic DNA mutations is a hallmark of aging, its relevance in cardiovascular disease or other age-related conditions has been, with the exception of cancer, largely unexplored. Here, we review recent clinical and preclinical studies that have identified acquired mutations in hematopoietic stem cells and subsequent clonal hematopoiesis as a new cardiovascular risk factor and a potential major driver of atherosclerosis. Understanding the mechanisms underlying the connection between somatic mutation-driven clonal hematopoiesis and cardiovascular disease will be highly relevant in the context of personalized medicine, as it may provide key information for the design of diagnostic, preventive or therapeutic strategies tailored to the effects of specific somatic mutations.
Keywords: Aging, cardiovascular disease, atherosclerosis, inflammation, somatic mutations, clonal hematopoiesis, CHIP
Aging and cardiovascular disease, an incompletely understood connection
Cardiovascular/cerebrovascular disease (CVD) is the leading cause of morbidity and mortality in both industrialized and low-income to middle-income countries, particularly in the elderly population. Furthermore, this situation is expected to worsen as the world population is aging and CVD incidence steeply increases with age, accounting for more than 40% of total deaths and more than 85% of chronic disease deaths in those older than 70 years.1 Although epidemiological studies clearly show that advanced age is the most important risk factor for CVD, we have an incomplete understanding of how it promotes disease progression, in particular in the context of atherosclerosis. While the effect of aging on atherosclerotic CVD reflects to a great extent the cumulative exposure to a number of traditional risk factors (e.g. hypercholesterolemia, diabetes, hypertension, smoking),2 clinical evidence suggest that additional, still unidentified age-related factors may contribute to atherosclerosis development. Indeed, age remains an independent risk factor for CVD even after statistical adjustment for traditional risk factors,3, 4 and most CVD events occur in elderly individuals who are at low or intermediate risk based on traditional risk factors. In one example, an international study that included more than 120,000 atherosclerotic CVD patients revealed that ~65% of patients older than 65 years old had either none or just one conventional CV risk factor.5 Consistent with this observation, imaging studies show that most individuals at low CV risk based on conventional risk algorithms exhibit substantial subclinical atherosclerosis.6–9 Additionally, emerging evidence suggests that the effects of some conventional risk factors on CV risk differ between young and elderly individuals. For example, the impact of hypercholesterolemia on CV risk has been shown to gradually decrease with aging in several studies,10–12 suggesting the existence of alternative non-conventional risk factors that contribute to the pathogenesis of atherosclerotic CVD in the elderly population. Consistent with these results, recent clinical trials of treatments with high intensity statin regimens or new cholesterol-lowering drugs (e.g. PCSK9 inhibitors) show that, despite massive reductions in blood cholesterol levels, atherosclerotic plaques progress in a significant proportion of individuals13, 14 and a substantial level of residual CV risk remains.15, 16 Therefore, although the major contribution to atherosclerosis of the cumulative exposure to conventional risk factors –particularly hypercholesterolemia- is undeniable,17 these aforementioned studies lead inevitably to one question: what else is driving atherosclerotic CVD in the elderly? Emerging evidence from human and mouse studies suggest that the acquisition of somatic mutations in hematopoietic cells may be part of the answer to this question.
Somatic mutations in the hematopoietic system and atherosclerotic CVD: evidence from human studies
Over the past two decades, research on the impact of human genetics on CVD has focused mostly on genetic variation transmitted through the germ line.18, 19 While these studies have unveiled a significant contribution of many inherited variants to CVD, recent evidence suggests that non-inherited, acquired mutations that affect somatic cells may also be important players in CVD and potentially other age-related disorders. These somatic mutations occur randomly, beginning in prenatal development.20–22 In most cases they have no or little phenotypic consequences, as most mutations have no effect on cellular function or in some cases are toxic or even lethal to the mutant cell, leading to its quick disappearance. However, in a few instances, a somatic mutation confers a competitive advantage to the cell, which leads to the progressive expansion of the mutant clone. Consequently, individuals become a mosaic of cells with different genotypes over time.21, 23, 24 Accumulating evidence reveals that this somatic genome mosaicism is indeed an inevitable consequence of the normal aging process in many tissues.25, 26 This is particularly true in highly proliferative tissues, such as the hematopoietic system, which generates >100 billion cells daily. Recent evidence show that individual hematopoietic stem/progenitor cells (HSPCs) accumulate somatic mutations as a function of age, even in healthy individuals. Human HSPCs have been calculated to develop 0.13 ± 0.02 exonic mutations per year of life, and thus it can be estimated that by age 50 an individual would accumulate an average of five coding gene mutations within each HSPC.24 This sets the stage for a robust Darwinian selection of mutations that provide a competitive advantage to the mutant HSPC by promoting its self-renewal, proliferation or survival. Such a competitive advantage eventually leads to the clonal expansion of the mutant HSPC in the hematopoietic system and, through its cellular progeny, in the blood and other tissues infiltrated by blood cells. Early studies documented the occurrence of age-related clonal hematopoiesis (frequently abbreviated as ARCH) based on the results of X-chromosome inactivation studies,27–30 and a substantial body of evidence has now demonstrated that this process of somatic mutation-driven clonal hematopoiesis (Figure 1A), is commonly observed in healthy elderly individuals.31–38 In the case of extreme age, there is sometimes a marked expansion of a few clones and a collapse of HSPC diversity caused by the acquisition of somatic mutations in key hematopoiesis regulators or other mechanisms of clonal expansion.39 For example, deep whole-genome sequencing of the peripheral blood cells from a 115 year old woman revealed that the majority of her white blood cells were derived from just two related HSPC clones.40
Figure 1. Somatic mutations in blood cells as a shared mechanism of hematological cancer and cardiovascular disease.
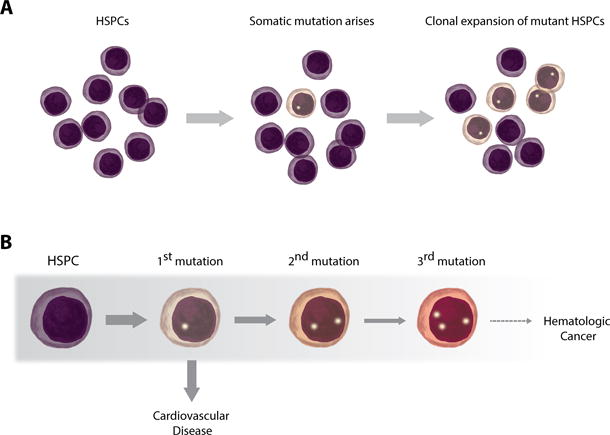
A. The accumulation of somatic mutations in hematopoietic progenitor and stem cells is an inevitable consequence of the process of aging. Some of these random mutations confer a competitive advantage to the mutant cells, leading to its clonal expansion. This phenomenon can be defined as somatic mutation-driven clonal hematopoiesis. B. Most individuals exhibiting somatic mutation-driven clonal hematopoiesis only carry one driver mutation (e.g. in DNMT3A, TET2, ASXL1 or JAK2). While this situation greatly increases the risk of acquiring additional driver mutations and eventually developing a hematologic cancer, this condition is infrequent, even in individuals with clonal hematopoiesis. The main cause of death in individuals exhibiting somatic mutation-driven clonal hematopoiesis is atherosclerotic cardiovascular disease. Grey shade in the figure indicates decreasing frequency.
