

Abstract
There is a growing interest in the use of lipid bilayer nanodiscs for various biochemical and biomedical applications. Among the different types of nanodiscs, the unique features of synthetic polymer-based nanodiscs have attracted additional interest. A styrene–maleic acid (SMA) copolymer demonstrated to form lipid nanodiscs has been used for structural biology related studies on membrane proteins. However, the application of SMA polymer based lipid nanodiscs is limited because of the strong absorption of the aromatic group interfering with various experimental measurements. Thus, there is considerable interest in the development of other molecular frameworks for the formation of polymer-based lipid nanodiscs. In this study, we report the first synthesis and characterization of a library of polymethacrylate random copolymers as alternatives to SMA polymer. In addition, we experimentally demonstrate the ability of these polymers to form lipid bilayer nanodiscs through the fragmentation of lipid vesicles by means of light scattering, electron microscopy, differential scanning calorimetry, and solution and solid-state NMR experiments. We further demonstrate a unique application of the newly developed polymer for kinetics and structural characterization of the aggregation of human islet amyloid polypeptide (also known as amylin) within the lipid bilayer of the polymer nanodiscs using thioflavin-T-based fluorescence and circular dichroism experiments. Our results demonstrate that the reported new styrene-free polymers can be used in high-throughput biophysical experiments. Therefore, we expect that the new polymer nanodiscs will be valuable in the structural studies of amyloid proteins and membrane proteins by various biophysical techniques.
Graphical Abstract
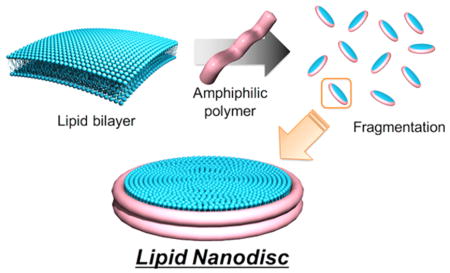
INTRODUCTION
Near-native cell membrane mimetics are highly important to study the structure and function of membrane proteins by a variety of biophysical techniques.1–3 Even though detergents have been used in the purification of membrane proteins and also in the form of micelles, studies have demonstrated that detergents can destabilize natively folded membrane protein structures and the intrinsic curvature of detergent micelles can distort the embedded protein structure.4,5 Studies have also reported the use of bicelles to study the structures of a variety of membrane proteins, particularly by solution and solid-state NMR techniques and crystallography, and also to investigate membrane active peptides such as antimicrobial peptides and amyloid proteins/peptides.6,7 However, the presence of detergents in bicelles and their diffusion into the planar lipid bilayer region of bicelles are concerns for detergent-sensitive membrane proteins.8 Therefore, there is a demand for the development of detergent-free membrane mimetics for various biophysical and biochemical studies on membrane proteins.
Recent studies have shown an increasing interest in the use of lipid bilayer nanodiscs formed by membrane scaffold proteins (MSPs), peptides, and synthetic polymers for various applications including isolation, purification, and high-resolution structural studies of membrane proteins.9–12 MPSs successfully form monodispersed lipid nanodiscs, which can incorporate membrane proteins; however, they require a preparation step using a detergent. Additionally, signals from MSPs are sometimes difficult to separate from that of a reconstituted membrane protein of interest.
Among the various types of nanodiscs reported in the literature, polymer-based nanodiscs are inexpensive, easy to produce, and chemically stable and enable detergent-free preparation. Therefore, there is considerable interest in the development of nanodisc-forming novel polymers that can be used to expand the applications of lipid nanodiscs for studies in structural biology and bionanotechnology. It is highly desirable to design polymers that can form stable nanodiscs and, at the same time, should contain a molecular framework to enable the application of various biophysical techniques typically employed in the structural studies of membrane proteins. For example, the previously reported styrene/maleic acid (SMA) copolymers can be used to form lipid bilayer nanodiscs for extraction, characterization, and structural studies of reconstituted membrane proteins and also for potential drug delivery.13–16 In spite of the rapidly increasing applications, unfortunately, SMA-based polymer nanodiscs pose challenges in the application of some of the most commonly used biophysical techniques such as circular dichroism (CD), UV/vis, and fluorescence spectroscopy due to the strong absorption of the styrene group of the polymer. The interference of the π-interaction of the styrene group with the embedded protein/peptide or lipid bilayer is also undesirable. In addition, the composition ratio of the two monomeric components of the SMA polymer cannot be varied in a wide range because the copolymerization of styrene and maleic acid typically produces an alternating copolymer. Recently, nonaromatic diisobutylene/maleic acid polymer has been found to form lipid nanodiscs; however, this combination of the monomer also produces alternating copolymer that does not allow structural variation.17
In this study, we report a systematic investigation of a library of new styrene-free nanodisc-forming polymers that are designed by employing a polymethacrylate framework for a spontaneous lipid nanodisc formation by solubilization of the lipid membrane. The polymethacrylate derivatives have several benefits to design a nanodisc-forming polymer: (a) they render an easy control of the synthesis by radical polymerization, (b) they allow for a variety of monomers with a variation in the side chains to be employed, and (c) they allow for the feasibility of a large-scale production at low cost. Here we report the strategies on the design, synthesis, and complete characterization of a library of amphiphilic polymethacrylate random copolymers. In addition, the ability to form lipid bilayer nanodiscs by these polymers is demonstrated using light scattering, electron microscopy, differential scanning calorimetry, and NMR experiments. We further demonstrate a unique application of the new polymer nanodiscs to monitor the structural transition of a 37-residue human islet amyloid polypeptide (IAPP, also known as amylin) in the course of its aggregation in a lipid bilayer environment.
