

Abstract
To elucidate the involvement of individual and inter-related pathological factors [i.e., amyloid-β (Aβ), metals, and oxidative stress] in the pathogenesis of Alzheimer’s disease (AD), chemical tools have been developed. Characteristics required for such tool construction, however, have not been clearly identified; thus, the optimization of available tools or new design has been limited. Here, key structural properties and mechanisms that can determine tools’ regulatory reactivities with multiple pathogenic features found in AD are reported. A series of small molecules was built up through rational structural selection and variations onto the framework of a tool useful for in vitro and in vivo metal–Aβ investigation. Variations include: (i) location and number of an Aβ interacting moiety; (ii) metal binding site; and (iii) denticity and structural flexibility. Detailed biochemical, biophysical, and computational studies were able to provide a foundation of how to originate molecular formulas to devise chemical tools capable of controlling the reactivities of various pathological components through distinct mechanisms. Overall, this multidisciplinary investigation illustrates a structure–mechanism-based strategy of tool invention for such a complicated brain disease.
Keywords: amyloid beta-peptides, chemical tools, metal ions, oxidative stress, structure–activity relationships
Introduction
Alzheimer’s disease (AD) is a progressive and fatal brain disorder that is defined by progressive neuronal loss and cognitive defects.[1] Due to the unclear and complicated etiology of AD, a cure for the disease has not been discovered. Amyloid-β (Aβ) peptides are suggested to be associated with AD pathogenesis because misfolded Aβ aggregates are primary components of senile plaques found in the AD-afflicted brain (amyloid cascade hypothesis).[1,2] Upon the proteolytic cleavage of amyloid precursor protein (APP) by β- and γ-secretases, Aβ peptides are produced. Two major isoforms, Aβ40 and Aβ42 (ca. 90 % and 9 % in the brain, respectively), are aggregation-prone and are able to form aggregates from monomers to various-sized oligomers and fibrils.[1a–c,2] Based on recent findings, soluble Aβ oligomers are observed to be toxic; however, a relationship between Aβ conformations and toxicity remains uncertain.[1c,2] Moreover, the AD-affected brain exhibits highly concentrated metal ions within senile plaques (e.g., ca. 0.4 mM for CuI/II, 1.0 mM for ZnII, 0.9 mM for FeII/III).[1d,3] Previous in vitro studies present that these metal ions (particularly, CuI/II and ZnII) can interact with Aβ peptides and facilitate peptide aggregation. Furthermore, complexes of Aβ and redox-active metal ions, such as CuI/II and FeII/III, are shown to overproduce reactive oxygen species (ROS) through Fenton-like reactions leading to oxidative stress.[1b–d,3] Thus, it has been proposed that individual or inter-related reactivities of metal-free Aβ, metal ions, and ROS may contribute to AD pathogenesis [specially, via an inter-communicator, metal-bound Aβ (metal–Aβ)] (Figure 1).[1c,d,3d,e]
Figure 1.

Structural investigations (i–iii) of small molecules to alter their ionization potentials (IPs) and reactivities with individual and inter-related AD pathological factors. Structural variations: (i) the different position and number of the dimethylamino functionality; (ii) the metal-binding sites with and without a dimethylamino group; (iii) the increased denticity and structural flexibility. Potential donor atoms for metal binding are indicated in blue. Isosurface plots of compounds’ SOMOs (blue, N; gray, C; white, H) are depicted underneath compound structures. The calculated IPs in both the gas and aqueous phases are summarized in the table (bottom). L2-b, N1,N1-dimethyl-N4-(pyridin-2-ylmethyl)benzene-1,4-diamine; L2-b1, N,N-dimethyl-6-((phenylamino)methyl)pyridin-3-amine; L2-b2, N1-((5-(dimethylamino)pyridin-2-yl)methyl)-N4,N4-dimethylbenzene-1,4-diamine; PMA1, pyridin-2-yl-methanamine; PMA2, 6-(aminomethyl)-N,N-dimethylpyridin-3-amine; DPA1, bis(pyridin-2-ylmethyl)amine; DPA2, 6-((((5-(dimethylamino)pyridin-2-yl)methyl)amino)methyl)-N,N-dimethylpyridin-3-amine.
To elucidate the molecular-level underpinnings of individual and inter-related risk features involved in AD pathogenesis, small molecules capable of targeting and modulating their reactivities have been developed as chemical tools.[4] Among them, L2-b (N1,N1-dimethyl-N4-(pyridin-2-ylmethyl)benzene-1,4-diamine; Figure 1) was recently developed for regulating metal–Aβ species, along with antioxidant activity, and its in vitro and in vivo efficacy toward metal–Aβ was demonstrated.[4b,f] Until now, however, it has not been determined how the molecular formulas and properties of such a tool could lead to specific reactivities for the desired target, which has restricted new or innovative tool invention. Here, we report our multidisciplinary studies employing a newly designed chemical library based on L2-b’s backbone (Figure 1). Our studies demonstrate the importance of rationally constructing and tuning structural features and mechanisms [e.g., peptide modifications, including degradation and covalent adduct formation, by oxidative transformations of small molecules based on their ionization potentials (IPs)] toward development of tools for regulating distinct and inter-related pathological features in AD. Moreover, through the design principle gained from structural and mechanistic details, a chemical tool for targeting and controlling multiple distinguishable factors (i.e., metals, metal-free Aβ, metal–Aβ, and oxidative stress) was successfully constructed. Overall, our studies illustrate how chemical tools can be devised for investigating individual or inter-related pathological factors in AD.
Results and Discussion
Rational selection and preparation of small molecules
To establish how structural characteristics can guide mechanistic directions of chemical tools for desired reactivities toward their distinct targets, a class of small molecules derived from the backbone of L2-b was rationally designed (Figure 1). In our chemical series, different structural variations or portions based on the framework of L2-b were applied or selected (Figure 1): (i) the position and number of the dimethylamino functionality, important for Aβ interaction,[4a–g,5] on the backbone of L2-b were altered, affording L2-b1 and L2-b2; (ii) the structural moieties of L2-b and L2-b1/L2-b2 for metal binding (i.e., PMA1 and PMA2, respectively) were included; (iii) the denticity and structural flexibility on L2-b’s structure were varied generating DPA1 and DPA2. Moreover, the blood–brain barrier (BBB) permeability of our compounds was also considered for their biological applications. The potential BBB penetration of small molecules was suggested based on Lipinski’s rules as well as the values obtained from logBB calculation and the parallel artificial membrane permeability assay adapted for the BBB (PAMPA-BBB)[4b–d,6] (Supporting Information, Table S1).