Clonal hematopoiesis is often driven by a single mutation in a cancer-related gene, and, accordingly, it is associated with increased risk of hematological malignancies.35, 36 Despite this, epidemiological studies show that most individuals carrying cancer-related somatic mutations that drive clonal expansion of the mutant cell will never develop blood cancer, since this is a rare condition requiring the accumulation of multiple additional oncogenic gene mutations (Figure 1B).35 Based on these observations, this phenomenon is frequently referred by hematologists as clonal hematopoiesis of indeterminate potential (CHIP).42, 43 Although large chromosomal rearrangements44–47 and point mutations in more than 100 cancer-related genes31–37, 41 have been identified as potential drivers of clonal hematopoiesis, most somatic mutations linked to this phenomenon reported to date affect a small subset of genes known to play important roles in hematopoiesis and/or hematological cancers (Table). Notably, most somatic mutations associated with clonal hematopoiesis occur in three genes (DNMT3A, TET2 and ASXL1) that encode for epigenetic regulators that have been implicated in the control of hematopoiesis.48–55 It can be estimated that at least 10-20% of individuals older than 65 years old exhibit a significant fraction of white blood cells (e.g. >4%) that harbor somatic mutations in these or other known driver genes.34–36 Furthermore, highly sensitive sequencing techniques that allow the detection of mutations in a smaller percentage of blood cells suggest that low levels of clonal hematopoiesis may be even more prevalent, trending towards inevitability in the very elderly.31, 33, 37 Despite these observations, the relevance of clonal hematopoiesis in the context of age-associated disorders other than cancer has been largely neglected. The scarce investigation of the clinical consequences of this phenomenon is somewhat surprising considering that mutations in HSPCs are likely to affect the function of their progeny –immune cells, red blood cells and platelets-, whose dysfunction is at the center of most age-related chronic disorders. This situation is bound to change as evidence has emerged supporting the concept that age-associated somatic mosaicism in the hematopoietic system and clonal hematopoiesis may have major pathophysiological implications in the setting of CVD and other chronic diseases.
Table.
Fifteen most frequently mutated genes identified as candidate drivers of clonal hematopoiesis in cancer-free individuals in several human studies
| GENE | REFERENCES |
|---|---|
| DNMT3A | 31–37, 41 |
| TET2 | 31, 32, 34–38, 41 |
| ASXL1 | 31, 32, 34–37, 41 |
| JAK2 | 31–37, 41 |
| TP53 | 31, 32, 34–37, 41 |
| PPM1D | 31, 34, 36, 41 |
| IDH2 | 31–37, 41 |
| CBLC | 31, 32, 34–37, 41 |
| SF3B1 | 31, 33–37, 41 |
| SRSF2 | 31, 33, 35–37, 41 |
| GNAS | 31, 34, 35, 37, 41 |
| KRAS | 32, 33, 35, 37, 41 |
| GNB1 | 31, 35, 37, 41 |
| NRAS | 31–33, 35, 37 |
| MYD88 | 31, 35–37 |
Early evidence of a connection between a specific somatic mutation in the hematopoietic system and the development of CVD in the absence of cancer can be found in some small studies that reported the presence of the JAK2 V617F mutation in blood cells of a higher than expected proportion of cancer-free patients with venous thrombosis,56, 57 stable coronary heart disease (CHD)58 or peripheral artery disease.59 While limited by their small sample size and retrospective nature, these studies, among others, opened the door to the possibility of a connection between somatic mutations in blood cells and CVD. This possibility was reinforced when whole-exome sequencing of blood cells of more than 5,000 cancer-free individuals in seven independent cohorts revealed that the presence of single-nucleotide variants and small insertions or deletions in any of 160 analyzed hematopoiesis-related genes is associated with a ~40% increase in all-cause mortality.35 Although the presence of one somatic mutation in any of these genes was associated with an ~11-fold increase in the future risk of hematologic cancer, this only affected 0.5-1% of mutation carriers each year and did not explain the marked increase in all-cause mortality. Unexpectedly, the increased mortality was associated with a substantial increase in deaths due to myocardial infarction or stroke among individuals exhibiting a relative frequency of the mutant allele (variant allele fraction or VAF) greater than 10%, typically indicative of the presence of more than 20% blood cells carrying the mutation in heterozygosis. Consistent with this finding, a significant association was found between the detection of somatic mutations in blood cells and the future incidence of atherosclerotic conditions (CHD and stroke):~45% of mutation carriers developed atherosclerotic CVD during the study, compared to ~20% of non-carriers. Thus, carrying somatic mutations in hematopoiesis-related genes increased >2-fold the risk of developing atherosclerotic CVD, an effect that was comparable or even greater than that of various conventional risk factors.35 While this study reported an association between CVD and a somatic mutation in any of 160 genes, mutations in just four genes, namely DNMT3A, TET2, ASXL1 and JAK2, accounted for more than 70% of the detected mutations. A more recent study has confirmed the existence of independent associations between CHD and mutations in each of these four genes with median VAF ranging from 10% to 20%.41 However, it is likely that larger studies with longer follow-up periods will identify additional mutated genes linked to CHD, and that significant associations will be observed even with lower VAF values. Indeed, this latter study also reported an association of CHD with any of 56 different somatic mutations in less-frequently mutated genes,41 supporting the possibility that other mutated genes may be connected to age-related CVD. Somatic mutations may also be linked to the premature development of CVD in young and middle age individuals, as a retrospective case-control analysis of individuals <50 years old revealed a remarkable 4-fold higher risk of early-onset myocardial infarction in individuals exhibiting somatic mutation-driven clonal hematopoiesis.41 Furthermore, the presence of somatic mutations in blood cells was also found to be associated with a 3-fold higher risk of exhibiting a high degree of coronary calcium calcification, a marker of subclinical atherosclerosis burden. Remarkably, the increased risk of coronary artery calcification was 12-fold in individuals exhibiting a VAF greater than 10%.41
Overall, these human data suggest a strong and unexpected connection between age-related somatic mutations in hematopoietic cells, clonal hematopoiesis and atherosclerosis. However, it must be noted that the descriptive nature of these types of epidemiological studies does not allow cause-effect relationships to be established. The associations between somatic mutation-driven clonal hematopoiesis and atherosclerotic CVD could simply reflect shared consequences of the aging process or be secondary to confounding factors. Furthermore, these studies do not fully address the issue of directionality; i.e. is clonal hematopoiesis a driver of CVD or are low-grade chronic inflammation and metabolic dysfunction, frequent in subjects at high cardiovascular risk, driving somatic mutagenesis and/or clonal hematopoiesis as suggested by some researchers?44, 60 Answering these questions requires the combination of additional human data and carefully designed experimental studies in animal models.
