

Abstract
The 2013 ACR–EULAR classification criteria for systemic sclerosis (SSc) have shifted the construct of SSc. The new reality is that patients recruited for trials may not be so severe and not so advanced. We can now look for therapeutics that might stop disease evolution and/or prevent organ involvement. This article highlights recent advances in research methodology, and broadens the potential range of design and analytic considerations when planning a SSc trial.
Keywords: Systemic sclerosis, scleroderma, randomized controlled trials, Bayesian, cross-over trial, enriched enrolment randomized withdrawal design, randomized placebo phase design
Our understanding of the vascular, immunologic and fibrotic domains in the pathogenesis of systemic sclerosis (SSc) is increasing in depth and scope. Clinical trials and observational studies in SSc are broadening to the vascular and immunologic domains, but continue to primarily study the fibrotic mechanism and manifestations of the disease. The 2013 American College of Rheumatology–European League Against Rheumatism classification criteria for SSc(1) have shifted the construct of SSc, and provides a new opportunity to look at different stages of disease evolution. These criteria allow classification of more patients with early manifestations and patients with limited skin involvement, thus allowing their inclusion into research studies.(1) Similarly, the Very Early Diagnosis of Systemic Sclerosis criteria facilitate the identification and diagnosis of patients with very early signs; some of whom have not yet been classified as SSc.(2) The new reality is that patients recruited for trials may not be so severe and not so advanced.
Therefore, we can now look for therapeutics that might stop disease evolution/progression and/or prevent internal organ involvement. Indeed, a recent position paper from the World Scleroderma Foundation outlined research priorities for future SSc clinical trials.(3) The SSc paradigm of treatment could include: reversal, stabilization, slowing down disease, prevention of new organ involvement and cure.(3) Clinically meaningful outcomes are preferred, including outcomes that matter to patients: fatigue, pain, quality of life, appearance, and hand function.(3) Yet, we still must address many research challenges.(4, 5) In fact, SSc remains a rare disease with limited numbers of patients for study recruitment. Unconscious bias and chance can still threaten the validity of estimates of treatment effect. Recent advances in research methodology can rise to some of these challenges and these novel strategies have been successfully applied in other areas of rheumatology.(4, 6–9) Moving forward, SSc investigators could consider these methodologic advances when designing SSc trials.
At the time that study hypotheses, patient populations and outcomes are considered, the study design and accompanying analysis, too, needs to be planned.(5) This short introduction is aimed at broadening the potential range of design and analytic considerations when planning a SSc trial.(4) Because some of these are designs which are not fully proven in SSc, they may best be initiated during Phase 2 or 4 of therapy development.
Sampling frame
This means rethinking the kinds of patients we include and exclude from trial participation. Past clinical trials for skin fibrosis have preferentially selected patients with current diffuse involvement. This was done as the disease is active in the early phase, and intervention may be effective in a shorter time period. The reality, however, is that patients affected by the limited subset have been completely excluded from clinical trials despite the fact that the majority of SSc patients have limited disease. A smaller proportion of new patients will have primarily fibrotic manifestations and more will succumb to complications of vasculopathic manifestations, gastrointestinal and/or lung involvement. Without ignoring diffuse disease, the classification of patients with earlier disease may require de-emphasizing the traditional concept of reversing fibrosis as the primary outcome and, instead, emphasize prevention of progression or emphasize other aspects like remission in SSc. Furthermore, sampling will still need to consider disease duration, prior disease course, disease acuity, prior internal organ involvement and differentiating early disease from longer duration disease. Increased rigor and attention to these factors may be needed. Innovative, yet untested, strategies to consider may include biomarker based patient selection.(10) Cohort enrichment strategies may be implemented to select patients who are most likely to respond to therapy.(11)
Outcome measures
A recent consensus report on ‘Points to Consider in Systemic Sclerosis Clinical Trials’ provides guidance on choice of outcome measures for clinical trials. Ideally, an outcome measure should be feasible, have demonstrable validity, reliability, and be responsive to change. Evaluation of the psychometric properties for some outcome measures in SSc trials have been evaluated.(12, 13) In addition, composite indices may provide more discriminatory power to separate an active agent from placebo and such efforts are underway in SSc.(12, 14) Likewise patient response outcomes will be required, and consideration should be given to including patient relevant outcomes in core sets.(14)
Study design: challenges
Fully statistically powered, randomized, controlled, double-blind trials (RCT), accounting for all appropriate confounders, are the acknowledged gold standard.(5) However there are problems when trying to use the traditional RCT for generating evidence in SSc. In rare diseases, a significant challenge relates to difficulties in accruing sufficient numbers of patients into clinical trials. The inability to recruit sufficient numbers of patients into clinical trials affects the ability to detect statistically significant, small-to-moderate treatment effects; and the ability to account for factors that may distort the estimated treatment effect. Current successful strategies, such as larger number of centers, and international collaborations, have overcome this issue. However, these strategies may be costly and require political will.