L2-b, L2-b1, L2-b2, and DPA2 were prepared following the previously reported methods with slight modifications (especially for L2-b1, L2-b2, and DPA2, the procedures are summarized Scheme 1).[4b,d] L2-b1 and L2-b2 were obtained in relatively high yield through the formation of imine followed by its reduction to amine using sodium borohydride (NaBH4).[4b,d] In the case of DPA2, the conversion of the primary amino group on picolinonitrile to the dimethylamino functionality was carried out, subsequently incorporating themselves to obtain the final product. Note that PMA1, PMA2, and DPA1 are commercially available.
Scheme 1.

Synthetic routes to (a) L2-b1, L2-b2 and (b) DPA2.
Influence on metal-free and metal-induced Aβ aggregation
The ability of our small molecules (Figure 1) to modulate Aβ aggregation in both the absence and presence of metal ions was monitored through inhibition and disaggregation experiments (reaction schemes of both studies shown in Figure 2a and Supporting Information, Figure S1 a, respectively). The experiments were performed using Aβ40 and Aβ42, two major Aβ isoforms found in the AD-affected brain.[1a–c] The molecular weight (MW) distributions and morphological changes of the resultant Aβ species were analyzed by gel electrophoresis with Western blotting (gel/Western blot) and transmission electron microscopy (TEM), respectively.[4a–h] If a compound could generate a variety of smaller Aβ species, the gel/Western blot would indicate significant smearing. The large aggregates produced upon treatment with a compound can be visualized by TEM, but are too large to penetrate into the gel matrix, thus presenting very little smearing on the gel/Western blot (Figure 2; Supporting Information, Figure S1).
Figure 2.

Effects of small molecules on formation of metal-free Aβ and metal–Aβ aggregates. (a) Scheme of inhibition experiments. Visualization of MW distributions of resultant (b) Aβ40 and (c) Aβ42 species by gel/Western blot with an anti-Aβ antibody (6E10). Conditions: [Aβ] = 25 μM; [CuCl2 or ZnCl2] = 25 μM; [compound] = 50 μM; pH 6.6 (for CuII experiments) or pH 7.4 (for metal-free and ZnII experiments); 37 °C; constant agitation. TEM images of the (d) Aβ40 and (e) Aβ42 samples from (b) and (c), respectively.
In inhibition experiments (analysis of compounds’ effect on formation of Aβ aggregates, Figure 2a), various MW distributions of both metal-free Aβ40 and metal–Aβ40 species were displayed to different extents from the samples containing L2-b2 (lane 2, Figure 2b) compared to compound-untreated peptides (lane C, Figure 2b). On the other hand, much less significant influence on Aβ aggregation was observed upon incubation with the other compounds (i.e., L2-b1, PMA1, PMA2, DPA1, and DPA2) with and without metal ions. Moreover, similar to Aβ40, both metal-free and metal-treated Aβ42 aggregation pathways were altered by treatment with L2-b2 (lane 2, Figure 2c), noticeably different from L2-b1, PMA1, PMA2, DPA1, and DPA2. In addition to gel/Western blot analyses, the morphologies of both metal-free Aβ40/Aβ42 and metal–Aβ40/Aβ42 aggregates produced upon incubation with L2-b1 or L2-b2 were monitored by TEM. The resultant Aβ40 and Aβ42 aggregates generated by treatment with L2-b2 were shown to be more amorphous aggregates and/or smaller fibrils than those obtained under compound-free and L2-b1-treated conditions (Figure 2d and e).
The results from disaggregation experiments (determination of the ability of compounds to disassemble preformed Aβ aggregates; Supporting Information, Figure S1 a) are similar to those from the inhibition studies (Supporting Information, Figure S1 b–e). Preformed metal-free Aβ40 and metal-treated Aβ40 aggregates incubated with L2-b2 displayed various-sized peptide aggregates to different degrees (lane 2, Supporting Information, Figure S1 b). Similar to inhibition experiments, L2-b1, PMA1, PMA2, DPA1, and DPA2 could not detectably disaggregate preformed Aβ40 aggregates or redirect their further aggregation under both metal-free and metal-treated conditions (Supporting Information, Figure S1 b). In the case of Aβ42, L2-b2 was also indicated to dismantle preformed metal-free and metal-treated Aβ42 aggregates, distinct from the other compounds (Supporting Information, Figure S1 c). Expected from the gel/Western blot studies, more noticeable morphological changes upon treatment of L2-b2 to preformed metal-free and metal-bound Aβ40/Aβ42 aggregates were visualized, indicating more amorphous aggregates or thinner fibrils than the resultant Aβ aggregates from compound-free and L2-b1-added samples (Supporting Information, Figure S1 d and e).
Collectively, our gel/Western blot and TEM results suggest that structural variations of small molecules govern their distinct reactivities toward both metal-free and metal-induced Aβ aggregation. L2-b2, which has the overall structure of L2-b with an additional dimethylamino group on the pyridine ring (Figure 1), is observed to redirect both metal-free Aβ and metal–Aβ aggregation pathways; however, L2-b1 with the dimethylamino functionality, differently positioned from the backbone of L2-b, is not able to alter peptide aggregation regardless of the presence of metal ions. PMA1 and PMA2, the metal chelating portions of L2-b and L2-b1/L2-b2, respectively (Figure 1), could not significantly control Aβ aggregation in both the absence and presence of metal ions. In addition, DPA1 and DPA2 (Figure 1), the small molecules with the greater metal binding denticity and structural flexibility than L2-b, are not capable of distinguishably impacting Aβ aggregation even in the presence of metal ions. Therefore, the results and observations from both the inhibition and disaggregation studies employing our chemical series validate that the overall framework of L2-b with the dimethylamino group(s) at proper position(s), instead of individual structural components, could achieve inhibitory reactivities of small molecules with metal-free Aβ and/or metal–Aβ.