RESULTS AND DISCUSSION
Design and Synthesis of Methacrylate Copolymers
The overall strategy used in the polymer design is to mimic the amphipathic helical structure of the natural apolipoprotein that forms a lipid bilayer nanodisc.18 Previous studies have shown that a truncated fragment of apolipoprotein and their oligomers can form a nanodisc by the interaction with lipids.19–23 Additionally, some amphipathic α-helical peptides have been found to form nanodiscs.24,25 These facts suggested that an amphipathic structure of these proteins and peptides is essential to form lipid nanodiscs. To mimic the amphiphilic nature of such proteins or peptides, we have employed amphiphilic polymethacrylate random copolymers consisting of hydrophobic and hydrophilic side chains to design a nanodisc-forming polymer. Although their monomer sequence is random, the amphiphilic polymethacrylate random copolymer was found to provide an amphiphipathic structure upon its interaction with a lipid bilayer.26 The hydrophobic butyl methacrylate and cationic methacroylcholine chloride of the resultant polymer are expected to interact with hydrophobic acyl chains and anionic phosphate headgroup of lipids, respectively, to enable the formation of a lipid nanodisc formation surrounded by the polymer. The copolymers were synthesized using the free radical polymerization initiated by azobis(isobutyronitrile) (AIBN) as shown in Scheme 1. The molecular weight of a polymer was adjusted by varying the amount of methyl 3-mercaptopropionate used as a chain-transfer agent. The hydrophobic/cationic ratio was varied by the feed ratio of two monomers. The resultant polymer was simply purified by reprecipitation in diethyl ether, which enabled the complete removal of unreacted monomers. The synthesized polymers were analyzed by 1H NMR experiments to calculate the fraction of hydrophobic unit (f), the degree of polymerization (DP), and the corresponding number-averaged molecular weight (Mn) as described in the experimental section. A library of amphiphilic polymethacrylates consisting of 25 polymers with a variation in f, DP, and Mn was established as listed in Table 1.
Scheme 1.
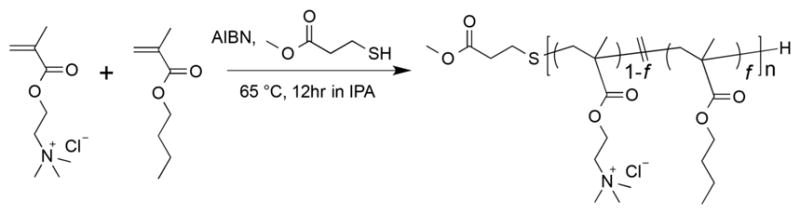
Synthesis of Amphiphilic Methacrylate Copolymers for Nanodisc Formation
Table 1.
Library of Polymers Synthesized for Investigation Presented in This Study
| entry | polymer | DP (−) | f (−) | Mn (kg mol−1) |
|---|---|---|---|---|
| 1 | N–C4–24–4.0 | 24 | 0.24 | 4.0 |
| 2 | N–C4–24–6.1 | 36 | 0.24 | 6.1 |
| 3 | N–C4–25–1.7 | 10 | 0.25 | 1.7 |
| 4 | N–C4–26–13 | 78 | 0.26 | 13 |
| 5 | N–C4–33–7.1 | 43 | 0.33 | 7.1 |
| 6 | N–C4–39–2.0 | 12 | 0.39 | 2.0 |
| 7 | N–C4–40–6.7 | 41 | 0.4 | 6.7 |
| 8 | N–C4–43–3.9 | 23 | 0.43 | 3.9 |
| 9 | N–C4–44–13 | 83 | 0.44 | 13 |
| 10 | N–C4–51–6.9 | 43 | 0.51 | 6.9 |
| 11 | N–C4–59–2.9 | 18 | 0.59 | 2.9 |
| 12 | N–C4–59–3.7 | 23 | 0.59 | 3.7 |
| 13 | N–C4–60–2.0 | 12 | 0.60 | 2.0 |
| 14 | N–C4–60–4.7 | 27 | 0.60 | 4.7 |
| 15 | N–C4–61–1.7 | 10 | 0.61 | 1.7 |
| 16 | N–C4–62–4.3 | 28 | 0.62 | 4.3 |
| 17 | N–C4–62–6.3 | 40 | 0.62 | 6.3 |
| 18 | N–C4–62–8.7 | 56 | 0.62 | 8.7 |
| 19 | N–C4–63–12 | 78 | 0.63 | 12 |
| 20 | N–C4–63–14 | 92 | 0.63 | 14 |
| 21 | N–C4–63–5.5 | 35 | 0.63 | 5.5 |
| 22 | N–C4–63–7.4 | 48 | 0.63 | 7.4 |
| 23 | N–C4–71–7.3 | 37 | 0.71 | 7.3 |
| 24 | N–C4–85–4.2 | 28 | 0.85 | 4.2 |
| 25 | N–C4–85–6.1 | 40 | 0.85 | 6.1 |
Copolymer-Induced Fragmentation of Lipid Bilayer and Nanodisc Formation
The ability of each synthesized polymer to solubilize lipids was examined by carrying out turbidity measurements on large unilamellar vesicles of DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) prepared by the extrusion method (LUVs of 100 nm in diameter). The addition of a polymer to DMPC vesicles resulted in a decrease of the solution turbidity in many cases, reflecting polymer-induced fragmentation of vesicles and resulting lipid nanodisc formation (Figure 1A). To further examine the effect of polymer concentration and hydrophobicity (f) of the polymer on the membrane fragmentation, the scattering intensity or turbidity was monitored by varying the polymer:lipid ratio (Figure 1B). Some polymers with moderate hydrophobicity fraction (f) around 0.3 to 0.6 induced a significant decrease in the scattering light intensity with increasing polymer concentration, reflecting the fragmentation of the vesicular membrane. On the other hand, the polymers with very high and low hydrophobicity (f = 0.85 and 0.24, respectively) were found to be ineffective in the solubilization of LUVs. Thus, it was realized that an optimization of the amphiphilic balance is important to obtain efficient nanodisc-forming polymers. Additionally, the vesicle solubilization was tested to screen the effect of the polymer’s molecular weight. Our experimental results suggested that a relatively low molecular weight polymer (~3000 g mol−1) was ineffective in the solubilization of LUVs when compared to large molecular weight polymers, reflecting that a short polymer cannot cover ~3 nm hydrophobic thickness of the lipid bilayer27 (Figure 1C)
Figure 1.