Study design: potential solutions
One solution is allowing standardized background medications and adding test medication or placebo, that is, use of an active comparator.(15) Physicians are more likely to encourage their patients to enroll into active-controlled trials.(16) This is often acceptable but requires additive effects with the background medication(s) (at least) and increased numbers of subjects because the effect size of treatment is decreased relative to the standard placebo-controlled trial. An example of an active comparator is the mycophenolate versus cyclophosphamide trial in SSc lung disease (ClinicalTrials.gov NCT00883129).
Patients may be more willing to participate in trials if they are offered post-trial provision of beneficial treatment (open label extension) when the trial is done compared to a previously proven treatment and the proven treatment is the offered post trial. This strategy is implemented in the clinical trial of tocilizumab compared to placebo in diffuse SSc for a year, followed by 1-year of open label therapy (ClinicalTrials.gov NCT01532869). When the treatment is offered at the end of the trial, patients may feel rewarded and recruitment will be enhanced.(15)
Cross-over trials compare two or more treatments by allocating each participant to all compared treatments in a randomly selected sequence. Since patients act as their own control, between patient variability is removed, increasing the precision of the estimated treatment effect and reducing the needed sample size. Cross-over trials have been widely used in pediatrics, oncology, pharmacology and psychiatry.(17) The carry-over effects from the first treatment may confound the trial and must be guarded against which generally requires between crossover washout periods. Appropriate outcomes must be selected where short-term changes are expected.
The enriched enrolment randomized withdrawal design enrolls study participants into an open label trial where all patients receive treatment (enrichment phase).(15) Once the responders are identified, they are enrolled into the randomized withdrawal phase where they are randomly allocated to continue receiving treatment or switch to control. The trial endpoints are the return of symptoms or the ability to continue receiving the treatment.(18) Subjects who have their therapy withdrawn can re-start treatment when they have flared. Thus, the time on placebo is limited. This study design was successfully used in the evaluation of etanercept in juvenile rheumatoid arthritis.(6) This design is hampered by the lack of validated endpoints of ‘flare’ in SSc, which are thus far not available.
The randomized placebo phase design (RPPD) is another design that reduces the time on control (often placebo) medication.(4) Patients in the RPPD are randomly allocated to either treatment or control. After a short, fixed time period (placebo phase) patients in the control group are blindly switched to the treatment. The design is based on the assumption that if the treatment is effective, patients who receive it sooner will respond, on average, sooner. At the end of the trial, average response times of the groups are compared, most often using time-to-event analysis.(19, 20) This design has been successfully used in the evaluation of rituximab in inflammatory myositis.(8)
Applied Bayesian methods are a solution that facilitates maximizing information from studies. Bayesian inference has 3 components: the prior, the likelihood and the posterior distributions. The prior is the probability of hypothesized treatment effects, based on information independent of the study (i.e. previous evidence). The likelihood summarizes the data from a new study. The posterior is the end result: the probability of hypothesized treatment effect based on data from the new study and prior evidence.(21) It is calculated using Bayes Theorem, which states that the posterior is directly proportional to the product of the likelihood and the prior. These are frequently illustrated using a Bayesian triplot. (Figure 1.)
Figure 1.