Biological activities
The capability of each compound to mediate cytotoxicity triggered by metal ions, metal-free Aβ, and metal–Aβ was examined. More than approximately 85 % of cell survival was exhibited when human neuroblastoma SK-N-BE(2)-M17 (M17) cells was treated with the different compounds (up to 50 μM without metal ions; up to 25 μM with metal ions; Supporting Information, Figure S2). Additionally, the regulating activity of L2-b2 against cytotoxicity induced by metal-free Aβ or metal–Aβ was further verified (Figure 3a). As shown in Figure 3a, our molecules in this study have relatively low toxicity in both the absence and presence of metal ions under conditions tested. Moreover, L2-b2, which has an additional dimethylamino group on L2-b’s backbone, is observed to possibly alleviate toxicity triggered by metal-free Aβ and metal–Aβ in living cells due to its abilities to modulate Aβ aggregation (vide supra) and scavenge free radicals (vide infra).
Figure 3.
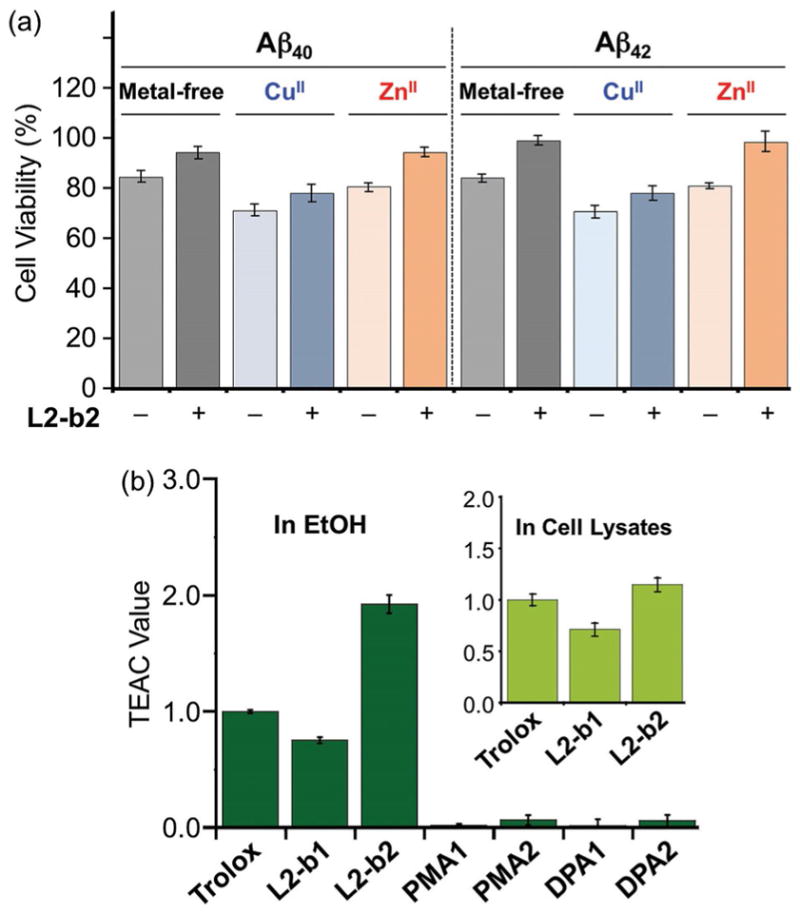
Biological activities of small molecules. (a) Viability of cells treated with L2-b2 and Aβ40 or Aβ42 in the absence and presence of CuCl2 or ZnCl2. SK-N-BE(2)-M17 (M17) cells were incubated with metal-free Aβ and metal–Aβ followed by the addition of L2-b2. Cell viability (%) was determined by the MTT assay compared to cells treated with DMSO only (0–1 %, v/v). Conditions: [Aβ] = 10 μM; [CuCl2 or ZnCl2] = 10 μM; [L2-b2] = 20 μM. (b) Free organic radical scavenging capability of L2-b1, L2-b2, PMA1, PMA2, DPA1, and DPA2, identified by the TEAC assay in EtOH or M17 cell lysates (inset). The TEAC values are relative to that of a vitamin E analogue, Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid). Error bars represent the standard error from three independent experiments (P < 0.05).
The scavenging ability of the different compounds against free radicals was measured by the Trolox (vitamin E analogue) equivalent antioxidant capacity (TEAC) assay, which can evaluate compounds’ capability of quenching ABTS cation radicals [ABTS•+; ABTS = 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)] in both an organic solution (i.e., EtOH) and a biologically relevant environment (i.e., cell lysates).[4c–g,7] As shown in Figure 3b, the TEAC values of L2-b1 and L2-b2 were determined (0.8 ± 0.1 and 1.9 ± 0.1 in EtOH; 0.7 ± 0.1 and 1.1 ± 0.1 in M17 lysates, respectively). Compound L2-b2 presents a greater ability to quench free organic radicals than Trolox in both media. The noticeable free organic radical scavenging ability of L2-b1 and L2-b2, compared to PMA1, PMA2, DPA1, and DPA2, is expected from their relative lower IP values (vide infra, Figure 1). Together, the entire framework of L2-b (with and without additional dimethylamino group(s)) over individual structural portions is responsible for relative lower IP values that could offer the distinct scavenging activity of small molecules with free radicals.
Mechanisms for modulating reactivities toward metal-free and metal-bound Aβ species
Ionization potentials
The IPs of our small molecules (Figure 1) were calculated to anticipate the possibility of their modulating ability toward Aβ aggregation and antioxidant capability. As depicted in Figure 1, L2-b and L2-b2 are shown to have relatively lower IP values than the other structures (i.e., L2-b2, PMA1, PMA2, DPA1, and DPA2) in both the gas and aqueous phases. Moreover, the singly occupied molecular orbitals (SOMOs) indicate that the structures of L2-b and L2-b2, composed of a dimethylamino group on the benzene ring, are observed to be more easily oxidized than the structure with the dimethylamino functionality only on the pyridine ring (i.e., L2-b1). Based on the IP values of our chemical series, oxidative transformations of the compounds (particularly, L2-b and L2-b2) could occur and subsequently direct their regulatory ability against Aβ peptides and free radicals (vide supra).