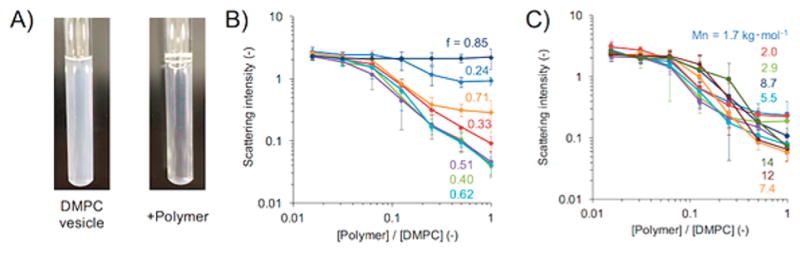
Formation of lipid nanodiscs by polymer-induced fragmentation of lipid membrane. (A) Dissolution of DMPC vesicles observed from the scattering measurement of DMPC vesicle solution in the presence of polymers. (B) Effect of the hydrophobic:hydrophilic ratio (f). (C) Effect of the number-averaged molecular weight (Mn) of the polymer.
Next, we examined the morphology of the polymer–lipid complex by negative-stain transmission electron microscopy (TEM). One of the polymers, which effectively solubilized the lipid membrane, N–C4–60–4.7, was mixed with DMPC liposome in a polymer:lipid molar ratio of 1:8. We observed an oval-shaped assembly with a homogeneous diameter, reflecting the lipid bilayer nanodisc formation (Figure 2A). We further used the TEM-based experimental observation to evaluate the ability to form lipid bilayer nanodiscs by all other synthesized polymers. Our results showed that the vesicular assembly of DMPC lipids was maintained in the presence of a relatively hydrophilic polymer (N–C4–24–6.1, Figure 2B), reflecting the polymer’s inability to induce fragmentation of DMPC vesicles. On the other hand, the addition of a hydrophobic polymer (N–C4–85–6.1) resulted in the formation of many small particles, which are likely to be spherical polymer micelles (Figure 2C). The molecular weight of the polymer also significantly affected the morphology of the lipid–polymer assembly. A small molecular weight polymer (N–C4–61–1.7) was found to partially disrupt lipid vesicles, as confirmed from the formation of a vesicle/nanodisc mixture (Figure 2D). In contrast, a large molecular weight polymer (N–C4–63–14) was found to form a heterogeneous mixture of small particles and large fragments of DMPC lipid vesicles (Figure 2E). The effects of the hydrophobic fraction (f) and molecular weight (Mn) of the amphiphilic polymethacrylate polymers on the lipid nanodisc formation, based on the solubilization assay as well as TEM observation, are summarized in Figure 2F. Based on the structure–activity relationship studies by TEM observation, an efficient nanodisc-forming polymer can be obtained by optimizing the hydrophobic fraction, f, in the range of 0.4 to 0.6, and molecular weight ranging from 3.0 to 9.0 kg mol−1 (indicated as orange diamonds in Figure 2F).
Figure 2.
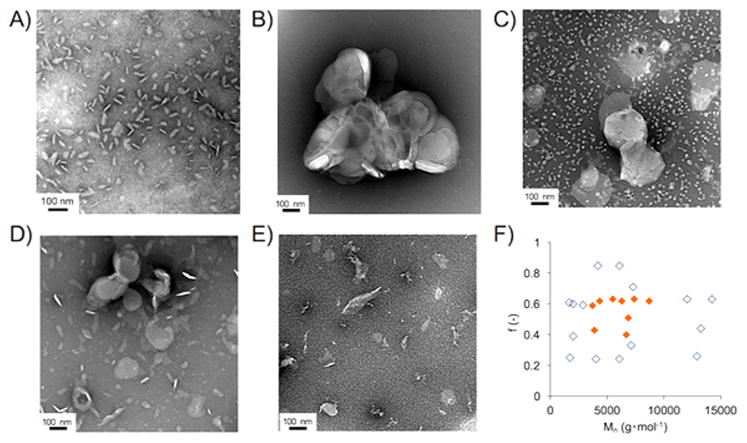
Negative-stain TEM observation of polymer–lipid complex (1:8 in molar ratio). (A) N–C4–60–4.7, (B) N–C4–24–6.1, (C) N–C4–85–6.1, (D) N–C4–61–1.7, (E) N–C4–63–14. (F) Effect of the hydrophobic fraction (f) and number-averaged molecular weight (Mn) of polymers on the nanodisc formation screened by negative-stain TEM. The ability of polymers to form nanodiscs is indicated by filled orange diamonds.