Somatic mutations in the hematopoietic system and atherosclerotic cardiovascular disease: evidence from mouse studies, the case of TET2
As described above, most reported somatic mutations associated with clonal hematopoiesis and CVD occur in a small number of genes, particularly in four genes (DNMT3A, TET2, ASXL1 and JAK2), which are known to modulate hematopoiesis,48–55, 61–63 but whose role in CVD remained mostly unexplored until recently. The first of these genes reported to exhibit somatic mutations in blood cells in individuals with clonal hematopoiesis without blood cancer was TET2 (ten eleven translocation 2).38 More than 130 different mutations have been reported in this gene in blood cells of cancer-free individuals,31, 32, 34–38, 41 which are considered to result in loss of gene function given that most are small insertions/deletions, non-sense mutations or missense mutations leading to the substitution of key amino acids in the catalytic domain. TET2 is a multifaceted transcriptional regulator that can facilitate both transcriptional activation and repression depending on the molecular/cellular context. It is able to catalyze the conversion of 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC), a process that facilitates DNA demethylation and transcriptional activation.64–66 Conversely, TET2 can also mediate transcriptional repression by recruiting histone deacetylases to gene promoters.67 Mouse studies have shown that TET2 deletion or haploinsufficiency result in increased HSPC self-renewal and a bias towards differentiation into the myeloid lineage,53–55, 68 consistent with human data linking TET2 mutations with clonal hematopoiesis. However, despite the broad expression pattern of TET2,69 the relevance of this protein in pathophysiological settings other than stem cell biology or cancer has just recently begun to be explored. In this regard, a recent study from our group demonstrated that TET2 loss of function-driven clonal hematopoiesis accelerates atherosclerosis in hyperlipidemic mice, providing experimental evidence in support of the causal contribution of somatic mutation-driven clonal hematopoiesis to atherosclerotic CVD.70 In this study, experimental conditions in mice recapitulated many features of clonal hematopoiesis linked to somatic TET2 mutations in humans. Competitive bone marrow (BM) transplantation experiments were employed to introduce a small number of TET2-deficient cells in atherosclerosis-prone Ldlr−/− mice and mimic the scenario of clonal hematopoiesis, where the mutation is initially present in a small clone that expands over time. In agreement with previous studies,53–55 TET2-deficient cells expanded progressively in BM, spleen and blood in this experimental setting, and they showed a mild myeloid bias, with a preferential expansion into the Ly-6C-high classical monocyte population.70 Importantly, this did not affect total numbers of white blood cells or their distribution in different subsets, consistent with human studies on cancer-free individuals carrying TET2 mutations.35, 38 The expansion ofTet2−/− cells in these conditions accelerated atherosclerosis substantially, leading to the formation of ~60% larger plaques. A similar, albeit milder, effect was observed in equivalent transplantation experiments with Tet2+/− cells, which may mimic better the human scenario given that somatic TET2 mutations are likely to be in heterozygosis in most individuals. Additionally, atherosclerotic plaque size was also increased when Tet2 ablation was restricted to myeloid cells.70 These findings were recently validated in independent studies in mice exhibiting full hematopoietic ablation of TET2.41 Collectively, these experimental studies provide strong support to the existence of causal connection between somatic mutations in this gene and the development of atherosclerotic CVD.
From a mechanistic point of view, in vitro and in vivo studies suggest that accelerated atherosclerosis in conditions of TET2 loss of function-driven clonal hematopoiesis is mainly due to the exacerbated expression of pro-inflammatory cytokines and chemokines in TET2-deficient macrophages,41, 70, 71 particularly due to the overproduction of the pro-inflammatory cytokine interleukin 1 beta (IL-1β).70 Interestingly, this mechanism is consistent with the possibility that a small number of TET2-mutant cells in the plaque may be sufficient to drive exacerbated vascular inflammation and atherosclerosis by promoting pro-inflammatory changes in both mutant and wild-type cells. In this regard, it was found that exacerbated IL-1β production by TET2-deficient plaque cells in mice promotes P-select in expression and endothelial cell activation in the plaque, leading to increased monocyte recruitment regardless of the Tet2 genotype of recruited monocytes.70 Furthermore, IL-1β has been reported to stimulate its own expression by an autoregulatory feedback loop72–76 and therefore it is likely that overproduction of IL-1β by TET2-deficient plaque cells promotes further expression of IL-1β in both TET2-deficient and wild-type cells. Hence, it can be speculated that a few TET2-mutant cells in the plaque act in a catalytic manner, amplifying and perpetuating vascular inflammation and atherosclerosis. Consistent with this “few bad apples spoil the barrel” scenario, the progressive expansion of Tet2−/− cells in a competitive BMT setting seems to have a comparable effect on atherosclerosis than that of the full ablation of TET2 in all BM cells.41, 70 This relatively surprising finding likely reflects the complexity of the actions of TET2 in the hematopoietic system and it suggests that the effects of TET2 deficiency on atherosclerosis may be dependent on cell type and/or stage of plaque development. It also provides valuable information on the variety of research strategies available to model clonal hematopoiesis in mice. While conventional full BM transplantation strategies are the most straightforward strategy to study the effects on atherosclerosis of clonal hematopoiesis-related mutations, the use of cell type-specific approaches or competitive BM transplantation strategies with low percentages of mutant cells may be advantageous in some instances. This may be the case when the mutant HSPCs exhibit a clear bias to differentiate into specific lineages or when the ubiquitous expression of the mutated gene in hematopoietic progenitors leads to hematological abnormalities or malignancies not present in most cancer-free individuals exhibiting clonal hematopoiesis (e.g. JAK2 gain-of-function mutations61–63).
The use of mouse studies tailored to the human scenario of clonal hematopoiesis may provide clinically-relevant information. For instance, the findings in our mouse model of TET2 mutation-mediated clonal hematopoiesis suggest that IL-1β blockade may be particularly effective for the prevention/treatment of CVD in individuals carrying somatic mutations in TET2. Supporting this possibility, pharmacological blockade of NLRP3 inflammasome-mediated IL-1β secretion had greater atheroprotective effects in conditions of TET2 loss of function than in wild-type controls, and completely suppressed differences in atherosclerosis development between genotypes.70 These findings are especially relevant in the context of the results of the CANTOS trial, which demonstrated the efficacy of a neutralizing antibody against IL-1β (Canakinumab) in the secondary prevention of atherosclerotic CVD in high-risk patients exhibiting evidence of systemic inflammation.77 Even though Canakinumab has shown efficacy against atherosclerotic CVD, its clinical use poses substantive challenges in the setting of chronic disorders, as it also increases the risk of infections.77 Therefore, this drug, and probably any anti-inflammatory drug, should probably be administered exclusively to those patients that are predicted to show the greatest response to this therapy and to obtain the most atheroprotection from it. Currently, the analysis of hsCRP, a marker of systemic inflammation, is the best available tool to identify these patients and distinguish between responders and non-responders to anti-inflammatory therapies.78 While no conclusive data has yet been provided, it can be speculated that clonal hematopoiesis driven by TET2 loss of function or other pre-leukemic mutations may be associated with increased systemic inflammation in some individuals, and that the genetic analysis of blood cells may be employed in the future to identify responders to anti-inflammatory therapies in the context of precision medicine strategies.79 In this regard, our preclinical data suggest that individuals with TET2 mutations may respond more favorably than the general population to IL-1β/NLRP3-targeted therapies, thus creating the basis for a personalized therapy for the prevention of accelerated atherosclerotic CVD in individuals carrying somatic mutations in this gene. This notion could be clinically validated through a genetic screening for somatic TET2 mutations in the CANTOS cohort. Furthermore, given that IL-1β plays a pivotal role in vascular inflammation80 and TET2 also affects the expression of other immunomodulatory molecules,41, 70, 71 it may be of interest to apply similar strategies in ongoing clinical trials with other broad spectrum anti-inflammatory compounds such as methotrexate81 or colchicine.82
Somatic mutation-driven clonal hematopoiesis and atherosclerotic cardiovascular disease beyond TET2: other genes and mechanisms?