Metal binding
CuII or ZnII binding of compounds was monitored by UV/Vis or 1H NMR spectroscopy. Changes of the UV/Vis spectra were observed upon addition of CuCl2 to the EtOH solution of all small molecules, indicative of their binding to CuII (Supporting Information, Figure S3 a–f). In case of L2-b1 and L2-b2, new optical bands were detected; for PMA1 and DPA1, the intensity of the absorption spectra was increased; the spectral shifts of PMA2 and DPA2 were observed upon treatment with CuII. Furthermore, ZnII binding of compounds was investigated by UV/Vis and 1H NMR spectroscopy. The addition of ZnII (1 equiv) to the CD3CN solution of L2-b1, PMA1, or PMA2 caused the variation of chemical shifts of the pyridyl protons suggesting the involvement of the N donor atoms on their pyridine ring in ZnII binding (Supporting Information, Figure S3 g–i). In addition, the optical spectra of L2-b2, DPA1, and DPA2 were altered upon introduction of ZnII to their EtOH solution (Supporting Information, Figure S3j–l). Together, our UV/Vis and NMR studies present that our molecules can interact with CuII and ZnII.
Interactions with metal-free and ZnII-treated Aβ40 monomers
For both metal-free Aβ and metal–Aβ aggregation pathways, L2-b2 is indicated to have a modulating ability, distinct from the other small molecules, particularly L2-b (reactivity only for metal–Aβ species)[4b,f] and L2-b1 (no noticeable reactivity for both metal-free Aβ and metal–Aβ) (Figure 2 and Supporting Information, Figure S1). In order to pinpoint the different reactivity of these small molecules (i.e., L2-b, L2-b1, L2-b2) toward targets, the interactions of L2-b1 and L2-b2 with monomeric Aβ40 in the absence of metal ions were first investigated by 2D band-selective optimized flip-angle short transient heteronuclear multiple quantum correlation (SOFAST-HMQC) NMR spectroscopy (Figure 4b, c and Supporting Information, Figure S4). Small but detectable chemical shift changes were observed upon titration with 10 equiv of the compounds to metal-free Aβ40 monomer (Supporting Information, Figure S4). To identify the amino acid residues potentially involved in binding of compounds to the peptide, their chemical shift perturbation (CSP) was calculated (Figure 4b and c), indicating that L2-b1 and L2-b2 triggered slightly noticeable CSP, though to different degrees, at the residues involved in self-recognition (E11; L17-A21)[1c,d] and at the hydrophobic C-terminal region (I31-G33, M35, G38, and V40), relatively similar to L2-b.[4e] L2-b1 and L2-b2 resulted in the chemical shift change of V40 at the C-terminus, like L2-b,[4e] which may reflect the rearrangement of the disordered C-terminus to pack against the compounds instead of direct or indirect interactions with Aβ40. Overall, L2-b1, L2-b2, and L2-b[4e] are observed to have weak interactions with metal-free Aβ.
Figure 4.

Interactions of L2-b, L2-b1, or L2-b2 with metal-free or ZnII-treated Aβ40 monomer. (a) Amino acid sequence of Aβ40. Plots of the chemical shift perturbation (CSP) determined through 2D 1H–15N SOFAST-HMQC NMR spectra of uniformly 15N-labeled monomeric Aβ40 upon titration with (b) L2-b1 or (c) L2-b2. The average CSP (dashed line) with standard deviation (dotted line) is presented. *Residues could not be resolved for analysis. Conditions: [Aβ40] = 80 μM; [L2-b1 or L2-b2] = 0 or 800 μM; 20 mM phosphate buffer, pH 7.4, 50 mM NaCl; 7 % D2O (v/v); 10 °C. Plots of the CSP obtained from 2D 1H–15N SOFAST-HMQC NMR spectra of uniformly 15N-labeled monomeric Aβ40 upon addition of ZnII without (blue) and with (black) (d) L2-b or (e) L2-b2. *Residues could not be resolved for analysis. Conditions: [Aβ40] = 80 μM; [ZnCl2] = 80 μM; [L2-b or L2-b2] = 80 μM; 20 mM phosphate buffer, pH 7.4, 50 mM NaCl; 7% v/v D2O. MD simulations showing interactions of (f) L2-b1 or (g) L2-b2 with monomeric Aβ40. Possible sites and energy of interactions of Aβ40 (PDB 1BA4) with L2-b1 or L2-b2 after all-atom MD simulations are summarized. The zoomed-in view (right, below) of each binding site with residues showing interaction distances labeled in & with dashed lines (additional MD simulations data in Supporting Information, Figure S5).
To visualize the interactions between L2-b, L2-b1, or L2-b2 and monomeric Aβ40 (PDB 1BA4[8]), studies using molecular docking and molecular dynamics (MD) simulations were conducted. Both rigid and flexible docking procedures were utilized using the Autodock Vina 1.5.6 program.[9] MD simulations were performed on the starting structure obtained from docking procedures on complexes of L2-b, L2-b1, or L2-b2 with Aβ40. These all-atom simulations were run by the GROMOS96 53a6 force field, as implemented in the GROMACS program.[10] Multiple interactions of compounds with Aβ40 (i.e., π–π interaction, C—H–π interaction, N—H–π interaction, and hydrogen bonding) were observed (Figure 4f, g and Supporting Information, Figure S5). First, L2-b may interact with both polar and non-polar residues through hydrogen bonding between its secondary amine and H6 and π–π interactions between its benzene/pyridine rings and F4 or H14, respectively (Supporting Information, Figure S5 a; left). Other amino acid residues of Aβ40 (e.g., L17 and F19) were also shown to be involved in interactions with L2-b through hydrogen bonding and a π–π interaction, respectively (Supporting Information, Figure S5a; right). As shown in Figure 4f, L2-b1 was held between two aromatic residues, F19 and F20, through π–π and C—H interactions, respectively. Additionally, hydrogen bonding between the backbone carbonyl O atom (between F19 and F20) and L2-b1’s secondary amine bridging the two aromatic rings could be generated. Three aromatic residues H6, Y10, and H14 might interact with L2-b1 through a π–π interaction (Y10), a C—H–π interaction (H14), and hydrogen bonding (H6 and D7) (Supporting Information, Figure S5 b; right).