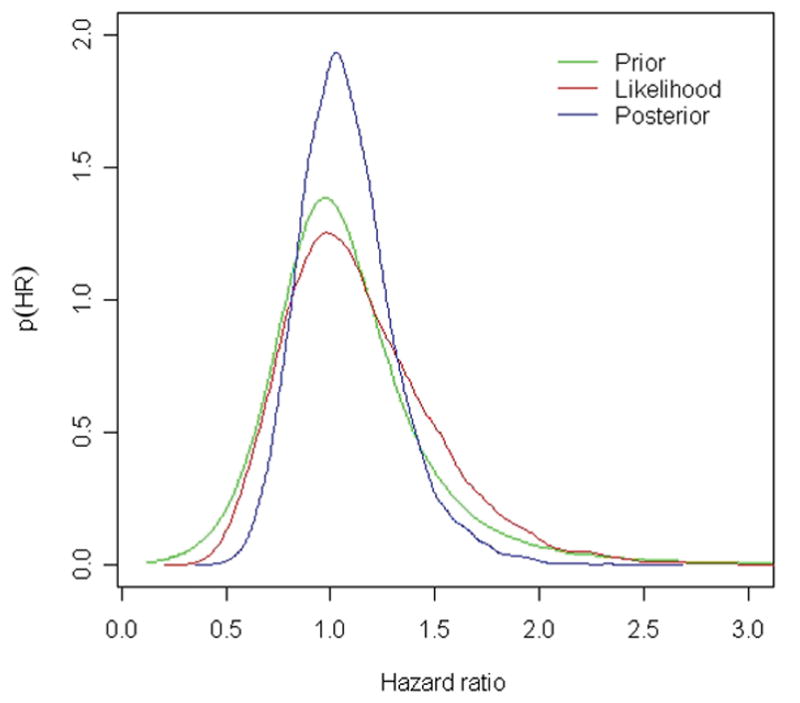
Bayesian triplot. The Bayesian triplot illustrates the prior probability distribution (pre-existing information, in green), the likelihood (new data, in red) and the posterior probability distribution (revised estimate based on pre-existing information and new data, in blue). Hazard ratio >1 indicates increased mortality, hazard ratio <1 indicates decreased mortality.
Reprinted with permission from The Journal of Rheumatology. JOHNSON, S.R. et al. J Rheumatol 2012;39(2). All rights reserved
The Bayesian approach confers a number of pragmatic advantages. First, applied Bayesian methods allow for the explicit incorporation of pre-existing knowledge/data into the estimation of treatment effect.(22) Previously published evidence and expert knowledge are usually ignored in the interpretation of a new trial. Bayesian inference quantitatively incorporates this external information when deriving the probability of a therapeutic benefit.(21) Using this approach, warfarin was demonstrated to have a low probability of improving survival in SSc-associated pulmonary arterial hypertension(23), and confirmed in a recent multicenter cohort study.(24) The ability to include external information improved the precision of the estimated treatment effect. The incorporation of experts’ beliefs about the treatment effect of an intervention may be incorporated in the analysis (statistical models) through applications such as www.UntappedKnowledge.org. This approach is currently being used to evaluate the effect of lung transplantation in SSc (Canadian Institutes of Health (CIHR) grant 301574).
Second, investigators can directly estimate of the probability of there being a meaningful treatment effect of a particular magnitude. This is true, irrespective of the sample size. More data will yield a more precise estimate of treatment. For example, in the cohort study evaluating the effect of warfarin in SSc-PAH, the probability of warfarin improving survival by 6 months or more was only 23.5%. Figure 2. A second example is the randomized controlled trial of methotrexate in SSc. Despite the trial’s inability to recruit patients to the trial’s targeted sample size, using this approach, methotrexate was demonstrated to have a high probability of a beneficial effect on skin score.(25)
Figure 2.