TET2 was the first gene reported to carry somatic mutations in cancer-free individuals exhibiting clonal hematopoiesis,38 and mutations in this gene have been the first to be causally linked to an age-associated disorder other than cancer, atherosclerotic CVD.35, 41, 70 However, human evidence suggest that many other mutated genes may contribute to this condition.35, 41 As discussed above, independent associations with atherosclerotic CVD have been reported for three additional genes, DNMT3A, ASXL1 and JAK2, and mutations in other less frequently mutated genes also seem to be associated with increased risk of atherosclerotic CVD.35, 41 It is reasonable to speculate that these additional mutated genes will differentially affect CVD due to their unique functions and mechanisms of action. Some mutations may induce qualitative changes in key immune cells, whereas others may affect the differentiation of HSPCs to specific lineages, leading to quantitative changes in specific immune cell subsets, particularly in individuals that exhibit large mutant clones and a reduced HSPC repertoire. Whether somatic mutations in clonal hematopoiesis-related genes other than TET2 contribute to atherosclerosis remains unexplored and additional experimental studies are warranted. However, previous research allows for some speculation on the different mechanisms by which some of these mutated genes may promote CVD.
Accumulating experimental evidence suggest that DNMT3A, the most frequently mutated gene in cancer-free individuals that exhibit clonal hematopoiesis, is an important modulator of both hematopoiesis and inflammatory responses. This gene encodes for a methyl transferase enzyme that catalyzes DNA methylation and modulates gene transcription in multiple scenarios. Supporting a major role of DNMT3A mutations in clonal hematopoiesis in humans, mouse HSPCs exhibiting heterozygotic DNMT3A loss of function develop a competitive advantage and myeloid skewing over time.83 Importantly, DNMT3A deficiency has also been reported to lead to several potentially pro-atherogenic phenotypes in immune cells, including exacerbated pro-inflammatory activation of mast cells,84 increased IFNγproduction by T cells85–87 and restrained immunosuppressive function in myeloid-derived suppressor cells.88 In contrast, DNMT3A inhibition has also been shown to increase the expression of interleukin 13 in T cells87 and to limit the production of type I interferons in macrophages,89 which could potentially protect against atherosclerosis development.90, 91 Therefore, given the complexity of the immunomodulatory functions of DNMT3A, carefully designed experimental studies will be required to determine the potential contribution of somatic mutations in this gene to CVD.
Another frequently mutated gene for which previous data support a role in CVD is JAK2, which encodes for an important signaling kinase. Indeed, early evidence linking somatic mutations to CVD was provided by the observation that essential thrombocytopenia and polycythemia vera patients carrying the JAK2 V617F gain-of-function mutation exhibit a higher risk of thrombosis.92–100 Increased blood counts of leukocytes, red blood cells and/or platelets are potential contributors to the increased CV risk in these patients. However, additional qualitative changes in blood cells are also likely to play a role, as increased CV risk has been observed in individuals carrying this mutation and exhibiting clonal hematopoiesis with little or no changes in blood cell counts.35, 101 Some mouse studies suggest that JAK2 V617F-driven clonal hematopoiesis exhibits a clear myeloid, particularly granulocytic, bias102 and thus it is possible that the effects of this somatic mutation on the CV system are mediated by phenotypic changes in myeloid cells. Given that findings in various experimental models support a role for JAK2 in the modulation of the pro-inflammatory activities of macrophages and neutrophils,103–105 it will be of interest to evaluate the impact of the JAK2 V617F mutation on CVD in the absence of hematological malignancies.
Beyond the above-mentioned genes, it must be noted that most other mutated genes associated with clonal hematopoiesis have never been evaluated for their role in CVD in any context. Furthermore, controversy exists on whether some of the observed mutations lead to loss or gain of gene function, as is the case of ASXL1 truncation mutations.51, 52, 106–108 On the other hand, it is tempting to speculate that some of the somatic mutations that lead to clonal hematopoiesis may be benign or exhibit protective rather than pathogenic effects on the CV system. In other words, some types of clonal hematopoiesis may be uncoupled from CVD or other age-associated chronic diseases, and these forms of clonal hematopoiesis might be overrepresented in individuals that exhibit exceptional longevity.
Somatic mutation-driven clonal hematopoiesis: manyopen questions
Beyond atherosclerosis, a role in other age-related conditions?
The link between acquired somatic mutation-driven clonal hematopoiesis and atherosclerosis development is now supported by both clinical and preclinical data. However, the possibility that mutations driving clonal hematopoiesis also modulate other forms of CVD or other age-related chronic conditions remains largely unexplored and deserves attention. Indeed, recent preclinical studies in mouse models suggest that TET2 loss of function-driven clonal hematopoiesis may accelerate heart failure, a condition that is common in the elderly.109 Furthermore, human studies have suggested a potential connection between various forms of somatic mutations in blood cells and type 2 diabetes, chronic pulmonary disease or psychiatric conditions.31, 32, 35, 44 Future experimental studies are warranted to examine the nature of these connections, especially given that many of the known mutations that drive clonal hematopoiesis affect immune cells and pathways that are at the center of many age-related chronic disorders.
Beyond mutations, what else modulates clonal hematopoiesis?
As indicated above, prior exome sequencing studies have suggested that 10-20% of individuals older than 65 years old exhibit somatic-mutation driven clonal hematopoiesis leading to a substantial percentage (e.g. >4%) of mutant cells in the blood.34–36 However, more recent studies using highly sensitive sequencing techniques that allow the detection of mutations in a smaller fraction of blood cells found that the prevalence of clonal hematopoiesis is probably at least double than that initially reported.31, 33, 37 Furthermore, the seeds for somatic-mutation driven clonal hematopoiesis may be present even in younger individuals, as one recent study employing targeted error-corrected sequencing found that somatic mutations in driver genes (most frequently DNMT3A and TET2) are present in a very small fraction of blood cells (e.g. 1/3000) in 95%of individuals between 50 and 60 years of age.110 Similarly, it has been estimated that almost half of healthy individuals at 50 years of age carry at least one HSPC with a randomly generated mutation in TP53, another potential driver of clonal hematopoiesis.111 Thus, the presence of low levels of hematopoietic cells with somatic mutations may be ubiquitous by middle age, but only a fraction of individuals may advance to a condition where the mutant clone undergoes marked expansion. Overall, these observations suggest the existence of additional factors, perhaps genetic or lifestyle factors, that regulate the clonal expansion of mutant HSPCs. For example, some germline variants, most notably in the JAK2 and TERT genes, have been associated with increased prevalence of clonal hematopoiesis associated with the JAK2 V617F mutation.112 Furthermore, smoking has also been found to be associated with clonal hematopoiesis.31, 36, 113 Additional investigations of the effect of these and other factors on somatic mutation-driven clonal hematopoiesis may lead to a better understanding of the complex molecular mechanisms that regulate the dynamics of this process, which could contribute to the development of therapies to prevent it.
Somatic mutations in bone marrow cells: a new factor to consider in the application of cell therapies?