The residues F19 and S26 were indicated to interact with L2-b2 through C—H–π interactions (between L2-b2’s benzene ring and the H atom from the aromatic ring of F19; between L2-b2’s pyridine ring and the H atom from βC of S26) (Figure 4g). Additionally, L2-b2’s secondary amine group and the backbone carbonyl O atom between F19 and F20 may form hydrogen bonding. Moreover, L2-b2 could be held between two aromatic residues F4 and F20 by means of C—H–π (between its pyridine ring and the H atom from the aromatic ring of F4) and N—H–π (between its secondary amine and F20) interactions, respectively. Hydrogen bonding between the N atom of dimethylamino group on L2-b2’s benzene ring and the backbone amine group of S8 could also be formed (Supporting Information, Figure S5 c; right). For all binding modes, binding energies and contributions of electrostatic or hydrophobic interactions were calculated and summarized in the table (Supporting Information, Figure S5). Together, through MD simulations, the potential interactions of L2-b, L2-b1, and L2-b2 with metal-free Aβ40 monomer could be envisioned. Based on our 2D NMR, MS, and MD simulations studies, the regulatory activity of molecules with metal-free Aβ may be achieved via the covalent adduct formation (observed by L2-b2; Figure 5b) rather than non-covalent interactions (e.g., π–π and C—H–π interactions, and hydrogen bonding).
Figure 5.

ESI-MS analysis of Aβ40 incubated with L2-b2 in the absence and presence of CuII. (a and b) The 3+ charge state of metal-free Aβ40 with and without L2-b2. When L2-b2 was treated with Aβ, the signal at m/z 1487.8 (132 Da increase from Aβ40) possibly corresponding to an adduct formed with Aβ and oxidized DMPD (cationic imine; cleaved from L2-b2) was observed. Conditions: [Aβ40] = 10 μM; [CuCl2] = 10 μM; [L2-b2] = 50 μM; 20 mM ammonium acetate, pH 7.5; 37 °C; 6 h incubation; no agitation. The 3 + charge state of Aβ40 incubated with (c) CuII or (d) both CuII and L2-b2. The signal highlighted in green corresponds to degraded Aβ by loss of 89.1 Da. Conditions: [Aβ40] = 20 μM; [CuCl2] = 20 μM; [L2-b2] = 120 μM; 100 mM ammonium acetate, pH 7.5; 37 °C; 30 min incubation; no agitation. (e) Amino acid sequence of Aβ40. (f) MS/MS analyses of Aβ40 with and without treatment of CuII and L2-b2. These data support that the amino acid sequence of Aβ is chemically modified within the first five residues (D1A2E3F4R5) of the peptide in the presence of both CuII and L2-b2. All the Aβ40 species containing the identified −89.1 Da covalent modification are highlighted in red, and are compared against control Aβ40 MS/MS sequencing data acquired under the same conditions.
The interaction of monomeric Aβ40 with L2-b2 capable of controlling metal-free Aβ aggregation (vide supra; Figure 2 and Supporting Information, Figure S1) was further monitored by electrospray ionization mass spectrometry (ESI-MS) (Figure 5a,b, and Supporting Information, Figure S6). L2-b2 or its degraded compounds, such as N,N-dimethyl-p-phenylenediamine (DMPD) and/or oxidized DMPD (i.e., cationic imine),[4g] were shown to have interactions with metal-free Aβ (Figure 5b), in contrast to L2-b that was unable to interact with metal-free Aβ.[4b,f] When L2-b2 was incubated with metal-free Aβ, a newly observed signal corresponding to the addition of 132 Da to Aβ, indicative of forming a covalent adduct of Aβ–cationic imine, was exhibited (magenta, Figure 5b). This adduct could be generated via primary amine-containing residues from Aβ (e.g., K16 and K28) (Figure 5b and Supporting Information, Figure S6), similar to the complex formation of benzoquinone (BQ) with Aβ (Aβ–BQ).[4g] Such distinct interactions of L2-b2 with Aβ (i.e., compounds’ degradation and transformation followed by covalent crosslinks with Aβ) could be associated with L2-b2-triggered alteration of metal-free Aβ aggregation, which was not observed from the samples of Aβ with L2-b.[4f]
For the interaction with ZnII–Aβ, 2D NMR spectroscopy was employed to analyze the samples containing L2-b or L2-b2 and ZnII-bound uniformly 15N-labeled Aβ40 monomer (Figure 4d, e and Supporting Information, Figure S7). L2-b induced relatively more CSP of R5 and H13 residues close to a metal binding site of Aβ40 (Figure 4d).[1b–d, 3d,e] As shown in Figure 4e, similar to L2-b, L2-b2 caused CSP of residues, such as R5, S8, and E11, within proximity of the metal binding region. Thus, L2-b and L2-b2 could interact with ZnII surrounded by Aβ40 possibly leading to mediation of ZnII–Aβ40 aggregation, as detected by gel/Western blot and TEM (Figure 2 and Supporting Information, Figure S1).
Interactions with metal-free and ZnII-treated Aβ fibrils
Along with Aβ monomer, to verify how L2-b or L2-b2 is able to disassemble preformed metal-free and/or metal-added Aβ aggregates to different extents, their interactions with metal-free and ZnII-treated Aβ42 fibrils were studied by saturation transfer difference (STD) NMR (Figure 6a–c). Signals in STD NMR spectroscopy are proportional to each atom of either L2-b or L2-b2 to its macromolecular binding partner, fibrils, which allows atomic-level mapping of ligand binding to fibrillar Aβ.[11] In the case of L2-b, the relatively strong saturation effect was observed at the pyridine ring with metal-free Aβ42 fibrils with the slight saturation effect at the dimethylamino group. In addition to the pyridine ring, upon treatment of ZnII–Aβ fibrils with L2-b, the relatively noticeable saturation effects on the molecule were also indicated at the methylene group between the pyridine ring and the secondary amine (Figure 6c; left). In contrast to L2-b, both of the dimethylamino groups and the pyridine ring of L2-b2 were found to have relatively significant saturation effects against metal-free Aβ42 fibrils (Figure 6c; right), suggesting that this molecule could be relatively packed into the fibrillar conformation of Aβ, as described by a previously reported compound.[4d] When ZnII was introduced to Aβ42 fibrils, the saturation effects on the dimethylamino group of the pyridine ring was observed to be relatively less than those on the benzene ring, along with the reduced saturation influence on the pyridine ring.
Figure 6.