Characterization of Lipid Nanodiscs Formed with the Copolymer
The size, shape, and homogeneity of the lipid nanodiscs formed using the polymethacrylate derivatives were further examined using cryo-TEM experiments. Cryo-TEM observation is a powerful technique to visualize the nanometer-sized molecular assembly in water.28 Our cryo-TEM experiments confirmed that the 1:8 mixture of the N–C4–60–4.7 polymers with DMPC vesicles formed homogeneous nanodiscs (Figure 3A). From the images of edge-on nanodiscs, the diameter of each disc was found to be 17 nm. The thickness of the nanodisc was estimated to be 5.5 nm, which corresponded to the thickness of a single DMPC lipid bilayer. The formation of a discoidal-shaped nanodisc assembly was further confirmed by tilting the specimen stage. Upon tilting the sample holder by 20°, a circular feature became rod-shaped, revealing the discoid structure of the nanodiscs (Figure 3B).29,30 Results obtained from dynamic light scattering (DLS) experiments confirmed that the addition of a polymer to DMPC vesicles resulted in a significant decrease in the hydrodynamic diameter (Dhy) and the formation of the monodispersed assembly in water (Figure 4A), reflecting that the vesicles in the solution were completely solubilized by the polymer to form lipid nanodiscs. Additionally, the hydrodynamic diameter of the polymer–lipid assembly was systematically examined by changing the lipid:polymer molar ratio (Figure 4B). The Dhy significantly dropped when the [lipid]/[polymer] was <16. This transition in the Dhy is likely to reflect the critical amount of a polymer needed to form naodiscs. Above this critical concentration, the Dhy slightly decreased with the increasing polymer concentration, reflecting that a higher concentration of the polymer results in the formation of smaller nanodiscs. This concentration dependence can be explained by assuming that the edge of the nanodisc is covered by several polymer molecules. The decrease of the [lipid]/[polymer] increases the fraction of the polymer bound to the nanodisc edge. As a result, smaller nanodiscs should be produced due to the increase in the total perimeter of the nanodiscs in the system.
Figure 3.

Cryo-TEM image of lipid nanodiscs formed by N–C4–60–4.7 and DMPC. (A) Wide-field cryo-TEM image of nanodisc solution and magnified images of edge-on and face-on nanodiscs. (B) Tilting of the specimen holder revealing the formation of discoidal structure.
Figure 4.
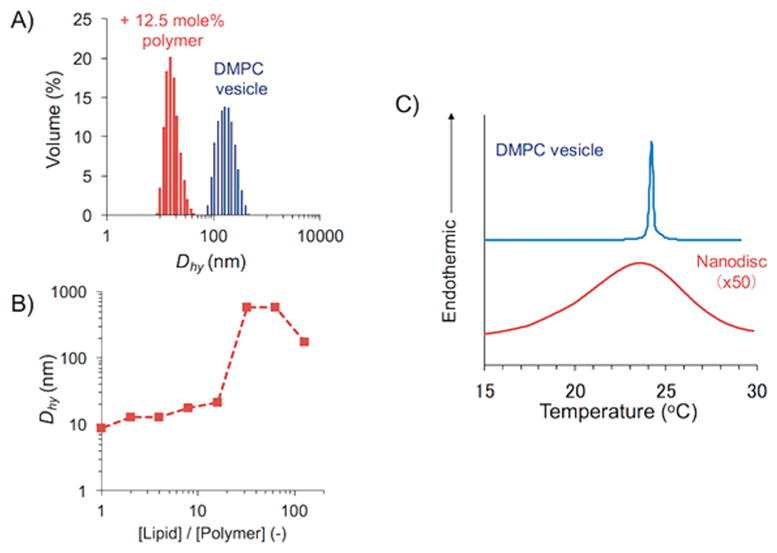
Characterization of DMPC nanodiscs formed with amphiphilic methacrylate copolymer: (A) DLS profile reveals monodispersed lipid nanodiscs in the presence of the polymer. (B) Variation of the size of nanodiscs depending on the lipid/polymer ratio. (C) DSC thermogram of DMPC nanodiscs.
We further confirmed the fragmentation of live E. coli cell membrane to demonstrate the potential of the nanodisc-forming polymer for the membrane protein purification as shown in Figure S1. The addition of the N–C4–60–4.7 polymer dramatically decreased the light scattering intensity of the E. coli suspension (Figure S1A). DLS experiments (Figure S1B) confirmed that the addition of polymer induced the formation of small particles, reflecting the polymer-induced disruption of E. coli cells. We observed the formation of discoidal particles in the negative-stain TEM images (Figure S1C and D). Thus, it is demonstrated that the polymethacrylate-based nanodisc-forming polymer is applicable not only to solubilized lipid vesicles but also to intact cell membranes.
The lipid bilayers encompassed in the copolymer nanodiscs were confirmed by measuring the gel to liquid crystalline main phase transition of DMPC lipids using differential scanning calorimetry (DSC). While the nanodiscs exhibited a gel to liquid crystalline phase transition, the interaction of polymer with lipids broadened (~5 °C range) and lowered (by ~2 °C) the phase transition temperature of DMPC (Figure 4C). It should be noted that the nanodisc-forming polymethacrylate random copolymer displayed a significantly smaller influence on the gel-to-liquid crystalline phase transition of the DMPC lipid bilayer as observed in the DSC thermogram compared to SMA nanodiscs.31 Since such a phase transition behavior is sensitive to the lipid packing in the membrane, the observed result indicates that the lipid bilayer was slightly influenced by the interaction with polymers. It is likely that the polymer perturbs the lipid packing, particularly close to the edge of the nanodisc, whereas the lipid bilayer structure in the naodisc center is well preserved, as further confirmed by solid-state NMR experiments (Figure 5). The observation of a single 31P peak for the lipid nanodiscs suggests that the polymer-induced edge effect on the lipid bilayer structure is likely to be negligible.
Figure 5.