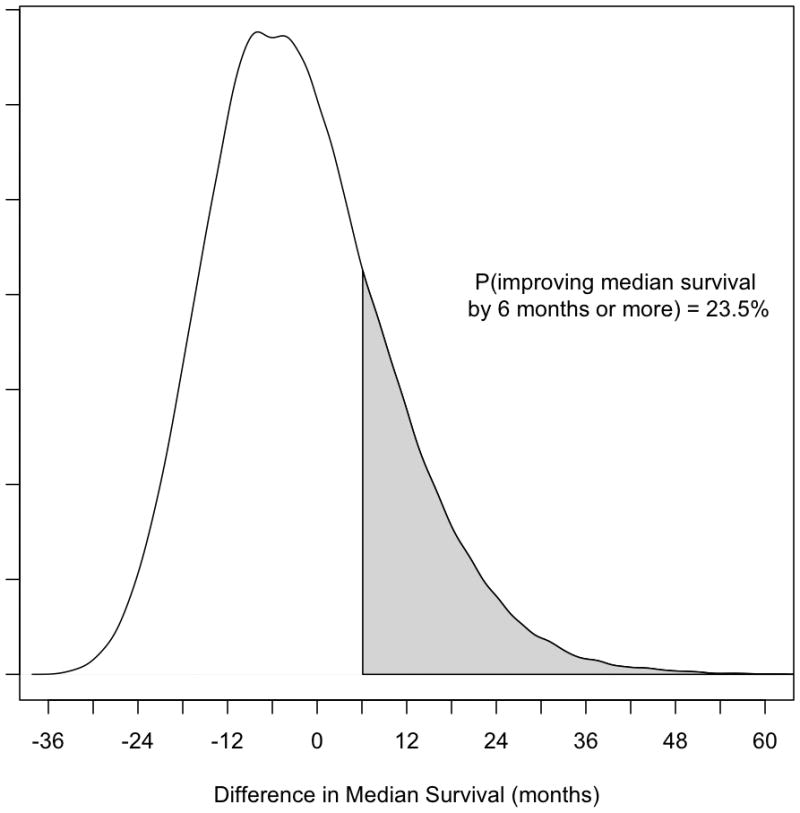
Density plot for difference in median survival times in patients with systemic sclerosis-associated pulmonary arterial hypertension untreated and treated with warfarin, using an informative group prior. Differences in median survival > 0 indicate improved survival associated with warfarin exposure. Differences in median survival < 0 indicate worsened survival associated with warfarin exposure. Y-axis indicates relative probability.
Reprinted with permission from The Journal of Rheumatology. JOHNSON, S.R. et al. J Rheumatol 2012;39(2). All rights reserved
Third, Bayesian meta-analysis of multiple n-of-1 studies can use small number of patients to generate estimates of the population effect and the likelihood of treatment benefit. Multiple n-of-1 studies are randomized clinical trials, with multiple crossovers, in single subjects.(26) By combining subjects who have had n-of-1 studies, it is possible to estimate the treatment effect in the population.(27) This approach has been used to evaluate the efficacy of amitriptyline for pain in juvenile idiopathic arthritis.(9) Using small numbers of patients (n=6), the investigators demonstrated a low probability of a beneficial treatment effect. This data was valuable to preventing the initiation of a large, potentially expensive, futile trial.
Conclusions
The paradigm for SSc research is shifting. Improved insight into the pathogenesis and manifestation of SSc, classification of patients at an earlier point in their disease, changing focus of the primary target of therapy, measuring the appropriate outcome, choice of the appropriate study design and analytic strategies, are all critical points to consider. Researchers will need to choose wisely and this article is meant to introduce the reader to a broader range of study designs than usually considered.
Acknowledgments
Dr. Johnson is supported by a Canadian Institutes of Health Research Clinician Scientist Award, the Oscar and Eleanor Markovitz Scleroderma Research Fund and the Freda Fejer Funds for Scleroderma Research. Dr. Khanna was supported by NIH/NIAMS K24 AR063120
Footnotes
Disclosures. Dr. Johnson has no conflicts of interest to disclose. Dr. Khanna has been a consultant or received grants from Actelion, Bayer, BMS, DIGNA, Genentech/Roche, Gilead, InterMune, Merck, NIH, Pulmonary Hypertension Association, Sanofi-Aventis, and Scleroderma Foundation. Dr. Allanore has been a consultant or received research grants from Actelion, Bayer, Genentech/Roche, Sanofi-Aventis, and Pfizer. Dr. Matucci-Cerinic has been a consultant or received grants from Actelion, Bayer, BMS, Roche, Gilead, and Pfizer. Dr. Furst has received grant/research support from AbbVie, Actelion, Amgen, BMS, Gilead, GSK, NIH, Novartis, Pfizer, Roche/Genentech, UCB; and has been a consultant to AbbVie, Actelion, Amgen, BMS, Janssen, Gilead, GSK, NIH, Novartis, Pfizer, Roche/Genentech, and UCB.