Interactions of L2-b or L2-b2 with metal-free and ZnII-treated Aβ fibrils. 1H STD NMR spectra of L2-b (left) or L2-b2 (right) in the presence (red) and absence (black) of (a) metal-free or (b) ZnII-added Aβ42 fibrils. Comparison of the STD signal intensities (red) to the STD reference (black) reflects the relative proximity of the corresponding proton from the ligand to Aβ42 fibrils. Conditions: [Aβ42] = 2 μM; [ZnCl2] = 2 μM; [L2-b or L2-b2] = 200 μM; 10 mM Tris-DCl, pD 7.4. (c) Normalized STD intensities mapped on to the structures of L2-b and L2-b2 against metal-free Aβ42 fibrils (left) and ZnII–Aβ42 fibrils (right). Yellow, orange, and blue circles indicate the STD effects of >75 %, 50–75 %, and <50 %, respectively. Gray circles indicate the absence of the STD effect. (d) MD simulations showing interactions of L2-b2 with metal-free Aβ40 fibrils. Two potential binding sites (i and ii) of interaction of L2-b2 with Aβ40 fibrils (PDB 2LMN) after all-atom MD simulations. Right: The zoomed-in view of each binding site with residues showing interaction distances labelled in & with dashed lines. Binding modes (for L2-b) and energies (for both L2-b and L2-b2) are presented in Supporting Information, Figure S8.
To gain a better understanding of the interactions between Aβ fibrils (PDB 2LMN[12]) and L2-b or L2-b2, MD simulations were conducted. As shown in Figure 6d and the Supporting Information, Figure S8, two binding modes (i.e., for both L2-b and L2-b2, alignment orthogonal to the surface of the β-strand; for L2-b2, intercalation into the loop of two β-strands) were observed. The complex of L2-b and fibrillar Aβ40 formed hydrogen bonds with the H atoms from the benzene ring, the pyridine ring, and the secondary amine bridging two aromatic rings of L2-b with the O atoms from the carboxyl groups of E22s, as well as the N atom from the pyridine ring of L2-b with the H atom from aromatic ring of F20 (Supporting Information, Figure S8 a). Additionally, a C—H–π interaction (between the H atom from the benzene ring of L2-b and the aromatic ring of F20) could stabilize the molecule to interact with Aβ fibrils (Supporting Information, Figure S8 a). In the case of L2-b2, this small molecule could be held on the fibril edge of the β-strand through a C—H–π interaction with F20 (between the pyridine/benzene rings of L2-b2 and the H atoms from the aromatic rings of F20s). E22 may further assist in L2-b2 binding to Aβ fibrils through hydrogen bonding formation between the H atom from the secondary amine between two aromatic rings of L2-b2 and the O atom from the carbonyl group of E22 (Figure 6d (i)). Furthermore, L2-b2 could be packed within the hydrophobic pocket of the fibril (intercalation into the loop of two β-strands) utilizing the interactions with A21 and F19 [C—H–π interaction (between the pyridine ring of L2-b2 and the H atom from the methyl group of A21) and π–π interaction (between the pyridine ring of L2-b2 and the aromatic ring of F19); Figure 6d(ii)]. This binding mode (packed by fibrils; intercalation) at the hydrophobic pocket, expected from STD NMR results (vide supra), may be linked to the relatively stronger direct interaction of L2-b2 with preformed metal-free Aβ aggregates, as shown in a previously reported compound.[4d]
Taken together, STD NMR and MD simulations suggest how L2-b and L2-b2 could interact with metal-free Aβ fibrils and ZnII–Aβ fibrils. Through STD NMR, the metal binding portion of L2-b (PMA1; Figure 1) was observed to be related to the contact with ZnII–Aβ fibrils. In addition, different structural portions of L2-b2 are indicated to have noticeable interactions with metal-free Aβ fibrils and ZnII–Aβ fibrils. Furthermore, MD simulations visualize how such structural features of L2-b and L2-b2 interact with Aβ fibrils, which suggests compounds’ binding modes against peptide fibrils (in particular, for L2-b2, alignment on the surface of the β-strand and intercalation into the loop that connects the two β-strands). Thus, the structural difference between L2-b and L2-b2 (i.e., additional dimethylamino group) is indicated to distinctly interact with metal-free and ZnII-treated Aβ fibrils.
Generation of degraded Aβ
To determine how L2-b2 is able to alter CuII–Aβ aggregation, nano-ESI-MS (nESI-MS) optimized for the detection of non-covalent protein complexes was applied.[13] When the peptide was incubated with L2-b2 in the presence of CuII, additional m/z signals corresponding to a mass loss of 89.1 (± 0.1) Da compared to apo Aβ40 were detected (green signal, Figure 5d), similar to the results of L2-b.[4f] Tandem MS (MS/MS) sequencing indicates that this signal represents a modified form of Aβ40, which lacks 89.1 Da from the first five residues of the N-terminus (D1A2E3F4R5) (Figure 5e and f). These MS/MS data indicate that L2-b2 likely binds to Aβ proximal to the binding site of CuII.[1b–d,3d,e] Neither L2-b2 nor CuII was directly detected in the complex with either the N-terminal cleavage product or apo Aβ40 supporting the formation of a transient ternary complex consistent with previously published results.[4f] These data support that, compared to L2-b,[4f] the additional dimethylamino functionality on the pyridine ring is shown to still generate N-terminally cleaved Aβ species (loss of 89.1 Da) that could redirect CuII–Aβ aggregation. Along with the MS data (Figure 5d and f), compared to L2-b, these observations suggest that the additional dimethylamino moiety enables L2-b2 to interact and react with both metal-free and CuII-bound Aβ.
Proposed mechanisms for reactivities of L2-b2 toward metal-free Aβ and metal–Aβ
Multiple mechanisms of L2-b2 to redirect Aβ aggregation in the absence and presence of metal ions are proposed on the basis of our NMR, MS, and computational results. L2-b2 could be cleaved through oxidative modifications generating transformed cationic imine[4g] that can be covalently bound to Aβ monomers to form an Aβ–cationic imine adduct (Figure 5b). Upon Aβ–cationic imine adduct formation, metal-free Aβ aggregation pathways could be redirected as previously reported.[4g] In addition, as shown in Figure 6, L2-b2 could be intercalated between β-sheets of Aβ fibrils, which could be associated with its regulatory activity with metal-free Aβ fibrils, possibly similar to a previously reported molecule.[4d] In the presence of ZnII, L2-b2 is indicated to interact with monomeric Aβ (close to the metal binding site of Aβ)[1b–d,3d,e] (Figure 4), which implies its potential contact with ZnII surrounded by Aβ subsequently modulating peptide aggregation. More detailed studies of L2-b2’s interaction with ZnII–Aβ are the subject of future studies. Lastly, toward CuII–Aβ, L2-b2 is able to lead to Aβ degradation (Figure 5), similar to L2-b.[4f] This observed Aβ degradation could be related to the formation of a transient ternary complex between Aβ, CuII, and L2-b2, subsequently followed by L2-b2’s oxidation and Aβ degradation of by well-known radical-mediated pathways.[14] Such degraded Aβ could lose aggregation propensity compared to full-length peptides.[4f] Based on analyses of Aβ products from both reactions of L2-b2 with metal-free Aβ and CuII–Aβ, the oxidation of this molecule occurs, which suggests that its oxidative transformation is required for the desired reactivities with an emphasis on importance of anticipating IP values for rational design (Figure 1). Collectively, L2-b2 is demonstrated to be a tool able to interact and react with all metal-free Aβ, CuII–Aβ, and ZnII–Aβ to different extents through several disparate mechanisms.