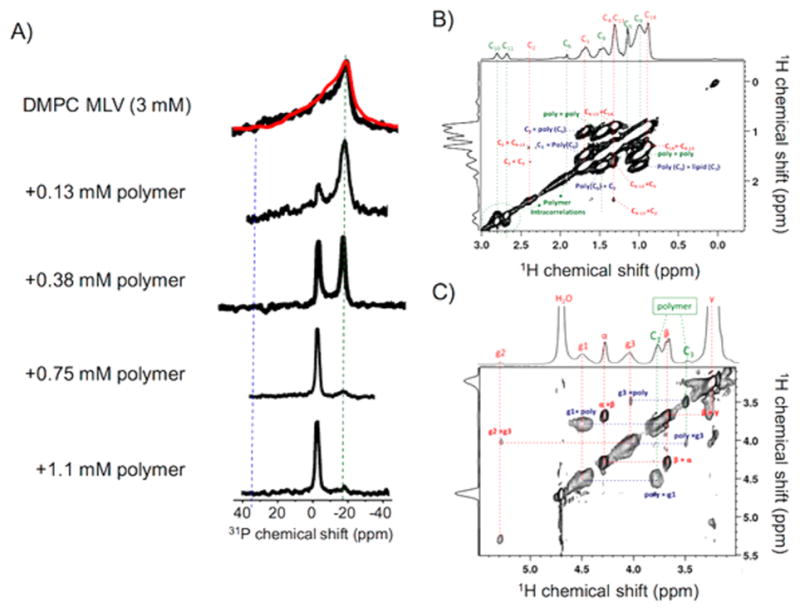
NMR experiments revealing the formation of nanodiscs. (A) 31P NMR spectra of 3 mM DMPC vesicles in the absence and presence of the indicated amount of polymer. The disappearance of the 31P chemical shift powder pattern and the increase in the intensity of a narrow isotropic peak (at −2.3 ppm) indicate the dissolution of MLVs and the formation of lipid bilayer nanodiscs. (B and C) 2D 1H–1H NOESY spectra of polymer nanodiscs containing DMPC lipids showing the interaction between the DMPC acyl chains and hydrophobic side chain of the methacrylate polymer, which provides evidence for the formation of an amphipathic belt by the polymers that surround the lipid bilayer; see the intermolecular contacts revealed by the cross peaks in the NOESY spectrum in Figure S4. A 2D 1H–1H NOESY spectrum of the lipid-free polymer solution is given in the Supporting Information (Figure S3).
NMR Characterization of Nanodiscs and Lipid–Polymer Interaction
Phosphorus-31 (31P) solid-state NMR experiments were performed under static conditions to examine the dissolution of DMPC multilamellar vesicles (MLVs) to form nanodiscs by a synthetic polymer. 31P NMR signal from the phosphate group of the lipid headgroup is sensitive to the lipid structure and can distinguish gel, lamellar, and other nonlamellar (such as cubic, hexagonal) phase structures. The 31P NMR spectrum of MLVs in the absence of the polymer (shown in Figure 5A) is a typical axially symmetric lamellar phase powder pattern with a span of ~45 ppm. 31P NMR spectra acquired after the addition of polymer to MLVs changed the powder pattern line shape observed for MLVs. A simulated chemical shift anisotropic (CSA) powder pattern of the 31P NMR spectrum (in red) is also shown in Figure 5A along with the powder pattern of DMPC MLVs. The parallel and perpendicular edge values of the DMPC MLV CSA powder pattern can be determined from the simulated spectrum. They are denoted as σ|| = 21 ppm (for parallel orientation) and σ⊥= −19 ppm (for perpendicular orientation). As shown in Figure 5A, upon the addition of polymer, an isotropic peak at −2.3 ppm appeared in the spectra, revealing the dissolution of MLVs to form fast-tumbling small lipid nanodiscs in the sample. The increasing intensity of the isotropic peak with the addition of an increasing amount of polymer further confirmed the efficient dissolution of the polymer to form isotropic lipid nanodiscs. Since a large amount of polymer is required to dissolve a large amount of MLVs, we used a small amount of lipids (or MLVs), which resulted in a poor signal-to-noise ratio particularly for the powder pattern line shapes of MLVs. Nevertheless, the observed signal-to-noise ratio was sufficient for the purposes of monitoring the formation of isotropic nanodiscs, as clearly indicated by the narrow isotropic peak observed at −2.3 ppm. Although the disappearance of the entire powder pattern line shape from MLVs cannot be quantified in the presence of the high-intensity isotropic peak, the disappearance of the perpendicular edge of the powder pattern (~−18 ppm) can be seen in the spectra shown in Figure 5A. The complete solubilization of MLVs, as seen from the complete disappearance of the perpendicular edge of the powder pattern, after the addition of a sufficient amount of polymer reveals the optimum amount of polymer needed to form a nanodisc sample. It is interesting to note that the presence of two narrow peaks at −2.3 and −18 ppm for the sample containing 12.5 mol % of polymer against DMPC indicates a heterogeneous mixture of isotropic and magnetically aligned nanodiscs, respectively. With the addition of more polymer, the sample becomes more homogeneous isotropic nanodiscs, as revealed by the single narrow peak at −2.3 ppm, while there can be a small variation in the size of nanodiscs as revealed by the DLS experiments.