Numerous “adult stem cell” therapies are being tested in clinical trials or are offered to the public by commercial entities.114–116 These therapies often involve the delivery of autologous or allogeneic BM cells or cells from tissues that contain a substantial fraction of BM-derived cells. In addition, some of these procedures employ cells that are selected based upon the expression of cell surface markers, such as CD34, that lead to the enrichment in HSPCs. Given the realization that somatic mutations in HSPCs can promote their clonal expansion and alter the function of their cellular progeny, questions can be raised about the long-term safety of some of these adult stem cell procedures. It is theoretically possible that the delivery of cell fractions from elderly individuals, who may exhibit clonal hematopoiesis, will nullify the therapeutic effect of the transplanted cells or even contribute to adverse outcomes due to the transplantation of hematopoietic cells that harbor somatic mutations. Furthermore, one cannot rule out the possibility that the adoptive transfer of mutant HSPCs could further accelerate their clonal expansion and contribute to disease processes as the patient ages. Future studies are therefore warranted to evaluate whether it might be appropriate to examine transplanted cell populations for their clonal hematopoiesis status prior to use.
Clonal hematopoiesis without known driver mutations: what are we missing?
Most research efforts to date in the context of clonal hematopoiesis have been focused on the identification of mutations in blood cancer-related genes previously known to modulate hematopoiesis. However, it is noteworthy that ~50%, or greater, of cases of clonal hematopoiesis occur without any known driver gene mutation.31, 36 While this may be related to technical issues in some cases (e.g. sequencing depth or specific blood cell populations being sequenced), these results strongly suggest the existence of additional, still unidentified somatic mutations that can drive clonal hematopoiesis and perhaps contribute to age-related disorders. The analysis of chromosomal rearrangements or mutations in non-protein coding genes may deserve special attention in this context. Indeed, a significant association between the presence of clonally-expanded blood cells carrying acquired large chromosomal rearrangements and the occurrence of vascular complications of diabetes has been reported.44 Alternatively, non-mutational mechanisms may also contribute to clonal hematopoiesis. Computer simulations suggest that, at least in some cases, clonal hematopoiesis can arise in the absence of a driver mutation simply due to stochastic neutral drift in the HSPC population.31 Among other non-mutational mechanisms,117 it is possible, for instance, that epigenetic alterations and the associated cell-to-cell variability in gene expression leads to the dominance of specific HSPC populations. Similarly, the heterogeneous distribution of extracellular signals in the BM niche could lead to the preferential expansion of some HSPCs due to their local environment. Interestingly, clonal hematopoiesis without candidate driver gene mutations has also been associated with an increase in all-cause mortality,31 although its potential association with CVD remains unexplored. Be it somatic mutations or non-mutational mechanisms, extensive investigation will be required to identify new drivers of clonal hematopoiesis and understand their clinical relevance.
Conclusions
The accumulation of random mutations in the hematopoietic system is an unavoidable consequence of the aging process. While the pathophysiologic relevance of these acquired somatic mutations has been traditionally restricted to the cancer field, emerging evidence suggest that these mutations are important drivers of CVD and potentially many other age-related disorders. A combination of clinical and preclinical studies will be required to understand the pathogenic effects (or lack of) of the various mutated genes, as well as the molecular mechanisms that govern the expansion of mutant hematopoietic stem cells. This information could eventually lead to the design of personalized therapies or preventive strategies for individuals carrying specific somatic mutations in blood cells.
Acknowledgments
The illustration was provided by Shraddha Nayak.
Sources of Funding
This work was funded by National Institutes of Health grants HL126141, HL131006, HL132564 and HL138014 to Kenneth Walsh and by American Heart Association Scientist Development Grant 17SDG33400213 to JoséJ. Fuster.
Non-standard Abbreviations and Acronyms
- ASXL1
additional sex combs like 1
- CHD
coronary heart disease
- CHIP
clonal hematopoiesis of indeterminate potential
- CVD
cardiovascular disease
- DNMT3A
DNA methyltransferase 3 alpha
- HSPC
hematopoietic stem/progenitor cell
- JAK2
Janus kinase 2
- TET2
tet methylcytosine dioxygenase 2
- TP53
tumor protein p53
Footnotes
Disclosures
We have no conflicts of interest to disclose.
References
- 1.Global Burden of Disease Study. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1459–1544. doi: 10.1016/S0140-6736(16)31012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sniderman AD, Furberg CD. Age as a modifiable risk factor for cardiovascular disease. Lancet. 2008;371:1547–9. doi: 10.1016/S0140-6736(08)60313-X. [DOI] [PubMed] [Google Scholar]
- 3.D’Agostino RB, Sr, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, Kannel WB. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117:743–53. doi: 10.1161/CIRCULATIONAHA.107.699579. [DOI] [PubMed] [Google Scholar]
- 4.Kannel WB, Vasan RS. Is age really a non-modifiable cardiovascular risk factor? Am J Cardiol. 2009;104:1307–10. doi: 10.1016/j.amjcard.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khot UN, Khot MB, Bajzer CT, Sapp SK, Ohman EM, Brener SJ, Ellis SG, Lincoff AM, Topol EJ. Prevalence of conventional risk factors in patients with coronary heart disease. JAMA. 2003;290:898–904. doi: 10.1001/jama.290.7.898. [DOI] [PubMed] [Google Scholar]
- 6.Baber U, Mehran R, Sartori S, Schoos MM, Sillesen H, Muntendam P, Garcia MJ, Gregson J, Pocock S, Falk E, Fuster V. Prevalence, impact, and predictive value of detecting subclinical coronary and carotid atherosclerosis in asymptomatic adults: the BioImage study. J Am Coll Cardiol. 2015;65:1065–74. doi: 10.1016/j.jacc.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez-Friera L, Penalvo JL, Fernandez-Ortiz A, et al. Prevalence, Vascular Distribution, and Multiterritorial Extent of Subclinical Atherosclerosis in a Middle-Aged Cohort: The PESA (Progression of Early Subclinical Atherosclerosis) Study. Circulation. 2015;131:2104–13. doi: 10.1161/CIRCULATIONAHA.114.014310. [DOI] [PubMed] [Google Scholar]
- 8.Laclaustra M, Casasnovas JA, Fernandez-Ortiz A, Fuster V, Leon-Latre M, Jimenez-Borreguero LJ, Pocovi M, Hurtado-Roca Y, Ordovas JM, Jarauta E, Guallar E, Ibanez B, Civeira F. Femoral and Carotid Subclinical Atherosclerosis Association With Risk Factors and Coronary Calcium: The AWHS Study. J Am Coll Cardiol. 2016;67:1263–74. doi: 10.1016/j.jacc.2015.12.056. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez-Friera L, Fuster V, Lopez-Melgar B, Oliva B, Garcia-Ruiz JM, Mendiguren J, Bueno H, Pocock S, Ibanez B, Fernandez-Ortiz A, Sanz J. Normal LDL-Cholesterol Levels Are Associated With Subclinical Atherosclerosis in the Absence of Risk Factors. J Am Coll Cardiol. 2017;70:2979–2991. doi: 10.1016/j.jacc.2017.10.024. [DOI] [PubMed] [Google Scholar]