Conclusions
Chemical tools capable of targeting and controlling individual or multiple pathogenic factors found in AD (i.e., metals, metal-free Aβ, metal-bound Aβ, and oxidative stress) have been developed to elucidate AD pathogenesis at the molecular level; however, such tool invention has been challenging. Unfortunately, a guideline of designing chemical tools for distinct targets (e.g., as the first step, selecting key structural and mechanistic properties of tools) has not been established. To contribute to this foundation, a new class of small molecules was constructed based on the structure of L2-b, known as a chemical regulator for metal–Aβ,[4b,f] with consideration of their BBB permeability and relatively low cytotoxicity. Employing our chemical series, the regulatory activities toward metal-free Aβ and metal–Aβ aggregation, along with free radical scavenging capability, are observed to be directed by compounds’ structures (e.g., functionality and entire backbone) and mechanistic characteristics (e.g., covalent adduct formation with peptides and peptide degradation through compounds’ transformations). Through our structure–mechanism-based design, a molecular multifunctional tool, L2-b2, was newly fashioned showing its abilities to regulate all of our desired targets (i.e., metals, metal-free Aβ, metal–Aβ, and oxidative stress). Taken together, our overall multidisciplinary studies through a chemical library present a design concept of chemical tools toward individual or multiple inter-related pathological factors in AD based on structural and mechanistic details. Our structure–mechanism-based concept could open new avenues for devising chemical tools capable of regulating the actions of diverse pathological factors in human diseases. In principle, depending on different targets, distinct mechanisms of chemical tools to regulate their actions should be taken into account.
Experimental Section
All reagents and solvents were purchased from commercial suppliers and used as received unless otherwise stated. PMA1, PMA2, and DPA1 were purchased from Sigma–Aldrich (St. Louis, MO, USA). L2-b, L2-b1, L2-b2, and DPA2 were synthesized as previously reported procedures.[4b] Aβ40 and Aβ42 were purchased from AnaSpec (Fremont, CA, USA) (Aβ42 = DAEFRHDSGYEVHHQKLVF-FAEDVGSNKGAIIGLMVGGVVIA). Double distilled H2O (ddH2O) was obtained from a Milli-Q Direct 16 system (Merck KGaA, Darmstadt, Germany). An Agilent 8453 UV-visible (UV/vis) spectrophotometer (Santa Clara, CA, USA) was used to measure optical spectra. TEM images were taken using a JEOL JEM-2100 transmission electron microscope (UNIST Central Research Facilities, Ulsan, Republic of Korea). Absorbance values for biological assays, including the MTT and TEAC assays, were measured on a SpectraMax M5e microplate reader (Molecular Devices, Sunnyvale, CA, USA). NMR studies of Aβ with compounds in both the absence and presence of ZnII were carried out on a 900 MHz Bruker spectrometer equipped with a cryogenic probe (Michigan State University in Lansing, MI, USA). A Waters (Milford, MA) Synapt G2 HDMS equipped with a nanoelectrospray ionization (nESI) or ESI source (Waters, Milford, MA, USA) was used to study complex formation between L2-b2 and Aβ40 with and without CuII.
Supplementary Material
Acknowledgments
This work was supported by the National Research Foundation (NRF) of Korea grant funded by the Korean government [NRF-2014R1A2A2A01004877 and 2016R1A5A1009405 (to M.H.L.); NRF-2014S1A2A2028270 (to M.H.L. and A.R.)]; the 2017 Research Fund (Project Number 1.170014.01) of Ulsan National Institute of Science and Technology (UNIST) (to M.H.L.); the University of Michigan Protein Folding Disease Initiative (to A.R., B.T.R., and M.H.L.); an NIH grant (to A.R.); the James and Esther King Biomedical Research Program of the Florida State Health Department Award [DOH grant number 08KN-11 to R.P.]; the National Honor Scientist Program (2010–0020414) of NRF of Korea (to K.S.K.). J.K. thanks the support from the Global Ph.D. fellowship program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2015HIA2A1030823). We thank Drs. Michael Beck, Shin Jung Lee, and Woo Jong Cho for valuable comments about L2-b, ESI-MS analysis, and IP calculations, respectively.
Footnotes
Supporting information, including full experimental details, Table S1, Figures S1–S8, and the ORCID identification number(s) for the author(s) of this article, can be found under http://dx.doi.org/10.1002/chem.201605401.
References
- 1.Jakob-Roetne R, Jacobsen H. Angew Chem Int Ed. 2009;48:3030–3059. doi: 10.1002/anie.200802808.Angew Chem. 2009;121:3074–3105.Kepp KP. Chem Rev. 2012;112:5193–5239. doi: 10.1021/cr300009x.Rodríguez-Rodríguez C, Telpoukhovskaia M, Orvig C. Coord Chem Rev. 2012;256:2308–2332.Beck MW, Pithadia AS, DeToma AS, Korshavn KJ, Lim MH. In: Ligand Design in Medicinal Inorganic Chemistry. Storr T, editor. Chapter 10. Wiley; Chichester: 2014. pp. 257–286.