The proton NMR spectrum of the lipid-free polymer solution shown in Figure S2 was acquired under static conditions using a 600 MHz solution NMR spectrometer and a cryoprobe, whereas the 1H NMR spectrum of a polymer nanodisc sample shown in Figure S4 was obtained under 8 kHz magic angle spinning (MAS) using a 500 MHz NMR spectrometer and CMP HXY probe. Two-dimensional 1H–1H NOESY experiments were performed on a solution of the polymer alone under static conditions using a cryoprobe at 600 MHz and also on a polymer nanodisc sample using a CMP HXY probe under 8 kHz MAS at 500 MHz. The 2D 1H–1H NOESY spectrum of the polymer solution is given in the Supporting Information (Figure S3), and that of the nanodisc sample is given in Figure 5 panels B and C. The NOESY spectrum of the polymer alone (Figure S3) in solution reveals the intramolecular correlations of side chain protons with backbone protons and also among the side chain protons. Since the lipid molecules are more mobile than most chemical groups of the polymer, narrow spectral lines are observed for lipids, while the polymer exhibits broad lines (Figure S4). Even though the difference in the time scale of mobility of these two different molecules in the nanodisc sample makes it difficult to observe cross peaks, the few cross peaks observed (in Figure 5, panels B and C) between the polymer side chain protons and the lipid acyl chain protons confirm the intermolecular structural interactions between the molecules to form discoidal lipid bilayers with the polymer likely forming the surrounding belt. The side chain of the polymer having the terminal carbon C9 is more flexible, and this dynamic behavior of this chain enables the lipid/polymer interaction to be stronger. This is evident from Figure 5B as the appearance of cross peaks between the C3 proton (1.6 ppm) of the lipid acyl chain and the hydrophobic side chain protons 8 (1.4 ppm) and 9 (0.9 ppm) of the polymer. The observation of these cross peaks in the 2D NOESY spectrum suggests that the hydrophobic side chain of the polymer is inserted into the hydrophobic region of the lipid bilayer, but the short side chain of the polymer may only reach down to around the C3 of the lipid acyl chain, as the NOESY spectrum does not show any cross peaks between the polymer side chain and the lower order carbons of the lipid acyl chain. In addition, cross peaks observed between the backbone protons of C1 (3.8 ppm) and C3 (3.5 ppm) of the polymer with the lipid’s glycerol backbone protons of g1 (4.5 ppm) and g3 (4 ppm), respectively, suggest a structural interaction between the terminal group of the polymer and the glycerol group in the lipid headgroup region, as shown in Figure 5C. A schematic representing the intermolecular interactions is shown in Figure S4.
Methacrylate Copolymer Nanodiscs Enable Real-Time Monitoring of Amyloid Aggregation in a Lipid Bilayer Environment
One of the most remarkable features of our polymer is that it is free of the styrene unit, which is indispensable in the previously used SMA polymer. The aromatic ring of the styrene is undesirable, as it interferes with commonly used biophysical experiments such as CD and fluorescence because of its strong absorption in the UV region. Additionally, the absence of an amide bond in the newly developed polymers enables a direct observation of CD signals originating from the reconstituted protein or peptide in the lipid nanodiscs. Therefore, to demonstrate the unique application of the reported methacrylate copolymers, we performed fluorescence and CD experiments that are commonly used to investigate the kinetics of amyloid aggregation and structural changes in the course of aggregation of an amyloid protein. For this demonstration, we chose the naturally occurring C-amidated form of human islet amyloid polypeptide (hIAPP), a 37-residue peptide whose amyloid aggregation property has been shown to play important roles in insulin-producing islet cell death in type-2 diabetics.32–34
We performed thioflavin T (ThT)-based fluorescence and CD experiments to demonstrate the ability to measure hIAPP–lipid interactions. As shown in Figure S5, the DLS profile reveals the formation of polymer nanodiscs with a lipid composition of 9:1 DMPC:DMPG, which was further used to investigate the lipid bilayer interaction of hIAPP. It is noteworthy that the polymethacrylate derivative did not affect the ThT fluorescence, whereas the conventional SMA polymer significantly enhanced the ThT fluorescence (Figure S6). The ThT fluorescence experimental results obtained in the absence of nanodiscs and in the presence of 9:1 DMPC:DMPG polymer nanodiscs for two different concentrations (10 and 20 μM) of hIAPP are shown in Figure 6 panels A and B, respectively. In the absence of nanodiscs, the sigmoidal ThT traces suggest the expected time- and concentration-dependent aggregation of hIAPP to form amyloid fibers, which is in agreement with results reported in the literature.34 On the other hand, the addition of nanodiscs significantly reduced and completely suppressed the ThT signal intensity. These observations suggest that the nanodiscs effectively suppressed the amyloid fiber formation of hIAPP. CD experiments were performed to further confirm the suppression of fiber formation by the nanodiscs. The hIAPP peptide forms a helical structure in the presence of nanodiscs that is indicated by the negative minima at 209 and 222 nm in the CD spectrum (Figure 6C). In contrast, the CD spectrum confirmed the formation of a beta-sheet structure as expected for amyloid fibers of hIAPP in the absence of nanodiscs (Figure 6D). We further confirmed that such a stabilization of the intermediate helical structure was not found when only the polymer was added to hIAPP (Figure S7). Thus, the ability of lipid nanodiscs to stabilize a helical intermediate of hIAPP is remarkable. Since hIAPP peptide is known to rapidly aggregate to form amyloid fibers in the presence of lipids, when used in the form of vesicles, it has been a challenge to probe the mechanism of hIAPP aggregation in a membrane environment.34–41 On the other hand, the feasibility of stabilizing a hIAPP helical intermediate species using lipid nanodiscs, reported for the first time, opens avenues for high-resolution structural studies as well as biophysical and biochemical investigations on the detailed roles of the lipid membrane.
Figure 6.