- 10.Gransbo K, Almgren P, Nilsson PM, Hedblad B, Engstrom G, Melander O. Risk factor exposure in individuals free from cardiovascular disease differs according to age at first myocardial infarction. Eur Heart J. 2016;37:1977–81. doi: 10.1093/eurheartj/ehw026. [DOI] [PubMed] [Google Scholar]
- 11.Sniderman AD, Islam S, McQueen M, Pencina M, Furberg CD, Thanassoulis G, Yusuf S. Age and Cardiovascular Risk Attributable to Apolipoprotein B, Low-Density Lipoprotein Cholesterol or Non-High-Density Lipoprotein Cholesterol. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.116.003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prospective Studies C. Lewington S, Whitlock G, Clarke R, Sherliker P, Emberson J, Halsey J, Qizilbash N, Peto R, Collins R. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007;370:1829–39. doi: 10.1016/S0140-6736(07)61778-4. [DOI] [PubMed] [Google Scholar]
- 13.Nicholls SJ, Ballantyne CM, Barter PJ, Chapman MJ, Erbel RM, Libby P, Raichlen JS, Uno K, Borgman M, Wolski K, Nissen SE. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365:2078–87. doi: 10.1056/NEJMoa1110874. [DOI] [PubMed] [Google Scholar]
- 14.Nicholls SJ, Puri R, Anderson T, et al. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV Randomized Clinical Trial. JAMA. 2016;316:2373–2384. doi: 10.1001/jama.2016.16951. [DOI] [PubMed] [Google Scholar]
- 15.Cholesterol Treatment Trialists C. Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–81. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR, Committee FS and Investigators Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 17.Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, Lisheng L, Investigators IS Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364:937–52. doi: 10.1016/S0140-6736(04)17018-9. [DOI] [PubMed] [Google Scholar]
- 18.Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18:331–344. doi: 10.1038/nrg.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Donnell CJ, Nabel EG. Genomics of cardiovascular disease. N Engl J Med. 2011;365:2098–109. doi: 10.1056/NEJMra1105239. [DOI] [PubMed] [Google Scholar]
- 20.Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science. 2013;341:1237758. doi: 10.1126/science.1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A, Lee S, Chittenden TW, D’Gama AM, Cai X, Luquette LJ, Lee E, Park PJ, Walsh CA. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015;350:94–98. doi: 10.1126/science.aab1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ju YS, Martincorena I, Gerstung M, et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 2017;543:714–718. doi: 10.1038/nature21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martincorena I, Roshan A, Gerstung M, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–6. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–78. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forsberg LA, Gisselsson D, Dumanski JP. Mosaicism in health and disease - clones picking up speed. Nat Rev Genet. 2017;18:128–142. doi: 10.1038/nrg.2016.145. [DOI] [PubMed] [Google Scholar]
- 26.Vijg J. Somatic mutations, genome mosaicism, cancer and aging. Curr Opin Genet Dev. 2014;26:141–9. doi: 10.1016/j.gde.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fey MF, Liechti-Gallati S, von Rohr A, Borisch B, Theilkas L, Schneider V, Oestreicher M, Nagel S, Ziemiecki A, Tobler A. Clonality and X-inactivation patterns in hematopoietic cell populations detected by the highly informative M27 beta DNA probe. Blood. 1994;83:931–8. [PubMed] [Google Scholar]
- 28.Gale RE, Fielding AK, Harrison CN, Linch DC. Acquired skewing of X-chromosome inactivation patterns in myeloid cells of the elderly suggests stochastic clonal loss with age. Br J Haematol. 1997;98:512–9. doi: 10.1046/j.1365-2141.1997.2573078.x. [DOI] [PubMed] [Google Scholar]
- 29.Busque L, Mio R, Mattioli J, Brais E, Blais N, Lalonde Y, Maragh M, Gilliland DG. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. 1996;88:59–65. [PubMed] [Google Scholar]
- 30.Shlush LI. Age related clonal hematopoiesis (ARCH) Blood. 2017 doi: 10.1182/blood-2017-07-746453. In press. [DOI] [PubMed] [Google Scholar]
- 31.Zink F, Stacey SN, Norddahl GL, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130:742–752. doi: 10.1182/blood-2017-02-769869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buscarlet M, Provost S, Zada YF, Barhdadi A, Bourgoin V, Lepine G, Mollica L, Szuber N, Dube MP, Busque L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. 2017;130:753–762. doi: 10.1182/blood-2017-04-777029. [DOI] [PubMed] [Google Scholar]
- 33.McKerrell T, Park N, Moreno T, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015;10:1239–45. doi: 10.1016/j.celrep.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–8. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Acuna-Hidalgo R, Sengul H, Steehouwer M, van de Vorst M, Vermeulen SH, Kiemeney L, Veltman JA, Gilissen C, Hoischen A. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am J Hum Genet. 2017;101:50–64. doi: 10.1016/j.ajhg.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Busque L, Patel JP, Figueroa ME, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44:1179–81. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodell MA, Rando TA. Stem cells and healthy aging. Science. 2015;350:1199–204. doi: 10.1126/science.aab3388. [DOI] [PubMed] [Google Scholar]
- 40.Holstege H, Pfeiffer W, Sie D, et al. Somatic mutations found in the healthy blood compartment of a 115-yr-old woman demonstrate oligoclonal hematopoiesis. Genome Res. 2014;24:733–42. doi: 10.1101/gr.162131.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaiswal S, Natarajan P, Silver AJ, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert BL. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bejar R. CHIP, ICUS, CCUS and other four-letter words. Leukemia. 2017;31:1869–1871. doi: 10.1038/leu.2017.181. [DOI] [PubMed] [Google Scholar]
- 44.Bonnefond A, Skrobek B, Lobbens S, Eury E, Thuillier D, Cauchi S, Lantieri O, Balkau B, Riboli E, Marre M, Charpentier G, Yengo L, Froguel P. Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nat Genet. 2013;45:1040–3. doi: 10.1038/ng.2700. [DOI] [PubMed] [Google Scholar]