Contributor Information
Sindhu R. Johnson, Toronto Scleroderma Program, Toronto Western Hospital, Mount Sinai Hospital, Department of Medicine; Institute of Health Policy, Management and Evaluation, University of Toronto, Toronto, Ontario, Canada.
Dinesh Khanna, Division of Rheumatology, University of Michigan Scleroderma Program, Ann Arbor, MI, USA.
Yannick Allanore, Rheumatology A Department, Paris Descartes University, Cochin Hospital, Paris, France.
Marco Matucci-Cerinic, Department of Rheumatology AVC, Department of BioMedicine, Division of Rheumatology AOUC, Department of Medicine & Denothecentre, University of Florence, Firenze, Italy.
Daniel E. Furst, Carl M Pearson Professor of Rheumatology, Division of Rheumatology, David Geffen School of Medicine, University of California Los Angeles, California, USA.
References
- 1.van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an american college of rheumatology/european league against rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737–47. doi: 10.1002/art.38098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matucci-Cerinic M, Bellando-Randone S, Lepri G, Bruni C, Guiducci S. Very early versus early disease: the evolving definition of the ‘many faces’ of systemic sclerosis. Ann Rheum Dis. 2013;72(3):319–21. doi: 10.1136/annrheumdis-2012-202295. [DOI] [PubMed] [Google Scholar]
- 3.Furst DE, Pope JE, Seibold JR, Bombardieri S, Denton CP, Distler O, et al. Progress and Priorities in Systemic Sclerosis: The Next Ten Years: Report from the World Scleroderma Foundation. JSRD. 2016 [Google Scholar]
- 4.Abrahamyan L, Diamond IR, Johnson SR, Feldman BM. A new toolkit for conducting clinical trials in rare disorders. J Popul Ther Clin Pharmacol. 2014;21(1):e66–78. [PubMed] [Google Scholar]
- 5.Pope JE, Khanna D, Johnson SR, Clements P. Disease modification and other trials in systemic sclerosis have come a long way, but have to go further. Arthritis Care Res (Hoboken) 2012;64(7):955–9. doi: 10.1002/acr.21673. [DOI] [PubMed] [Google Scholar]
- 6.Lovell DJ, Giannini EH, Reiff A, Cawkwell GD, Silverman ED, Nocton JJ, et al. Etanercept in children with polyarticular juvenile rheumatoid arthritis. Pediatric Rheumatology Collaborative Study Group. N Engl J Med. 2000;342(11):763–9. doi: 10.1056/NEJM200003163421103. [DOI] [PubMed] [Google Scholar]
- 7.Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–44. doi: 10.1056/NEJMoa067659. [DOI] [PubMed] [Google Scholar]
- 8.Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65(2):314–24. doi: 10.1002/art.37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huber AM, Tomlinson GA, Koren G, Feldman BM. Amitriptyline to relieve pain in juvenile idiopathic arthritis: a pilot study using Bayesian metaanalysis of multiple N-of-1 clinical trials. J Rheumatol. 2007;34(5):1125–32. [PubMed] [Google Scholar]
- 10.Streicher K, Morehouse CA, Groves CJ, Rajan B, Pilataxi F, Lehmann KP, et al. The plasma cell signature in autoimmune disease. Arthritis Rheumatol. 2014;66(1):173–84. doi: 10.1002/art.38194. [DOI] [PubMed] [Google Scholar]
- 11.Jordan S, Distler JHW, Maurer B, Walker UA, Huscher D, Allanore Y, et al. Effects of endothelin-1 receptor antagonists on skin fibrosis in systemic sclerosis patients from the EUSTAR database. JSRD. 2016
- 12.Furst D, Khanna D, Matucci-Cerinic M, Clements P, Steen V, Pope J, et al. Systemic sclerosis - continuing progress in developing clinical measures of response. J Rheumatol. 2007;34(5):1194–200. [PubMed] [Google Scholar]
- 13.Johnson SR, Hawker GA, Davis AM. The health assessment questionnaire disability index and scleroderma health assessment questionnaire in scleroderma trials: an evaluation of their measurement properties. Arthritis Rheum. 2005;53(2):256–62. doi: 10.1002/art.21084. [DOI] [PubMed] [Google Scholar]