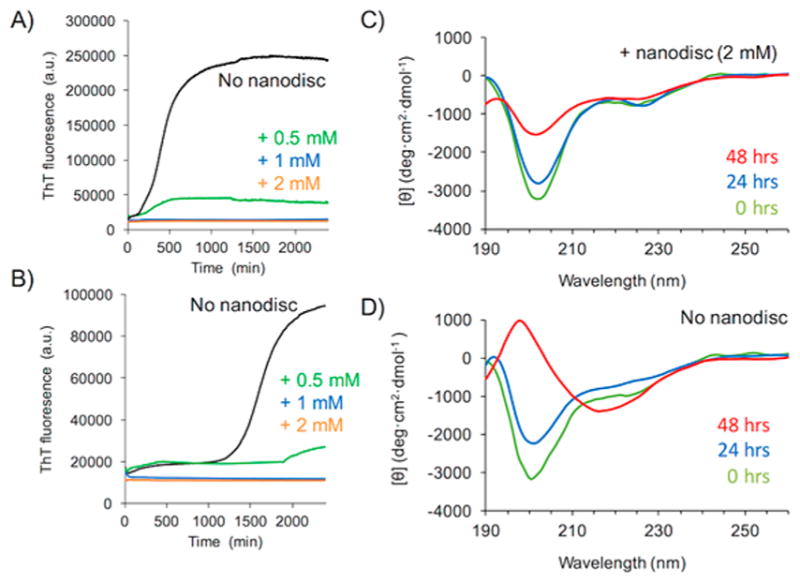
Polymer nanodiscs inhibit human-IAPP aggregation by stabilizing a helical intermediate. Thioflavin T (ThT) fluorescence experiments showing the aggregation of human-IAPP (at 20 μM (A) and 10 μM (B)) in the absence (black traces) and in the presence of polymer nanodiscs containing 0.5 mM (green), 1.0 mM (blue), and 2.0 mM (orange) of 9:1 DMPC:DMPG lipids. CD spectra of human-IAPP (at 20 μM) in the presence (C) and absence (D) of polymer nanodiscs containing 2.0 mM of 9:1 DMPC:DMPG lipids. ThT experiments reveal that the nanodiscs inhibit the aggregation of human-IAPP to form amyloid fibers, and the CD spectra show the formation of a helical intermediate of the peptide in 9:1 DMPC:DMPG lipid nanodiscs.
CONCLUSIONS
We have successfully designed and synthesized amphiphilic polymethacrylate copolymers that have been demonstrated to spontaneously form lipid nanodiscs through the fragmentation of a lipid bilayer. A structural search in the polymer library revealed that it is important to optimize the hydrophobicity as well as the molecular weight to obtain a nanodisc-forming polymer. DLS and electron microscopy and NMR experiments confirmed that the polymer is able to produce monodispersed homogeneous polymer lipid nanodiscs. In addition to lipid vesicles, the polymer was found to induce the fragmentation of intact E. coli cell membrane. Solid-state NMR measurements demonstrated the dissolution of large lipid aggregates to form rapidly tumbling isotropic nanodiscs. The 2D NOESY experiments provided insights into the intermolecular interactions that play a role in the formation of lipid nanodiscs in which a lipid bilayer is wrapped by several polymers. The experimental results obtained from the kinetics and structural changes of hIAPP aggregation to form amyloid fibers demonstrate the use of the newly developed methacrylate copolymers for studies using fluorescence and CD experiments and, therefore, their use in the investigation of amyloid proteins. Therefore, we believe that the reported polymer-based nanodiscs can be used for structural and functional studies on a variety of amyloid and membrane proteins.
Supplementary Material
Acknowledgments
This work was supported in part by Grants-in-Aid for Challenging Research (Exploratory, No. 17K19353) from the Japan Society for the Promotion of Science (JSPS) (to K.Y.) and by the National Institutes of Health (AG048934 to A.R.). The authors deeply thank Ms. S. Fujita, Graduate School of Materials Science, NAIST, for the technical assistance in TEM observation.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b10591.
Experimental section, additional NMR data, DLS data, and fluorescence spectra including Figures S1–S7 (PDF)
References
- 1.Phillips R, Ursell T, Wiggins P, Sens P. Nature. 2009;459:379–385. doi: 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Denisov IG, Sligar SG. Chem Rev. 2017;117:4669–4713. doi: 10.1021/acs.chemrev.6b00690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Das N, Murray DT, Cross TA. Nat Protoc. 2013;8:2256–2270. doi: 10.1038/nprot.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denisov IG, Sligar SG. Nat Struct Mol Biol. 2016;23:481–486. doi: 10.1038/nsmb.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miao Y, Cross TA. Curr Opin Struct Biol. 2013;23:919–928. doi: 10.1016/j.sbi.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanders CR, Hare BJ, Howard KP, Prestegard JH. Prog Nucl Magn Reson Spectrosc. 1994;26:421–444. [Google Scholar]
- 7.Dürr UH, Gildenberg M, Ramamoorthy A. Chem Rev. 2012;112:6054–6074. doi: 10.1021/cr300061w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang M, Huang R, Ackermann R, Im SC, Waskell L, Schwendeman A, Ramamoorthy A. Angew Chem, Int Ed. 2016;128:4497–4499. doi: 10.1002/anie.201600073. [DOI] [PubMed] [Google Scholar]
- 9.Bayburt TH, Grinkova YV, Sligar SG. Nano Lett. 2002;2:853–856. [Google Scholar]
- 10.Bayburt TH, Grinkova YV, Sligar SG. Arch Biochem Biophys. 2006;450:215–222. doi: 10.1016/j.abb.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 11.Hagn F, Etzkorn M, Raschle T, Wagner G. J Am Chem Soc. 2013;135:1919–1925. doi: 10.1021/ja310901f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ritchie TK, Grinkova YV, Bayburt TH, Denisov IG, Zolnerciks JK, Atkins WM, Sligar SG. Methods Enzymol. 2009;464:211–231. doi: 10.1016/S0076-6879(09)64011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orwick-Rydmark M, Lovett JE, Graziadei A, Lindholm L, Hicks MR, Watts A. Nano Lett. 2012;12:4687–4692. doi: 10.1021/nl3020395. [DOI] [PubMed] [Google Scholar]