- 2.a) Ono K, Condron MM, Teplow DB. Proc Natl Acad Sci USA. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Haass C, Selkoe DJ. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]; c) Kepp KP. J Alzheimers Dis. 2017;55:447–457. doi: 10.3233/JAD-160550. [DOI] [PubMed] [Google Scholar]
- 3.a) Duce JA, Bush AI. Prog Neurobiol. 2010;92:1–18. doi: 10.1016/j.pneurobio.2010.04.003. [DOI] [PubMed] [Google Scholar]; b) Que EL, Domaille DW, Chang CJ. Chem Rev. 2008;108:1517–1549. doi: 10.1021/cr078203u. [DOI] [PubMed] [Google Scholar]; c) Zatta P, Drago D, Bolognin S, Sensi SL. Trends Pharmacol Sci. 2009;30:346–355. doi: 10.1016/j.tips.2009.05.002. [DOI] [PubMed] [Google Scholar]; d) Faller P. Chem Bio Chem. 2009;10:2837–2845. doi: 10.1002/cbic.200900321. [DOI] [PubMed] [Google Scholar]; e) Chassaing S, Collin F, Dorlet P, Gout J, Hureau C, Faller P. Curr Top Med Chem. 2012;12:2573–2595. doi: 10.2174/1568026611212220011. [DOI] [PubMed] [Google Scholar]; f) Belaidi AA, Bush AI. J Neurochem. 2016;139:179–197. doi: 10.1111/jnc.13425. [DOI] [PubMed] [Google Scholar]
- 4.a) Hindo SS, Mancino AM, Braymer JJ, Liu Y, Vivekanandan S, Ramamoorthy A, Lim MH. J Am Chem Soc. 2009;131:16663–16665. doi: 10.1021/ja907045h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Choi JS, Braymer JJ, Nanga RPR, Ramamoorthy A, Lim MH. Proc Natl Acad Sci USA. 2010;107:21990–21995. doi: 10.1073/pnas.1006091107. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Derrick JS, Kerr RA, Korshavn KJ, McLane MJ, Kang J, Nam E, Ramamoorthy A, Ruotolo BT, Lim MH. Inorg Chem. 2016;55:5000–5013. doi: 10.1021/acs.inorgchem.6b00525. [DOI] [PubMed] [Google Scholar]; d) Lee S, Zheng X, Krishnamoorthy J, Savelieff MG, Park HM, Brender JR, Kim JH, Derrick JS, Kochi A, Lee HJ, Kim C, Ramamoorthy A, Bowers MT, Lim MH. J Am Chem Soc. 2014;136:299–310. doi: 10.1021/ja409801p. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Savelieff MG, Liu Y, Senthamarai RRP, Korshavn KJ, Lee HJ, Ramamoorthy A, Lim MH. Chem Commun. 2014;50:5301–5303. doi: 10.1039/c3cc48473d. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Beck MW, Oh SB, Kerr RA, Lee HJ, Kim SH, Kim S, Jang M, Ruotolo BT, Lee JY, Lim MH. Chem Sci. 2015;6:1879–1886. doi: 10.1039/c4sc03239j. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Derrick JS, Kerr RA, Nam Y, Oh SB, Lee HJ, Earnest KG, Suh N, Peck KL, Ozbil M, Korshavn KJ, Ramamoorthy A, Prabhakar R, Merino EJ, Shearer J, Lee JY, Ruotolo BT, Lim MH. J Am Chem Soc. 2015;137:14785–14797. doi: 10.1021/jacs.5b10043. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Sharma AK, Pavlova ST, Kim J, Finkelstein D, Hawco NJ, Rath NP, Kim J, Mirica LM. J Am Chem Soc. 2012;134:6625–6636. doi: 10.1021/ja210588m. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Rodríguez-Rodríguez C, Sanchez de Groot N, Rimola A, Alvarez-Larena A, Lloveras V, Vidal-Gancedo J, Ventura S, Vendrell J, Sodupe M, Gonzalez-Duarte P. J Am Chem Soc. 2009;131:1436–1451. doi: 10.1021/ja806062g. [DOI] [PubMed] [Google Scholar]
- 5.Kung HF, Lee C-W, Zhuang Z-P, Kung M-P, Hou C, Plossl K. J Am Chem Soc. 2001;123:12740–12741. doi: 10.1021/ja0167147. [DOI] [PubMed] [Google Scholar]
- 6.a) Avdeef A, Bendels S, Di L, Faller B, Kansy M, Sugano K, Yamauchi Y. J Pharm Sci. 2007;96:2893–2909. doi: 10.1002/jps.21068. [DOI] [PubMed] [Google Scholar]; b) BBB Protocol and Test Compounds. pION Inc; Billerica, MA, USA: 2009. [Google Scholar]
- 7.a) Re R, Pellegrini N, Proteggente A, Pannala A, Yang M, Rice-Evans C. Free Radical Biol Med. 1999;26:1231–1237. doi: 10.1016/s0891-5849(98)00315-3. [DOI] [PubMed] [Google Scholar]; b) Schugar H, Green DE, Bowen ML, Scott LE, Storr T, Bohmerle K, Thomas F, Allen DD, Lockman PR, Merkel M, Thompson KH, Orvig C. Angew Chem Int Ed. 2007;46:1716–1718. doi: 10.1002/anie.200603866. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2007;119:1746–1748. [Google Scholar]
- 8.Coles M, Bicknell W, Watson AA, Fairlie DP, Craik DJ. Biochemistry. 1998;37:11064–11077. doi: 10.1021/bi972979f. [DOI] [PubMed] [Google Scholar]
- 9.Trott O, Olson AJ. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF. J Comput Chem. 2004;25:1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]; b) Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lindahl E, Hess B, van der Spoel D. J Mol Model. 2001;7:306–317. [Google Scholar]
- 11.Mayer M, Meyer B. J Am Chem Soc. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 12.Petkova AT, Yau W-M, Tycko R. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Hernández H, Robinson CV. Nat Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]; b) Hilton GR, Benesch JLP. J R Soc Interface. 2012;9:801–816. doi: 10.1098/rsif.2011.0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Hawkins CL, Davies MJ. Biochim Biophys Acta Bioenerg. 2001;1504:196–219. doi: 10.1016/s0005-2728(00)00252-8. [DOI] [PubMed] [Google Scholar]; b) Garrison WM. Chem Rev. 1987;87:381–398. [Google Scholar]; c) Porter MR, Kochi A, Karty JA, Lim MH, Zaleski JM. Chem Sci. 2015;6:1018–1026. doi: 10.1039/c4sc01979b. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Beck MW, Derrick JS, Kerr RA, Oh SB, Cho WJ, Lee SJC, Ji Y, Han J, Tehrani ZA, Suh N, Kim S, Larsen SD, Kim KS, Lee JY, Ruotolo BT, Lim MH. Nat Commun. 2016;7:13115. doi: 10.1038/ncomms13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.