- 14.Khanna D, Lovell DJ, Giannini E, Clements PJ, Merkel PA, Seibold JR, et al. Development of a provisional core set of response measures for clinical trials of systemic sclerosis. Ann Rheum Dis. 2008;67(5):703–9. doi: 10.1136/ard.2007.078923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abrahamyan L, Diamond IR, Johnson SR, Feldman B. A new toolkit for conducting clinical trials in rare disorders. J Popul Ther Clin Pharmacol. 2014 In press. [PubMed] [Google Scholar]
- 16.Halpern SD, Ubel PA, Berlin JA, Townsend RR, Asch DA. Physicians’ preferences for active-controlled versus placebo-controlled trials of new antihypertensive drugs. J Gen Intern Med. 2002;17(9):689–95. doi: 10.1046/j.1525-1497.2002.11024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta-analyses involving cross-over trials: methodological issues. Int J Epidemiol. 2002;31(1):140–9. doi: 10.1093/ije/31.1.140. [DOI] [PubMed] [Google Scholar]
- 18.Straube S, Derry S, McQuay HJ, Moore RA. Enriched enrollment: definition and effects of enrichment and dose in trials of pregabalin and gabapentin in neuropathic pain. A systematic review. Br J Clin Pharmacol. 2008;66(2):266–75. doi: 10.1111/j.1365-2125.2008.03200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feldman B, Wang E, Willan A, Szalai JP. The randomized placebo-phase design for clinical trials. J Clin Epidemiol. 2001;54(6):550–7. doi: 10.1016/s0895-4356(00)00357-7. [DOI] [PubMed] [Google Scholar]
- 20.Abrahamyan L, Li CS, Beyene J, Willan AR, Feldman BM. Survival distributions impact the power of randomized placebo-phase design and parallel groups randomized clinical trials. J Clin Epidemiol. 2011;64(3):286–92. doi: 10.1016/j.jclinepi.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 21.Wijeysundera DN, Austin PC, Hux JE, Beattie WS, Laupacis A. Bayesian statistical inference enhances the interpretation of contemporary randomized controlled trials. J Clin Epidemiol. 2009;62(1):13–21. e5. doi: 10.1016/j.jclinepi.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 22.Johnson SR, Tomlinson GA, Hawker GA, Granton JT, Feldman BM. Methods to elicit beliefs for Bayesian priors: a systematic review. J Clin Epidemiol. 2010;63(4):355–69. doi: 10.1016/j.jclinepi.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 23.Johnson SR, Granton JT, Tomlinson GA, Grosbein HA, Le T, Lee P, et al. Warfarin in systemic sclerosis-associated and idiopathic pulmonary arterial hypertension. A Bayesian approach to evaluating treatment for uncommon disease. J Rheumatol. 2012;39(2):276–85. doi: 10.3899/jrheum.110765. [DOI] [PubMed] [Google Scholar]
- 24.Olsson KM, Delcroix M, Ghofrani HA, Tiede H, Huscher D, Speich R, et al. Anticoagulation and Survival in Pulmonary Arterial Hypertension: Results From the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA) Circulation. 2014;129(1):57–65. doi: 10.1161/CIRCULATIONAHA.113.004526. [DOI] [PubMed] [Google Scholar]
- 25.Johnson SR, Feldman BM, Pope JE, Tomlinson GA. Shifting our thinking about uncommon disease trials: the case of methotrexate in scleroderma. J Rheumatol. 2009;36(2):323–9. doi: 10.3899/jrheum.071169. [DOI] [PubMed] [Google Scholar]
- 26.Guyatt GH, Heyting A, Jaeschke R, Keller J, Adachi JD, Roberts RS. N of 1 randomized trials for investigating new drugs. Control Clin Trials. 1990;11(2):88–100. doi: 10.1016/0197-2456(90)90003-k. [DOI] [PubMed] [Google Scholar]
- 27.Zucker DR, Ruthazer R, Schmid CH. Individual (N-of-1) trials can be combined to give population comparative treatment effect estimates: methodologic considerations. J Clin Epidemiol. 2010;63(12):1312–23. doi: 10.1016/j.jclinepi.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]