- 14.Dorr JM, Koorengevel MC, Schafer M, Prokofyev AV, Scheidelaar S, van der Cruijsen EA, Dafforn TR, Baldus M, Killian JA. Proc Natl Acad Sci U S A. 2014;111:18607–18612. doi: 10.1073/pnas.1416205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SC, Knowles TJ, Postis VL, Jamshad M, Parslow RA, Lin YP, Goldman A, Sridhar P, Overduin M, Muench SP, Dafforn TR. Nat Protoc. 2016;11:1149–1162. doi: 10.1038/nprot.2016.070. [DOI] [PubMed] [Google Scholar]
- 16.Dorr JM, Scheidelaar S, Koorengevel MC, Dominguez JJ, Schafer M, van Walree CA, Killian JA. Eur Biophys J. 2016;45:3–21. doi: 10.1007/s00249-015-1093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oluwole AO, Danielczak B, Meister A, Babalola JO, Vargas C, Keller S. Angew Chem, Int Ed. 2017;56:1919–1924. doi: 10.1002/anie.201610778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frank PG, Marcel YL. J Lipid Res. 2000;41:853–872. [PubMed] [Google Scholar]
- 19.Tufteland M, Pesavento JB, Bermingham RL, Hoeprich PD, Jr, Ryan RO. Peptides. 2007;28:741–746. doi: 10.1016/j.peptides.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondo H, Ikeda K, Nakano M. Colloids Surf, B. 2016;146:423–430. doi: 10.1016/j.colsurfb.2016.06.040. [DOI] [PubMed] [Google Scholar]
- 21.Imura T, Tsukui Y, Taira T, Aburai K, Sakai K, Sakai H, Abe M, Kitamoto D. Langmuir. 2014;30:4752–4759. doi: 10.1021/la500267b. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Y, Imura T, Leman LJ, Curtiss LK, Maryanoff BE, Ghadiri MR. J Am Chem Soc. 2013;135:13414–13424. doi: 10.1021/ja404714a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nasr ML, Baptista D, Strauss M, Sun ZJ, Grigoriu S, Huser S, Pluckthun A, Hagn F, Walz T, Hogle JM, Wagner G. Nat Methods. 2017;14:49–52. doi: 10.1038/nmeth.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subbarao NK, Fielding CJ, Hamilton RL, Szoka FC. Proteins: Struct, Funct, Genet. 1988;3:187–198. doi: 10.1002/prot.340030307. [DOI] [PubMed] [Google Scholar]
- 25.Park SH, Berkamp S, Cook GA, Chan MK, Viadiu H, Opella SJ. Biochemistry. 2011;50:8983–5. doi: 10.1021/bi201289c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palermo EF, Vemparala S, Kuroda K. Biomacromolecules. 2012;13:1632–1641. doi: 10.1021/bm300342u. [DOI] [PubMed] [Google Scholar]
- 27.Tristram-Nagle S, Liu Y, Legleiter J, Nagle JF. Biophys J. 2002;83:3324–3335. doi: 10.1016/S0006-3495(02)75333-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kikuchi J, Yasuhara K. In: Supramolecular Chemistry: from Molecules to Nanomaterials. Steed JW, Gale PA, editors. Vol. 2. John Wiley & Sons Ltd; Chichester: 2012. pp. 633–646. [Google Scholar]
- 29.Yasuhara K, Miki S, Nakazono H, Ohta A, Kikuchi J. Chem Commun. 2011;47:4691–4693. doi: 10.1039/c1cc10254k. [DOI] [PubMed] [Google Scholar]
- 30.Beck P, Liebi M, Kohlbrecher J, Ishikawa T, Rüegger H, Fischer P, Walde P, Windhab E. Langmuir. 2010;26:5382–5387. doi: 10.1021/la903806a. [DOI] [PubMed] [Google Scholar]
- 31.Orwick MC, Judge PJ, Procek J, Lindholm L, Graziadei A, Engel A, Grobner G, Watts A. Angew Chem, Int Ed. 2012;51:4653–4657. doi: 10.1002/anie.201201355. [DOI] [PubMed] [Google Scholar]
- 32.Westermark P, Li ZC, Westermark GT, Leckström A, Steiner DF. FEBS Lett. 1996;379:203–206. doi: 10.1016/0014-5793(95)01512-4. [DOI] [PubMed] [Google Scholar]
- 33.Paulsson JF, Westermark GT. Diabetes. 2005;54:2117–2125. doi: 10.2337/diabetes.54.7.2117. [DOI] [PubMed] [Google Scholar]; Wiltzius JJ, Sievers SA, Sawaya MR, Eisenberg D. Protein Sci. 2009;18:1521–1530. doi: 10.1002/pro.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brender JR, Salamekh S, Ramamoorthy A. Acc Chem Res. 2012;45:454–462. doi: 10.1021/ar200189b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khemtemourian L, Gazit E, Miranker A. J Diabetes Res. 2016;2016:2535878. doi: 10.1155/2016/2535878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brender JR, Dürr UH, Heyl D, Budarapu MB, Ramamoorthy A. Biochim Biophys Acta, Biomembr. 2007;1768:2026–2029. doi: 10.1016/j.bbamem.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Last NB, Rhoades E, Miranker AD. Proc Natl Acad Sci U S A. 2011;108:9460–9465. doi: 10.1073/pnas.1102356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khemtémourian L, Doménech E, Doux JP, Koorengevel MC, Killian JA. J Am Chem Soc. 2011;133:15598–15604. doi: 10.1021/ja205007j. [DOI] [PubMed] [Google Scholar]
- 39.Nath A, Miranker AD, Rhoades E. Angew Chem, Int Ed. 2011;50:10859–10862. doi: 10.1002/anie.201102887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pithadia A, Brender JR, Fierke CA, Ramamoorthy A. J Diabetes Res. 2016;2016:2046327. doi: 10.1155/2016/2046327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sciacca MF, Brender JR, Lee DK, Ramamoorthy A. Biochemistry. 2012;51:7676–7684. doi: 10.1021/bi3009888. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.