- 45.Forsberg LA, Rasi C, Razzaghian HR, et al. Age-related somatic structural changes in the nuclear genome of human blood cells. Am J Hum Genet. 2012;90:217–28. doi: 10.1016/j.ajhg.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jacobs KB, Yeager M, Zhou W, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44:651–8. doi: 10.1038/ng.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laurie CC, Laurie CA, Rice K, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44:642–50. doi: 10.1038/ng.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guryanova OA, Lieu YK, Garrett-Bakelman FE, et al. Dnmt3a regulates myeloproliferation and liver-specific expansion of hematopoietic stem and progenitor cells. Leukemia. 2016;30:1133–42. doi: 10.1038/leu.2015.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fisher CL, Pineault N, Brookes C, Helgason CD, Ohta H, Bodner C, Hess JL, Humphries RK, Brock HW. Loss-of-function Additional sex combs like 1 mutations disrupt hematopoiesis but do not cause severe myelodysplasia or leukemia. Blood. 2010;115:38–46. doi: 10.1182/blood-2009-07-230698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abdel-Wahab O, Gao J, Adli M, et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med. 2013;210:2641–59. doi: 10.1084/jem.20131141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J, Li Z, He Y, et al. Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood. 2014;123:541–53. doi: 10.1182/blood-2013-05-500272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, Rao A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A. 2011;108:14566–71. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quivoron C, Couronne L, Della Valle V, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 56.De T, Prabhakar P, Nagaraja D, Christopher R. Janus kinase (JAK) 2 V617F mutation in Asian Indians with cerebral venous thrombosis and without overt myeloproliferative disorders. J Neurol Sci. 2012;323:178–82. doi: 10.1016/j.jns.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 57.De Stefano V, Fiorini A, Rossi E, Za T, Farina G, Chiusolo P, Sica S, Leone G. Incidence of the JAK2 V617F mutation among patients with splanchnic or cerebral venous thrombosis and without overt chronic myeloproliferative disorders. J Thromb Haemost. 2007;5:708–14. doi: 10.1111/j.1538-7836.2007.02424.x. [DOI] [PubMed] [Google Scholar]
- 58.Muendlein A, Gasser K, Kinz E, Stark N, Leiherer A, Rein P, Saely CH, Grallert H, Peters A, Drexel H, Lang AH. Evaluation of the prevalence and prospective clinical impact of the JAK2 V617F mutation in coronary patients. Am J Hematol. 2014;89:295–301. doi: 10.1002/ajh.23632. [DOI] [PubMed] [Google Scholar]
- 59.Muendlein A, Kinz E, Gasser K, Leiherer A, Rein P, Saely CH, Grallert H, Peters A, Fraunberger P, Drexel H, Lang AH. Occurrence of the JAK2 V617F mutation in patients with peripheral arterial disease. Am J Hematol. 2015;90:E17–21. doi: 10.1002/ajh.23874. [DOI] [PubMed] [Google Scholar]
- 60.Hasselbalch HC. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood. 2012;119:3219–25. doi: 10.1182/blood-2011-11-394775. [DOI] [PubMed] [Google Scholar]
- 61.Li J, Spensberger D, Ahn JS, et al. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F-positive essential thrombocythemia. Blood. 2010;116:1528–38. doi: 10.1182/blood-2009-12-259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mullally A, Lane SW, Ball B, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010;17:584–96. doi: 10.1016/j.ccr.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood. 2010;115:3589–97. doi: 10.1182/blood-2009-04-215848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ko M, Huang Y, Jankowska AM, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He YF, Li BZ, Li Z, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Q, Zhao K, Shen Q, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–93. doi: 10.1038/nature15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, Yang FC, Xu M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118:4509–18. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nagase T, Kikuno R, Nakayama M, Hirosawa M, Ohara O. Prediction of the coding sequences of unidentified human genes. XVIII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2000;7:273–81. doi: 10.1093/dnares/7.4.271. [DOI] [PubMed] [Google Scholar]
- 70.Fuster JJ, MacLauchlan S, Zuriaga MA, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. doi: 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol. 2017;55:56–70 e13. doi: 10.1016/j.exphem.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 72.Manson JC, Symons JA, Di Giovine FS, Poole S, Duff GW. Autoregulation of interleukin 1 production. Eur J Immunol. 1989;19:261–5. doi: 10.1002/eji.1830190207. [DOI] [PubMed] [Google Scholar]
- 73.Hiscott J, Marois J, Garoufalis J, D’Addario M, Roulston A, Kwan I, Pepin N, Lacoste J, Nguyen H, Bensi G, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. 1993;13:6231–40. doi: 10.1128/mcb.13.10.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Warner SJ, Auger KR, Libby P. Interleukin 1 induces interleukin 1. II. Recombinant human interleukin 1 induces interleukin 1 production by adult human vascular endothelial cells. J Immunol. 1987;139:1911–7. [PubMed] [Google Scholar]
- 75.Dinarello CA, Ikejima T, Warner SJ, Orencole SF, Lonnemann G, Cannon JG, Libby P. Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol. 1987;139:1902–10. [PubMed] [Google Scholar]
- 76.Warner SJ, Auger KR, Libby P. Human interleukin 1 induces interleukin 1 gene expression in human vascular smooth muscle cells. J Exp Med. 1987;165:1316–31. doi: 10.1084/jem.165.5.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 78.Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, Group CT Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet. 2017 doi: 10.1016/S0140-6736(17)32814-3. In press. [DOI] [PubMed] [Google Scholar]
- 79.Baylis RA, Gomez D, Mallat Z, Pasterkamp G, Owens GK. The CANTOS Trial: One Important Step for Clinical Cardiology but a Giant Leap for Vascular Biology. Arterioscler Thromb Vasc Biol. 2017;37:e174–e177. doi: 10.1161/ATVBAHA.117.310097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ridker PM. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream To Identify Novel Targets for Atheroprotection. Circ Res. 2016;118:145–56. doi: 10.1161/CIRCRESAHA.115.306656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Everett BM, Pradhan AD, Solomon DH, Paynter N, Macfadyen J, Zaharris E, Gupta M, Clearfield M, Libby P, Hasan AA, Glynn RJ, Ridker PM. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166:199–207 e15. doi: 10.1016/j.ahj.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Colchicine Cardiovascular Outcomes Trial (COLCOT) (COLCOT) available at https://www.clinicaltrials.gov/ct2/show/NCT02551094?term=colcot&rank=1.
- 83.Cole CB, Russler-Germain DA, Ketkar S, et al. Haploinsufficiency for DNA methyltransferase 3A predisposes hematopoietic cells to myeloid malignancies. J Clin Invest. 2017;127:3657–3674. doi: 10.1172/JCI93041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leoni C, Montagner S, Rinaldi A, Bertoni F, Polletti S, Balestrieri C, Monticelli S. Dnmt3a restrains mast cell inflammatory responses. Proc Natl Acad Sci U S A. 2017;114:E1490–E1499. doi: 10.1073/pnas.1616420114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gamper CJ, Agoston AT, Nelson WG, Powell JD. Identification of DNA methyltransferase 3a as a T cell receptor-induced regulator of Th1 and Th2 differentiation. J Immunol. 2009;183:2267–76. doi: 10.4049/jimmunol.0802960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pham D, Yu Q, Walline CC, Muthukrishnan R, Blum JS, Kaplan MH. Opposing roles of STAT4 and Dnmt3a in Th1 gene regulation. J Immunol. 2013;191:902–11. doi: 10.4049/jimmunol.1203229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu Q, Zhou B, Zhang Y, Nguyen ET, Du J, Glosson NL, Kaplan MH. DNA methyltransferase 3a limits the expression of interleukin-13 in T helper 2 cells and allergic airway inflammation. Proc Natl Acad Sci U S A. 2012;109:541–6. doi: 10.1073/pnas.1103803109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rodriguez-Ubreva J, Catala-Moll F, Obermajer N, Alvarez-Errico D, Ramirez RN, Company C, Vento-Tormo R, Moreno-Bueno G, Edwards RP, Mortazavi A, Kalinski P, Ballestar E. Prostaglandin E2 Leads to the Acquisition of DNMT3A-Dependent Tolerogenic Functions in Human Myeloid-Derived Suppressor Cells. Cell Rep. 2017;21:154–167. doi: 10.1016/j.celrep.2017.09.018. [DOI] [PubMed] [Google Scholar]
- 89.Li X, Zhang Q, Ding Y, Liu Y, Zhao D, Zhao K, Shen Q, Liu X, Zhu X, Li N, Cheng Z, Fan G, Wang Q, Cao X. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat Immunol. 2016;17:806–15. doi: 10.1038/ni.3464. [DOI] [PubMed] [Google Scholar]
- 90.Cardilo-Reis L, Gruber S, Schreier SM, Drechsler M, Papac-Milicevic N, Weber C, Wagner O, Stangl H, Soehnlein O, Binder CJ. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol Med. 2012;4:1072–86. doi: 10.1002/emmm.201201374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goossens P, Gijbels MJ, Zernecke A, Eijgelaar W, Vergouwe MN, van der Made I, Vanderlocht J, Beckers L, Buurman WA, Daemen MJ, Kalinke U, Weber C, Lutgens E, de Winther MP. Myeloid type I interferon signaling promotes atherosclerosis by stimulating macrophage recruitment to lesions. Cell Metab. 2010;12:142–53. doi: 10.1016/j.cmet.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 92.Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet. 2005;366:1945–53. doi: 10.1016/S0140-6736(05)67785-9. [DOI] [PubMed] [Google Scholar]
- 93.Cheung B, Radia D, Pantelidis P, Yadegarfar G, Harrison C. The presence of the JAK2 V617F mutation is associated with a higher haemoglobin and increased risk of thrombosis in essential thrombocythaemia. Br J Haematol. 2006;132:244–5. doi: 10.1111/j.1365-2141.2005.05858.x. [DOI] [PubMed] [Google Scholar]
- 94.Finazzi G, Rambaldi A, Guerini V, Carobbo A, Barbui T. Risk of thrombosis in patients with essential thrombocythemia and polycythemia vera according to JAK2 V617F mutation status. Haematologica. 2007;92:135–6. doi: 10.3324/haematol.10634. [DOI] [PubMed] [Google Scholar]
- 95.Hsiao HH, Yang MY, Liu YC, Lee CP, Yang WC, Liu TC, Chang CS, Lin SF. The association of JAK2V617F mutation and leukocytosis with thrombotic events in essential thrombocythemia. Exp Hematol. 2007;35:1704–7. doi: 10.1016/j.exphem.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 96.Kittur J, Knudson RA, Lasho TL, Finke CM, Gangat N, Wolanskyj AP, Li CY, Wu W, Ketterling RP, Pardanani A, Tefferi A. Clinical correlates of JAK2V617F allele burden in essential thrombocythemia. Cancer. 2007;109:2279–84. doi: 10.1002/cncr.22663. [DOI] [PubMed] [Google Scholar]
- 97.Vannucchi AM, Antonioli E, Guglielmelli P, Longo G, Pancrazzi A, Ponziani V, Bogani C, Ferrini PR, Rambaldi A, Guerini V, Bosi A, Barbui T, Consortium MPDR Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia. 2007;21:1952–9. doi: 10.1038/sj.leu.2404854. [DOI] [PubMed] [Google Scholar]
- 98.Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110:840–6. doi: 10.1182/blood-2006-12-064287. [DOI] [PubMed] [Google Scholar]
- 99.Dahabreh IJ, Zoi K, Giannouli S, Zoi C, Loukopoulos D, Voulgarelis M. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res. 2009;33:67–73. doi: 10.1016/j.leukres.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 100.Xavier SG, Gadelha T, Rezende SM, Zalcberg IR, Spector N. JAK2V617F mutation in patients with thrombosis: to screen or not to screen? Int J Lab Hematol. 2011;33:117–24. doi: 10.1111/j.1751-553X.2010.01275.x. [DOI] [PubMed] [Google Scholar]
- 101.Jaiswal S, Natarajan P, Ebert BL. Clonal Hematopoiesis and Atherosclerosis. N Engl J Med. 2017;377:1401–1402. doi: 10.1056/NEJMc1710381. [DOI] [PubMed] [Google Scholar]
- 102.Lundberg P, Takizawa H, Kubovcakova L, Guo G, Hao-Shen H, Dirnhofer S, Orkin SH, Manz MG, Skoda RC. Myeloproliferative neoplasms can be initiated from a single hematopoietic stem cell expressing JAK2-V617F. J Exp Med. 2014;211:2213–30. doi: 10.1084/jem.20131371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Okugawa S, Ota Y, Kitazawa T, Nakayama K, Yanagimoto S, Tsukada K, Kawada M, Kimura S. Janus kinase 2 is involved in lipopolysaccharide-induced activation of macrophages. Am J Physiol Cell Physiol. 2003;285:C399–408. doi: 10.1152/ajpcell.00026.2003. [DOI] [PubMed] [Google Scholar]
- 104.Oku S, Takenaka K, Kuriyama T, et al. JAK2 V617F uses distinct signalling pathways to induce cell proliferation and neutrophil activation. Br J Haematol. 2010;150:334–44. doi: 10.1111/j.1365-2141.2010.08249.x. [DOI] [PubMed] [Google Scholar]
- 105.Desai HR, Sivasubramaniyam T, Revelo XS, et al. Macrophage JAK2 deficiency protects against high-fat diet-induced inflammation. Sci Rep. 2017;7:7653. doi: 10.1038/s41598-017-07923-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Abdel-Wahab O, Adli M, LaFave LM, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22:180–93. doi: 10.1016/j.ccr.2012.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Balasubramani A, Larjo A, Bassein JA, Chang X, Hastie RB, Togher SM, Lahdesmaki H, Rao A. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat Commun. 2015;6:7307. doi: 10.1038/ncomms8307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang H, Kurtenbach S, Guo Y, et al. Gain-of-function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood. 2017 doi: 10.1182/blood-2017-06-789669. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, Zuriaga MA, Yoshiyama M, Goukassian D, Cooper MA, Fuster JJ, Walsh K. Tet2-mediated clonal hematopoiesis accelerates experimental heart failure through an IL-1β/NLRP3 inflammasome mechanism. J Am Coll Cardiol. 2018 doi: 10.1016/j.jacc.2017.12.037. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484. doi: 10.1038/ncomms12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518:552–555. doi: 10.1038/nature13968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hinds DA, Barnholt KE, Mesa RA, Kiefer AK, Do CB, Eriksson N, Mountain JL, Francke U, Tung JY, Nguyen HM, Zhang H, Gojenola L, Zehnder JL, Gotlib J. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood. 2016;128:1121–8. doi: 10.1182/blood-2015-06-652941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Coombs CC, Zehir A, Devlin SM, et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell. 2017;21:374–382 e4. doi: 10.1016/j.stem.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Behfar A, Crespo-Diaz R, Terzic A, Gersh BJ. Cell therapy for cardiac repair–lessons from clinical trials. Nat Rev Cardiol. 2014;11:232–46. doi: 10.1038/nrcardio.2014.9. [DOI] [PubMed] [Google Scholar]
- 115.Turner L, Knoepfler P. Selling Stem Cells in the USA: Assessing the Direct-to-Consumer Industry. Cell Stem Cell. 2016;19:154–157. doi: 10.1016/j.stem.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 116.Afzal MR, Samanta A, Shah ZI, Jeevanantham V, Abdel-Latif A, Zuba-Surma EK, Dawn B. Adult Bone Marrow Cell Therapy for Ischemic Heart Disease: Evidence and Insights From Randomized Controlled Trials. Circ Res. 2015;117:558–75. doi: 10.1161/CIRCRESAHA.114.304792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Klein AM, Simons BD. Universal patterns of stem cell fate in cycling adult tissues. Development. 2011;138:3103–11. doi: 10.1242/dev.060103. [DOI] [PubMed] [Google Scholar]